
Diverse Effects of Cilostazol on Proprotein Convertase Subtilisin/Kexin Type 9 between Obesity and Non-Obesity
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Cilostazol Induces PPARγ Upregulation and PCSK9 Expression in HepG2 Cells
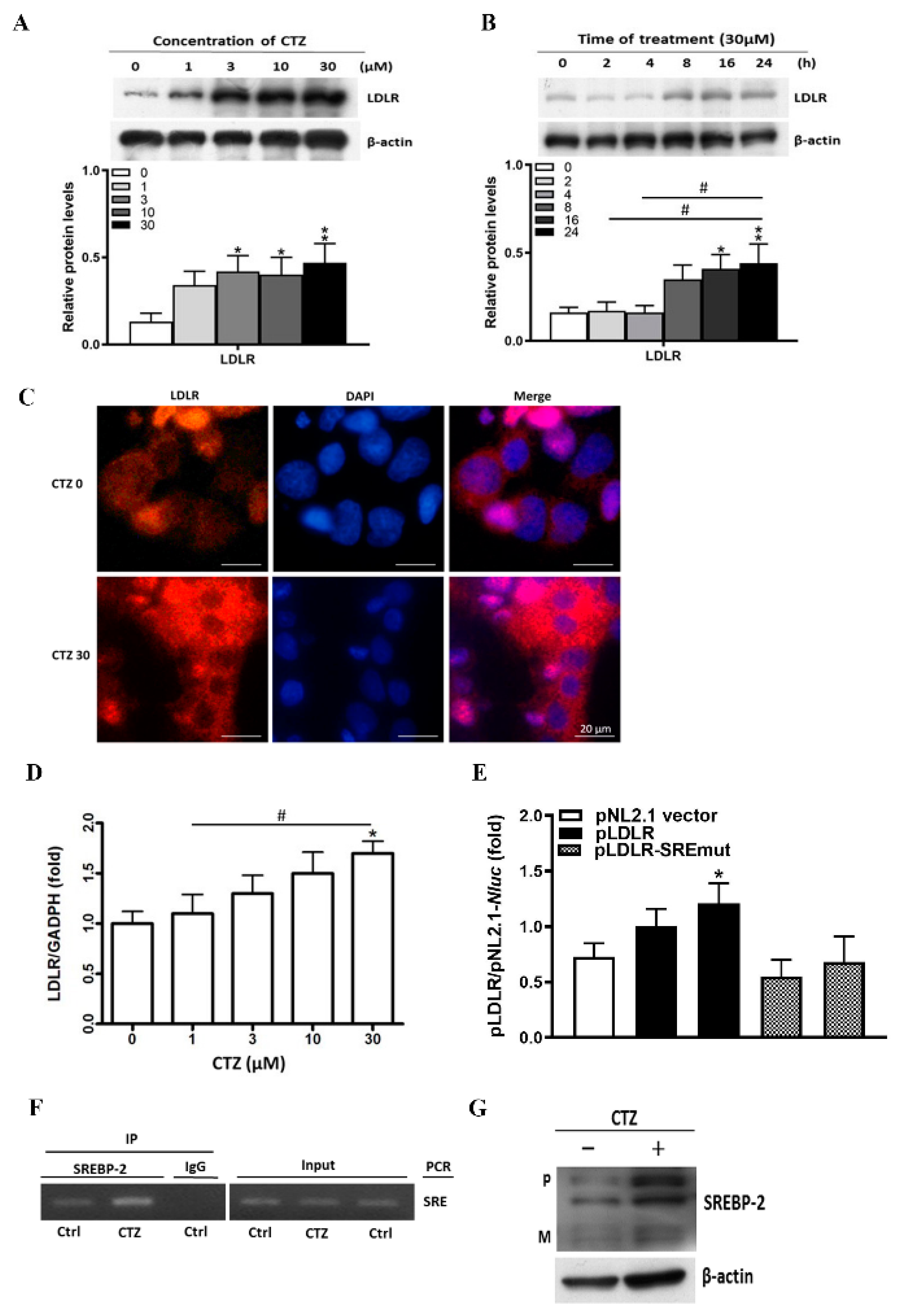
2.2. Cilostazol Induces LDLR Expression in HepG2 Cells
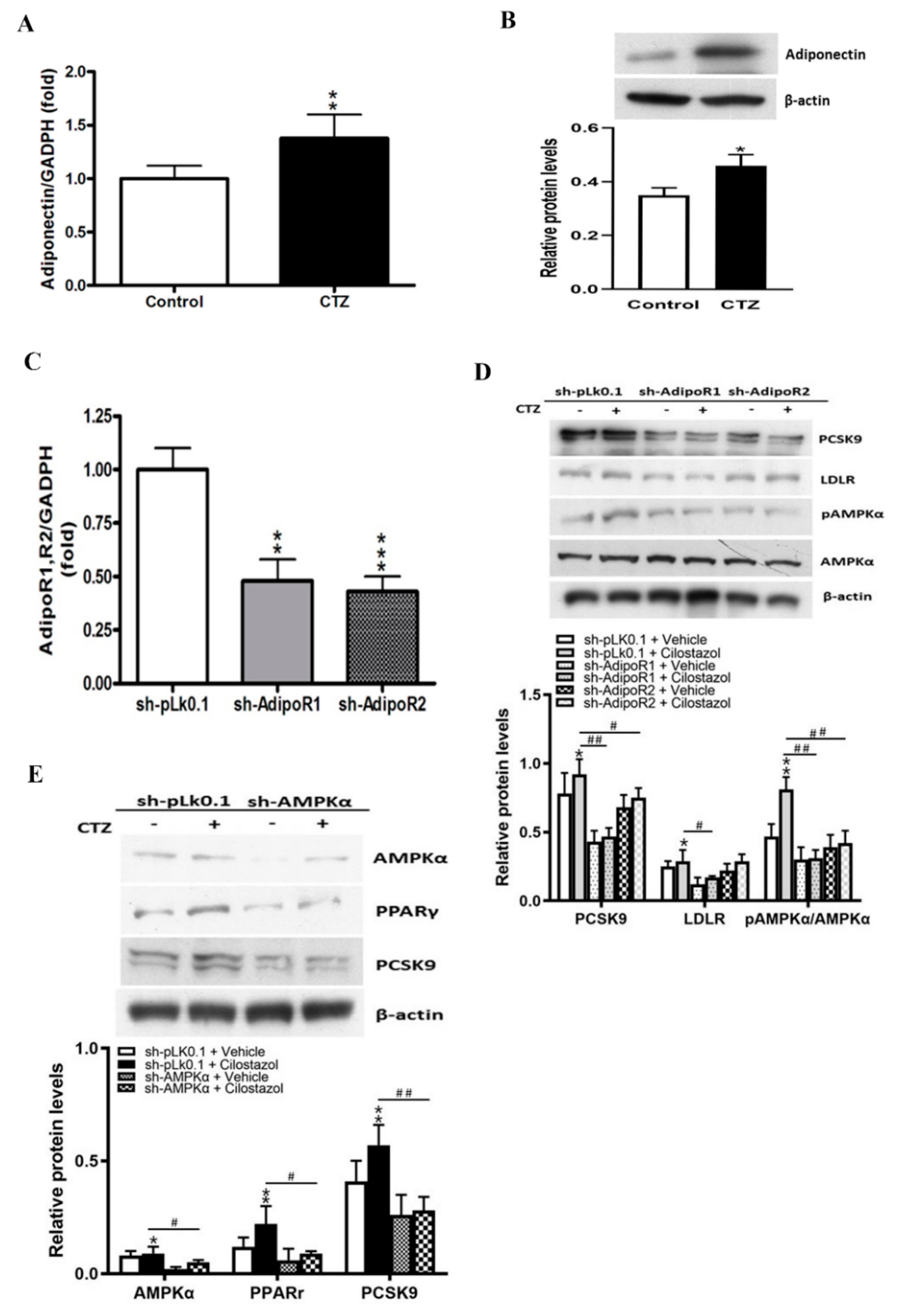
2.3. Cilostazol Mediates the Expression of Adiponectin and the Subsequent Activation of AMPKα/PPARγ Signaling Molecules in HepG2 Cells
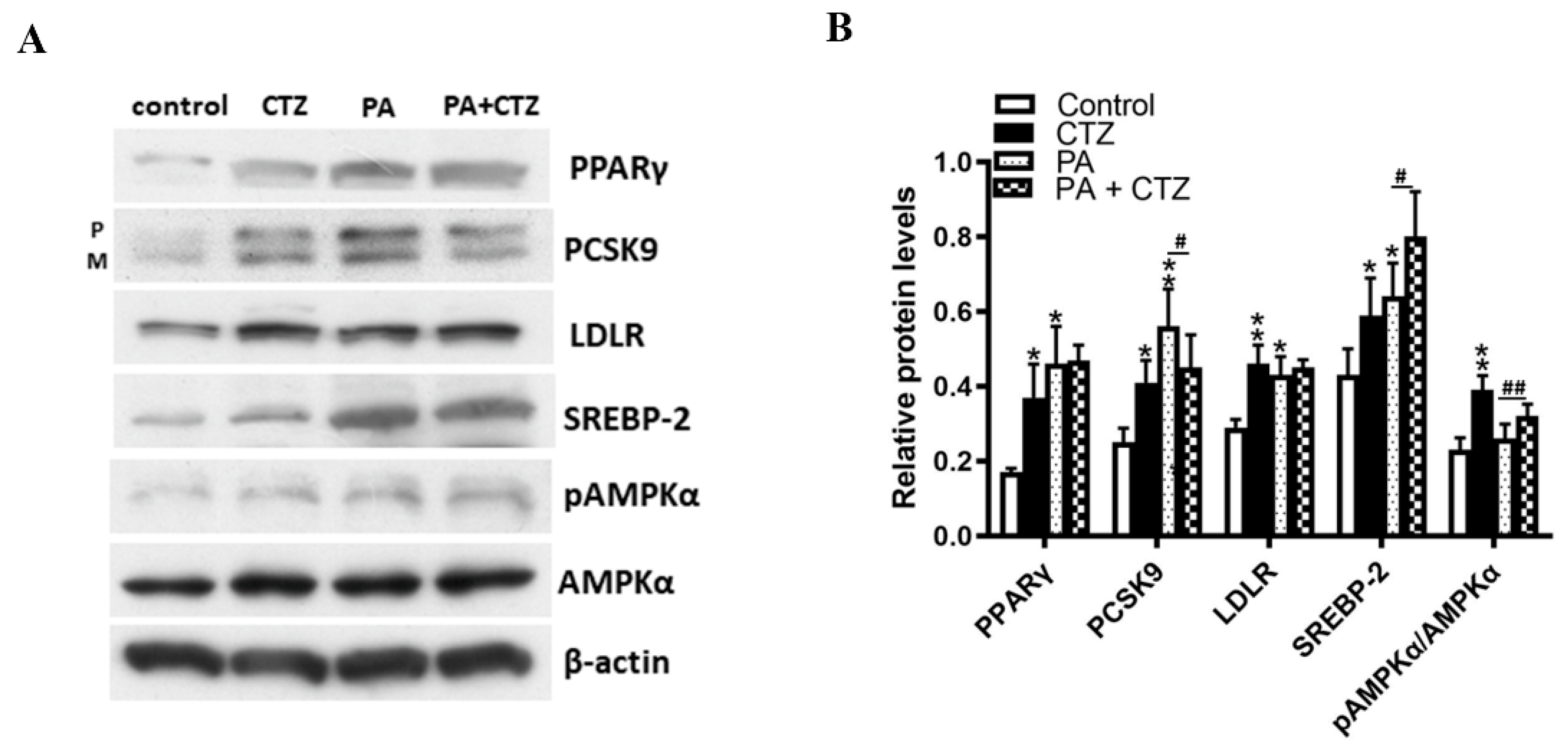
2.4. Diverse Effects of Cilostazol on PCSK9 Levels in HepG2 Cells Treated with Palmitic Acid (PA) or without
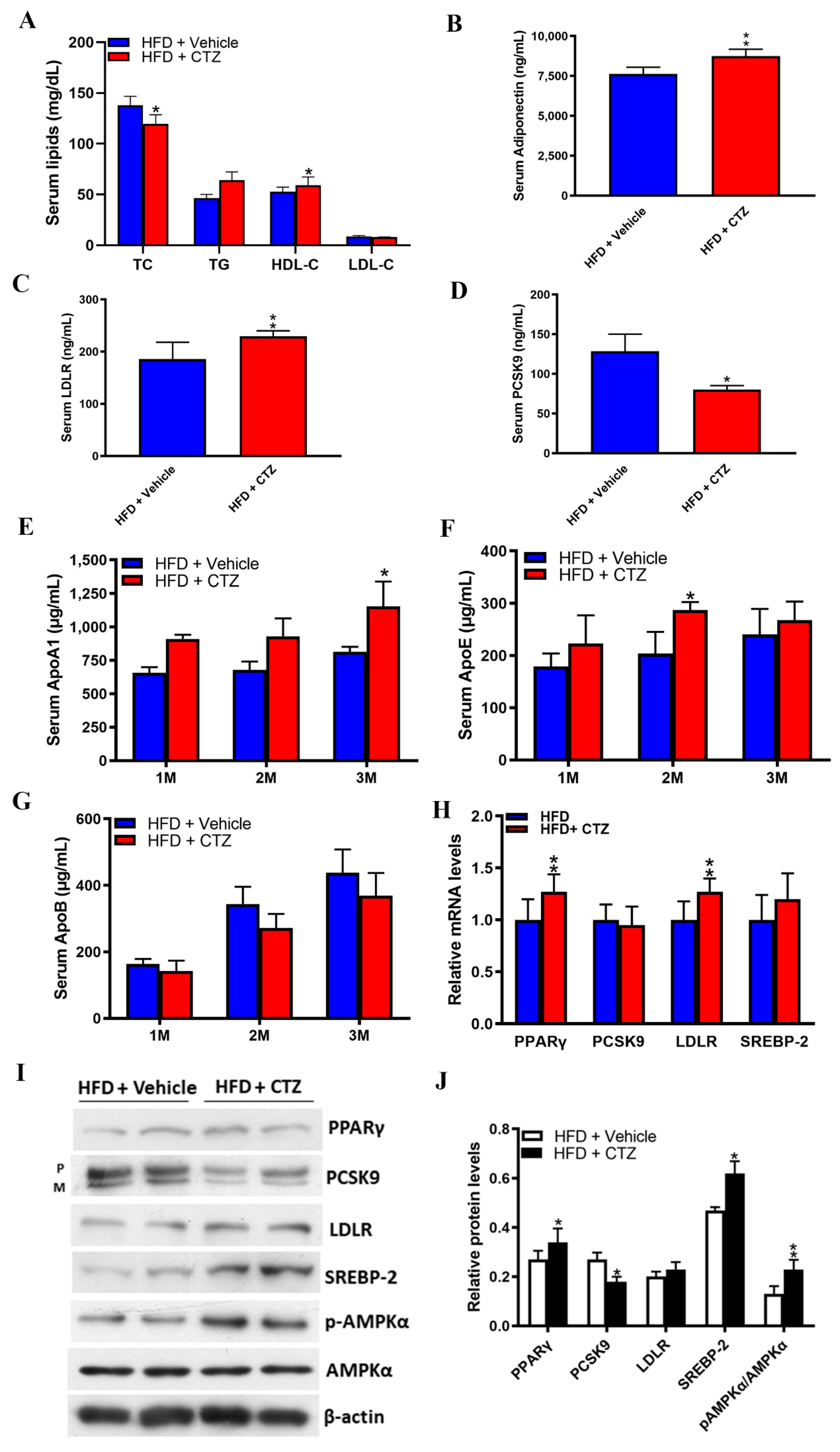
2.5. Cilostazol Attenuates Lipid Accumulation in the Livers of Obese Mice
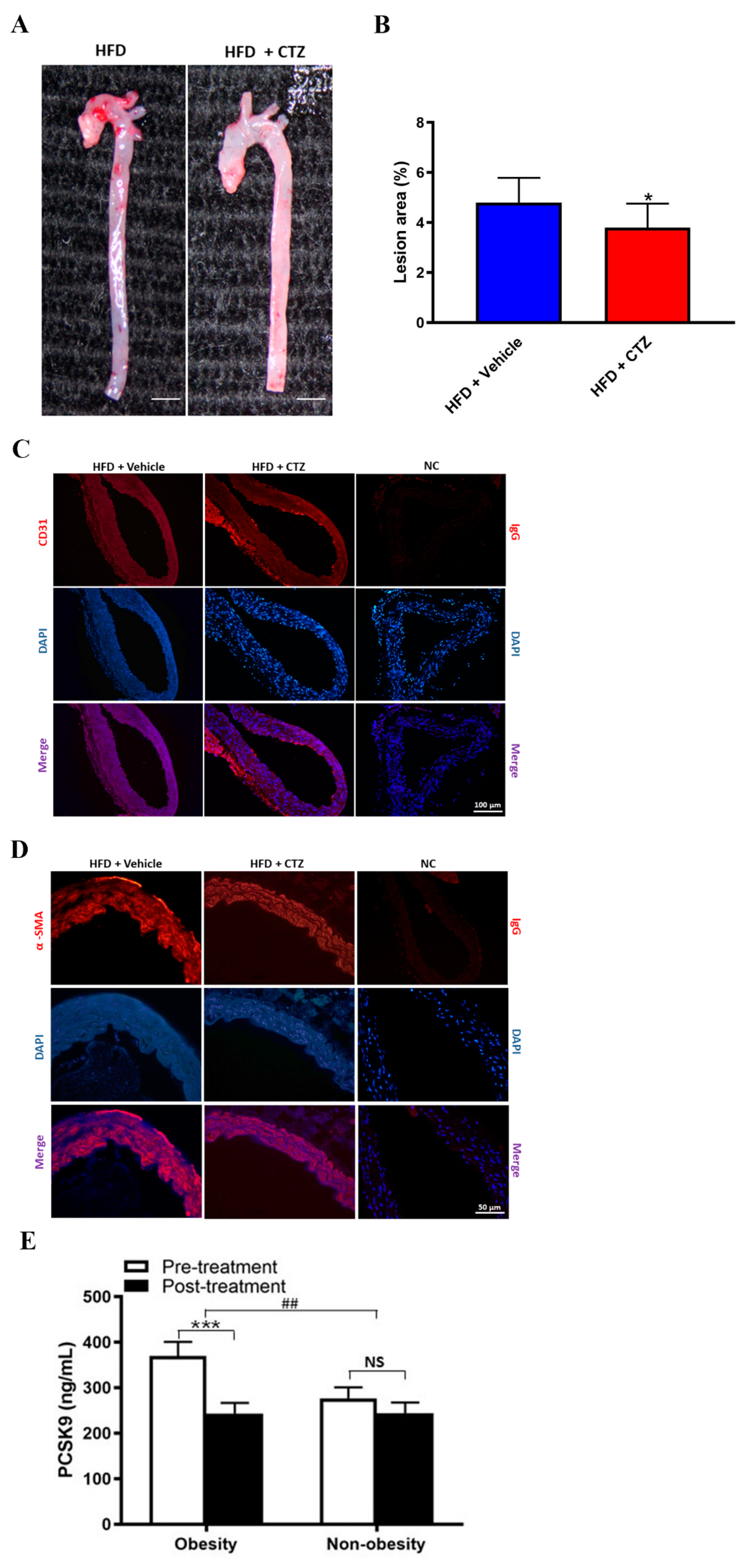
2.6. Cilostazol Treatment Provides Anti-Atherosclerosis Effects in Mice with HFD-Induced Obesity and Shows Diverse Effects on PCSK9 Expression in Obese and Non-Obese Participants
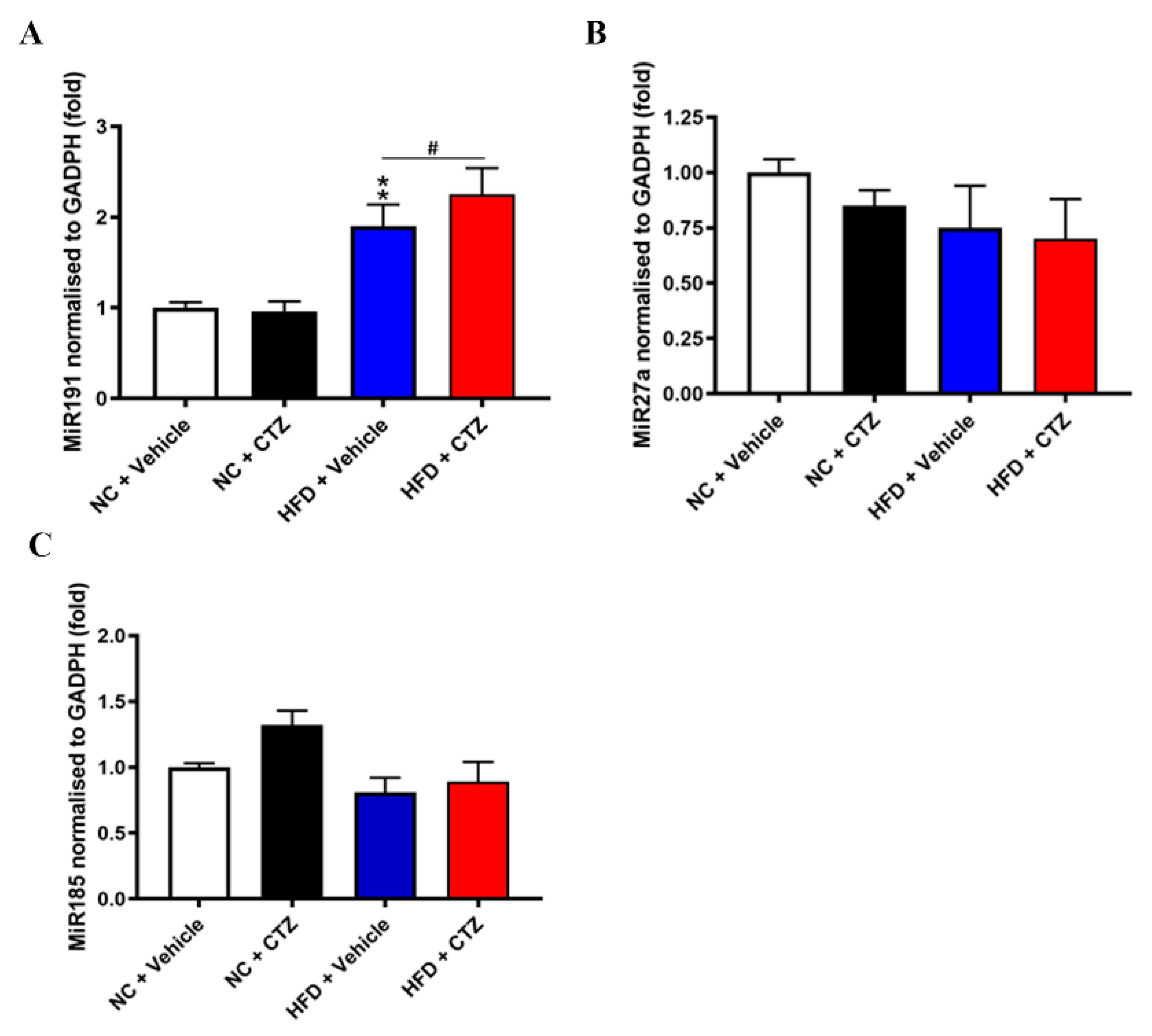
2.7. Diverse Effects of Cilostazol on PCSK9-Relevant miRNAs in the Liver of the Mice Fed with NC and HFD
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Preparation
4.2. Western Blotting Assay
4.3. Immunofluorescence Staining
4.4. Quantitative Real-Time Polymerase Chain Reaction (RT-PCR)
4.5. siRNA Transfection
4.6. Effects of Cilostazol on Promoter Activity with or without PPRE or SRE Mutation
4.7. ChIP Assay
4.8. Animal Model
4.9. Serum Biochemical Analysis
4.10. Histology and Oil Red O Staining
4.11. Clinical Data
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, S.; Li, J.-J. PCSK9: A key factor modulating atherosclerosis. J. Atheroscler. Thromb. 2015, 22, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Sabatine, M.S. PCSK9 inhibitors: Clinical evidence and implementation. Nat. Rev. Cardiol. 2019, 16, 155–165. [Google Scholar] [CrossRef]
- Cohen, J.C.; Boerwinkle, E.; Mosley, T.H., Jr.; Hobbs, H.H. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N. Engl. J. Med. 2006, 354, 1264–1272. [Google Scholar] [CrossRef] [PubMed]
- Baass, A.; Dubuc, G.; Tremblay, M.; Delvin, E.E.; O’Loughlin, J.; Levy, E.; Davignon, J.; Lambert, M. Plasma PCSK9 is associated with age, sex, and multiple metabolic markers in a population-based sample of children and adolescents. Clin. Chem. 2009, 55, 1637–1645. [Google Scholar] [CrossRef] [PubMed]
- Cui, Q.; Ju, X.; Yang, T.; Zhang, M.; Tang, W.; Chen, Q.; Hu, Y.; Haas, J.V.; Troutt, J.S.; Pickard, R.T.; et al. Serum PCSK9 is associated with multiple metabolic factors in a large Han Chinese population. Atherosclerosis 2010, 213, 632–636. [Google Scholar] [CrossRef]
- Feingold, K.R.; Moser, A.H.; Shigenaga, J.K.; Patzek, S.M.; Grunfeld, C. Inflammation stimulates the expression of PCSK9. Biochem. Biophys. Res. Commun. 2008, 374, 341–344. [Google Scholar] [CrossRef]
- Urban, D.; Pöss, J.; Böhm, M.; Laufs, U. Targeting the proprotein convertase subtilisin/kexin type 9 for the treatment of dyslipidemia and atherosclerosis. J. Am. Coll. Cardiol. 2013, 62, 1401–1408. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhu, C.-G.; Xu, R.-X.; Li, S.; Guo, Y.-L.; Sun, J.; Li, J.-J. Relation of circulating PCSK9 concentration to fibrinogen in patients with stable coronary artery disease. J. Clin. Lipidol. 2014, 8, 494–500. [Google Scholar] [CrossRef]
- Li, S.; Guo, Y.-L.; Xu, R.-X.; Zhang, Y.; Zhu, C.-G.; Sun, J.; Qing, P.; Wu, N.-Q.; Jiang, L.-X.; Li, J.-J. Association of plasma PCSK9 levels with white blood cell count and its subsets in patients with stable coronary artery disease. Atherosclerosis 2014, 234, 441–445. [Google Scholar] [CrossRef]
- Ding, Z.; Liu, S.; Wang, X.; Deng, X.; Fan, Y.; Shahanawaz, J.; Reis, R.J.S.; Varughese, K.I.; Sawamura, T.; Mehta, J.L. Cross-talk between LOX-1 and PCSK9 in vascular tissues. Cardiovasc. Res. 2015, 107, 556–567. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Guo, Y.-L.; Xu, R.-X.; Zhang, Y.; Zhu, C.-G.; Sun, J.; Qing, P.; Wu, N.-Q.; Li, J.-J. Plasma PCSK9 levels are associated with the severity of coronary stenosis in patients with atherosclerosis. Int. J. Cardiol. 2014, 174, 863–864. [Google Scholar] [CrossRef] [PubMed]
- Chao, T.H.; Chen, I.C.; Li, Y.H.; Lee, P.T.; Tseng, S.Y. Plasma Levels of Proprotein Convertase Subtilisin/Kexin Type 9 Are Elevated in Patients with Peripheral Artery Disease and Associated with Metabolic Disorders and Dysfunction in Circulating Progenitor Cells. J. Am. Heart Assoc. 2016, 5, e003497. [Google Scholar] [CrossRef] [PubMed]
- Sahebkar, A.; Simental-Mendía, L.E.; Guerrero-Romero, F.; Golledge, J.; Watts, G.F. Effect of statin therapy on plasma proprotein convertase subtilisin kexin 9 (PCSK9) concentrations: A systematic review and meta-analysis of clinical trials. Diabetes Obes. Metab. 2015, 17, 1042–1055. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Korstanje, R. Proprotein convertases in high-density lipoprotein metabolism. Biomark. Res. 2013, 1, 27. [Google Scholar] [CrossRef]
- Druce, I.; Abujrad, H.; Ooi, T.C. PCSK9 and triglyceride-rich lipoprotein metabolism. J. Biomed. Res. 2015, 29, 429–436. [Google Scholar] [CrossRef]
- Duan, Y.; Chen, Y.; Hu, W.; Li, X.; Yang, X.; Zhou, X.; Yin, Z.; Kong, D.; Yao, Z.; Hajjar, D.P.; et al. Peroxisome proliferator-activated receptor γ activation by ligands and dephosphorylation induces proprotein convertase subtilisin kexin type 9 and low density lipoprotein receptor expression. J. Biol. Chem. 2012, 287, 23667–23677. [Google Scholar] [CrossRef]
- Sun, L.; Yang, X.; Li, Q.; Zeng, P.; Liu, Y.; Liu, L.; Chen, Y.; Yu, M.; Ma, C.; Li, X.; et al. Activation of Adiponectin Receptor Regulates Proprotein Convertase Subtilisin/Kexin Type 9 Expression and Inhibits Lesions in ApoE-Deficient Mice. Arter. Thromb. Vasc. Biol. 2017, 37, 1290–1300. [Google Scholar] [CrossRef]
- Chao, T.-H.; Tseng, S.-Y.; Li, Y.-H.; Liu, P.-Y.; Cho, C.-L.; Shi, G.-Y.; Wu, H.-L.; Chen, J.-H. A novel vasculo-angiogenic effect of cilostazol mediated by cross-talk between multiple signalling pathways including the ERK/p38 MAPK signalling transduction cascade. Clin. Sci. 2012, 123, 147–159. [Google Scholar] [CrossRef]
- Chao, T.-H.; Tseng, S.-Y.; Chen, I.-C.; Tsai, Y.-S.; Huang, Y.-Y.; Liu, P.-Y.; Ou, H.-Y.; Li, Y.-H.; Wu, H.-L.; Cho, C.-L.; et al. Cilostazol enhances mobilization and proliferation of endothelial progenitor cells and collateral formation by modifying vasculo-angiogenic biomarkers in peripheral arterial disease. Int. J. Cardiol. 2014, 172, e371–e374. [Google Scholar] [CrossRef]
- Chao, T.-H.; Chen, I.-C.; Lee, C.-H.; Chen, J.-Y.; Tsai, W.-C.; Li, Y.-H.; Tseng, S.-Y.; Tsai, L.-M.; Tseng, W.-K. Cilostazol Enhances Mobilization of Circulating Endothelial Progenitor Cells and Improves Endothelium-Dependent Function in Patients at High Risk of Cardiovascular Disease. Angiology 2016, 67, 638–646. [Google Scholar] [CrossRef]
- Tseng, S.-Y.; Chao, T.-H.; Li, Y.-H.; Liu, P.-Y.; Lee, C.-H.; Cho, C.-L.; Wu, H.-L.; Chen, J.-H. Cilostazol improves high glucose-induced impaired angiogenesis in human endothelial progenitor cells and vascular endothelial cells as well as enhances vasculoangiogenesis in hyperglycemic mice mediated by the adenosine monophosphate-activated protein kinase pathway. J. Vasc. Surg. 2016, 63, 1051–1062.e3. [Google Scholar] [CrossRef] [PubMed]
- Tseng, S.-Y.; Chao, T.-H.; Li, Y.-H.; Cho, C.-L. Cilostazol Improves Proangiogenesis Functions in Human Early Endothelial Progenitor Cells through the Stromal Cell-Derived Factor System and Hybrid Therapy Provides a Synergistic Effect In Vivo. BioMed Res. Int. 2016, 2016, 3639868. [Google Scholar] [CrossRef] [PubMed]
- Biscetti, F.; Pecorini, G.; Straface, G.; Arena, V.; Stigliano, E.; Rutella, S.; Locatelli, F.; Angelini, F.; Ghirlanda, G.; Flex, A. Cilostazol promotes angiogenesis after peripheral ischemia through a VEGF-dependent mechanism. Int. J. Cardiol. 2013, 167, 910–916. [Google Scholar] [CrossRef] [PubMed]
- Kawabe-Yako, R.; Masaaki, I.; Masuo, O.; Asahara, T.; Itakura, T. Cilostazol activates function of bone marrow-derived endothelial progenitor cell for re-endothelialization in a carotid balloon injury model. PLoS ONE 2011, 6, e24646. [Google Scholar] [CrossRef]
- Hicks, C.W.; Yang, C.; Ndumele, C.E.; Folsom, A.R.; Heiss, G.; Black, J.H.; Selvin, E.; Matsushita, K. Associations of Obesity with Incident Hospitalization Related to Peripheral Artery Disease and Critical Limb Ischemia in the ARIC Study. J. Am. Heart Assoc. 2018, 7, e008644. [Google Scholar] [CrossRef] [PubMed]
- Mba, C.M.; Mbacham, W.; Sobngwi, E.; Mbanya, J.C. Is PCSK9 Associated with Plasma Lipid Levels in a Sub-Saharan African Population of Patients with Obesity and Type 2 Diabetes? Diabetes Metab. Syndr. Obes. Targets Ther. 2019, 12, 2791–2797. [Google Scholar] [CrossRef]
- Achari, A.E.; Jain, S.K. Adiponectin, a Therapeutic Target for Obesity, Diabetes, and Endothelial Dysfunction. Int. J. Mol. Sci. 2017, 18, 1321. [Google Scholar] [CrossRef]
- Wang, J.-F.; Yu, M.-L.; Yu, G.; Bian, J.-J.; Deng, X.-M.; Wan, X.-J.; Zhu, K.-M. Serum miR-146a and miR-223 as potential new biomarkers for sepsis. Biochem. Biophys. Res. Commun. 2010, 394, 184–188. [Google Scholar] [CrossRef]
- Aryal, B.; Singh, A.K.; Rotllan, N.; Price, N.; Fernández-Hernando, C. MicroRNAs and lipid metabolism. Curr. Opin. Lipidol. 2017, 28, 273–280. [Google Scholar] [CrossRef]
- Momtazi, A.A.; Banach, M.; Pirro, M.; Stein, E.A.; Sahebkar, A. MicroRNAs: New Therapeutic Targets for Familial Hypercholesterolemia? Clin. Rev. Allergy Immunol. 2018, 54, 224–233. [Google Scholar] [CrossRef] [Green Version]
- Naeli, P.; Azad, F.M.; Malakootian, M.; Seidah, N.; Mowla, S.J. Post-transcriptional Regulation of PCSK9 by miR-191, miR-222, and miR-224. Front. Genet. 2017, 8, 189. [Google Scholar] [CrossRef] [PubMed]
- Sanada, F.; Kanbara, Y.; Taniyama, Y.; Otsu, R.; Carracedo, M.; Ikeda-Iwabu, Y.; Muratsu, J.; Sugimoto, K.; Yamamoto, K.; Rakugi, H.; et al. Induction of Angiogenesis by a Type III Phosphodiesterase Inhibitor, Cilostazol, Through Activation of Peroxisome Proliferator-Activated Receptor-gamma and cAMP Pathways in Vascular Cells. Arter. Thromb. Vasc. Biol. 2016, 36, 545–552. [Google Scholar] [CrossRef]
- Yanai, H.; Yoshida, H. Beneficial Effects of Adiponectin on Glucose and Lipid Metabolism and Atherosclerotic Progression: Mechanisms and Perspectives. Int. J. Mol. Sci. 2019, 20, 1190. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.-C.; Tseng, W.-K.; Li, Y.-H.; Tseng, S.-Y.; Liu, P.-Y.; Chao, T.-H. Effect of cilostazol on plasma levels of proprotein convertase subtilisin/kexin type 9. Oncotarget 2017, 8, 108042–108053. [Google Scholar] [CrossRef] [PubMed]
- Elam, M.B.; Heckman, J.; Crouse, J.R.; Hunninghake, D.B.; Herd, J.A.; Davidson, M.; Gordon, I.L.; Bortey, E.B.; Forbes, W.P. Effect of the novel antiplatelet agent cilostazol on plasma lipoproteins in patients with intermittent claudication. Arter. Thromb. Vasc. Biol. 1998, 18, 1942–1947. [Google Scholar] [CrossRef] [PubMed]
- Tani, T.; Uehara, K.; Sudo, T.; Marukawa, K.; Yasuda, Y.; Kimura, Y. Cilostazol, a selective type III phosphodiesterase inhibitor, decreases triglyceride and increases HDL cholesterol levels by increasing lipoprotein lipase activity in rats. Atherosclerosis 2000, 152, 299–305. [Google Scholar] [CrossRef]
- Ikewaki, K.; Mochizuki, K.; Iwasaki, M.; Nishide, R.; Mochizuki, S.; Tada, N. Cilostazol, a potent phosphodiesterase type III inhibitor, selectively increases antiatherogenic high-density lipoprotein subclass LpA-I and improves postprandial lipemia in patients with type 2 diabetes mellitus. Metabolism 2002, 51, 1348–1354. [Google Scholar] [CrossRef]
- Oh, Y.J.; Kim, H.Y.; Lee, M.H.; Suh, S.H.; Choi, Y.; Nam, T.-G.; Kwon, W.Y.; Lee, S.Y.; Yoo, Y.H. Cilostazol Improves HFD-Induced Hepatic Steatosis by Upregulating Hepatic STAMP2 Expression through AMPK. Mol. Pharmacol. 2018, 94, 1401–1411. [Google Scholar] [CrossRef]
- Lin, J.-L.; Tseng, W.-K.; Lee, P.-T.; Lee, C.-H.; Tseng, S.-Y.; Chen, P.-W.; Chang, H.-Y.; Chao, T.-H. A Randomized Controlled Trial Evaluating Outcome Impact of Cilostazol in Patients with Coronary Artery Disease or at a High Risk of Cardiovascular Disease. J. Pers. Med. 2022, 12, 938. [Google Scholar] [CrossRef]
- Hwang, L.-C.; Bai, C.-H.; Chen, C.-J. Prevalence of Obesity and Metabolic Syndrome in Taiwan. J. Formos. Med. Assoc. 2006, 105, 626–635. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, P.-W.; Tseng, S.-Y.; Chang, H.-Y.; Lee, C.-H.; Chao, T.-H. Diverse Effects of Cilostazol on Proprotein Convertase Subtilisin/Kexin Type 9 between Obesity and Non-Obesity. Int. J. Mol. Sci. 2022, 23, 9768. https://doi.org/10.3390/ijms23179768
Chen P-W, Tseng S-Y, Chang H-Y, Lee C-H, Chao T-H. Diverse Effects of Cilostazol on Proprotein Convertase Subtilisin/Kexin Type 9 between Obesity and Non-Obesity. International Journal of Molecular Sciences. 2022; 23(17):9768. https://doi.org/10.3390/ijms23179768
Chicago/Turabian StyleChen, Po-Wei, Shih-Ya Tseng, Hsien-Yuan Chang, Cheng-Han Lee, and Ting-Hsing Chao. 2022. "Diverse Effects of Cilostazol on Proprotein Convertase Subtilisin/Kexin Type 9 between Obesity and Non-Obesity" International Journal of Molecular Sciences 23, no. 17: 9768. https://doi.org/10.3390/ijms23179768
APA StyleChen, P.-W., Tseng, S.-Y., Chang, H.-Y., Lee, C.-H., & Chao, T.-H. (2022). Diverse Effects of Cilostazol on Proprotein Convertase Subtilisin/Kexin Type 9 between Obesity and Non-Obesity. International Journal of Molecular Sciences, 23(17), 9768. https://doi.org/10.3390/ijms23179768