Abstract
Some novel fluorinated quinazolines (5a–j) were designed and synthesized to be evaluated for their anticonvulsant activity and their neurotoxicity. Structures of all newly synthesized compounds were confirmed by their infrared (IR), mass spectrometry (MS) spectra, 1H nuclear magnetic resonance (NMR), 13C-NMR, and elemental analysis (CHN). The anticonvulsant activity was evaluated by a subcutaneous pentylenetetrazole (scPTZ) test and maximal electroshock (MES)-induced seizure test, while neurotoxicity was evaluated by a rotorod test. The molecular docking was performed for all newly-synthesized compounds to assess their binding affinities to the GABA-A receptor in order to rationalize their anticonvulsant activities in a qualitative way. The data obtained from the molecular modeling was correlated with that obtained from the biological screening. These data showed considerable anticonvulsant activity for all newly-synthesized compounds. Compounds 5b, 5c, and 5d showed the highest binding affinities toward the GABA-A receptor, along with the highest anticonvulsant activities in experimental mice. These compounds also showed low neurotoxicity and low toxicity in the median lethal dose test compared to the reference drugs. A GABA enzymatic assay was performed for these highly active compounds to confirm the obtained results and explain the possible mechanism for anticonvulsant action. The most active compounds might be used as leads for future modification and optimization.
1. Introduction
Epilepsy is a neurological disorder characterized by recurrent seizures [1,2,3,4,5]. It represents a major health problem and inflicts millions of people worldwide [6,7,8,9,10]. Approximately, one third of epilepsy patients have a multidrug resistance (MDR) syndrome and refractory epilepsy (RE). The processes of (MDR) and (RE) result from enhanced expression and up-regulation of p-glycoprotein (P-GP) into the brain. This leads to defective access of the anticonvulsant drugs to their target in the central nervous system (CNS) [8].
Despite the non-stop discovery of new antiepileptic drugs (AEDs), the treatment process is still inadequate and unsatisfactory due to several side effects, such as neurotoxicity, depression, drowsiness, ataxia, gastrointestinal disturbances, megaloblastic anemia, hirsutism, and other CNS disorders [5].
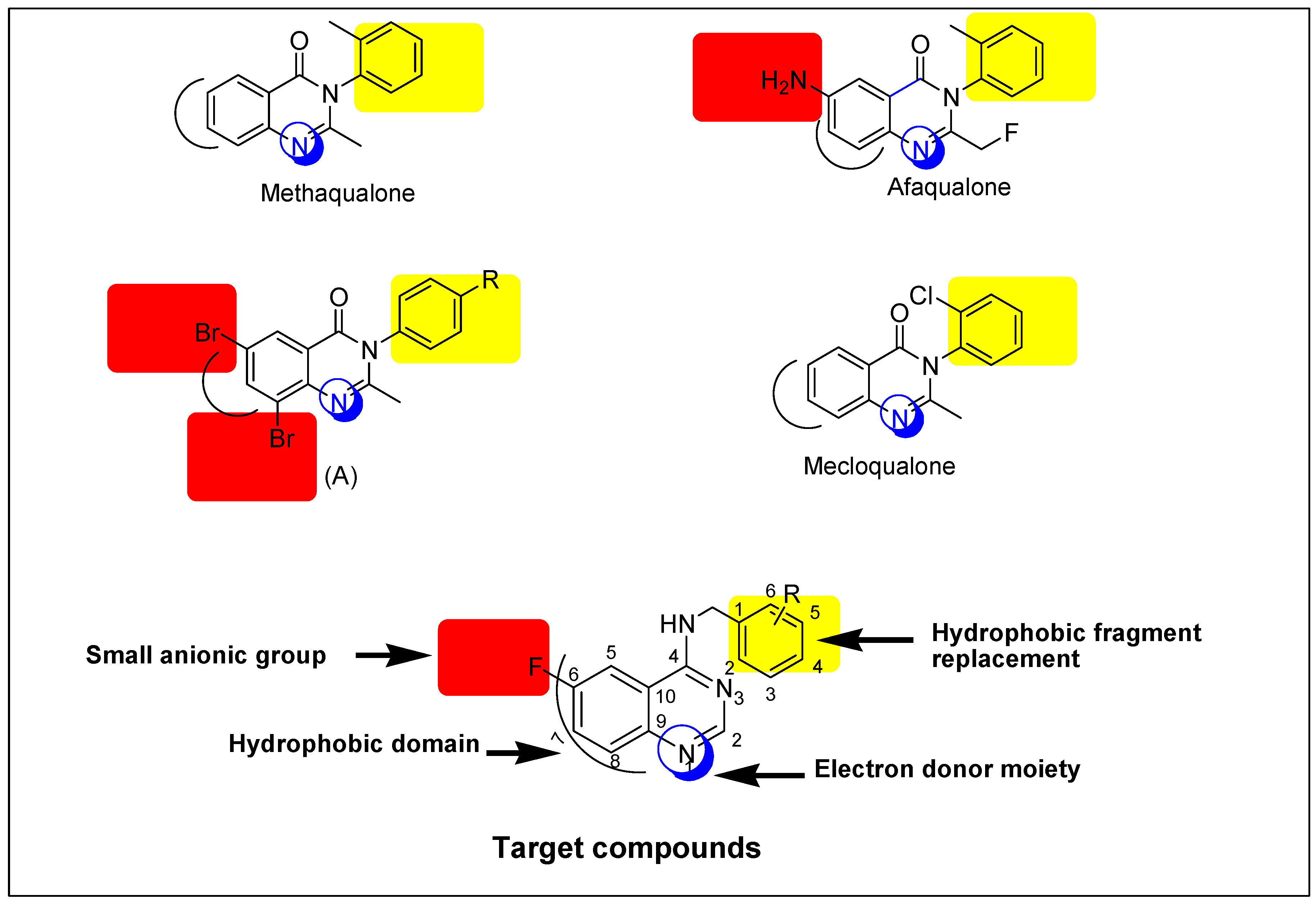
The quinazoline ring represents a very important advantaged substructure in organic synthesis for preparing different biologically active molecules [4,5,6]. Typically, the most important pharmacological effects that have been reported for quinazoline derivatives are sedative-hypnotic and anticonvulsant activities [9]. Many quinazoline derivatives were reported as anticonvulsants but none of these derivatives are clinically approved because these derivatives had high neurotoxicity values (TD50) and low protective index values (PI) [5]. Therefore, we need to search for safer and more effective anticonvulsants. Based on that, some novel quinazoline derivatives were designed and prepared to be biologically tested as anticonvulsants. The general structure of target compounds is shown in Figure 1.
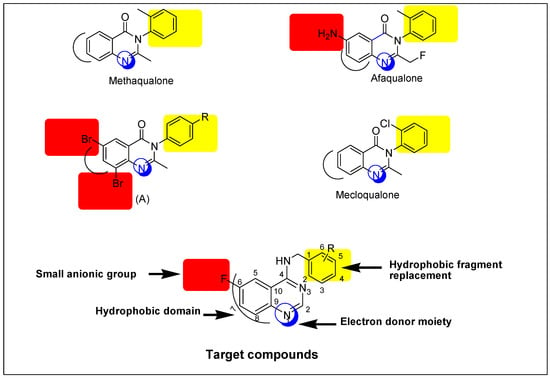
Figure 1.
Structural similarities and pharmacophoric features of reported and designed quinazolines (5a–j) as anticonvulsant agents.
A literature study explains that the existence of quinazoline nucleus in the molecule is a necessary requirement for the anticonvulsant activity. This nucleus can be substituted on the heteroatom or the aromatic ring, as shown in Figure 1. Moreover, the quinazoline moiety with a suitable substituent, mostly amine or a substituted amine at the 4-position, and either a halogen or electron-rich substituent at the 6-position or 8-position, is known to improve antiepileptic activity [5,6]. Based on the previous rationale, it was believed to be valuable to study the effects of two pharmacophoric groups, like quinazoline and aromatic amine, in one molecule for anticonvulsant activity. The quinazoline nucleus is attached to an aromatic unit via a (CH2) linker. This design formed new quinazoline derivatives that possess essential features of the lead structure methaqualone. Selection of the aromatic amines was following the Hansch manual method of drug design which was proposed by Topliss [11]. It is a technique in which the substituents are nominated, ordered, and tested based on the effectiveness. A fluorine atom was selected as a substituent on the quinazoline moiety owing to its safety, small size, low steric interference, and ready availability [12,13]. Additionally, the presence of fluorine at the 6-position could enhance the pharmacokinetic and pharmacodynamic properties of the newly-synthesized compounds [6,13]. The title compounds were synthesized as an attempt to find new efficient and safe anticonvulsants. Similarities between the reported anticonvulsants methaqualone, afaqualone, mecloqualone, compound (A) and our newly-designed compounds are shown in Figure 1.
2. Results and Discussion
2.1. Chemistry
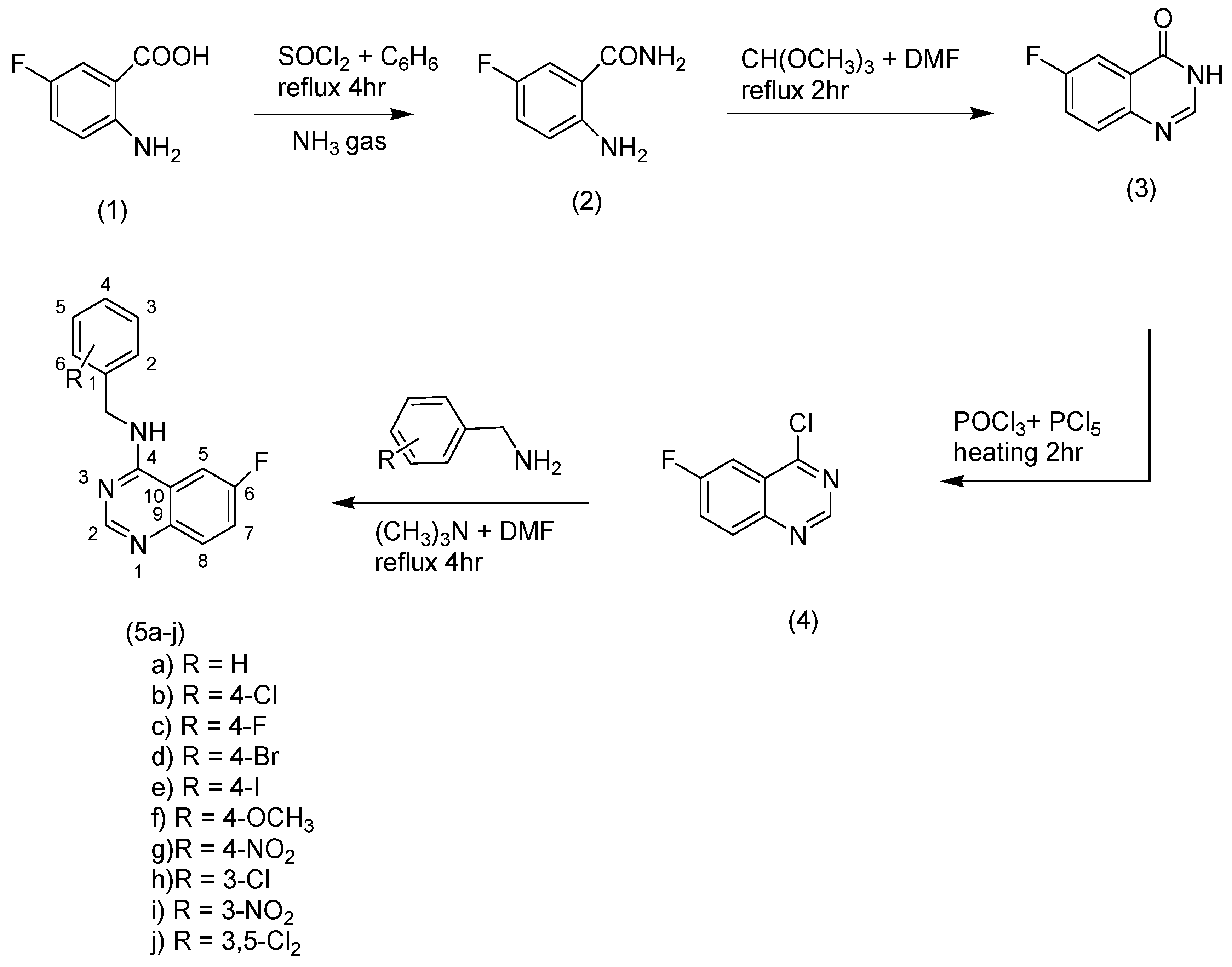
Substituted-6-fluoro-quinazoline-4-amine (5a–j) derivatives were synthesized according to the methods outlined in Scheme 1. Synthesis of 2-amino-5-fluorobenzamide (2) was accomplished by refluxing of thionylchloride and 2-amino-5-fluorobenzoic acid in benzene, then the mixture was exposed to dry ammonia gas. 6-fluroquinazolin-4(3H)-one (3) was prepared by reaction of the amide compound (2) with trimethoxymethane in dimethylformamide (DMF). Chlorination of compound (3) was performed by heating (3) with a mixture of phosphorous oxychloride and phosphorous pentachloride. A nucleophilic displacement reaction for the chloroquinazoline (4) was accomplished through treating of compound (4) with different substituted aromatic amines in the presence of trimethylamine to obtain the title compounds (5a–j). Infrared, mass spectrometry, 1H nuclear magnetic resonance (NMR), 13C-NMR, and elemental analyses were performed to confirm all structures. The title compounds contained different substituents with different electronic environments, as shown in Scheme 1. These substituents are joined to the fixed moiety 6-fluroquinazoline. They included different hydrophilic and hydrophobic groups, either electron-deficient or electron-rich groups, to study and compare their anticonvulsant activity.
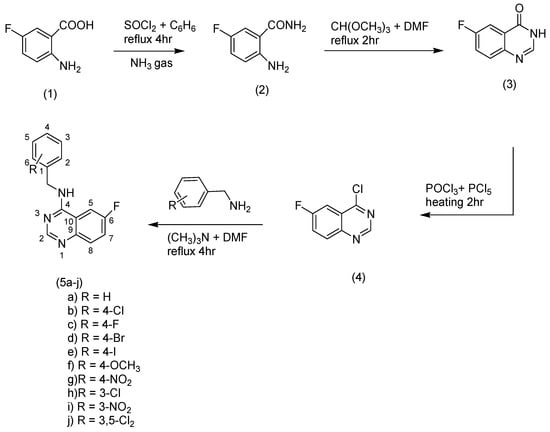
Scheme 1.
Synthesis of the target compounds (5a–j).
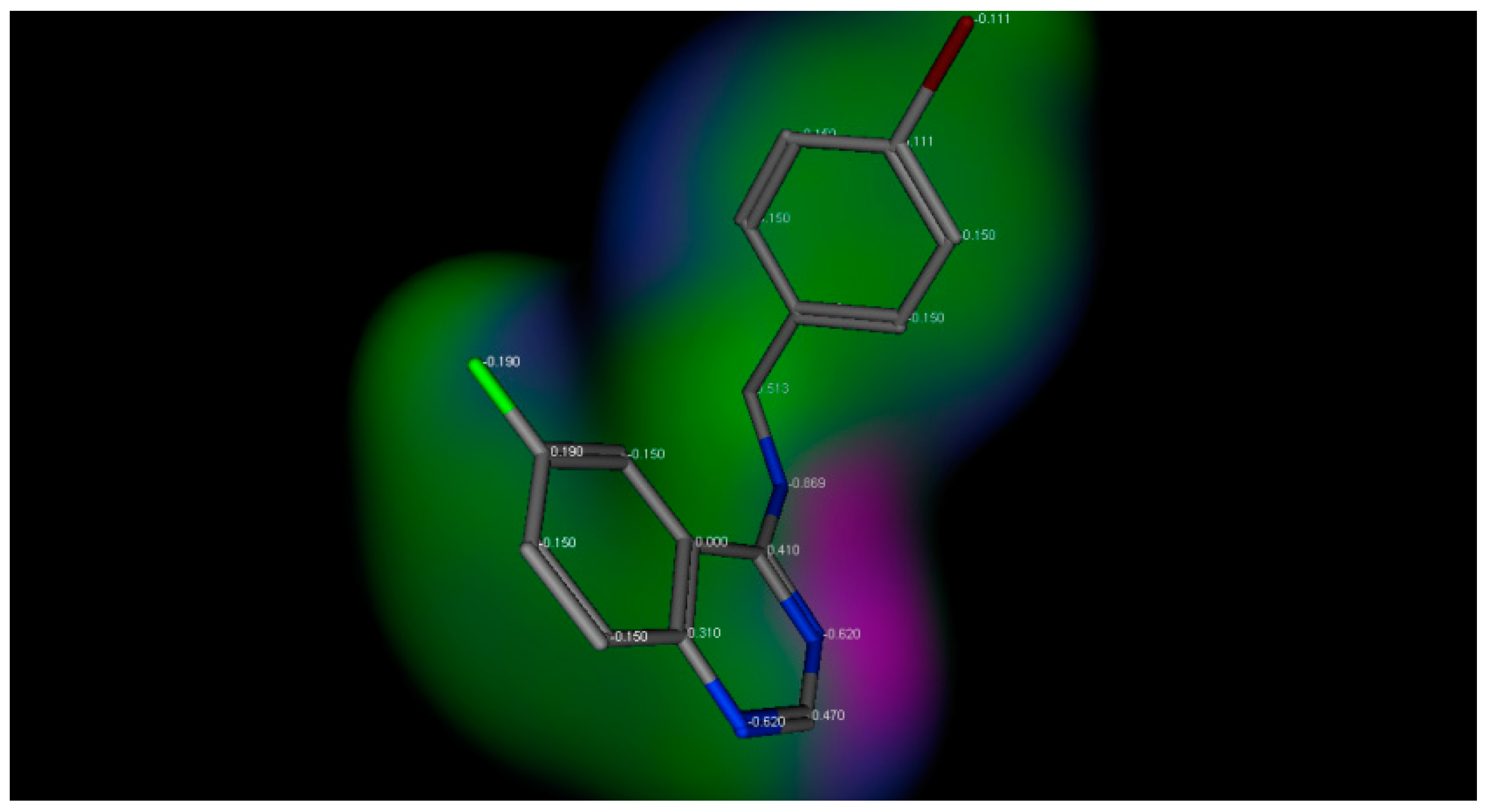
The title compounds contain different substituents with different electronic environments, as shown in Figure 2. These substituents are joined to the fixed moiety 6-fluroquinazoline. They included different hydrophilic and hydrophobic groups, either electron-deficient or electron-rich groups, to study and compare their anticonvulsant activity. Figure 2 shows the electronic environment of the title compounds represented by compound 3d.
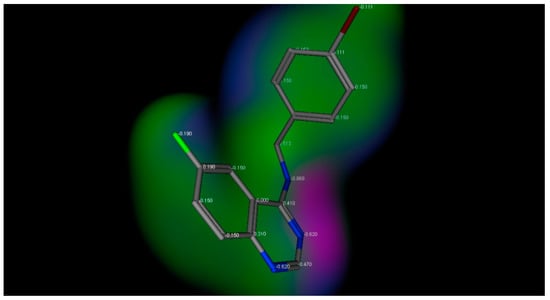
Figure 2.
Electronic environment of the title compounds exemplified by compound 3d. Green color indicates hydrophobic area (low electron density), pink color indicates the hydrogen bond area (high electron density), and the blue color indicates the mild polar area (mild electron density).
2.2. Anticonvulsant Screening
The anticonvulsant screening of the title compounds was performed according to standard procedures [14,15,16]. The initial screening was performed at 0.5 mmol/kg of all synthesized compounds (5a–j) by chemical induction of the convulsions using pentylenetetrazole (PTZ) and electrical induction of convulsions using maximal electroshock (MES) models of seizures [17,18]. The scPTZ model has been reported to be a good predictor of those anticonvulsants to protect against generalized spike-wave epilepsies which are known as absence seizures [19,20]. These anticonvulsants are functionally related to methaqualone. The MES model is used to predict the ability of anticonvulsants to protect against generalized tonic-clonic seizures which are functionally similar to phenytoin [5,6]. Thus, scPTZ and MES screenings became the best broadly applicable seizure models for preliminary detection of antiepileptic agents.
The primary anticonvulsant screening for the newly synthesized compounds displayed that all of them were active in PTZ test at 0.5 mmol/kg. Compounds 5b, 5c, 5d, and 5e showed 100% protection, compounds 5a and 5f showed 83% protection, while compounds 5g, 5h, and 5j showed 66% protection. For the MES test, compound 5f showed 66% protection, compounds 5a and 5g showed 50% protection, while compounds 5h, 5i, and 5j showed 33% protection. These results are elucidated in Table 1.

Table 1.
Preliminary anticonvulsant screening of the newly-synthesized compounds in comparing with standard compounds methaqualone (0.5 mmol/kg) and valproate (1.8 mmol/kg).
The most active compounds 5b, 5c, and 5d, were exposed to further investigations in mice to determine and quantify their anticonvulsant activity. Further investigations included the study of the effective dose that caused protection for 50% of mice (ED50) and that produce minimal neurological toxicity in 50% of mice (TD50) to determine their neurotoxicity, as shown in Table 2.
Table 2.
ED50, TD50, LD50, therapeutic index (TI), and protective index (PI) for the highest active compounds 5b, 5c, and 5d compared to the reference drugs methaqualone and valproate.
Compounds 5b, 5c, and 5d had (ED50) values of 152, 165, and 140 mg/kg, respectively (see Table 2). The reference drugs methaqualone and valproate produced (ED50) values of 200 and 300 mg/kg, respectively. The ED50 values of the test compounds were smaller than the reference drugs at molar doses. The protective index (PI = TD50/ED50) is an index representing the margin of safety between anticonvulsant doses and doses of anticonvulsant drugs producing acute adverse effects, like motor coordination impairment, ataxia, or any other neurotoxic symptoms [9,10]. The highest active compounds 5b, 5c, and 5d, had TD50 values of 270, 350, and 320 mg/kg. These values revealed that these compounds exerted low neurological disorders. From the results of ED50 and TD50, compounds 5b, 5c, and 5d had PI values of 1.78, 2.12, and 2.28, while methaqualone and valproate had PI values of 2 and 1.5. These results showed a high safety margin of the title compounds compared to the reference drugs. The therapeutic index (TI = LD50/ED50) is a comparison of the amount of a therapeutic agent that causes the therapeutic effect to the amount that causes death in mice [5,9]. Compounds 5b, 5c, and 5d had LD50 values of 580, 430, and 550 mg/kg, and TI values of 3.82, 2.6, and 3.93, while methaqualone and valproate had TI values of 2.5 and 1.67.
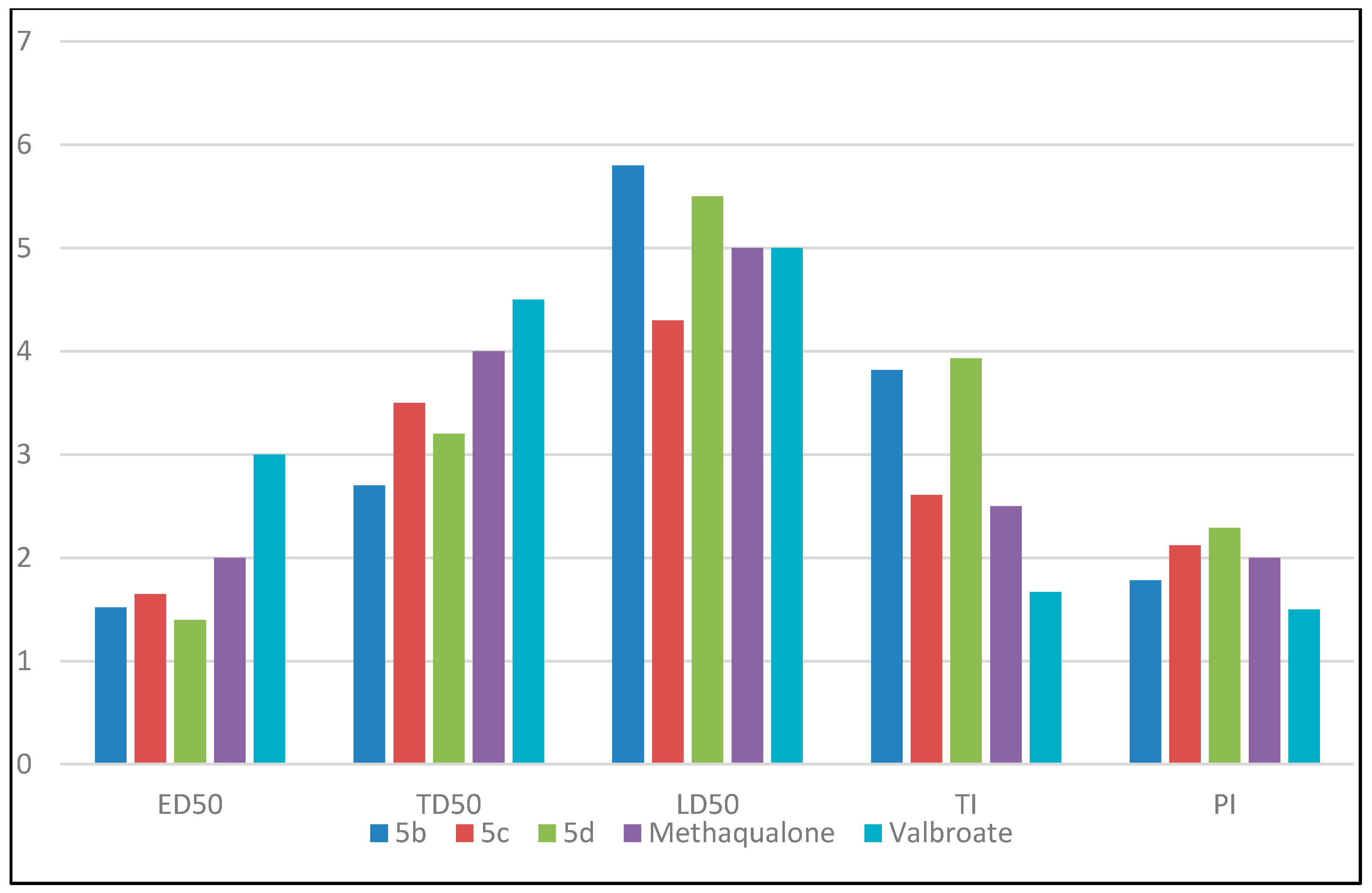
Figure 2 represents a comparison between the values of ED50, TD50, LD50, TI, and PI for the highest active compounds 5b, 5c, and 5d, against reference drugs methaqualone and valproate. The results of the anticonvulsant screening explained the broad spectrum activity of the title compounds against partial and generalized epilepsy. Structure activity relationship (SAR) studies explained that different substitutions on the aromatic ring of the aromatic amine exerted varied anticonvulsant activity. A significant variation in the anticonvulsant activity was obtained by changing the electronic nature and the position of the substituent group attached to the aromatic ring. Based on the data shown above, all para-substituted analogs had higher activity than meta- or di-substituted analogs. The electron withdrawing groups attached to the aromatic ring at the para position produced more anticonvulsant activity than that containing electron-donating groups or remained unsubstituted. For example, electronic withdrawing groups, like bromo substitution at the para position from the aromatic ring, improved the anticonvulsant activity, while other derivatives showed less activity. Compounds activity was arranged as the following: 4Br (5d) > 4Cl (5b) > 4F (5c) > 4I (5e) > 4OCH3 (5f) > H (5a) > 4NO2 (5g) > 3Cl (5h) > 3,5Cl2 (5j) > 3NO2 (5i). A comparison between ED50, TD50, LD50, TI, and PI for the highest active compounds 5b, 5c, and 5d, is shown in Figure 3.
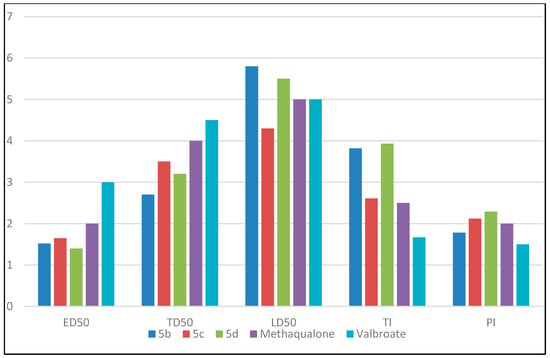
Figure 3.
Comparison between ED50, TD50, LD50, TI, and PI for the highest active compounds 5b, 5c and 5d. The values of ED50, TD50, are LD50 are divided by 100 for simplicity.
2.3. Docking Studies
Many quinazoline derivatives were reported as GABA-A receptor stimulants [21,22]. The GABA-A receptor is an ionotropic receptor and a ligand-gated ion channel. Its endogenous ligand is γ-aminobutyric acid (GABA), the major inhibitory neurotransmitter in the central nervous system. Upon activation, the GABA-A receptor selectively conducts Cl− through its pore, resulting in hyperpolarization of the neuron. This causes an inhibitory effect on neurotransmission by diminishing the chance of a successful action potential [21]. The active site of the GABA-A receptor is the binding site for GABA and several drugs, such as muscimol. The protein also contains a number of different allosteric binding sites which modulate the activity of the receptor indirectly. These allosteric sites are the targets of various other drugs, including benzodiazepines, barbiturates, and ethanol [23,24]. Methaqualone is a positive allosteric GABA-A receptor modulator. It binds to allosteric sites on the GABA-A receptor complex and affects it in a positive manner, causing increased efficiency of the main site and, therefore, an indirect increase in Cl− conductance [23,24].
Our target compounds were designed to replace the carbonyl group (hydrogen bond acceptor) at the 4-position of methaqualone by an amino group (hydrogen bond donor), which occupy the same position at the active site. On the other hand, this amino group was joined to a distal-substituted phenyl moiety through a CH2 linker which imparts a good hydrophobic domain which occupies the same position at the active site occupied by 2-chlorophenyl at the 3-position of methaqualone. The phenyl group of methaqualone was substituted at the 2-position while, in our designed compounds, it was substituted at different positions. All derivatives were substituted with fluorine atoms to increase hydrophobic interactions. These modifications were made in order to increase hydrogen bonding at the 4-position and also increase hydrophobic bonding to study their effects on SAR, hoping to obtain new compounds with higher GABA affinity and consequently higher anticonvulsant activity.
In order to qualify the docking results in terms of accuracy of the predicted binding conformations in comparison with the experimental procedure, the reported GABA-A receptor stimulant drug (Methaqualone) was used as a reference ligand. The docking study has been conducted to predict the binding mode and to rationalize the observed biological activity.
The obtained results indicated that all studied ligands have similar position and orientation inside the putative binding site of GABA-A receptor (PDB code 4COF) which reveals a large space bounded by a membrane-binding domain that serves as an entry channel for substrate to the active site (see Figure 4). In addition, the affinity of any small molecule can be considered as a unique tool in the field of drug design. There is a relationship between the affinity of organic molecules and the free energy of binding. This relationship can contribute in prediction and interpretation of the activity of the organic compounds toward the specific target protein [12,15]. The obtained results of the free energy of binding (∆G) explained that most of these compounds had good binding affinity toward the receptor and the computed values reflected the overall trend (see Table 3).
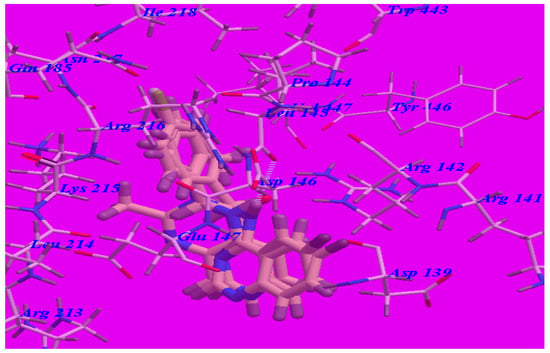
Figure 4.
Docking and superimposition of compounds 5d, 5b, 5c, and methaqualone in the vicinity of the GABA-A receptor (4COF).
Table 3.
The calculated ∆G (free energy of binding) and binding affinities for the ligands.
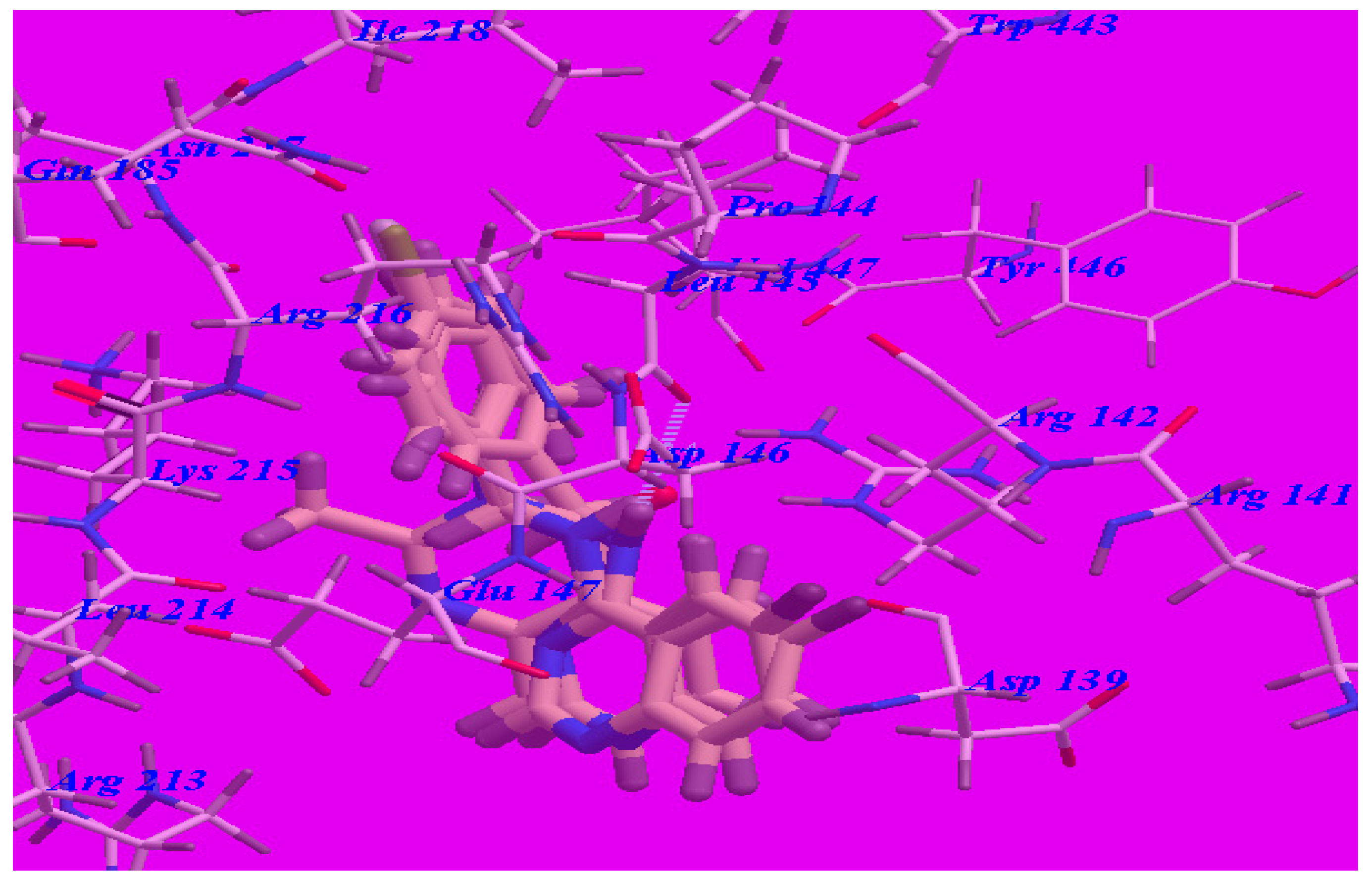
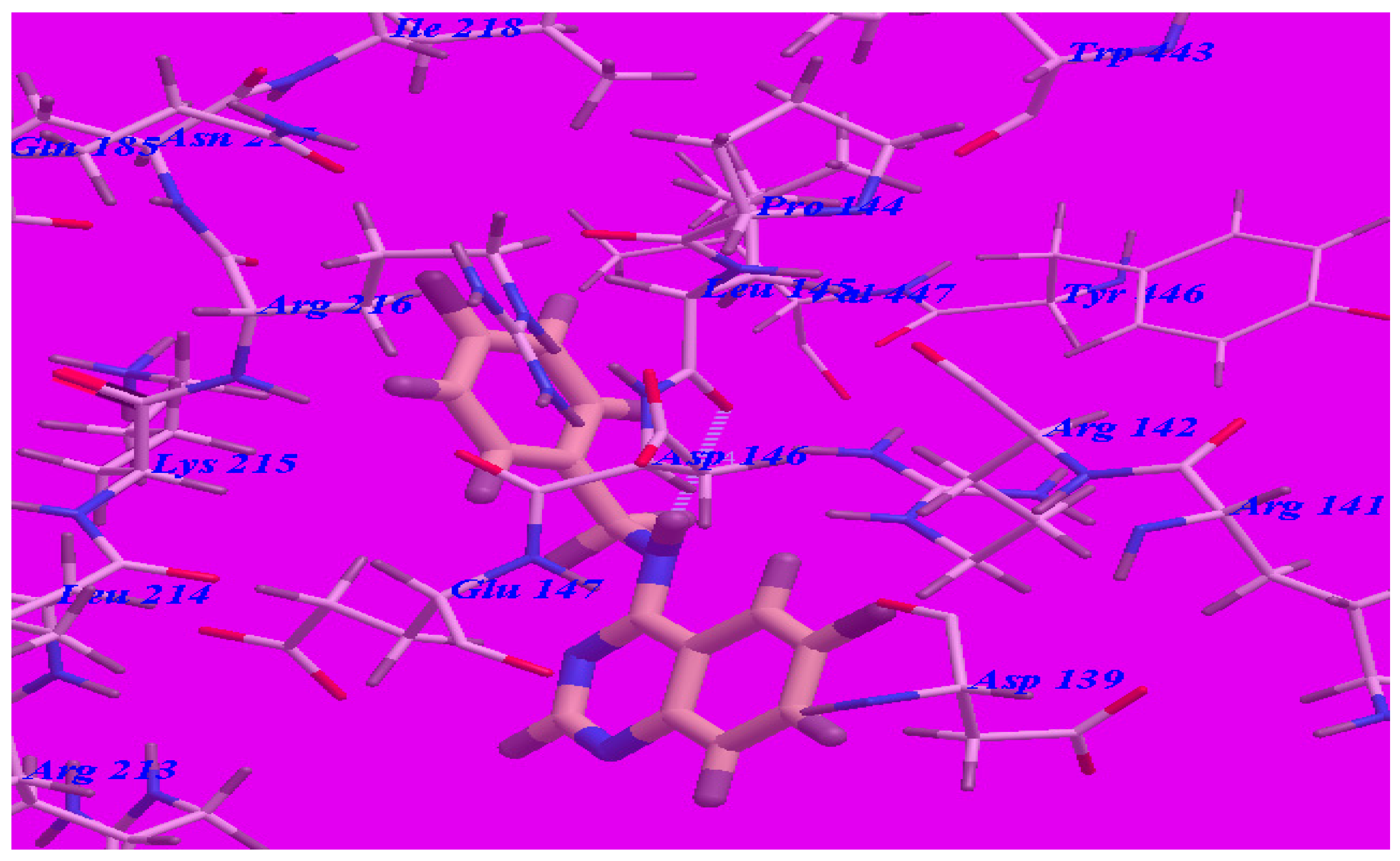
The proposed binding mode of methaqualone revealed an affinity value of –50.69 kcal/mol and the carbonyl group formed two H-bonds with Arg142 (–NH groups) with distances of 1.65 Å and 2.38 Å (see Figure 5). The quinazolinone moiety occupied the hydrophobic pocket formed by Asp139, Arg142, Asp146, Glu147, and Arg213. The phenyl ring at the 3-position was oriented in the hydrophobic cleft formed by Leu145, Asp146, Lys215, Arg216, and Ile218. The methyl group at 2-position was oriented in the hydrophobic cleft formed by Arg142, Leu145, and Val447.
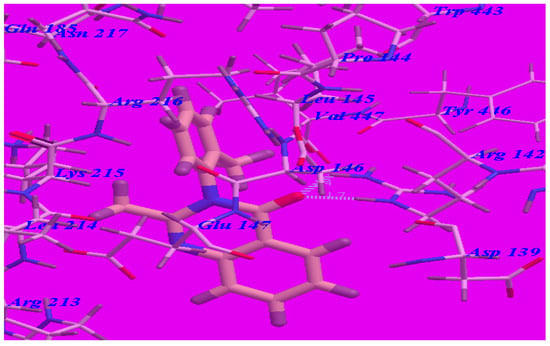
Figure 5.
Predicted binding mode for methaqualone with the GABA-A receptor (4COF). H-bonds are indicated by dotted lines.
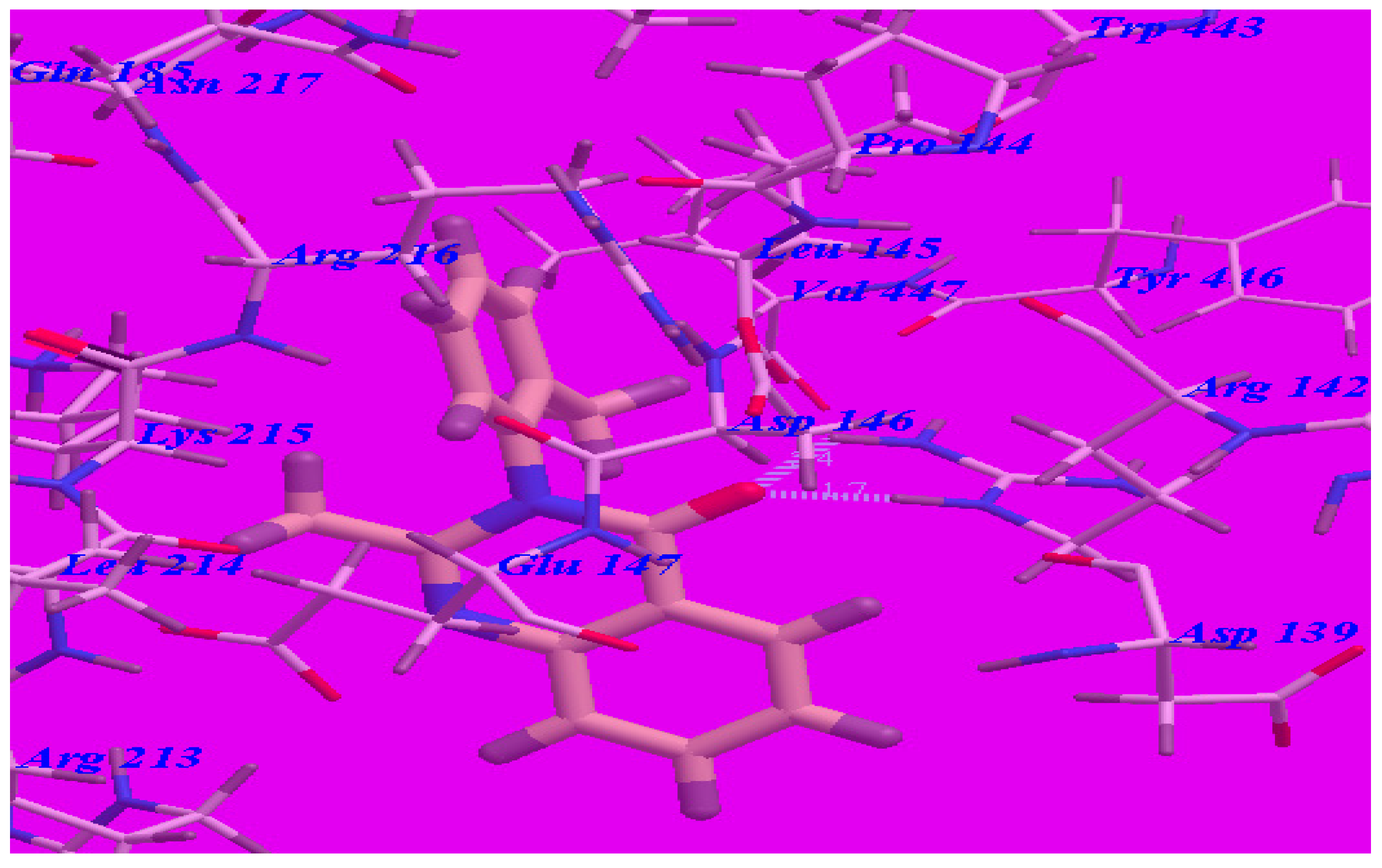
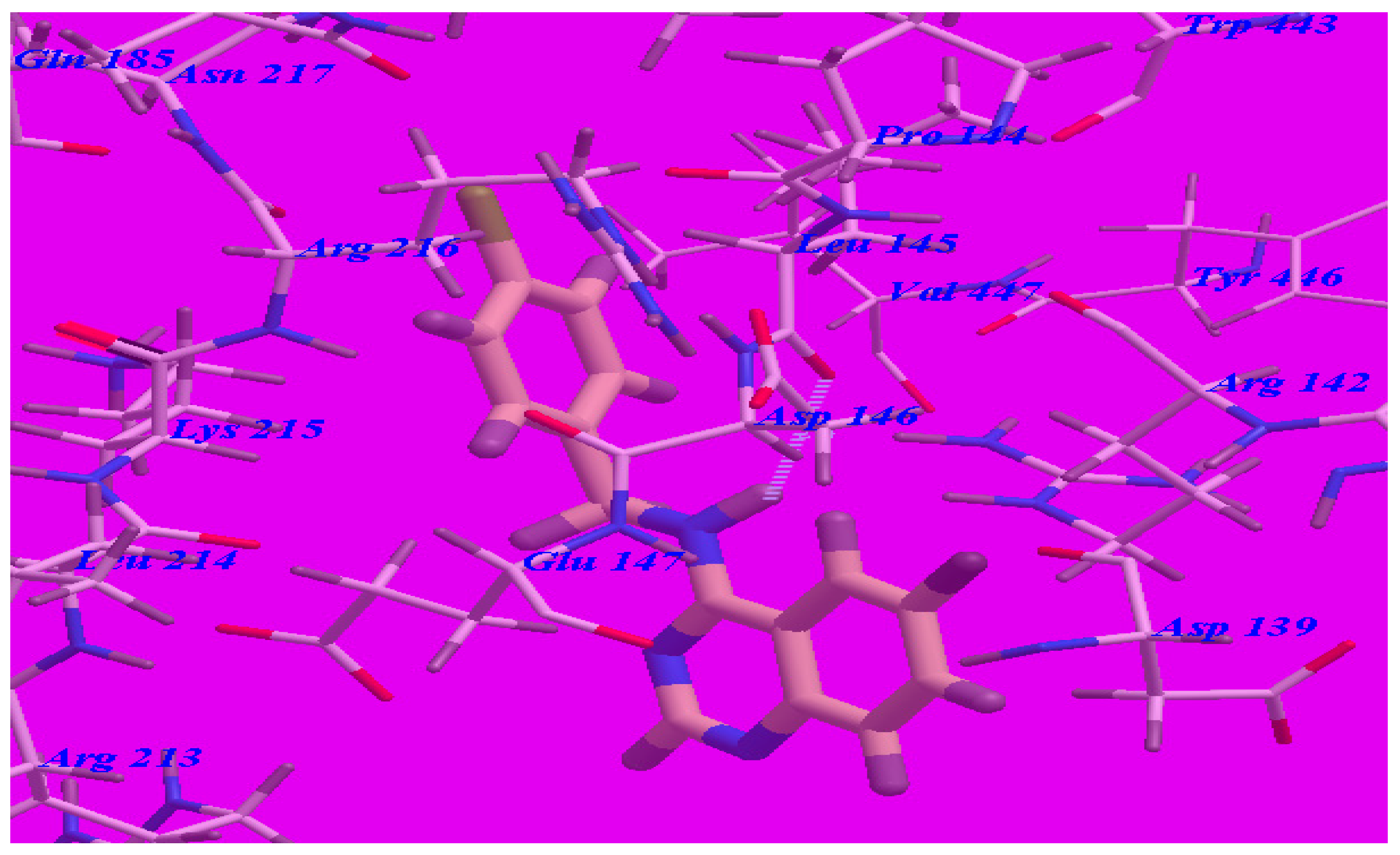
The proposed binding mode of compound 5d (affinity value of –69.68 kcal/mol and one H-bond) (see Figure 6) is virtually the same as that of methaqualone, where the NH group at the 4-position formed a hydrogen bond with Leu145 (–O atom) with a distance of 2.43 Å. The 6-fluoroquinazoline moiety occupied the hydrophobic pocket formed by Asp139, Arg141, Arg142, Asp146, Glu147, and Arg213. The distal phenyl ring was oriented in the hydrophobic cleft formed by Leu145, Asp146, Glu147, Lys215, Arg216, and Ile218. These interactions of compound 5d may explain the highest binding free energy and anticonvulsant activity.
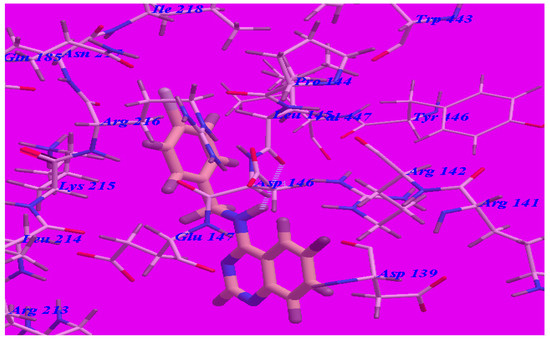
Figure 6.
Predicted binding mode for compound 5d with a GABA-A receptor.
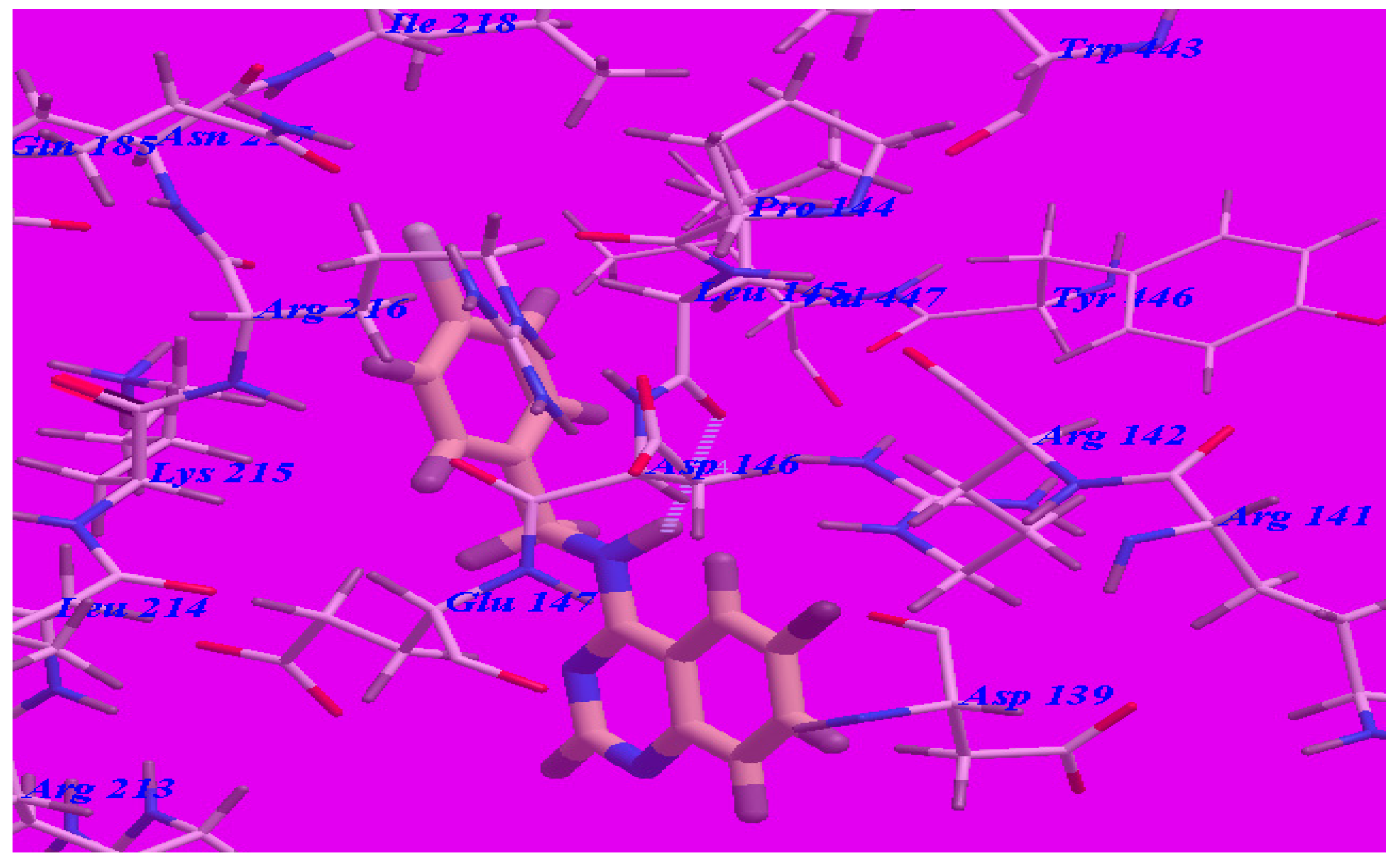
Furthermore, the proposed binding modes of compounds 5b (affinity value of −68.97 kcal/mol and one H-bond) (see Figure 7) and 5c (affinity value of −67.50 kcal/mol and one H-bond) (see Figure 8) are virtually the same as that of 5d where the three derivatives were superimposed. These interactions may explain the high binding free energy and anticonvulsant activity.
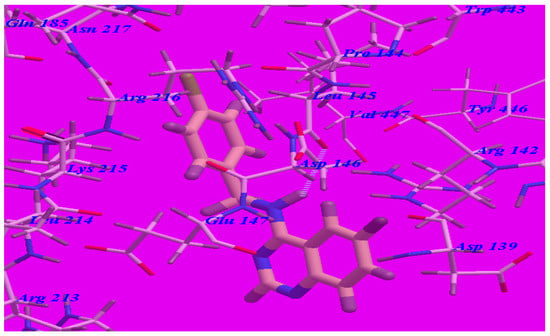
Figure 7.
Predicted binding mode for compound 5b with a GABA-A receptor.
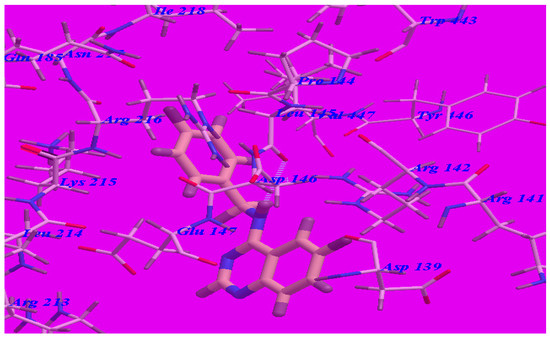
Figure 8.
Predicted binding mode for compounds 5c with a GABA-A receptor.
2.4. GABA Enzymatic Assay
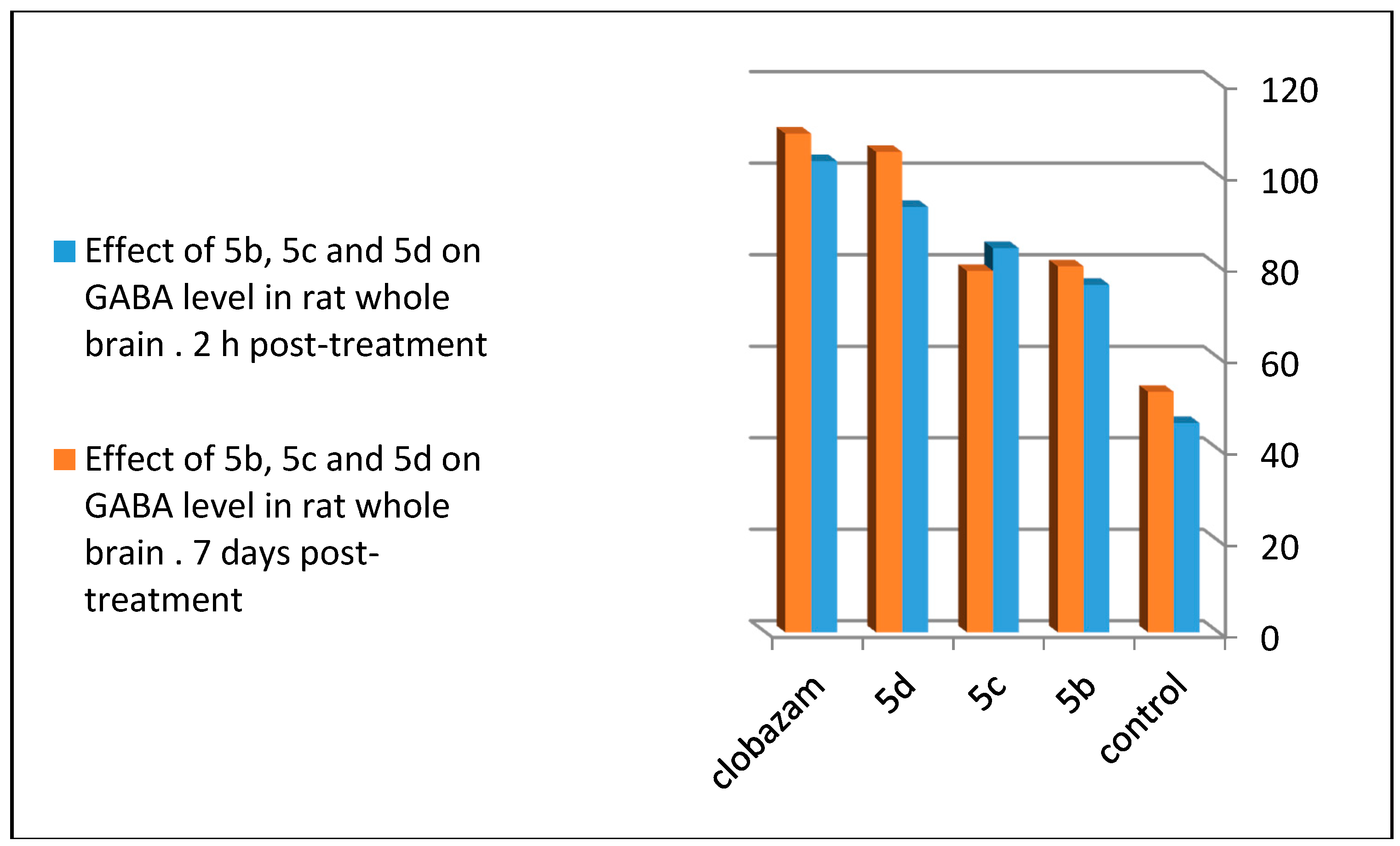
To follow the effect on the GABA receptor, 5b, 5c, and 5d were subjected to neurochemical examination for the determination of the GABA level in different areas of whole rat brain. The three tested compounds statistically increased the GABA concentration compared to the control with 5d (p < 0.001 level of significance), and 5b and 5c (p < 0.004, significant after 2 h of administration). Chronic oral administration for seven days showed a non-significant result for 5d compared to 5b and 5c (p < 0.01 significant). Tested compounds revealed lesser or closely equal concentrations of GABA to clobazam, which was used as the standard anticonvulsant drug [16] (Table 4, Figure 9). From these result, one can conclude that the para bromo-substituted quinazoline ring in 5d, para chloro-substituted quinazoline ring in 5b, and para fluoro-substituted quinazoline ring in 5c increased the effectiveness of the compound at the GABA receptor through increasing its level of concentration in the rat brain assay.
Table 4.
The values of GABA level in different areas of whole rat brain.
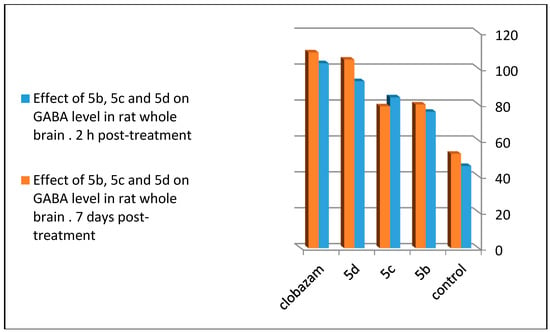
Figure 9.
Comparison between the effects of 5b, 5c, and 5d on the GABA level in rat whole brain 2 h post-treatment and seven days post-treatment.
3. Materials and Methods
3.1. General
Chemicals were obtained from Sigma-Aldrich (Aldrich chemical Co., Milwaukee, WI, USA). A Griffin melting point apparatus (Griffin, Valdosta, GA, USA) was used to measure melting points (m.p.). A Nicolet IR 300 (Thermo Fisher Scientific, Barrington, RI, USA) was used to measure the infrared spectra. 1H-NMR and 13C-NMR (Varian, Palo Alto, CA, USA) were performed in deuterated dimethyl sulfoxide (DMSO-d6) using a Varian 300 MHz instrument. Tetramethylsilane (TMS) was used as internal standard. MS spectra were measured using a JEOL-SX-102 electron impact ionization instrument (Joel, Peabody, MA, USA). Evaporation processes were performed under reduced pressure by using a Buchi rotary evaporator (Buchi, Postfach, Flawil, Switzerland). Elemental analyses (C, H, N) were accomplished using a Perkin-Elmer 240C analyzer (Perkin-Elmer, Norwalk, CT, USA). The synthesized compounds were within ±0.4% of the theoretical values. Purity of the newly-synthesized compounds was above 99.6%.
3.2. General Method of the Synthesis of 2-Amino-5-Fluorobenzamide (2)
A suspension of 2-amino-5-fluorobenzoic acid (1) (1.0 g, 6.3 mmol) in benzene (30 mL) under reflux was mixed with thionyl chloride (1.5 g, 12.6 mmol). The resulting mixture was stirred under reflux for 4 h, and then evaporated. The residue was dissolved in 200 mL of benzene and treated with anhydrous ammonia gas at room temperature. After removing the solvent, compound 2 was crystalized from ethanol and obtained as brown powder [25]. Yield 75%; m.p.: 144–146 °C; 1H-NMR (DMSO-d6, 300 MHz) δ: 5.72(s, 2H, NH2,), 6.54(s, CONH2, 2H) 7.63 (d, 1H, J = 8.6 Hz, Ar-H), 7.92 (d, 1H, J = 7.4 Hz, Ar-H), 8.14 (s, 1H, Ar-H). C7H7FN2O (154.05): MS (ESI) m/z 155.05 [M + 1].
3.3. General Method of the Synthesis of 6-Fluroquinazolin-4(3H)-one (3)
This compound was previously reported [25] but we used another method for preparation of this compound to improve the yield. In a 10 mL volume stainless steel pressure-resistant vessel were placed (1.54 g, 0.01 mol) of 2-amino-5-fluorobenzamide 2, (1.06 g, 0.01 mol) of trimethoxymethane, and 5 mL of DMF. The reaction was heated at 170 °C for 2 h. After the reaction was completed the product was cooled, collected, and crystallized from ethyl alcohol. Yield 70%; mp: 252–254 °C; 1H-NMR (DMSO-d6, 300 MHz) δ: 7.67 (d, 1H, J = 8.6 Hz, ArH), 7.85 (d, 1H, J = 9.7 Hz, ArH), 8.12 (s, 1H, ArH), 8.73 (dd, 1H, J = 8.4 Hz and 4.8 Hz, ArH), 11.83(s, 1H, NH), C8H5FN2O (164.04): MS (ESI) m/z 165.04 [M + 1].
3.4. General Method of the Synthesis of 4-Chloro-6-Fluoro-Quinazoline (4)
A mixture of compound 3 (1.64 g, 0.01 mol) in phosphorus oxychloride (0.05 mol, 7.5 mL) and phosphorous pentachloride (0.03 mol, 6 g) was heated on a water bath for 2 hr. The reaction mixture was cooled and poured slowly onto crushed ice. The resulted precipitate was filtered off, washed with water, dried, and purified by column chromatography with hexanes/ethyl acetate to afford the desired product. Yield 65%; mp: 140–142 °C; 1H-NMR (DMSO-d6, 300 MHz) δ 8.12 (s, 1H, ArH), 7.47 (dd, 1H, J = 7.9 Hz and 2.8 Hz, ArH), 7.53 (dd, 1H, J = 8.6 Hz and 5.1 Hz, ArH), 7.62 (s, 1H, ArH); C8H4ClFN2 (182): MS (ESI) m/z 183 [M + 1]
3.5. General Method of the Synthesis of Quinazoline Derivatives (5a–j)
Compound 4 (1.84 g, 0.01 mol) was mixed with (1.06 g, 0.01 mol) of trimethylamine and (0.01 mol) of the appropriate aromatic amine in 5 mL of dimethylformamide (DMF). The mixture was refluxed for 4 h, cooled, and poured on crushed ice. The crystals were collected and crystallized from ethyl alcohol. The melting points for all synthesized compounds were above 300 °C.
N-Benzyl-6-fluoroquinazoline-4-amine (5a): Yield 68%; white solid in a purity higher than 98%, 1H-NMR (DMSO-d6): δ 3.95 (s, 2H, CH2), 5.12 (s, 1H, NH), 6.21–7.93 (m, 9H, Ar-H); 13C-NMR (DMSO-d6): δ 48.57, 57.18, 112.48, 114.16, 124.3, 125, 126.22, 127.14, 128.82, 128.84, 135.21, 136, 140.37, 153.4, 162.3. MS (ESI) m/z 254.1 [M + 1]. Anal. Calcd. For C15H12FN3 (253.1): C, 71.13; H, 4.78; N, 16.59. Found C, 71.34; H, 5.01; N, 16.54.
N-(4-Chlorobenzyl)-6-fluoroquinazoline-4-amine (5b): Yield 65%; white solid in a purity higher than 98%, 1H-NMR (DMSO-d6): δ 3.93 (s, 2H, CH2), 5.34 (s, 1H, NH), 6.31–7.73 (m, 8H, Ar-H); 13C-NMR (DMSO-d6): δ 47.39, 57.78, 111.2, 113.86, 122.93, 124.14, 125.72, 127.34, 128, 128.87, 135, 136.21, 140.87, 151.24, 161.43. MS (ESI) m/z 288.06 [M + 1]. Anal. Calcd. For C15H11ClFN3 (287.06): C, 62.62; H, 3.85; N, 14.60. Found C, 62.33; H, 4.01; N, 14.53.
N-(4-Fluorobenzyl)-6-Fluoroquinazoline-4-Amine (5c): Yield 62%; white solid in a purity higher than 99%, 1H-NMR (DMSO-d6): δ 3.87 (s, 2H, CH2), 5.29 (s, 1H, NH), 6.52-7.82 (m, 8H, Ar-H); 13C-NMR (DMSO-d6): δ 46.89, 57.41, 112.32, 114.16, 121.97, 122.26, 125.12, 126.74, 127.45, 128.52, 134.37, 136, 139.87, 151.36, 162.13. MS (ESI) m/z 272.09 [M + 1]. Anal. Calcd. For C15H11F2N3 (271.09): C, 66.42; H, 4.09; N, 15.49. Found C, 66.23; H, 4.12; N, 15.51.
N-(4-Bromobenzyl)-6-fluoroquinazoline-4-amine (5d): Yield 65%; white solid in a purity higher than 97%, 1H-NMR (DMSO-d6): δ 3.79 (s, 2H, CH2), 5.41 (s, 1H, NH), 6.64-7.91 (m, 8H, Ar-H); 13C-NMR (DMSO-d6): δ 44.19, 55.81, 111.62, 114.86, 120.87, 122.52, 124.32, 126, 128.25, 131.24, 135.27, 136.81, 138.27, 152.46, 160.73. MS (ESI) m/z 332.01 [M + 1]. Anal. Calcd. For C15H11BrFN3 (331.01): C, 54.24; H, 3.34; N, 12.65. Found C, 54.43; H, 3.52; N, 12.41.
N-(4-Iodobenzyl)-6-fluoroquinazoline-4-amine (5e): Yield 67%; white solid in a purity higher than 96%, 1H-NMR (DMSO-d6): δ 3.89 (s, 2H, CH2), 5.41 (s, 1H, NH), 6.64–7.91 (m, 8H, Ar-H); 13C-NMR (DMSO-d6): δ 44.28, 54.92, 110.52, 112, 120.17, 122.47, 123.52, 125.47, 127.15, 130.21, 136.37, 137.71, 138.52, 152, 160.24. MS (ESI) m/z 380 [M + 1]. Anal. Calcd. For C15H11FIN3 (379): C, 47.51; H, 2.92; N, 11.08. Found C, 47.41; H, 3.22; N, 11.21.
N-(4-Methoxybenzyl)-6-fluoroquinazoline-4-amine (5f): Yield 60%; white solid in a purity higher than 98%, 1H-NMR (DMSO-d6): δ 3.81 (s, 2H, CH2), 4.05 (s, 3H, OCH3), 5.36 (s, 1H, NH), 6.57–7.79 (m, 8H, Ar-H); 13C-NMR (DMSO-d6): δ 45, 56.12, 58.72, 112.62, 114.31, 118.27, 122.47, 122.72, 125.17, 128.18, 131.61, 137.47, 139.71, 142.52, 152.21, 163.34. MS (ESI) m/z 284.11 [M + 1]. Anal. Calcd. For C16H14FN3O (283.11): C, 67.83; H, 4.98; N, 14.83. Found C, 67.75; H, 5.23; N, 14.91.
N-(4-Nitrobenzyl)-6-fluoroquinazoline-4-amine (5g): Yield 62%; white solid in a purity higher than 97%, 1H-NMR (DMSO-d6): δ 3.91 (s, 2H, CH2), 5.41 (s, 1H, NH), 6.82–8.11 (m, 8H, Ar-H); 13C-NMR (DMSO-d6): δ 42.58, 56.72, 111.72, 114.61, 118.57, 122, 123.52, 125.47, 127.15, 130.21, 136.37, 137.71, 138.52, 152, 161.54. MS (ESI) m/z 299.08 [M + 1]. Anal. Calcd. For C15H11FN4O2 (298.08): C, 60.40; H, 3.72; N, 18.78. Found C, 60.37; H, 4.01; N, 18.65.
N-(3-Chlorobenzyl)-6-fluoroquinazoline-4-amine (5h): Yield 65%; white solid in a purity higher than 98%, 1H-NMR (DMSO-d6): δ 3.94 (s, 2H, CH2), 5.61 (s, 1H, NH), 6.64–7.94 (m, 8H, Ar-H); 13C-NMR (DMSO-d6): δ 40.28, 54.78, 114.12, 116.92, 118.53, 120, 122.12, 126.57, 128.21, 131.41, 136.64, 137.01, 139.42, 152, 160.54. MS (ESI) m/z 288.06 [M + 1]. Anal. Calcd. For C15H11ClFN3 (287.06): C, 62.62; H, 3.85; N, 14.60. Found C, 62.51; H, 3.96; N, 14.43.
N-(3-Nitrobenzyl)-6-fluoroquinazoline-4-amine (5i): Yield 62%; white solid in a purity higher than 98%, 1H-NMR (DMSO-d6): δ 3.89 (s, 2H, CH2), 5.31 (s, 1H, NH), 6.74–8.21 (m, 8H, Ar-H); 13C-NMR (DMSO-d6): δ 42.76, 54.82, 111.82, 114.91, 116.78, 122.32, 123, 124.62, 127.81, 132.41, 136.57, 138.32, 141.42, 150.6, 162.31. MS (ESI) m/z 299.09 [M + 1]. Anal. Calcd. For C15H11FN4O2 (298.09): C, 60.40; H, 3.72; N, 18.78. Found C, 60.21; H, 3.61; N, 18.95.
N-(3,5-Dichlorobenzyl)-6-fluoroquinazoline-4-amine (5j): Yield 67%; white solid in a purity higher than 97%, 1H-NMR (DMSO-d6): δ 3.91 (s, 1H, NH), 5.48 (s, 2H, 2NH), 6.74–7.97 (m, 7H, Ar-H); 13C-NMR (DMSO-d6): δ 40.63, 52.78, 112.98, 116, 116.93, 121.24, 122.76, 124.85, 128.71, 130.61, 134.84, 138.21, 140.02, 152.41, 160.04. MS (ESI) m/z 322.02 [M + 1]. Anal. Calcd. For C15H10Cl2FN3 (321.02): C, 55.92; H, 3.13; N, 13.04. Found C, 56.07; H, 3.31; N, 13.22.
3.6. Docking Studies
In the present work, all the target compounds were subjected to docking study to explore their binding mode to the GABA-A receptor. All modeling experiments were performed using the molsoft program which provides a unique set of tools for the modeling of protein/ligand interactions. It predicts how small flexible molecules, such as substrates or drug candidates, bind to a protein of a known 3D structure represented by grid interaction potentials. Each experiment used the biological target GABA-A receptor downloaded from the Brookhaven Protein Databank (PDB) (http://www.rcsb.org/pdb/explore/explore.do?structureId=4COF) [26].
3.7. Pharmacological Tests
Swiss albino mice 25–40 g (12–15 weeks old) were used in the experiments. These mice were obtained from the Animal Centre, King Abdul-Aziz University. The mice were kept under typical conditions of humidity, temperature (23 ± 2 °C), and light (12 h light/12 h dark). The animals were fed with a standard mouse pellet diet and had free access to water. The protocol used for the testing of animals was approved by the Institutional Animal Ethics Committee (approval number 6063/1435). All of the biological experiments were carried out according to the respective internationally valid guidelines. The test group and vehicle control group included six animals. Median toxic dose (TD50) is the dose required to get 50% of the animals reporting specific toxic effects. It was determined by a rotorod test in mice [5,9]. Protective index is the ratio between TD50 and ED50. This was determined from the equation PI = TD50/ED50. The median lethal dose (LD50) is the amount of therapeutic agent that leads to 50% death in mice. LD50 was calculated from the dose-response curves with at least four doses [9]. Therapeutic index (TI) is the ratio between the dosage of a drug that causes a lethal effect and the dosage that causes a therapeutic effect. It was determined from the equation TI = LD50/ED50 [5].
3.8. Anticonvulsant Screening
Anticonvulsant screening was evaluated by the pentylentetrazole (PTZ) model and maximal electroshock (MES) model according to the reported procedures [20]. Test compounds were dissolved in 10% DMSO and injected intraperitoneal (i.p.) at a dose of 0.5 mmol/kg one half hour before seizure induction. Sodium valproate (1.8 mmol/kg) and methaqualone (0.8 mmol/kg) were used as two reference drugs. Seizure induction in the MES test was obtained by an electrical stimulus (50 mA, 60 Hz) from an electroconvulsiometer apparatus. The electrical current was applied for 0.2 s via euricular electrodes. Protection against these seizures was identified by the abolition of the hind leg and tonic maximal extension component of the seizure [17,18]. Seizure induction in the PTZ test was obtained by the i.p. injection of a convulsing dose of PTZ (100 mg/kg). Tonic-clonic convulsions, Seizures, hypnosis, and death were recorded. The highest active compounds 5b, 5c, and 5d were further evaluated by using different doses in the PTZ model in order to determine their protective and therapeutic indexes. Three groups of six mice were used. Each group was given a range of i.p. doses of the test drug until at least four points were established in the range of 10%–90% from seizure protection or minimal observed neurotoxicity. The value of (ED50), slopes and standard errors were determined by using a computer program based on the method described by Finney [19,20].
3.9. Neurotoxicity Screening
Neurological toxicity (NT) was determined by a rotorod test in mice [14,15]. Groups of animals (mice) were trained to balance on a revolving rod (3 cm diameter and 6 rpm speed). The mice were allowed three trials to remain on the revolving rod for 20 s. The trained mice were given the test compounds i.p. at different dose levels. When the trained animals show lack of rolling roller performance then the test compound was considered neurotoxic. The taught animals were examined at 0.5 h and 4 h after the drug administration and then the neurotoxic symptoms were recorded as the median toxic dose (TD50). The acute neurotoxicity was tested following the method described by Dunham [14].
3.10. GABA Assay
This assay was accomplished in brain tissue extracts enzymatically. Adult Wistar rats were separated into three groups of six animals each. After 2 h of i.p. drug administration (100 mg/kg), the rats were decapitated and the brains were put into separate vials containing 4–6 mL of ice-cold 80% ethanol and processed further following the reported procedures [27]. The chronic study was also performed through oral administration of the test compounds (30 mg/kg) for seven days. The data of GABA level concentration (μgm/100 mg of brain tissue) was collected as the minimum concentration that caused at least 50% inhibition established at one or more time points.
4. Conclusions
Some novel derivatives of N-substituted-6-fluoro-quinazoline-4-amine were designed, synthesized, and biologically evaluated as anticonvulsant agents by using a subcutaneous pentylenetetrazole (scPTZ) test and a maximal electroshock (MES) test. Their neurotoxicity was also tested by using a rotorod test. All of the derivatives induced significant anticonvulsant activity. Three compounds 5b, 5c, and 5d, showed the highest anticonvulsant activities in experimental mice with ED50 of 152, 165, and 140 mg/kg, while the reference drugs methaqualone and valproate had ED50 of 200 and 300 mg/kg. Structure activity relationship (SAR) studies explained that different substitutions on the phenyl ring of the aromatic amine exerted varied anticonvulsant activity. Changing the electronic nature and the position of the substituent group attached to the aromatic ring led to changing anticonvulsant activity. The three most active compounds had electron withdrawing groups at the 4-position of the phenyl ring. A 4-bromo substitution in 5d showed higher activity than 4-chloro in 5b, and this showed higher activity than a 4-fluoro in 5c. These three compounds also showed a high safety margin and low neurotoxicity compared to the reference drugs. The molecular docking study was performed to evaluate the binding mode of the proposed compounds with the GABA-A receptor. The resulting data from the docking studies explained that all of the newly-synthesized derivatives had significantly high affinity towards the GABA-A receptor compared to methaqualone as a reference ligand. The data obtained from the biological screening integrated with that obtained from the modelling study. The enzymatic assay proved that these highly active compounds significantly increased the GABA level. The most active compounds 5b–d could be used as a pattern for future design, optimization, and investigation to produce more effective analogs.
Acknowledgments
The authors extend their appreciation to the Deanship of Scientific Research at Taibah University for funding the work through the research group project No (6063/1435). The authors would also like to thank Pharmacology Department, College of Pharmacy, Taibah University for helping in the pharmacological screening.
Author Contributions
M.F. Zayed designed the idea, and the protocol of the study, synthesized the starting and intermediate compounds and wrote the manuscript. S.K. Ihmaid and H.E.A. Ahmed synthesized the final compounds, wrote their experimental parts and interpreted their spectral data. Khaled El-Adl performed and interpreted the modeling study helped in interpretation of the biological studies. A.M. Asiri performed the biological studies and wrote their experimental data. A.M. Omar helped in the enzymatic assay. All the authors revised the whole manuscript and responded to reviewers’ comments.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zayed, M.F.; Ayyad, R.R. Some novel anticonvulsant agents derived from Phthalazinedione. Arzneimittelforschung 2012, 62, 532–536. [Google Scholar] [CrossRef] [PubMed]
- Jatav, V.; Mishra, P.; Kashaw, S.; Stables, J.P. CNS depressant and anticonvulsant activities of some novel 3-[5-substituted1,3,4-thiadiazole-2-yl]-2-styryl quinazoline-4(3H)-ones. Eur. J. Med. Chem. 2008, 43, 1945–1954. [Google Scholar] [CrossRef] [PubMed]
- El-Naser Ossman, A.R.; El-Sayed Barakat, S. Synthesis and Anticonvulsant Activity of Some new 3-(p-Sulfamoylphenyl)-4(3H)-Quinazolinones. Arzneimittelforsch 1994, 44, 915–919. [Google Scholar] [PubMed]
- Jin, H.G.; Sun, X.Y.; Chai, K.Y.; Piao, H.R.; Quan, Z.S. Anticonvulsant and toxicity evaluation of some 7-alkoxy-4,5-dihydro-[1,2,4]triazolo[4,3-a]quinoline-1(2H)-ones. Bioorg. Med. Chem. 2006, 14, 6868–6873. [Google Scholar] [CrossRef] [PubMed]
- Zayed, M.F.; Ahmed, E.A.; Omar, A.M.; Abdelrahim, A.S.; El-Adl, K. Design, synthesis and biological evaluation studies of novel quinazolinone derivatives as anticonvulsant agents. Med. Chem. Res. 2013, 22, 5823–5831. [Google Scholar] [CrossRef]
- Zayed, M.F. New fluorinated quinazolinone derivatives as anticonvulsant agents. J. Taibah Univ. Med. Sci. 2014, 9, 104–109. [Google Scholar] [CrossRef]
- Elhelby, A.A.; Ayyad, R.R.; Zayed, M.F. Synthesis and biological evalution of some novel quinoxaline derivatives as anticonvulsant agents. Arzneimittelforschung 2011, 61, 379–381. [Google Scholar] [PubMed]
- Scharfman, H. Upregulation of Multidrug Resistance Transporters in the Epileptic Brain. Epilepsy Curr. 2002, 2, 200–202. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.K.; El-Adl, K.; Zayed, M.F.; Mahdy, H.A. Design, synthesis, docking and biological evaluation of some novel 5-chloro-2-substituted sulfanylbenzoxazole derivatives as anticonvulsant agents. Med. Chem. Res. 2014, 24, 99–114. [Google Scholar] [CrossRef]
- Krall, R.L.; Penry, J.K.; White, B.G.; Kupferberg, H.J.; Swinyard, E.A. Antiepileptic Drug Development: II. Anticonvulsant Drug Screening. Epilepsia 1968, 19, 409–428. [Google Scholar] [CrossRef]
- Topliss, J.G. A manual method for applying the Hansch approach to drug design. J. Med. Chem. 1977, 20, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Englert, L.; Biela, A.; Zayed, M.F.; Heine, A.; Hangauer, D.; Klebe, G. Displacement of disordered water molecules from hydrophobic pocket creates enthalpic signature: Binding of phosphonamidate to the S1’-pocket of thermolysin. Biochim. Biophys. Acta 2010, 1800, 1192–1202. [Google Scholar] [CrossRef] [PubMed]
- Kirk, L.K. Fluorine in medicinal chemistry: Recent therapeutic applications of fluorinated small molecules. J. Fluorine. Chem. 2006, 127, 1013–1029. [Google Scholar] [CrossRef]
- Dunham, N.W.; Miya, T.S. A note on a simple apparatus for detecting neurological deficit in rats and mice. J. Am. Pharm. Assoc. 1957, 46, 208–209. [Google Scholar] [CrossRef]
- Crivori, P.; Cruciani, G.; Carrupt, P.A.; Testa, B. Predicting blood-brain barrier permeation from three-dimensional molecular structure. J. Med. Chem. 2000, 11, 2204–2216. [Google Scholar] [CrossRef]
- Yogeeswari, P.; Sriram, D.; Thirumurugan, R.; Raghavendran, J.V.; Sudhan, K.; Pavana, R.K.; Stables, J. Discovery of N-(2,6-dimethylphenyl)-substituted semicarbazones as anticonvulsants: hybrid pharmacophore-based design. J. Med. Chem. 2005, 48, 6202–6211. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, J.F.; Rathman, T.L.; Sleevi, M.C.; Campbell, J.A.; Greenwood, T.D.J. Synthesis and anticonvulsant activity of some new 2-substituted 3-aryl-4(3H)-quinazolinone. J. Med. Chem. 1990, 33, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Swinyard, E.A.; Brown, W.C.; Goodman, L.S. Comparative Assays of Antiepileptic Drugs in Mice and Rats. J. Pharmacol. Exp. Ther. 1952, 106, 319–330. [Google Scholar] [PubMed]
- Chen, L.; Sun, Y.X.; Chai, Y.K.; Lee, J.S.; Song, M.S.; Quan, Z.S. Synthesis and anticonvulsant evaluation of 4-(4-alkoxylphenyl)-3-ethyl-4H-1,2,4-triazoles as open-chain analogues of 7-alkoxyl-4,5-dihydro[1,2,4]triazolo[4,3-a]quinolines. Bioorg. Med. Chem. 2007, 15, 6775–6781. [Google Scholar] [CrossRef] [PubMed]
- Zappalà, M.; Grasso, S.; Micale, N.; Zuccalà, G.; Menniti, F.S.; Ferreri, G.; Desarro, G.; Micheli, C. 1-Aryl-6,7-methylenedioxy-3H-quinazolin-4-ones as Anticonvulsant Agents. Bioorg. Med. Chem. Lett. 2003, 13, 4427–4430. [Google Scholar] [CrossRef] [PubMed]
- Ajeet, K.A. Designing of hybrid form of benzothiazole-quinazoline as GABA-A inhibitor with anticonvulsant profile: An in-silico approach. Am. J. Pharm. Sci. 2013, 1, 116–120. [Google Scholar] [CrossRef]
- Kumar, P.; Shrivastava, B.; Pandeya, S.N.; Stables, J.P. Design, synthesis and potential 6 Hz psychomotor seizure test activity of some novel 2-(substituted)-3-{[substituted]amino}quinazolin-4(3H)-one. Eur. J. Med. Chem. 2011, 46, 1006–1018. [Google Scholar] [CrossRef] [PubMed]
- Johnston, G.A. GABAA Receptor Pharmacology. Pharmacol. Ther. 1996, 69, 173–198. [Google Scholar] [CrossRef]
- Ibrahim, M.K.; El-Adl, K.; Al-Karmalawy, A.A. Design, synthesis, molecular docking and anticonvulsant evaluation of novel 6-iodo-2-phenyl-3-substituted-quinazolin-4(3H)-ones. Bull. Fac. Pharm. Cairo Univ. 2015, 53, 101–116. [Google Scholar] [CrossRef]
- Kuo, S.; Hour, M.; Huang, L.; Lee, K. Preparation of 2-Phenyl-4-quinazolinones and 2-Phenyl-4-alkoxyquinazolines. U.S. Patent 6,479,499, 12 December 2002. [Google Scholar]
- Protein Data Bank. Crystal Structure of a Human Gamma-Aminobutyric Acid Receptor, the GABA(A)R-beta3 Homopentamer. Available online: http://www.rcsb.org/pdb/explore/explore.do?structureId=4COF (accessed on 20 December 2016).
- Roberts, E. Methods in Enzymology; Academic Press: New York, NY, USA, 1962; Volume VI, p. 612. [Google Scholar]
- Sample Availability: Samples of the compounds are currently not available from the authors.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).