
A Fun-Guide to Innate Immune Responses to Fungal Infections


Abstract
:1. Introduction
2. Innate Immune Control of Fungal Infections
2.1. Barriers to Fungal Entry
2.2. Host Recognition of Fungi
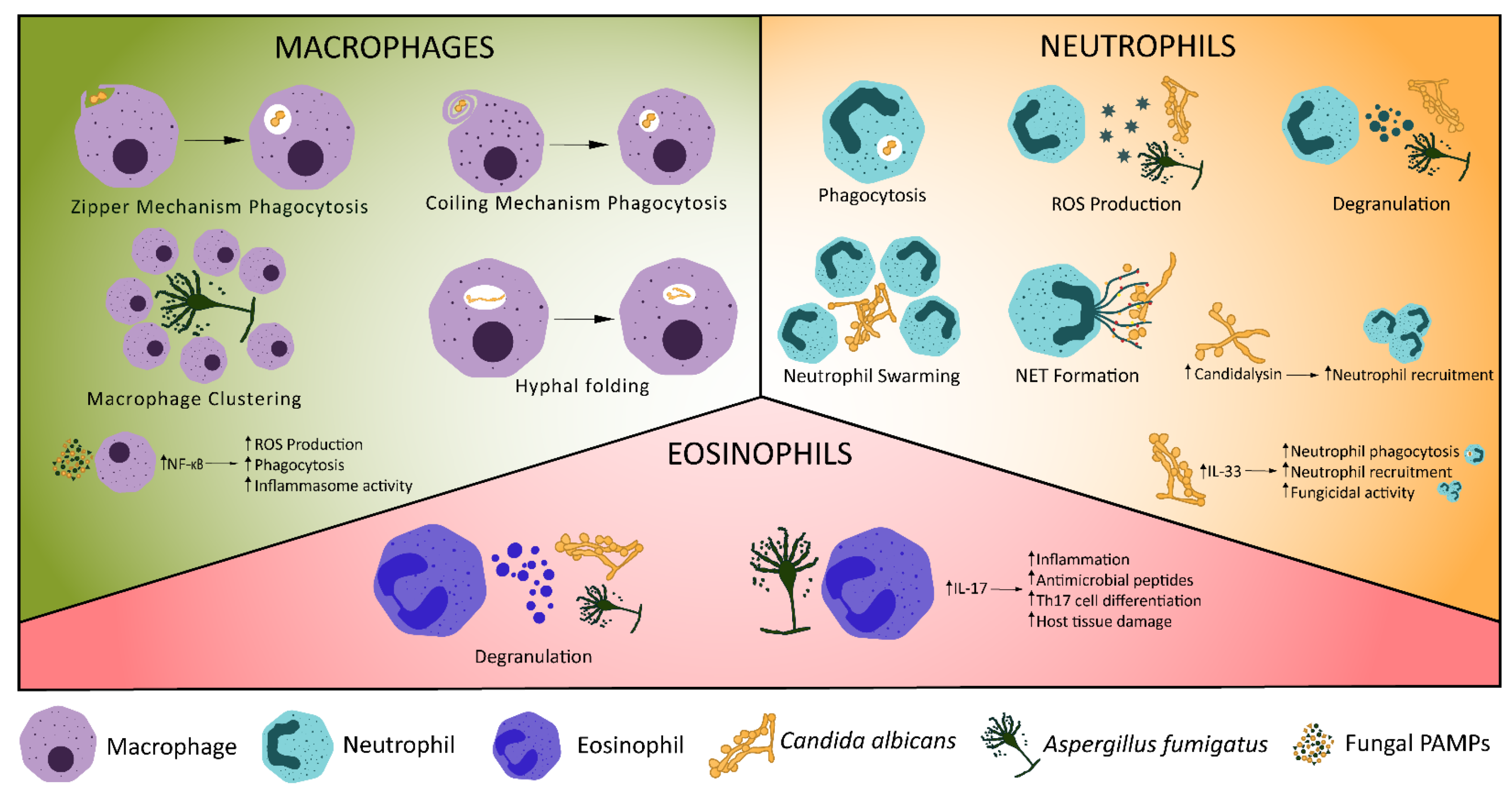
2.3. Macrophages
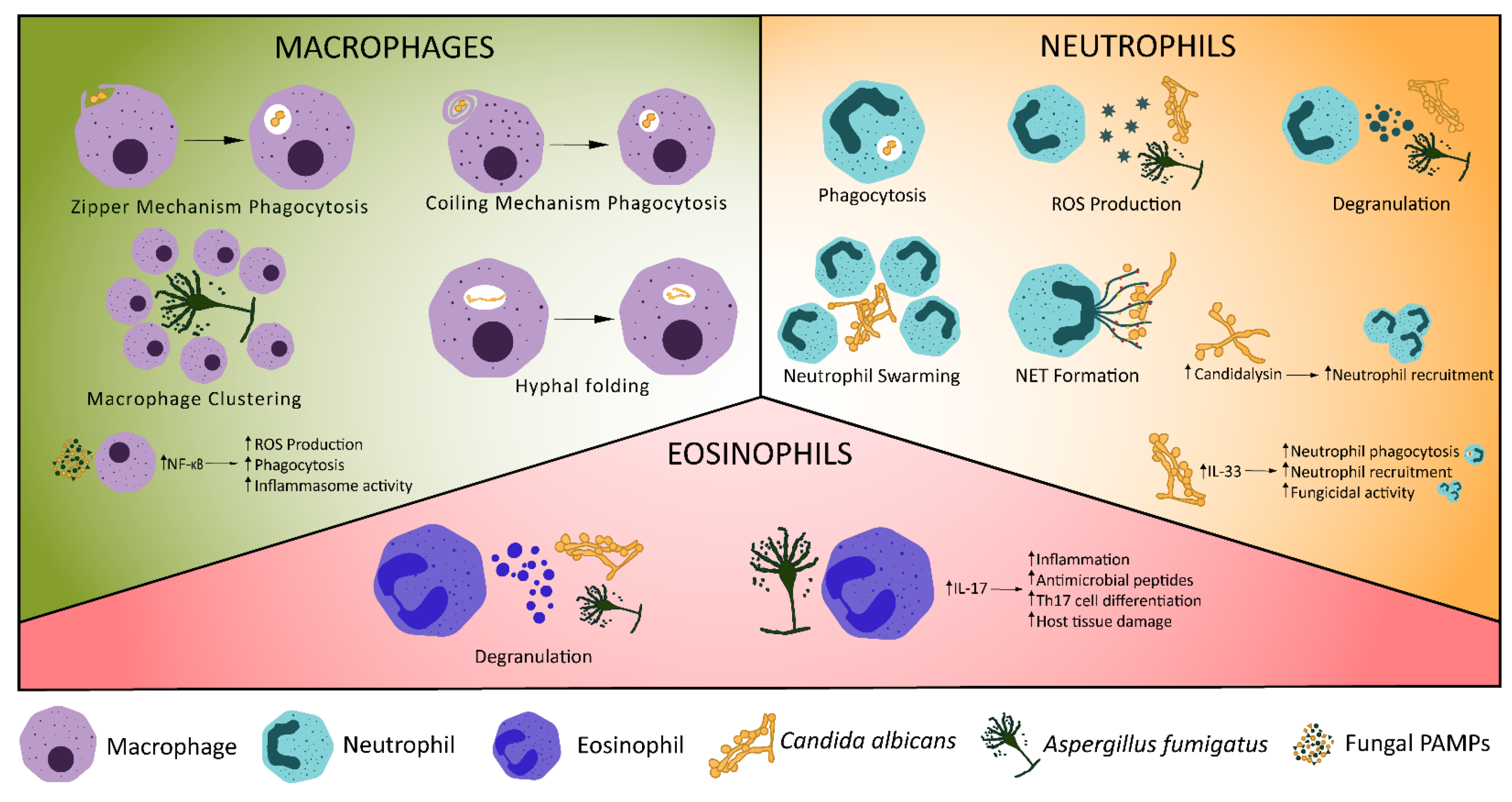
2.4. Neutrophils
2.5. Other Innate Immune Components of Fungal Protection
3. Failures of Innate Immunity in Disease
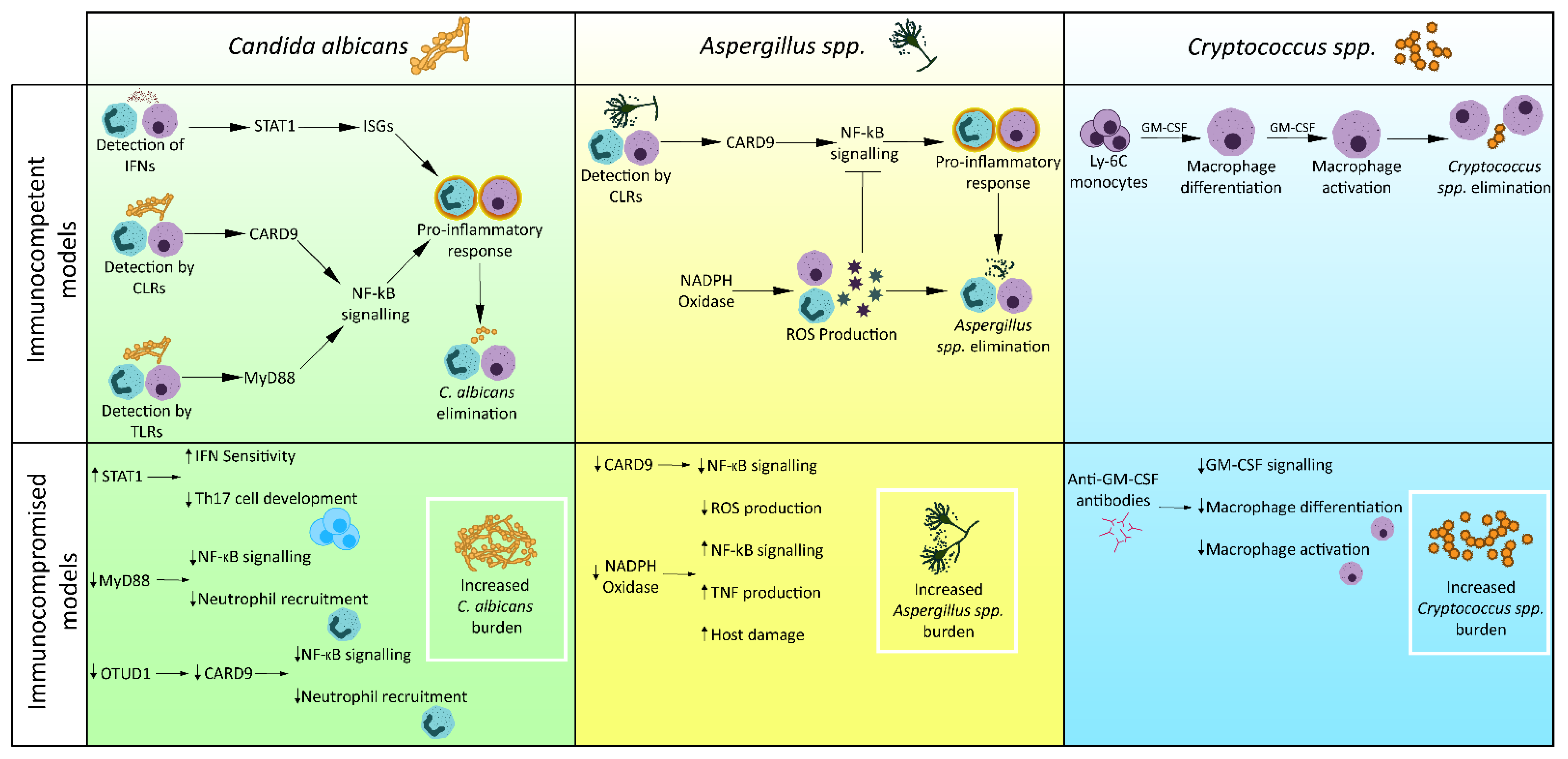
3.1. Candida albicans
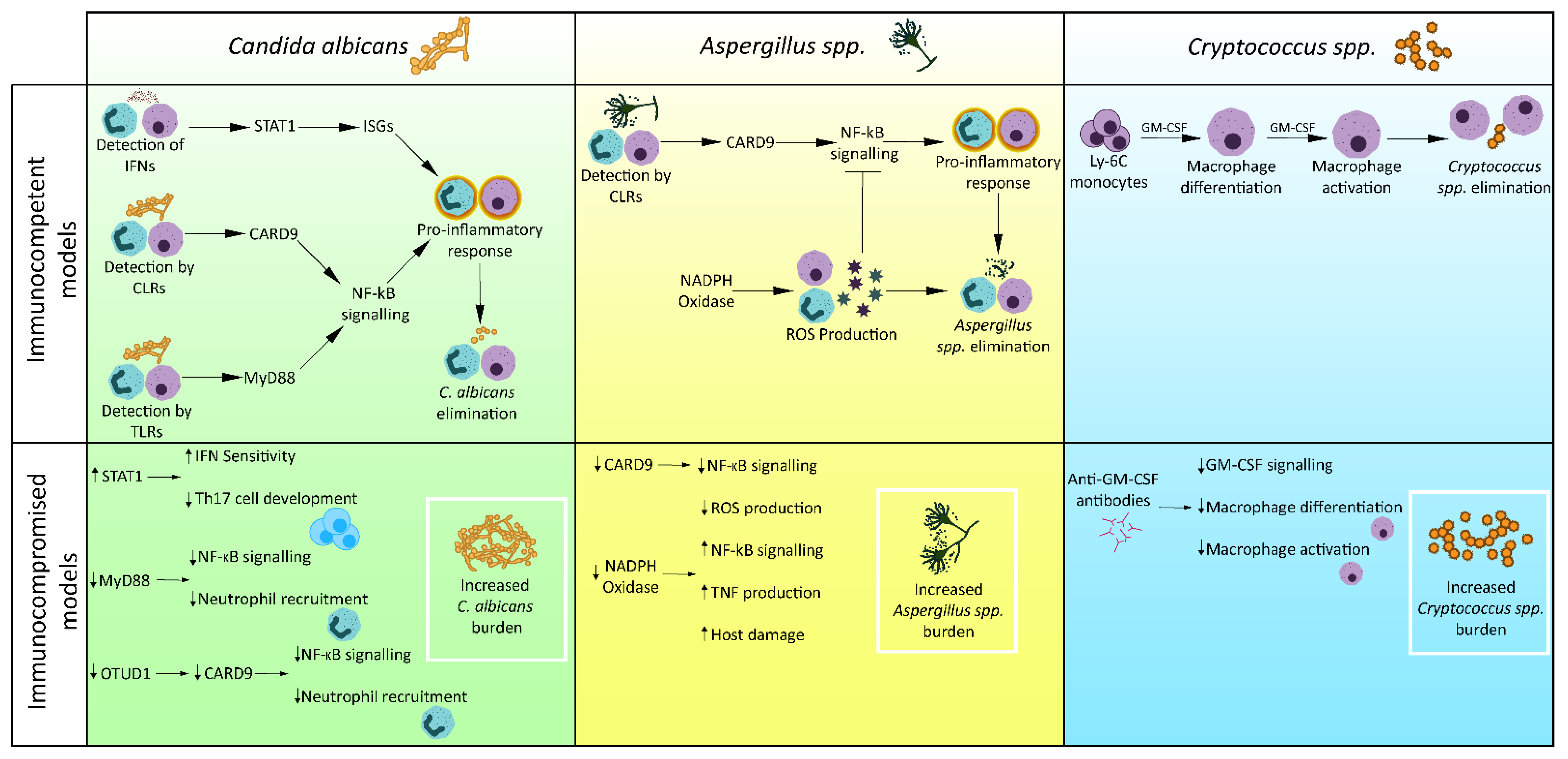
3.2. Cryptococcus spp.
3.3. Aspergillus spp.
4. Host-Directed Therapies
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kainz, K.; Bauer, M.A.; Madeo, F.; Carmona-Gutierrez, D. Fungal Infections in Humans: The Silent Crisis. Microb. Cell 2020, 7, 143–145. [Google Scholar] [CrossRef] [PubMed]
- Bongomin, F.; Gago, S.; Oladele, R.; Denning, D. Global and Multi-National Prevalence of Fungal Diseases—Estimate Precision. J. Fungi 2017, 3, 57. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, M.L.; Albuquerque, P.C. Searching for a Change: The Need for Increased Support for Public Health and Research on Fungal Diseases. PLoS Negl. Trop. Dis. 2018, 12, e0006479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benedict, K.; Molinari, N.A.M.; Jackson, B.R. Public Awareness of Invasive Fungal Diseases—United States, 2019. Morb. Mortal. Wkly. Rep. 2020, 69, 1343. [Google Scholar] [CrossRef]
- Enoch, D.A.; Yang, H.; Aliyu, S.H.; Micallef, C. The Changing Epidemiology of Invasive Fungal Infections. Methods Mol. Biol. 2017, 1508, 17–65. [Google Scholar] [CrossRef]
- Friedman, D.Z.P.; Schwartz, I.S. Emerging Fungal Infections: New Patients, New Patterns, and New Pathogens. J. Fungi 2019, 5, 67. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Pei, X.; Luo, Y.; Tan, Y.; Tie, R.; He, J.; Zheng, W.; Zhang, J.; Cai, Z.; Lin, M.; et al. Invasive Fungal Infection in Allogeneic Hematopoietic Stem Cell Transplant Recipients: Single Center Experiences of 12 Years. J. Zhejiang Univ. Sci. B 2015, 16, 796–804. [Google Scholar] [CrossRef]
- Maskarinec, S.A.; Johnson, M.D.; Perfect, J.R. Genetic Susceptibility to Fungal Infections: What Is in the Genes? Curr. Clin. Microbiol. Rep. 2016, 3, 81–91. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.-L.; Yu, S.-J.; Heitman, J.; Wellington, M.; Chen, Y.-L. New Facets of Antifungal Therapy. Virulence 2017, 8, 222–236. [Google Scholar] [CrossRef] [Green Version]
- Daele, R.V.; Spriet, I.; Wauters, J.; Maertens, J.; Mercier, T.; Hecke, S.V.; Brüggemann, R. Antifungal Drugs: What Brings the Future? Med. Mycol. 2019, 57, S328–S343. [Google Scholar] [CrossRef] [Green Version]
- Perfect, J.R. The Antifungal Pipeline: A Reality Check. Nat. Rev. Drug Discov. 2017, 16, 603–616. [Google Scholar] [CrossRef] [Green Version]
- Rauseo, A.M.; Coler-Reilly, A.; Larson, L.; Spec, A. Hope on the Horizon: Novel Fungal Treatments in Development. Open Forum Infect. Dis. 2020, 7, ofaa016. [Google Scholar] [CrossRef] [Green Version]
- Lockhart, S.R.; Etienne, K.A.; Vallabhaneni, S.; Farooqi, J.; Chowdhary, A.; Govender, N.P.; Colombo, A.L.; Calvo, B.; Cuomo, C.A.; Desjardins, C.A.; et al. Simultaneous Emergence of Multidrug-Resistant Candida Auris on 3 Continents Confirmed by Whole-Genome Sequencing and Epidemiological Analyses. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2017, 64, 134–140. [Google Scholar] [CrossRef] [Green Version]
- van der Linden, J.W.M.; Snelders, E.; Kampinga, G.A.; Rijnders, B.J.A.; Mattsson, E.; Debets-Ossenkopp, Y.J.; Van Tiel, F.H.; Melchers, W.J.G.; Verweij, P.E. Clinical Implications of Azole Resistance in Aspergillus Fumigatus, the Netherlands, 2007–2009. Emerg. Infect. Dis. 2011, 17, 1846–1854. [Google Scholar] [CrossRef]
- Hernandez, H.; Martinez, L.R. Relationship of Environmental Disturbances and the Infectious Potential of Fungi. Microbiology 2018, 164, 233–241. [Google Scholar] [CrossRef]
- Figueiredo, R.; Carneiro, L.; Bozza, M. Fungal Surface and Innate Immune Recognition of Filamentous Fungi. Front. Microbiol. 2011, 2, 248. [Google Scholar] [CrossRef] [Green Version]
- Drummond, R.A.; Gaffen, S.L.; Hise, A.G.; Brown, G.D. Innate Defense against Fungal Pathogens. Cold Spring Harb. Perspect. Med. 2015, 5, a019620. [Google Scholar] [CrossRef] [Green Version]
- Heung, L.J. Monocytes and the Host Response to Fungal Pathogens. Front. Cell. Infect. Microbiol. 2020, 10, 34. [Google Scholar] [CrossRef]
- Findley, K.; Oh, J.; Yang, J.; Conlan, S.; Deming, C.; Meyer, J.A.; Schoenfeld, D.; Nomicos, E.; Park, M.; Kong, H.H.; et al. Human Skin Fungal Diversity. Nature 2013, 498, 367–370. [Google Scholar] [CrossRef]
- de Hoog, S.; Monod, M.; Dawson, T.; Boekhout, T.; Mayser, P.; Gräser, Y. Skin Fungi from Colonization to Infection. Microbiol. Spectr. 2017, 5, 5.4.05. [Google Scholar] [CrossRef]
- Igyártó, B.Z.; Haley, K.; Ortner, D.; Bobr, A.; Gerami-Nejad, M.; Edelson, B.T.; Zurawski, S.M.; Malissen, B.; Zurawski, G.; Berman, J.; et al. Skin-Resident Murine Dendritic Cell Subsets Promote Distinct and Opposing Antigen-Specific T Helper Cell Responses. Immunity 2011, 35, 260–272. [Google Scholar] [CrossRef] [Green Version]
- Hamilos, D.L. Allergic Fungal Rhinitis and Rhinosinusitis. Proc. Am. Thorac. Soc. 2010, 7, 245–252. [Google Scholar] [CrossRef]
- Bartemes, K.R.; Kita, H. Innate and Adaptive Immune Responses to Fungi in the Airway. J. Allergy Clin. Immunol. 2018, 142, 353–363. [Google Scholar] [CrossRef] [Green Version]
- Marshall, J.S.; Warrington, R.; Watson, W.; Kim, H.L. An Introduction to Immunology and Immunopathology. Allergy Asthma Clin. Immunol. 2018, 14, 49. [Google Scholar] [CrossRef] [Green Version]
- Vautier, S.; MacCallum, D.M.; Brown, G.D. C-Type Lectin Receptors and Cytokines in Fungal Immunity. Cytokine 2012, 58, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Caffrey-Carr, A.K.; Liu, K.; Espinosa, V.; Croteau, W.; Dhingra, S.; Rivera, A.; Cramer, R.A.; Obar, J.J. MDA5 Is an Essential Sensor of a Pathogen-Associated Molecular Pattern Associated with Vitality That Is Necessary for Host Resistance against Aspergillus Fumigatus. J. Immunol. 2020, 205, 3058–3070. [Google Scholar] [CrossRef]
- Espinosa, V.; Dutta, O.; McElrath, C.; Du, P.; Chang, Y.-J.; Cicciarelli, B.; Pitler, A.; Whitehead, I.; Obar, J.J.; Durbin, J.E.; et al. Type III Interferon Is a Critical Regulator of Innate Antifungal Immunity. Sci. Immunol. 2017, 2, eaan5357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Netea, M.G.; Brown, G.D.; Kullberg, B.J.; Gow, N.A.R. An Integrated Model of the Recognition of Candida Albicans by the Innate Immune System. Nat. Rev. Microbiol. 2008, 6, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Chai, L.Y.A.; Kullberg, B.J.; Vonk, A.G.; Warris, A.; Cambi, A.; Latgé, J.-P.; Joosten, L.A.B.; van der Meer, J.W.M.; Netea, M.G. Modulation of Toll-Like Receptor 2 (TLR2) and TLR4 Responses by Aspergillus Fumigatus. Infect. Immun. 2009, 77, 2184–2192. [Google Scholar] [CrossRef] [Green Version]
- Patin, E.C.; Thompson, A.; Orr, S.J. Pattern Recognition Receptors in Fungal Immunity. Semin. Cell Dev. Biol. 2019, 89, 24–33. [Google Scholar] [CrossRef]
- Jannuzzi, G.P.; de Almeida, J.R.F.; Paulo, L.N.M.; de Almeida, S.R.; Ferreira, K.S. Intracellular PRRs Activation in Targeting the Immune Response Against Fungal Infections. Front. Cell. Infect. Microbiol. 2020, 10, 562. [Google Scholar] [CrossRef]
- Bourgeois, C.; Majer, O.; Frohner, I.E.; Lesiak-Markowicz, I.; Hildering, K.-S.; Glaser, W.; Stockinger, S.; Decker, T.; Akira, S.; Müller, M.; et al. Conventional Dendritic Cells Mount a Type I IFN Response against Candida Spp. Requiring Novel Phagosomal TLR7-Mediated IFN-β Signaling. J. Immunol. 2011, 186, 3104–3112. [Google Scholar] [CrossRef] [Green Version]
- Ramirez-Ortiz, Z.G.; Specht, C.A.; Wang, J.P.; Lee, C.K.; Bartholomeu, D.C.; Gazzinelli, R.T.; Levitz, S.M. Toll-Like Receptor 9-Dependent Immune Activation by Unmethylated CpG Motifs in Aspergillus Fumigatus DNA. Infect. Immun. 2008, 76, 2123–2129. [Google Scholar] [CrossRef] [Green Version]
- Kasperkovitz, P.V.; Khan, N.S.; Tam, J.M.; Mansour, M.K.; Davids, P.J.; Vyas, J.M. Toll-like Receptor 9 Modulates Macrophage Antifungal Effector Function during Innate Recognition of Candida Albicans and Saccharomyces Cerevisiae. Infect. Immun. 2011, 79, 4858–4867. [Google Scholar] [CrossRef] [Green Version]
- Richardson, M.B.; Williams, S.J. MCL and Mincle: C-Type Lectin Receptors That Sense Damaged Self and Pathogen-Associated Molecular Patterns. Front. Immunol. 2014, 5, 288. [Google Scholar] [CrossRef]
- Fradin, C.; Poulain, D.; Jouault, T. β-1,2-Linked Oligomannosides from Candida Albicans Bind to a 32-Kilodalton Macrophage Membrane Protein Homologous to the Mammalian Lectin Galectin-3. Infect. Immun. 2000, 68, 4391–4398. [Google Scholar] [CrossRef] [Green Version]
- Rajaram, M.V.S.; Arnett, E.; Azad, A.K.; Guirado, E.; Ni, B.; Gerberick, A.D.; He, L.-Z.; Keler, T.; Thomas, L.J.; Lafuse, W.P.; et al. M. Tuberculosis-Initiated Human Mannose Receptor Signaling Regulates Macrophage Recognition and Vesicle Trafficking by FcRγ-Chain, Grb2, and SHP-1. Cell Rep. 2017, 21, 126–140. [Google Scholar] [CrossRef] [Green Version]
- Rosowski, E.E.; Raffa, N.; Knox, B.P.; Golenberg, N.; Keller, N.P.; Huttenlocher, A. Macrophages Inhibit Aspergillus Fumigatus Germination and Neutrophil-Mediated Fungal Killing. PLoS Pathog. 2018, 14, e1007229. [Google Scholar] [CrossRef] [Green Version]
- Inglesfield, S.; Jasiulewicz, A.; Hopwood, M.; Tyrrell, J.; Youlden, G.; Mazon-Moya, M.; Millington, O.R.; Mostowy, S.; Jabbari, S.; Voelz, K. Robust Phagocyte Recruitment Controls the Opportunistic Fungal Pathogen Mucor Circinelloides in Innate Granulomas In Vivo. mBio 2018, 9, e02010-17. [Google Scholar] [CrossRef] [Green Version]
- Uderhardt, S.; Martins, A.J.; Tsang, J.S.; Lämmermann, T.; Germain, R.N. Resident Macrophages Cloak Tissue Microlesions to Prevent Neutrophil-Driven Inflammatory Damage. Cell 2019, 177, 541–555.e17. [Google Scholar] [CrossRef] [Green Version]
- Cagnina, R.E.; Michels, K.R.; Bettina, A.M.; Burdick, M.D.; Scindia, Y.; Zhang, Z.; Braciale, T.J.; Mehrad, B. Neutrophil-Derived Tumor Necrosis Factor Drives Fungal Acute Lung Injury in Chronic Granulomatous Disease. J. Infect. Dis. 2021, 224, 1225–1235. [Google Scholar] [CrossRef]
- Gilbert, A.S.; Wheeler, R.T.; May, R.C. Fungal Pathogens: Survival and Replication within Macrophages. Cold Spring Harb. Perspect. Med. 2015, 5, a019661. [Google Scholar] [CrossRef] [Green Version]
- Griffin, F.M.; Griffin, J.A.; Leider, J.E.; Silverstein, S.C. Studies on the Mechanism of Phagocytosis. I. Requirements for Circumferential Attachment of Particle-Bound Ligands to Specific Receptors on the Macrophage Plasma Membrane. J. Exp. Med. 1975, 142, 1263–1282. [Google Scholar] [CrossRef] [Green Version]
- Rittig, M.G.; Schröppel, K.; Seack, K.-H.; Sander, U.; N’Diaye, E.-N.; Maridonneau-Parini, I.; Solbach, W.; Bogdan, C. Coiling Phagocytosis of Trypanosomatids and Fungal Cells. Infect. Immun. 1998, 66, 4331–4339. [Google Scholar] [CrossRef]
- Bryan, A.M.; You, J.K.; Li, G.; Kim, J.; Singh, A.; Morstein, J.; Trauner, D.; Pereira de Sá, N.; Normile, T.G.; Farnoud, A.M.; et al. Cholesterol and Sphingomyelin Are Critical for Fcγ Receptor–Mediated Phagocytosis of Cryptococcus Neoformans by Macrophages. J. Biol. Chem. 2021, 297, 101411. [Google Scholar] [CrossRef]
- Urso, K.; Charles, J.F.; Shull, G.E.; Aliprantis, A.O.; Balestrieri, B. Anion Exchanger 2 Regulates Dectin-1-Dependent Phagocytosis and Killing of Candida Albicans. PLoS ONE 2016, 11, e0158893. [Google Scholar] [CrossRef]
- Schuit, K.E. Phagocytosis and Intracellular Killing of Pathogenic Yeasts by Human Monocytes and Neutrophils. Infect. Immun. 1979, 24, 932–938. [Google Scholar] [CrossRef] [Green Version]
- Brown, G.D. Innate Antifungal Immunity: The Key Role of Phagocytes. Annu. Rev. Immunol. 2011, 29, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Hatinguais, R.; Pradhan, A.; Brown, G.D.; Brown, A.J.P.; Warris, A.; Shekhova, E. Mitochondrial Reactive Oxygen Species Regulate Immune Responses of Macrophages to Aspergillus Fumigatus. Front. Immunol. 2021, 12, 641495. [Google Scholar] [CrossRef]
- Shlezinger, N.; Hohl, T.M. Mitochondrial Reactive Oxygen Species Enhance Alveolar Macrophage Activity against Aspergillus Fumigatus but Are Dispensable for Host Protection. mSphere 2021, 6, e00260-21. [Google Scholar] [CrossRef] [PubMed]
- Dragotakes, Q.; Fu, M.S.; Casadevall, A. Dragotcytosis: Elucidation of the Mechanism for Cryptococcus Neoformans Macrophage-to-Macrophage Transfer. J. Immunol. 2019, 202, 2661–2670. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.; Kannambath, S.; Herbst, S.; Rogers, A.; Soresi, S.; Carby, M.; Reed, A.; Mostowy, S.; Fisher, M.C.; Shaunak, S.; et al. Calcineurin Orchestrates Lateral Transfer of Aspergillus Fumigatus during Macrophage Cell Death. Am. J. Respir. Crit. Care Med. 2016, 194, 1127–1139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, H.; Croudace, J.E.; Lammas, D.A.; May, R.C. Expulsion of Live Pathogenic Yeast by Macrophages. Curr. Biol. 2006, 16, 2156–2160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bain, J.M.; Lewis, L.E.; Okai, B.; Quinn, J.; Gow, N.A.R.; Erwig, L.-P. Non-Lytic Expulsion/Exocytosis of Candida Albicans from Macrophages. Fungal Genet. Biol. 2012, 49, 677–678. [Google Scholar] [CrossRef] [Green Version]
- Seoane, P.I.; Taylor-Smith, L.M.; Stirling, D.; Bell, L.C.K.; Noursadeghi, M.; Bailey, D.; May, R.C. Viral Infection Triggers Interferon-Induced Expulsion of Live Cryptococcus Neoformans by Macrophages. PLoS Pathog. 2020, 16, e1008240. [Google Scholar] [CrossRef]
- Gilbert, A.S.; Seoane, P.I.; Sephton-Clark, P.; Bojarczuk, A.; Hotham, R.; Giurisato, E.; Sarhan, A.R.; Hillen, A.; Velde, G.V.; Gray, N.S.; et al. Vomocytosis of Live Pathogens from Macrophages Is Regulated by the Atypical MAP Kinase ERK5. Sci. Adv. 2017, 3, e1700898. [Google Scholar] [CrossRef] [Green Version]
- Lewis, L.E.; Bain, J.M.; Lowes, C.; Gillespie, C.; Rudkin, F.M.; Gow, N.A.R.; Erwig, L.-P. Stage Specific Assessment of Candida Albicans Phagocytosis by Macrophages Identifies Cell Wall Composition and Morphogenesis as Key Determinants. PLoS Pathog. 2012, 8, e1002578. [Google Scholar] [CrossRef] [Green Version]
- Okagaki, L.H.; Nielsen, K. Titan Cells Confer Protection from Phagocytosis in Cryptococcus Neoformans Infections. Eukaryot. Cell 2012, 11, 820–826. [Google Scholar] [CrossRef] [Green Version]
- Knox, B.P.; Deng, Q.; Rood, M.; Eickhoff, J.C.; Keller, N.P.; Huttenlocher, A. Distinct Innate Immune Phagocyte Responses to Aspergillus Fumigatus Conidia and Hyphae in Zebrafish Larvae. Eukaryot. Cell 2014, 13, 1266–1277. [Google Scholar] [CrossRef] [Green Version]
- Oghiso, Y.; Kubota, Y. Enhanced Interleukin 1 Production by Alveolar Macrophages and Increase in Ia-Positive Lung Cells in Silica-Exposed Rats. Microbiol. Immunol. 1986, 30, 1189–1198. [Google Scholar] [CrossRef] [Green Version]
- Takemura, R.; Stenberg, P.E.; Bainton, D.F.; Werb, Z. Rapid Redistribution of Clathrin onto Macrophage Plasma Membranes in Response to Fc Receptor-Ligand Interaction during Frustrated Phagocytosis. J. Cell Biol. 1986, 102, 55–69. [Google Scholar] [CrossRef] [Green Version]
- Uwamahoro, N.; Verma-Gaur, J.; Shen, H.-H.; Qu, Y.; Lewis, R.; Lu, J.; Bambery, K.; Masters, S.L.; Vince, J.E.; Naderer, T.; et al. The Pathogen Candida Albicans Hijacks Pyroptosis for Escape from Macrophages. Am. Soc. Microbiol. 2014, 5, e00003-14. [Google Scholar] [CrossRef] [Green Version]
- Bain, J.M.; Alonso, M.F.; Childers, D.S.; Walls, C.A.; Mackenzie, K.; Pradhan, A.; Lewis, L.E.; Louw, J.; Avelar, G.M.; Larcombe, D.E.; et al. Immune Cells Fold and Damage Fungal Hyphae. Proc. Natl. Acad. Sci. USA 2021, 118, e2020484118. [Google Scholar] [CrossRef]
- Atri, C.; Guerfali, F.Z.; Laouini, D. Role of Human Macrophage Polarization in Inflammation during Infectious Diseases. Int. J. Mol. Sci. 2018, 19, 1801. [Google Scholar] [CrossRef] [Green Version]
- Subramani, A.; Griggs, P.; Frantzen, N.; Mendez, J.; Tucker, J.; Murriel, J.; Sircy, L.M.; Millican, G.E.; McClelland, E.E.; Seipelt-Thiemann, R.L.; et al. Intracellular Cryptococcus Neoformans Disrupts the Transcriptome Profile of M1- and M2-Polarized Host Macrophages. PLoS ONE 2020, 15, e0233818. [Google Scholar] [CrossRef]
- Reales-Calderón, J.A.; Aguilera-Montilla, N.; Corbí, Á.L.; Molero, G.; Gil, C. Proteomic Characterization of Human Proinflammatory M1 and Anti-Inflammatory M2 Macrophages and Their Response to Candida Albicans. Proteomics 2014, 14, 1503–1518. [Google Scholar] [CrossRef]
- Hardison, S.E.; Herrera, G.; Young, M.L.; Hole, C.R.; Wozniak, K.L.; Wormley, F.L. Protective Immunity against Pulmonary Cryptococcosis Is Associated with STAT1-Mediated Classical Macrophage Activation. J. Immunol. 2012, 189, 4060–4068. [Google Scholar] [CrossRef] [Green Version]
- Gao, Q.; Zhang, J.; Chen, C.; Chen, M.; Sun, P.; Du, W.; Zhang, S.; Liu, Y.; Zhang, R.; Bai, M.; et al. In Situ Mannosylated Nanotrinity-Mediated Macrophage Remodeling Combats Candida Albicans Infection. ACS Nano 2020, 14, 3980–3990. [Google Scholar] [CrossRef]
- Luvanda, M.K.; Posch, W.; Vosper, J.; Zaderer, V.; Noureen, A.; Lass-Flörl, C.; Wilflingseder, D. Dexamethasone Promotes Aspergillus Fumigatus Growth in Macrophages by Triggering M2 Repolarization via Targeting PKM2. J. Fungi 2021, 7, 70. [Google Scholar] [CrossRef]
- Drummond, R.A.; Muthulekha, S.; Vasileios, O.; Bing, Z.; Dambuza, I.M.; Schaefer, B.C.; Bohrer, A.C.; Mayer-Barber, K.D.; Lira, S.A.; Yoichiro, I.; et al. CARD9+ Microglia Promote Antifungal Immunity via IL-1β- and CXCL1-Mediated Neutrophil Recruitment. Nat. Immunol. 2019, 20, 559–570. [Google Scholar] [CrossRef]
- Nguyen, N.Z.N.; Tran, V.G.; Baek, J.; Kim, Y.; Youn, E.H.; Na, S.W.; Park, S.J.; Seo, S.-K.; Kwon, B. IL-33 Coordinates Innate Defense to Systemic Candida Albicans Infection by Regulating IL-23 and IL-10 in an Opposite Way. J. Immunol. 2022, 208, 660–671. [Google Scholar] [CrossRef]
- Lilly, L.M.; Scopel, M.; Nelson, M.P.; Burg, A.R.; Dunaway, C.W.; Steele, C. Eosinophil Deficiency Compromises Lung Defense against Aspergillus Fumigatus. Infect. Immun. 2014, 82, 1315–1325. [Google Scholar] [CrossRef] [Green Version]
- Yadav, B.; Specht, C.A.; Lee, C.K.; Pokrovskii, M.; Huh, J.R.; Littman, D.R.; Levitz, S.M. Lung Eosinophils Elicited during Allergic and Acute Aspergillosis Express RORγt and IL-23R but Do Not Require IL-23 for IL-17 Production. PLoS Pathog. 2021, 17, e1009891. [Google Scholar] [CrossRef]
- de Oliveira Malacco, N.L.S.; Rachid, M.A.; da Silva Gurgel, I.L.; Moura, T.R.; Sucupira, P.H.F.; de Sousa, L.P.; da Glória de Souza, D.; de Castro Russo, R.; Teixeira, M.M.; Soriani, F.M. Eosinophil-Associated Innate IL-17 Response Promotes Aspergillus Fumigatus Lung Pathology. Front. Cell. Infect. Microbiol. 2019, 8, 453. [Google Scholar] [CrossRef] [Green Version]
- Yamasaki, S.; Matsumoto, M.; Takeuchi, O.; Matsuzawa, T.; Ishikawa, E.; Sakuma, M.; Tateno, H.; Uno, J.; Hirabayashi, J.; Mikami, Y.; et al. C-Type Lectin Mincle Is an Activating Receptor for Pathogenic Fungus, Malassezia. Proc. Natl. Acad. Sci. USA 2009, 106, 1897–1902. [Google Scholar] [CrossRef] [Green Version]
- Swidergall, M.; Khalaji, M.; Solis, N.V.; Moyes, D.L.; Drummond, R.A.; Hube, B.; Lionakis, M.S.; Murdoch, C.; Filler, S.G.; Naglik, J.R. Candidalysin Is Required for Neutrophil Recruitment and Virulence During Systemic Candida Albicans Infection. J. Infect. Dis. 2019, 220, 1477–1488. [Google Scholar] [CrossRef] [PubMed]
- Richardson, J.P.; Willems, H.M.E.; Moyes, D.L.; Shoaie, S.; Barker, K.S.; Tan, S.L.; Palmer, G.E.; Hube, B.; Naglik, J.R.; Peters, B.M. Candidalysin Drives Epithelial Signaling, Neutrophil Recruitment, and Immunopathology at the Vaginal Mucosa. Infect. Immun. 2018, 86, e00645-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, W.; Wang, Q.; Deng, Z.; Li, T.; Xiao, H.; Wu, Z. TRAF1 Suppresses Antifungal Immunity through CXCL1-Mediated Neutrophil Recruitment during Candida Albicans Intradermal Infection. Cell Commun. Signal. 2020, 18, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carpentier, I.; Beyaert, R. TRAF1 Is a TNF Inducible Regulator of NF-ΚB Activation. FEBS Lett. 1999, 460, 246–250. [Google Scholar] [CrossRef] [Green Version]
- Ho, J.; Yang, X.; Nikou, S.-A.; Kichik, N.; Donkin, A.; Ponde, N.O.; Richardson, J.P.; Gratacap, R.L.; Archambault, L.S.; Zwirner, C.P.; et al. Candidalysin Activates Innate Epithelial Immune Responses via Epidermal Growth Factor Receptor. Nat. Commun. 2019, 10, 2297. [Google Scholar] [CrossRef] [Green Version]
- Swidergall, M.; Solis, N.V.; Millet, N.; Huang, M.Y.; Lin, J.; Phan, Q.T.; Lazarus, M.D.; Wang, Z.; Yeaman, M.R.; Mitchell, A.P.; et al. Activation of EphA2-EGFR Signaling in Oral Epithelial Cells by Candida Albicans Virulence Factors. PLoS Pathog. 2021, 17, e1009221. [Google Scholar] [CrossRef]
- Le, H.T.; Tran, V.G.; Kim, W.; Kim, J.; Cho, H.R.; Kwon, B. IL-33 Priming Regulates Multiple Steps of the Neutrophil-Mediated Anti-Candida Albicans Response by Modulating TLR and Dectin-1 Signals. J. Immunol. 2012, 189, 287–295. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Rueda, N.; Rouges, C.; Touahri, A.; Misme-Aucouturier, B.; Albassier, M.; Pape, P.L. In Vitro Immune Responses of Human PBMCs against Candida Albicans Reveals Fungal and Leucocyte Phenotypes Associated with Fungal Persistence. Sci. Rep. 2020, 10, 6211. [Google Scholar] [CrossRef]
- Nur, S.; Sparber, F.; Lemberg, C.; Guiducci, E.; Schweizer, T.A.; Zwicky, P.; Becher, B.; LeibundGut-Landmann, S. IL-23 Supports Host Defense against Systemic Candida Albicans Infection by Ensuring Myeloid Cell Survival. PLoS Pathog. 2019, 15, e1008115. [Google Scholar] [CrossRef] [Green Version]
- Lämmermann, T.; Afonso, P.V.; Angermann, B.R.; Wang, J.M.; Kastenmüller, W.; Parent, C.A.; Germain, R.N. Neutrophil Swarms Require LTB4 and Integrins at Sites of Cell Death in Vivo. Nature 2013, 498, 371–375. [Google Scholar] [CrossRef]
- Sun, D.; Shi, M. Neutrophil Swarming toward Cryptococcus Neoformans Is Mediated by Complement and Leukotriene B4. Biochem. Biophys. Res. Commun. 2016, 477, 945–951. [Google Scholar] [CrossRef] [Green Version]
- Hopke, A.; Scherer, A.; Kreuzburg, S.; Abers, M.S.; Zerbe, C.S.; Dinauer, M.C.; Mansour, M.K.; Irimia, D. Neutrophil Swarming Delays the Growth of Clusters of Pathogenic Fungi. Nat. Commun. 2020, 11, 2031. [Google Scholar] [CrossRef]
- Hind, L.E.; Giese, M.A.; Schoen, T.J.; Beebe, D.J.; Keller, N.; Huttenlocher, A. Immune Cell Paracrine Signaling Drives the Neutrophil Response to A. Fumigatus in an Infection-on-a-Chip Model. Cell. Mol. Bioeng. 2021, 14, 133–145. [Google Scholar] [CrossRef]
- Lämmermann, T. In the Eye of the Neutrophil Swarm—Navigation Signals That Bring Neutrophils Together in Inflamed and Infected Tissues. J. Leukoc. Biol. 2016, 100, 55–63. [Google Scholar] [CrossRef]
- Lee, E.K.S.; Gillrie, M.R.; Li, L.; Arnason, J.W.; Kim, J.H.; Babes, L.; Lou, Y.; Sanati-Nezhad, A.; Kyei, S.K.; Kelly, M.M.; et al. Leukotriene B4-Mediated Neutrophil Recruitment Causes Pulmonary Capillaritis during Lethal Fungal Sepsis. Cell Host Microbe 2018, 23, 121–133.e4. [Google Scholar] [CrossRef]
- Lacy, P. Mechanisms of Degranulation in Neutrophils. Allergy Asthma Clin. Immunol. 2006, 2, 98–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diamond, R.D.; Krzesicki, R.; Jao, W. Damage to Pseudohyphal Forms of Candida Albicans by Neutrophils in the Absence of Serum In Vitro. J. Clin. Investig. 1978, 61, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Hartl, D.; Latzin, P.; Hordijk, P.; Marcos, V.; Rudolph, C.; Woischnik, M.; Krauss-Etschmann, S.; Koller, B.; Reinhardt, D.; Roscher, A.A.; et al. Cleavage of CXCR1 on Neutrophils Disables Bacterial Killing in Cystic Fibrosis Lung Disease. Nat. Med. 2007, 13, 1423–1430. [Google Scholar] [CrossRef] [PubMed]
- Swamydas, M.; Gao, J.-L.; Break, T.J.; Johnson, M.D.; Jaeger, M.; Rodriguez, C.A.; Lim, J.K.; Green, N.M.; Collar, A.L.; Fischer, B.G.; et al. CXCR1-Mediated Neutrophil Degranulation and Fungal Killing Promotes Candida Clearance and Host Survival. Sci. Transl. Med. 2016, 8, 322ra10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winterbourn, C.C.; Hampton, M.B.; Livesey, J.H.; Kettle, A.J. Modeling the Reactions of Superoxide and Myeloperoxidase in the Neutrophil Phagosome: Implications for Microbial Killing. J. Biol. Chem. 2006, 281, 39860–39869. [Google Scholar] [CrossRef] [Green Version]
- Dupré-Crochet, S.; Erard, M.; Nüβe, O. ROS Production in Phagocytes: Why, When, and Where? J. Leukoc. Biol. 2013, 94, 657–670. [Google Scholar] [CrossRef]
- Urban, C.F.; Ermert, D.; Schmid, M.; Abu-Abed, U.; Goosmann, C.; Nacken, W.; Brinkmann, V.; Jungblut, P.R.; Zychlinsky, A. Neutrophil Extracellular Traps Contain Calprotectin, a Cytosolic Protein Complex Involved in Host Defense against Candida Albicans. PLoS Pathog. 2009, 5, e1000639. [Google Scholar] [CrossRef] [Green Version]
- Urban, C.F.; Reichard, U.; Brinkmann, V.; Zychlinsky, A. Neutrophil Extracellular Traps Capture and Kill Candida Albicans Yeast and Hyphal Forms. Cell. Microbiol. 2006, 8, 668–676. [Google Scholar] [CrossRef]
- Bianchi, M.; Hakkim, A.; Brinkmann, V.; Siler, U.; Seger, R.A.; Zychlinsky, A.; Reichenbach, J. Restoration of NET Formation by Gene Therapy in CGD Controls Aspergillosis. Blood 2009, 114, 2619–2622. [Google Scholar] [CrossRef] [Green Version]
- Isles, H.M.; Loynes, C.A.; Alasmari, S.; Kon, F.C.; Henry, K.M.; Kadochnikova, A.; Hales, J.; Muir, C.F.; Keightley, M.-C.; Kadirkamanathan, V.; et al. Pioneer Neutrophils Release Chromatin within in Vivo Swarms. eLife 2021, 10, e68755. [Google Scholar] [CrossRef]
- Wilson, A.S.; Randall, K.L.; Pettitt, J.A.; Ellyard, J.I.; Blumenthal, A.; Enders, A.; Quah, B.J.; Bopp, T.; Parish, C.R.; Brüstle, A. Neutrophil Extracellular Traps and Their Histones Promote Th17 Cell Differentiation Directly via TLR2. Nat. Commun. 2022, 13, 528. [Google Scholar] [CrossRef]
- Mengesha, B.G.; Conti, H.R. The Role of IL-17 in Protection against Mucosal Candida Infections. J. Fungi 2017, 3, 52. [Google Scholar] [CrossRef]
- Taylor, P.R.; Leal, S.M.; Sun, Y.; Pearlman, E. Aspergillus and Fusarium Corneal Infections Are Regulated by Th17 Cells and IL-17–Producing Neutrophils. J. Immunol. 2014, 192, 3319–3327. [Google Scholar] [CrossRef] [Green Version]
- Rohrbach, A.; Slade, D.; Thompson, P.; Mowen, K. Activation of PAD4 in NET Formation. Front. Immunol. 2012, 3, 360. [Google Scholar] [CrossRef] [Green Version]
- Guiducci, E.; Lemberg, C.; Küng, N.; Schraner, E.; Theocharides, A.P.A.; LeibundGut-Landmann, S. Candida Albicans-Induced NETosis Is Independent of Peptidylarginine Deiminase 4. Front. Immunol. 2018, 9, 1573. [Google Scholar] [CrossRef]
- Tatsiy, O.; McDonald, P.P. Physiological Stimuli Induce PAD4-Dependent, ROS-Independent NETosis, With Early and Late Events Controlled by Discrete Signaling Pathways. Front. Immunol. 2018, 9, 2036. [Google Scholar] [CrossRef] [Green Version]
- Thiam, H.R.; Wong, S.L.; Qiu, R.; Kittisopikul, M.; Vahabikashi, A.; Goldman, A.E.; Goldman, R.D.; Wagner, D.D.; Waterman, C.M. NETosis Proceeds by Cytoskeleton and Endomembrane Disassembly and PAD4-Mediated Chromatin Decondensation and Nuclear Envelope Rupture. Proc. Natl. Acad. Sci. USA 2020, 117, 7326–7337. [Google Scholar] [CrossRef] [Green Version]
- Clark, H.L.; Abbondante, S.; Minns, M.S.; Greenberg, E.N.; Sun, Y.; Pearlman, E. Protein Deiminase 4 and CR3 Regulate Aspergillus Fumigatus and β-Glucan-Induced Neutrophil Extracellular Trap Formation, but Hyphal Killing Is Dependent Only on CR3. Front. Immunol. 2018, 9, 1182. [Google Scholar] [CrossRef]
- Silva, J.C.; Rodrigues, N.C.; Thompson-Souza, G.A.; de Muniz, V.S.; Neves, J.S.; Figueiredo, R.T. Mac-1 Triggers Neutrophil DNA Extracellular Trap Formation to Aspergillus Fumigatus Independently of PAD4 Histone Citrullination. J. Leukoc. Biol. 2020, 107, 69–83. [Google Scholar] [CrossRef]
- Karkowska-Kuleta, J.; Smolarz, M.; Seweryn-Ozog, K.; Satala, D.; Zawrotniak, M.; Wronowska, E.; Bochenska, O.; Kozik, A.; Nobbs, A.H.; Gogol, M.; et al. Proteinous Components of Neutrophil Extracellular Traps Are Arrested by the Cell Wall Proteins of Candida Albicans during Fungal Infection, and Can Be Used in the Host Invasion. Cells 2021, 10, 2736. [Google Scholar] [CrossRef]
- Alflen, A.; Lopez, P.A.; Hartmann, A.-K.; Maxeiner, J.; Bosmann, M.; Sharma, A.; Platten, J.; Ries, F.; Beckert, H.; Ruf, W.; et al. Neutrophil Extracellular Traps Impair Fungal Clearance in a Mouse Model of Invasive Pulmonary Aspergillosis. Immunobiology 2020, 225, 151867. [Google Scholar] [CrossRef] [PubMed]
- Pazhakh, V.; Ellett, F.; Croker, B.A.; O’Donnell, J.A.; Pase, L.; Schulze, K.E.; Greulich, R.S.; Gupta, A.; Reyes-Aldasoro, C.C.; Andrianopoulos, A.; et al. β-Glucan–Dependent Shuttling of Conidia from Neutrophils to Macrophages Occurs during Fungal Infection Establishment. PLoS Biol. 2019, 17, e3000113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, Y.; Matsuwaki, Y.; Shin, S.-H.; Ponikau, J.U.; Kita, H. Nonpathogenic, Environmental Fungi Induce Activation and Degranulation of Human Eosinophils. J. Immunol. 2005, 175, 5439–5447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gladiator, A.; Wangler, N.; Trautwein-Weidner, K.; LeibundGut-Landmann, S. Cutting Edge: IL-17–Secreting Innate Lymphoid Cells Are Essential for Host Defense against Fungal Infection. J. Immunol. 2013, 190, 521–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reeder, K.M.; Mackel, J.J.; Godwin, M.S.; Dunaway, C.W.; Blackburn, J.P.; Patel, R.P.; Steele, C. Role of Common γ-Chain Cytokines in Lung Interleukin-22 Regulation after Acute Exposure to Aspergillus Fumigatus. Infect. Immun. 2018, 86, e00157-18. [Google Scholar] [CrossRef] [Green Version]
- Di Luccia, B.; Gilfillan, S.; Cella, M.; Colanna, M.; Huang, S.C.-C. ILC3s Integrate Glycolysis and Mitochondrial Production of Reactive Oxygen Species to Fulfill Activation Demands | Journal of Experimental Medicine|Rockefeller University Press. J. Exp. Med. 2019, 216, 2231–2241. [Google Scholar] [CrossRef]
- Böttger, E.C.; Metzger, S.; Bitter-Suermann, D.; Stevenson, G.; Kleindienst, S.; Burger, R. Impaired Humoral Immune Response in Complement C3-Deficient Guinea Pigs: Absence of Secondary Antibody Response. Eur. J. Immunol. 1986, 16, 1231–1235. [Google Scholar] [CrossRef]
- Tsoni, S.V.; Kerrigan, A.M.; Marakalala, M.J.; Srinivasan, N.; Duffield, M.; Taylor, P.R.; Botto, M.; Steele, C.; Brown, G.D. Complement C3 Plays an Essential Role in the Control of Opportunistic Fungal Infections. Infect. Immun. 2009, 77, 3679–3685. [Google Scholar] [CrossRef] [Green Version]
- Teschner, D.; Cholaszczyńska, A.; Ries, F.; Beckert, H.; Theobald, M.; Grabbe, S.; Radsak, M.; Bros, M. CD11b Regulates Fungal Outgrowth but Not Neutrophil Recruitment in a Mouse Model of Invasive Pulmonary Aspergillosis. Front. Immunol. 2019, 10, 123. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, X.M.; Heflin, K.E.; Lavigne, L.M.; Yu, K.; Kim, M.; Salomon, A.R.; Reichner, J.S. Lectin Site Ligation of CR3 Induces Conformational Changes and Signaling*. J. Biol. Chem. 2012, 287, 3337–3348. [Google Scholar] [CrossRef] [Green Version]
- van Bruggen, R.; Drewniak, A.; Jansen, M.; van Houdt, M.; Roos, D.; Chapel, H.; Verhoeven, A.J.; Kuijpers, T.W. Complement Receptor 3, Not Dectin-1, Is the Major Receptor on Human Neutrophils for β-Glucan-Bearing Particles. Mol. Immunol. 2009, 47, 575–581. [Google Scholar] [CrossRef]
- Johnson, C.M.; O’Brien, X.M.; Byrd, A.S.; Parisi, V.E.; Loosely, A.J.; Li, W.; Witt, H.; Faridi, H.M.; Lefort, C.T.; Gupta, V.; et al. Integrin Cross-Talk Regulates the Human Neutrophil Response to Fungal β-Glucan in the Context of the Extracellular Matrix: A Prominent Role for VLA3 in the Antifungal Response. J. Immunol. 2017, 198, 318–334. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Li, Z.; Wang, S.; Zhang, C.; Han, L.; Sun, Q.; Han, X. Aspergillus Fumigatus Induces the Release of IL-8 and MCP-1 by Activating Nuclear Transcription Through Dectin-1 and CR3 Receptors in Alveolar Epithelial Cells. Curr. Microbiol. 2021, 78, 3474–3482. [Google Scholar] [CrossRef]
- Nobile, C.J.; Johnson, A.D. Candida Albicans Biofilms and Human Disease. Annu. Rev. Microbiol. 2015, 69, 71–92. [Google Scholar] [CrossRef] [Green Version]
- Carolus, H.; Van Dyck, K.; Van Dijck, P. Candida Albicans and Staphylococcus Species: A Threatening Twosome. Front. Microbiol. 2019, 10, 2162. [Google Scholar] [CrossRef]
- Yapar, N. Epidemiology and Risk Factors for Invasive Candidiasis. Ther. Clin. Risk Manag. 2014, 10, 95–105. [Google Scholar] [CrossRef] [Green Version]
- Okada, S.; Puel, A.; Casanova, J.-L.; Kobayashi, M. Chronic Mucocutaneous Candidiasis Disease Associated with Inborn Errors of IL-17 Immunity. Clin. Transl. Immunol. 2016, 5, e114. [Google Scholar] [CrossRef]
- Humbert, L.; Cornu, M.; Proust-Lemoine, E.; Bayry, J.; Wemeau, J.-L.; Vantyghem, M.-C.; Sendid, B. Chronic Mucocutaneous Candidiasis in Autoimmune Polyendocrine Syndrome Type 1. Front. Immunol. 2018, 9, 2570. [Google Scholar] [CrossRef] [Green Version]
- Bassetti, M.; Giacobbe, D.R.; Vena, A.; Trucchi, C.; Ansaldi, F.; Antonelli, M.; Adamkova, V.; Alicino, C.; Almyroudi, M.-P.; Atchade, E.; et al. Incidence and Outcome of Invasive Candidiasis in Intensive Care Units (ICUs) in Europe: Results of the EUCANDICU Project. Crit. Care 2019, 23, 219. [Google Scholar] [CrossRef] [Green Version]
- Maródi, L.; Cypowyj, S.; Tóth, B.; Chernyshova, L.; Puel, A.; Casanova, J.-L. Molecular Mechanisms of Mucocutaneous Immunity against Candida and Staphylococcus Species. J. Allergy Clin. Immunol. 2012, 130, 1019–1027. [Google Scholar] [CrossRef] [Green Version]
- Lanternier, F.; Cypowyj, S.; Picard, C.; Bustamante, J.; Lortholary, O.; Casanova, J.-L.; Puel, A. Primary Immunodeficiencies Underlying Fungal Infections. Curr. Opin. Pediatrics 2013, 25, 736–747. [Google Scholar] [CrossRef] [Green Version]
- Corvilain, E.; Casanova, J.-L.; Puel, A. Inherited CARD9 Deficiency: Invasive Disease Caused by Ascomycete Fungi in Previously Healthy Children and Adults. J. Clin. Immunol. 2018, 38, 656–693. [Google Scholar] [CrossRef]
- Drummond, R.A.; Saijo, S.; Iwakura, Y.; Brown, G.D. The Role of Syk/CARD9 Coupled C-Type Lectins in Antifungal Immunity. Eur. J. Immunol. 2011, 41, 276–281. [Google Scholar] [CrossRef]
- Drummond, R.A.; Collar, A.L.; Swamydas, M.; Rodriguez, C.A.; Lim, J.K.; Mendez, L.M.; Fink, D.L.; Hsu, A.P.; Zhai, B.; Karauzum, H.; et al. CARD9-Dependent Neutrophil Recruitment Protects against Fungal Invasion of the Central Nervous System. PLoS Pathog. 2015, 11, e1005293. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhang, H.; Wang, X.; Shao, Z.; Li, Y.; Zhao, G.; Liu, F.; Liu, B.; Zheng, Y.; Chen, T.; et al. OTUD1 Regulates Antifungal Innate Immunity through Deubiquitination of CARD9. J. Immunol. 2021, 206, 1832–1843. [Google Scholar] [CrossRef] [PubMed]
- Picard, C.; Casanova, J.-L.; Puel, A. Infectious Diseases in Patients with IRAK-4, MyD88, NEMO, or IκBα Deficiency. Clin. Microbiol. Rev. 2011, 24, 490–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villamón, E.; Gozalbo, D.; Roig, P.; Murciano, C.; O’Connor, J.E.; Fradelizi, D.; Gil, M.L. Myeloid Differentiation Factor 88 (MyD88) Is Required for Murine Resistance to Candida Albicans and Is Critically Involved in Candida-Induced Production of Cytokines. Eur. Cytokine Netw. 2004, 15, 263–271. [Google Scholar] [PubMed]
- Hu, W.; van Steijn, L.; Li, C.; Verbeek, F.J.; Cao, L.; Merks, R.M.H.; Spaink, H.P. A Novel Function of TLR2 and MyD88 in the Regulation of Leukocyte Cell Migration Behavior during Wounding in Zebrafish Larvae. Front. Cell Dev. Biol. 2021, 9, 210. [Google Scholar] [CrossRef]
- Huppler, A.R.; Conti, H.R.; Hernández-Santos, N.; Darville, T.; Biswas, P.S.; Gaffen, S.L. Role of Neutrophils in IL-17-Dependent Immunity to Mucosal Candidiasis. J. Immunol. 2014, 192, 1745–1752. [Google Scholar] [CrossRef]
- Kao, C.-Y.; Chen, Y.; Thai, P.; Wachi, S.; Huang, F.; Kim, C.; Harper, R.W.; Wu, R. IL-17 Markedly up-Regulates Beta-Defensin-2 Expression in Human Airway Epithelium via JAK and NF-KappaB Signaling Pathways. J. Immunol. 2004, 173, 3482–3491. [Google Scholar] [CrossRef]
- Davidson, L.; van den Reek, J.M.P.A.; Bruno, M.; van Hunsel, F.; Herings, R.M.C.; Matzaraki, V.; Boahen, C.K.; Kumar, V.; Groenewoud, H.M.M.; van de Veerdonk, F.L.; et al. Risk of Candidiasis Associated with Interleukin-17 Inhibitors: A Real-World Observational Study of Multiple Independent Sources. Lancet Reg. Health Eur. 2022, 13, 10266. [Google Scholar] [CrossRef]
- Chimenz, R.; Tropeano, A.; Chirico, V.; Ceravolo, G.; Salpietro, C.; Cuppari, C. IL-17 Serum Level in Patients with Chronic Mucocutaneous Candidiasis Disease. Pediatric Allergy Immunol. 2022, 33, 77–79. [Google Scholar] [CrossRef]
- Puel, A.; Cypowyj, S.; Maródi, L.; Abel, L.; Picard, C.; Casanova, J.-L. Inborn Errors of Human IL-17 Immunity Underlie Chronic Mucocutaneous Candidiasis. Curr. Opin. Allergy Clin. Immunol. 2012, 12, 616–622. [Google Scholar] [CrossRef] [Green Version]
- Puel, A.; Döffinger, R.; Natividad, A.; Chrabieh, M.; Barcenas-Morales, G.; Picard, C.; Cobat, A.; Ouachée-Chardin, M.; Toulon, A.; Bustamante, J.; et al. Autoantibodies against IL-17A, IL-17F, and IL-22 in Patients with Chronic Mucocutaneous Candidiasis and Autoimmune Polyendocrine Syndrome Type I. J. Exp. Med. 2010, 207, 291–297. [Google Scholar] [CrossRef] [Green Version]
- Kisand, K.; Bøe Wolff, A.S.; Podkrajšek, K.T.; Tserel, L.; Link, M.; Kisand, K.V.; Ersvaer, E.; Perheentupa, J.; Erichsen, M.M.; Bratanic, N.; et al. Chronic Mucocutaneous Candidiasis in APECED or Thymoma Patients Correlates with Autoimmunity to Th17-Associated Cytokines. J. Exp. Med. 2010, 207, 299–308. [Google Scholar] [CrossRef]
- Bader, O.; Weig, M.S.; Gross, U.; Schön, M.P.; Mempel, M.; Buhl, T. Photo Quiz. A 32-Year-Old Man with Ulcerative Mucositis, Skin Lesions, and Nail Dystrophy. Chronic Mucocutaneous Candidiasis by Multidrug-Resistant Candida Albicans. Clin. Infect. Dis. 2012, 54, 972, 1035–1036. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, N.; Watanabe, J.; Kitamura, T.; Yamada, Y.; Kanegasaki, S.; Nakata, K. Lungs of Patients with Idiopathic Pulmonary Alveolar Proteinosis Express a Factor Which Neutralizes Granulocyte-Macrophage Colony Stimulating Factor. FEBS Lett. 1999, 442, 246–250. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.-H.; Teitz-Tennenbaum, S.; Neal, L.M.; Murdock, B.J.; Malachowski, A.N.; Dils, A.J.; Olszewski, M.A.; Osterholzer, J.J. Local GM-CSF–Dependent Differentiation and Activation of Pulmonary Dendritic Cells and Macrophages Protect against Progressive Cryptococcal Lung Infection in Mice. J. Immunol. 2016, 196, 1810–1821. [Google Scholar] [CrossRef] [Green Version]
- Rajasingham, R.; Smith, R.M.; Park, B.J.; Jarvis, J.N.; Govender, N.P.; Chiller, T.M.; Denning, D.W.; Loyse, A.; Boulware, D.R. Global Burden of Disease of HIV-Associated Cryptococcal Meningitis: An Updated Analysis. Lancet Infect. Dis. 2017, 17, 873–881. [Google Scholar] [CrossRef] [Green Version]
- de Araújo, G.R.S.; de Alcantara, C.L.; Rodrigues, N.; de Souza, W.; Pontes, B.; Frases, S. Ultrastructural Study of Cryptococcus Neoformans Surface During Budding Events. Front. Microbiol. 2021, 12, 405. [Google Scholar] [CrossRef]
- Esher, S.K.; Zaragoza, O.; Alspaugh, J.A. Cryptococcal Pathogenic Mechanisms: A Dangerous Trip from the Environment to the Brain. Mem. Inst. Oswaldo Cruz 2018, 113, e180057. [Google Scholar] [CrossRef] [Green Version]
- Chayakulkeeree, M.; Perfect, J.R. Cryptococcosis. Infect. Dis. Clin. N. Am. 2006, 20, 507–544. [Google Scholar] [CrossRef]
- Bartlett, K.H.; Kidd, S.E.; Kronstad, J.W. The Emergence of Cryptococcus Gattii in British Columbia and the Pacific Northwest. Curr. Infect. Dis. Rep. 2008, 10, 58–65. [Google Scholar] [CrossRef]
- May, R.C.; Stone, N.R.H.; Wiesner, D.L.; Bicanic, T.; Nielsen, K. Cryptococcus: From Environmental Saprophyte to Global Pathogen. Nat. Rev. Microbiol. 2016, 14, 106–117. [Google Scholar] [CrossRef]
- Rosen, L.B.; Freeman, A.F.; Yang, L.M.; Jutivorakool, K.; Olivier, K.N.; Angkasekwinai, N.; Suputtamongkol, Y.; Bennett, J.E.; Pyrgos, V.; Williamson, P.R.; et al. Anti-GM-CSF Autoantibodies in Patients with Cryptococcal Meningitis. J. Immunol. 2013, 190, 3959–3966. [Google Scholar] [CrossRef] [Green Version]
- Saijo, T.; Chen, J.; Chen, S.C.-A.; Rosen, L.B.; Yi, J.; Sorrell, T.C.; Bennett, J.E.; Holland, S.M.; Browne, S.K.; Kwon-Chung, K.J. Anti-Granulocyte-Macrophage Colony-Stimulating Factor Autoantibodies Are a Risk Factor for Central Nervous System Infection by Cryptococcus Gattii in Otherwise Immunocompetent Patients. mBio 2014, 5, e00912-14. [Google Scholar] [CrossRef] [Green Version]
- Perrineau, S.; Guery, R.; Monnier, D.; Puel, A.; Lanternier, F. Anti-GM-CSF Autoantibodies and Cryptococcus Neoformans Var. Grubii CNS Vasculitis. J. Clin. Immunol. 2020, 40, 767–769. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, T.; Tanaka, N.; Watanabe, J.; Uchida; Kanegasaki, S.; Yamada, Y.; Nakata, K. Idiopathic Pulmonary Alveolar Proteinosis as an Autoimmune Disease with Neutralizing Antibody against Granulocyte/Macrophage Colony-Stimulating Factor. J. Exp. Med. 1999, 190, 875–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandres, M.V.; Modi, P.; Sharma, S. Aspergillus fumigatus. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Kolwijck, E.; van de Veerdonk, F.L. The Potential Impact of the Pulmonary Microbiome on Immunopathogenesis of Aspergillus-Related Lung Disease. Eur. J. Immunol. 2014, 44, 3156–3165. [Google Scholar] [CrossRef] [PubMed]
- Roth, R.S.; Masouridi-Levrat, S.; Chalandon, Y.; Mamez, A.-C.; Giannotti, F.; Riat, A.; Fischer, A.; Poncet, A.; Glampedakis, E.; Van Delden, C.; et al. Invasive Mold Infections in Allogeneic Hematopoietic Cell Transplant Recipients in 2020: Have We Made Enough Progress? Open Forum Infect. Dis. 2021, 9, ofab596. [Google Scholar] [CrossRef] [PubMed]
- Ferdjallah, A.; Young, J.-A.H.; MacMillan, M.L. A Review of Infections After Hematopoietic Cell Transplantation Requiring PICU Care: Transplant Timeline Is Key. Front. Pediatrics 2021, 9, 634449. [Google Scholar] [CrossRef]
- Luo, X.-L.; Li, J.-X.; Huang, H.-R.; Duan, J.-L.; Dai, R.-X.; Tao, R.-J.; Yang, L.; Hou, J.; Jia, X.-M.; Xu, J.-F. LL37 Inhibits Aspergillus Fumigatus Infection via Directly Binding to the Fungus and Preventing Excessive Inflammation. Front. Immunol. 2019, 10, 283. [Google Scholar] [CrossRef]
- Fonseca, M.T.; Moretti, E.H.; Marques, L.M.M.; Machado, B.F.; Brito, C.F.; Guedes, J.T.; Komegae, E.N.; Vieira, T.S.; Festuccia, W.T.; Lopes, N.P.; et al. A Leukotriene-Dependent Spleen-Liver Axis Drives TNF Production in Systemic Inflammation. Sci. Signal. 2021, 14, eabb0969. [Google Scholar] [CrossRef]
- Dyugovskaya, L.; Polyakov, A.; Ginsberg, D.; Lavie, P.; Lavie, L. Molecular Pathways of Spontaneous and TNF-α–Mediated Neutrophil Apoptosis under Intermittent Hypoxia. Am. J. Respir. Cell Mol. Biol. 2011, 45, 154–162. [Google Scholar] [CrossRef]
- Blaser, H.; Dostert, C.; Mak, T.W.; Brenner, D. TNF and ROS Crosstalk in Inflammation. Trends Cell Biol. 2016, 26, 249–261. [Google Scholar] [CrossRef]
- Rieber, N.; Gazendam, R.P.; Freeman, A.F.; Hsu, A.P.; Collar, A.L.; Sugui, J.A.; Drummond, R.A.; Rongkavilit, C.; Hoffman, K.; Henderson, C.; et al. Extrapulmonary Aspergillus Infection in Patients with CARD9 Deficiency. JCI Insight 2016, 1, e89890. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Huang, C.; Song, Y.; Ma, Y.; Wan, Z.; Zhu, X.; Wang, X.; Li, R. Primary Cutaneous Aspergillosis in a Patient with CARD9 Deficiency and Aspergillus Susceptibility of Card9 Knockout Mice. J. Clin. Immunol. 2021, 41, 427–440. [Google Scholar] [CrossRef]
- Ademe, M. Immunomodulation for the Treatment of Fungal Infections: Opportunities and Challenges. Front. Cell. Infect. Microbiol. 2020, 10, 469. [Google Scholar] [CrossRef]
- Zumla, A.; Rao, M.; Wallis, R.S.; Kaufmann, S.H.E.; Rustomjee, R.; Mwaba, P.; Vilaplana, C.; Yeboah-Manu, D.; Chakaya, J.; Ippolito, G.; et al. Host-Directed Therapies for Infectious Diseases: Current Status, Recent Progress, and Future Prospects. Lancet. Infect. Dis. 2016, 16, e47–e63. [Google Scholar] [CrossRef] [Green Version]
- Armstrong-James, D.; Brown, G.D.; Netea, M.G.; Zelante, T.; Gresnigt, M.S.; van de Veerdonk, F.L.; Levitz, S.M. Immunotherapeutic Approaches to Treatment of Fungal Diseases. Lancet Infect. Dis. 2017, 17, e393–e402. [Google Scholar] [CrossRef]
- Mehta, H.M.; Malandra, M.; Corey, S.J. G-CSF and GM-CSF in Neutropenia. J. Immunol. 2015, 195, 1341–1349. [Google Scholar] [CrossRef] [PubMed]
- Damiani, G.; McCormick, T.S.; Leal, L.O.; Ghannoum, M.A. Recombinant Human Granulocyte Macrophage-Colony Stimulating Factor Expressed in Yeast (Sargramostim): A Potential Ally to Combat Serious Infections. Clin. Immunol. 2020, 210, 108292. [Google Scholar] [CrossRef]
- Kullberg, B.J.; Netea, M.G.; Vonk, A.G.; van der Meer, J.W. Modulation of Neutrophil Function in Host Defense against Disseminated Candida Albicans Infection in Mice. FEMS Immunol. Med. Microbiol. 1999, 26, 299–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez, C.E.; Lyman, C.A.; Lee, S.; Del Guercio, C.; Roilides, E.; Bacher, J.; Gehrt, A.; Feuerstein, E.; Tsokos, M.; Walsh, T.J. recombinant human macrophage colony-stimulating factor augments pulmonary host defences against aspergillus fumigatus. Cytokine 2001, 15, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Dongari-Bagtzoglou, A.; Kashleva, H. Granulocyte-Macrophage Colony-Stimulating Factor Responses of Oral Epithelial Cells to Candida Albicans. Oral Microbiol. Immunol. 2003, 18, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Gavino, C.; Cotter, A.; Lichtenstein, D.; Lejtenyi, D.; Fortin, C.; Legault, C.; Alirezaie, N.; Majewski, J.; Sheppard, D.C.; Behr, M.A.; et al. CARD9 Deficiency and Spontaneous Central Nervous System Candidiasis: Complete Clinical Remission With GM-CSF Therapy. Clin. Infect. Dis. 2014, 59, 81–84. [Google Scholar] [CrossRef] [Green Version]
- Celmeli, F.; Oztoprak, N.; Turkkahraman, D.; Seyman, D.; Mutlu, E.; Frede, N.; Köksoy, S.; Grimbacher, B. Successful Granulocyte Colony-Stimulating Factor Treatment of Relapsing Candida Albicans Meningoencephalitis Caused by CARD9 Deficiency. Pediatric Infect. Dis. J. 2016, 35, 428–431. [Google Scholar] [CrossRef]
- Du, B.; Shen, N.; Hu, J.; Tao, Y.; Mo, X.; Cao, Q. Complete Clinical Remission of Invasive Candida Infection with CARD9 Deficiency after G-CSF Treatment. Comp. Immunol. Microbiol. Infect. Dis. 2020, 70, 101417. [Google Scholar] [CrossRef]
- Sam, Q.H.; Yew, W.S.; Seneviratne, C.J.; Chang, M.W.; Chai, L.Y.A. Immunomodulation as Therapy for Fungal Infection: Are We Closer? Front. Microbiol. 2018, 9, 1612. [Google Scholar] [CrossRef]
- Uchida, K.; Beck, D.C.; Yamamoto, T.; Berclaz, P.-Y.; Abe, S.; Staudt, M.K.; Carey, B.C.; Filippi, M.-D.; Wert, S.E.; Denson, L.A.; et al. GM-CSF Autoantibodies and Neutrophil Dysfunction in Pulmonary Alveolar Proteinosis. N. Engl. J. Med. 2007, 356, 567–579. [Google Scholar] [CrossRef]
- Shiomi, A.; Usui, T. Pivotal Roles of GM-CSF in Autoimmunity and Inflammation. Mediat. Inflamm. 2015, 2015, e568543. [Google Scholar] [CrossRef] [Green Version]
- Cook, A.D.; Braine, E.L.; Hamilton, J.A. Stimulus-Dependent Requirement for Granulocyte-Macrophage Colony-Stimulating Factor in Inflammation. J. Immunol. 2004, 173, 4643–4651. [Google Scholar] [CrossRef] [Green Version]
- Fleetwood, A.J.; Lawrence, T.; Hamilton, J.A.; Cook, A.D. Granulocyte-Macrophage Colony-Stimulating Factor (CSF) and Macrophage CSF-Dependent Macrophage Phenotypes Display Differences in Cytokine Profiles and Transcription Factor Activities: Implications for CSF Blockade in Inflammation. J. Immunol. 2007, 178, 5245–5252. [Google Scholar] [CrossRef] [Green Version]
- Negoro, P.E.; Xu, S.; Dagher, Z.; Hopke, A.; Reedy, J.L.; Feldman, M.B.; Khan, N.S.; Viens, A.L.; Alexander, N.J.; Atallah, N.J.; et al. Spleen Tyrosine Kinase Is a Critical Regulator of Neutrophil Responses to Candida Species. mBio 2020, 11, e02043-19. [Google Scholar] [CrossRef]
- Zajta, E.; Csonka, K.; Tóth, A.; Tiszlavicz, L.; Németh, T.; Orosz, A.; Novák, Á.; Csikós, M.; Vágvölgyi, C.; Mócsai, A.; et al. Signaling through Syk or CARD9 Mediates Species-Specific Anti-Candida Protection in Bone Marrow Chimeric Mice. mBio 2021, 12, e01608-21. [Google Scholar] [CrossRef]
- Miller, Y.I.; Choi, S.-H.; Wiesner, P.; Bae, Y.S. The SYK Side of TLR4: Signalling Mechanisms in Response to LPS and Minimally Oxidized LDL. Br. J. Pharmacol. 2012, 167, 990. [Google Scholar] [CrossRef] [Green Version]
- Markham, A. Fostamatinib: First Global Approval. Drugs 2018, 78, 959–963. [Google Scholar] [CrossRef]
- Basso, V.; Garcia, A.; Tran, D.Q.; Schaal, J.B.; Tran, P.; Ngole, D.; Aqeel, Y.; Tongaonkar, P.; Ouellette, A.J.; Selsted, M.E. Fungicidal Potency and Mechanisms of θ-Defensins against Multidrug-Resistant Candida Species. Antimicrob. Agents Chemother. 2018, 62, e00111-18. [Google Scholar] [CrossRef] [Green Version]
- Basso, V.; Tran, D.Q.; Schaal, J.B.; Tran, P.; Eriguchi, Y.; Ngole, D.; Cabebe, A.E.; Park, A.Y.; Beringer, P.M.; Ouellette, A.J.; et al. Rhesus Theta Defensin 1 Promotes Long Term Survival in Systemic Candidiasis by Host Directed Mechanisms. Sci. Rep. 2019, 9, 16905. [Google Scholar] [CrossRef]
- Tongaonkar, P.; Trinh, K.K.; Schaal, J.B.; Tran, D.; Gulko, P.S.; Ouellette, A.J.; Selsted, M.E. Rhesus Macaque θ-Defensin RTD-1 Inhibits Proinflammatory Cytokine Secretion and Gene Expression by Inhibiting the Activation of NF-ΚB and MAPK Pathways. J. Leukoc. Biol. 2015, 98, 1061–1070. [Google Scholar] [CrossRef]
- Beringer, P.M.; Bensman, T.J.; Ho, H.; Agnello, M.; Denovel, N.; Nguyen, A.; Wong-Beringer, A.; She, R.; Tran, D.Q.; Moskowitz, S.M.; et al. Rhesus θ-Defensin-1 (RTD-1) Exhibits in Vitro and in Vivo Activity against Cystic Fibrosis Strains of Pseudomonas Aeruginosa. J. Antimicrob. Chemother. 2016, 71, 181–188. [Google Scholar] [CrossRef] [Green Version]
- Bensman, T.J.; Jayne, J.G.; Sun, M.; Kimura, E.; Meinert, J.; Wang, J.C.; Schaal, J.B.; Tran, D.; Rao, A.P.; Akbari, O.; et al. Efficacy of Rhesus Theta-Defensin-1 in Experimental Models of Pseudomonas Aeruginosa Lung Infection and Inflammation. Antimicrob. Agents Chemother. 2017, 61, e00154-17. [Google Scholar] [CrossRef] [Green Version]
- Jayne, J.G.; Bensman, T.J.; Schaal, J.B.; Park, A.Y.J.; Kimura, E.; Tran, D.; Selsted, M.E.; Beringer, P.M. Rhesus θ-Defensin-1 Attenuates Endotoxin-Induced Acute Lung Injury by Inhibiting Proinflammatory Cytokines and Neutrophil Recruitment. Am. J. Respir. Cell Mol. Biol. 2018, 58, 310–319. [Google Scholar] [CrossRef]
- Group, T.I.C.G.D.C.S. A Controlled Trial of Interferon Gamma to Prevent Infection in Chronic Granulomatous Disease. N. Engl. J. Med. 1991, 324, 509–516. [Google Scholar] [CrossRef]
- Monk, E.J.M.; Harris, C.; Döffinger, R.; Hayes, G.; Denning, D.W.; Kosmidis, C. Interferon Gamma Replacement as Salvage Therapy in Chronic Pulmonary Aspergillosis: Effects on Frequency of Acute Exacerbation and All-Cause Hospital Admission. Thorax 2020, 75, 513–516. [Google Scholar] [CrossRef] [Green Version]
- Joint Formulary Committee. British National Formulary (online) London: BMJ Group and Pharmaceutical Press. Available online: http://www.medicinescomplete.com (accessed on 28 June 2022).
- Stevenhagen, A.; van Furth, R. Interferon-Gamma Activates the Oxidative Killing of Candida Albicans by Human Granulocytes. Clin. Exp. Immunol. 1993, 91, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Hertzog, P.J.; Ravasi, T.; Hume, D.A. Interferon-γ: An Overview of Signals, Mechanisms and Functions. J. Leukoc. Biol. 2004, 75, 163–189. [Google Scholar] [CrossRef]
- Colombo, S.A.P.; Hashad, R.; Denning, D.W.; Kumararatne, D.S.; Ceron-Gutierrez, L.; Barcenas-Morales, G.; MacDonald, A.S.; Harris, C.; Doffinger, R.; Kosmidis, C. Defective Interferon-Gamma Production Is Common in Chronic Pulmonary Aspergillosis. J. Infect. Dis. 2021, 225, jiab583. [Google Scholar] [CrossRef] [PubMed]
- Elks, P.M.; Brizee, S.; van der Vaart, M.; Walmsley, S.R.; van Eeden, F.J.; Renshaw, S.A.; Meijer, A.H. Hypoxia Inducible Factor Signaling Modulates Susceptibility to Mycobacterial Infection via a Nitric Oxide Dependent Mechanism. PLoS Pathog. 2013, 9, e1003789. [Google Scholar] [CrossRef] [PubMed]
- Ogryzko, N.V.; Lewis, A.; Wilson, H.L.; Meijer, A.H.; Renshaw, S.A.; Elks, P.M. Hif-1α–Induced Expression of Il-1β Protects against Mycobacterial Infection in Zebrafish. J. Immunol. 2019, 202, 494–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Wang, Y.; Li, Y.; Yu, Q.; Jin, X.; Wang, X.; Jia, A.; Hu, Y.; Han, L.; Wang, J.; et al. HIF1α-Dependent Glycolysis Promotes Macrophage Functional Activities in Protecting against Bacterial and Fungal Infection. Sci. Rep. 2018, 8, 3603. [Google Scholar] [CrossRef]
- Netea, M.G.; Quintin, J.; Van Der Meer, J.W. Trained immunity: A memory for innate host defense. Cell Host Microbe 2011, 9, 355–361. [Google Scholar] [CrossRef] [Green Version]
- Netea, M.G.; Domínguez-Andrés, J.; Barreiro, L.B.; Chavakis, T.; Divangahi, M.; Fuchs, E.; Joosten, L.A.; van der Meer, J.W.; Mhlanga, M.M.; Mulder, W.J.; et al. Defining trained immunity and its role in health and disease. Nat. Rev. Immunol. 2020, 20, 375–388. [Google Scholar] [CrossRef] [Green Version]
- Quintin, J.; Saeed, S.; Martens, J.H.; Giamarellos-Bourboulis, E.J.; Ifrim, D.C.; Logie, C.; Jacobs, L.; Jansen, T.; Kullberg, B.J.; Wijmenga, C.; et al. Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe 2012, 12, 223–232. [Google Scholar] [CrossRef] [Green Version]
- Lilly, E.A.; Yano, J.; Esher, S.K.; Hardie, E.; Fidel, P.L., Jr.; Noverr, M.C. Spectrum of trained innate immunity induced by low-virulence Candida species against lethal polymicrobial intra-abdominal infection. Infect. Immun. 2019, 87, e00348-19. [Google Scholar] [CrossRef] [Green Version]
- Lilly, E.A.; Bender, B.E.; Esher Righi, S.; Fidel, P.L., Jr.; Noverr, M.C. Trained Innate Immunity Induced by Vaccination with Low-Virulence Candida Species Mediates Protection against Several Forms of Fungal Sepsis via Ly6G+ Gr-1+ Leukocytes. mBio 2021, 12, e0254821. [Google Scholar] [CrossRef]
- Quintin, J. Fungal mediated innate immune memory, what have we learned? Semin. Cell Dev. Biol. 2019, 89, 71–77. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Pattern Recognition Receptor | Localisation | Cell Expression | Adaptor Proteins | Effectors | Pathogen-/Damage-Associated Molecular Patterns Recognised | Fungal Species | References |
|---|---|---|---|---|---|---|---|
| TLR2 | Plasma membrane | Monocytes, macrophages, dendritic cells, mast cells, neutrophils | MyD88, Mal | NF-κB, TNF, TGFβ, IL-10, IL-12, IFNγ | Phospholipomannan, β-glucans | C. albicans, A. fumigatus, P. brasiliensis | [28,29,30,31] |
| TLR4 | Plasma membrane, endosome membrane | Monocytes, macrophages, dendritic cells, mast cells, neutrophils, B cells, intestinal epithelium | MyD88, Mal, TRIF, TRAM | NF-κB, TNF, IL-8, Type I IFN | O-linked mannosyl, Mannan, Glucuronoxylomannan | C. albicans, A. fumigatus | [28,29,30,31] |
| TLR7 | Endosome membrane | Monocytes, macrophages, dendritic cells, B cells | MyD88 | IFN-β, Type I IFN | ssRNA | C. albicans | [28,30,31,32] |
| TLR9 | Endosome membrane | Monocytes, macrophages, dendritic cells, B cells | MyD88 | NF-κB, IL-12, TNFα | Unmethylated DNA with CpG motif | Candida spp., C. neoformans, A. fumigatus, P. brasiliensis, M. furfur | [28,30,31,33,34] |
| Dectin-1 | Plasma membrane | Monocytes, macrophages, dendritic cells, neutrophils, mast cells, some T cells | hemITAM | IL-2, IL-6, IL-10, IL-23 | β-1,3-glucans | Candida spp., C. neoformans, A. fumigatus, H. capsulatum, S. cerevisiae, P. brasiliensis | [28,30,31] |
| Dectin-2 | Plasma membrane | Monocytes, macrophages, dendritic cells, neutrophils | ITAM-FcRγ | TNFα | Mannose | C. albicans, C. glabrata, C. neoformans, A. fumigatus, H. capsulatum | [28,30,31] |
| Mincle | Plasma membrane | Monocytes, macrophages, dendritic cells, neutrophils, mast cells, some B cells | ITAM-FcRγ | NF-κB, IL-1, IL-6, IL-10 IL-12, IL-23 | α-mannose, glyceroglycolipid, mannosyl fatty acids, MSG/gpA | A. fumigatus, C. albicans, P. carinii, Malassezia spp. | [30,31,35] |
| DC-SIGN | Plasma membrane | Macrophages, dendritic cells, activated B cells | LSP1 | IL-10 | Mannose, N-linked mannans, galactomannans | C. albicans, C. neoformans, A. fumigatus, S. cerevisiae | [28,30,31] |
| Mannose Receptor | Plasma membrane | Macrophages, Kupffer cells, endothelial cells | Associated with FcRγ and GBR2, exact mechanism unknown | TNF, IL-1β | Mannose, α-glucans, chitin | C. albicans, C. neoformans, A. fumigatus, H. capsulatum, S. cerevisiae, P. brasiliensis | [28,30,36,37] |
| MDA5 | Cytoplasm | Monocytes, macrophages, dendritic cells, B cells, epithelial cells, endothelial cells, fibroblasts | CARDs, MAVs | NF-κB, Type I IFN, Type III IFN, TNFα, IL-12, | dsRNA | C. albicans, A. fumigatus | [26,30] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burgess, T.B.; Condliffe, A.M.; Elks, P.M. A Fun-Guide to Innate Immune Responses to Fungal Infections. J. Fungi 2022, 8, 805. https://doi.org/10.3390/jof8080805
Burgess TB, Condliffe AM, Elks PM. A Fun-Guide to Innate Immune Responses to Fungal Infections. Journal of Fungi. 2022; 8(8):805. https://doi.org/10.3390/jof8080805
Chicago/Turabian StyleBurgess, Thomas B., Alison M. Condliffe, and Philip M. Elks. 2022. "A Fun-Guide to Innate Immune Responses to Fungal Infections" Journal of Fungi 8, no. 8: 805. https://doi.org/10.3390/jof8080805
APA StyleBurgess, T. B., Condliffe, A. M., & Elks, P. M. (2022). A Fun-Guide to Innate Immune Responses to Fungal Infections. Journal of Fungi, 8(8), 805. https://doi.org/10.3390/jof8080805