



Remodeling of the Neurovascular Unit Following Cerebral Ischemia and Hemorrhage
 ,
,  and
and
Abstract
:1. Introduction
2. The Structural Components of the Neurovascular Unit (NVU)
2.1. Neurons
2.2. Endothelial Cells
2.3. Pericytes
2.4. Astrocytes
2.5. Microglia and Macrophages
2.6. Junctional Complexes
2.7. Basement Membrane
3. Processes and Signaling Pathways Involved in the Development and Remodeling of NVU
3.1. Formation of Blood Vessels (Vasculogenesis and Angiogenesis)
3.2. Maturation of Blood Vessels (Barriergenesis)
3.3. Arteriogenesis
4. Remodeling of NVU after Ischemic Stroke
4.1. The Effect of Stroke on Endothelial Cells in the NVU
4.2. The Effect of Stroke on Pericytes in the NVU
4.3. The Effect of Stroke on Astrocytes in the NVU
4.4. The Effect of Stroke on Microglia and Macrophages in the NVU
4.5. The Effect of Stroke on the Basement Membrane
5. Predisposition of NVU to Vascular Risk Factors
5.1. Diabetes and Hyperglycemia
5.2. Hypertension
5.3. Hyperlipidemia
5.4. Aging
5.5. Sex
6. Remodeling of NVU after Cerebral Hemorrhage
6.1. Structural NVU Remodeling following Cerebral Hemorrhage
6.1.1. Endothelial Cells/Tight Junctions (TJs) Disruption/BBB
6.1.2. Pericytes
6.1.3. Astrocytes
6.1.4. Microglia
6.1.5. Neurons
6.1.6. ECM
6.2. Risk Factors for Hemorrhagic Stroke
6.2.1. Hypertension
6.2.2. Vascular Malformation
AVM
CCM
DAVFs
6.2.3. Intracranial Aneurysms (IAs)
Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AJ | Adherens Junction |
| ANG II | Angiotensin II |
| Ang-1 | Angiopoietin-1 |
| ApoE | Apolipoprotein E |
| AVM | Arteriovenous Malformation |
| bAVM | Brain Arteriovenous Malformation |
| BBB | Blood Brain Barrier |
| BM | Basement Membrane |
| Cav-1 | Caveolin-1 |
| CBF | Cerebral Blood Flow |
| CCM | Cerebral Cavernous Malformation |
| CNS | Central Nervous System |
| CSD | Cortical Spreading Depolarization |
| CSFs | Colony-Stimulating Factors |
| DAMP | Damage-associated Molecular-pattern |
| DAVF | Dural Arteriovenous Fistulae |
| EC | Endothelial Cell |
| ECM | Extracellular Matrix |
| eNOS | Endothelial NO Synthase |
| FSS | Fluid Sheer Stress |
| GFAP | Glial Fibrillary Acidic Protein |
| GJ | Gap Junction |
| GPCR124 | G-Protein Coupled Receptor 124 |
| HIF-1α | Hypoxia-inducible Faction 1-alpha |
| HMGB1 | High-mobility Group Box 1 protein |
| HSPG | Heparan Sulfate Proteoglycan |
| IA | Intracranial Aneurysm |
| ICH | Intracerebral Hemorrhage |
| ICP | Intracranial Pressure |
| IL | Interleukin |
| IVH | Intraventricular Hemorrhage |
| LAP | Latency Associated Peptide |
| MCAO | Middle Cerebral Artery Occlusion |
| MFSD2A | Major Facilitator Superfamily Domain Containing 2A |
| MHC-II | Major Histocompatibility Complex II |
| MMP | Metalloproteinase |
| NF-kB | Nuclear Factor-kB |
| NGF | Nerve Growth Factor |
| NO | Nitric Oxide |
| NVC | Neurovascular Coupling |
| NVU | Neurovascular Unit |
| P2RY12 | Purinergic Receptor P2Y, G-protein Coupled 12 |
| PDGF | Platelet-Derived Growth Factor |
| PGE2 | Prostaglandin E2 |
| PTN | Pleiotrophin |
| PVM | Perivascular Macrophage |
| ROCK | Rho-Associated Coiled-coil Kinase |
| ROS | Reactive Oxygen Species |
| SAH | Subarachnoid Hemorrhage |
| Shh | Sonic Hedgehog |
| SHR | Spontaneously Hypertensive Rat |
| SSeCKS | SRc-suppressed C-Kinase Substrate |
| TGF-β | Transforming Growth Factor-β |
| TJ | Tight Junction |
| tMCAO | transient Middle Cerebral Artery Occlusion |
| TNF-α | Tumor Necrosis Factor-α |
| TRPV4 | Transient Receptor Potential Vanilloid-type 4 |
| VEGF | Vascular Endothelial Growth Factor |
| VH | Venous Hypertension |
| VSMC | Vascular Smooth Muscle Cell |
| ZO | Zona Occludins |
References
- Iadecola, C. The Neurovascular Unit Coming of Age: A Journey through Neurovascular Coupling in Health and Disease. Neuron 2017, 96, 17–42. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic, B.V. The Blood-Brain Barrier in Health and Chronic Neurodegenerative Disorders. Neuron 2008, 57, 178–201. [Google Scholar] [CrossRef] [PubMed]
- Obermeier, B.; Daneman, R.; Ransohoff, R.M. Development, maintenance and disruption of the blood-brain barrier. Nat. Med. 2013, 19, 1584–1596. [Google Scholar] [CrossRef]
- Ben-Menachem, E.; Johansson, B.B.; Svensson, T.H. Increased vulnerability of the blood-brain barrier to acute hypertension following depletion of brain noradrenaline. J. Neural Transm. 1982, 53, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Cohen, Z.; Molinatti, G.; Hamel, E. Astroglial and Vascular Interactions of Noradrenaline Terminals in the Rat Cerebral Cortex. J. Cereb. Blood Flow Metab. 1997, 17, 894–904. [Google Scholar] [CrossRef] [PubMed]
- Cohen, Z.; Bonvento, G.; Lacombe, P.; Hamel, E. Serotonin in the regulation of brain microcirculation. Prog. Neurobiol. 1996, 50, 335–362. [Google Scholar] [CrossRef]
- Vaucher, E.; Hamel, E. Cholinergic basal forebrain neurons project to cortical microvessels in the rat: Electron microscopic study with anterogradely transported Phaseolus vulgaris leucoagglutinin and choline acetyltransferase immunocytochemistry. J. Neurosci. 1995, 15, 7427–7441. [Google Scholar] [CrossRef]
- Tong, X.K.; Hamel, E. Regional cholinergic denervation of cortical microvessels and nitric oxide synthase-containing neurons in Alzheimer’s disease. Neuroscience 1999, 92, 163–175. [Google Scholar] [CrossRef]
- Vaucher, E.; Tong, X.K.; Cholet, N.; Lantin, S.; Hamel, E. GABA neurons provide a rich input to microvessels but not nitric oxide neurons in the rat cerebral cortex: A means for direct regulation of local cerebral blood flow. J. Comp. Neurol. 2000, 421, 161–171. [Google Scholar] [CrossRef]
- Buxton, R.B.; Frank, L.R. A Model for the Coupling between Cerebral Blood Flow and Oxygen Metabolism during Neural Stimulation. J. Cereb. Blood Flow Metab. 1997, 17, 64–72. [Google Scholar] [CrossRef]
- Chow, B.W.; Gu, C. The Molecular Constituents of the Blood–Brain Barrier. Trends Neurosci. 2015, 38, 598–608. [Google Scholar] [CrossRef] [PubMed]
- Delaney, C.; Campbell, M. The blood brain barrier: Insights from development and ageing. Tissue Barriers 2017, 5, e1373897. [Google Scholar] [CrossRef]
- De Bock, M.; Leybaert, L.; Giaume, C. Connexin Channels at the Glio-Vascular Interface: Gatekeepers of the Brain. Neurochem. Res. 2017, 42, 2519–2536. [Google Scholar] [CrossRef] [PubMed]
- Nagasawa, K.; Chiba, H.; Fujita, H.; Kojima, T.; Saito, T.; Endo, T.; Sawada, N. Possible involvement of gap junctions in the barrier function of tight junctions of brain and lung endothelial cells. J. Cell. Physiol. 2006, 208, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Reese, T.S.; Karnovsky, M.J. Fine structural localization of a blood-brain barrier to exogenous peroxidase. J. Cell Biol. 1967, 34, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Tso, M.K.; Macdonald, R.L. Subarachnoid hemorrhage: A review of experimental studies on the microcirculation and the neurovascular unit. Transl. Stroke Res. 2014, 5, 174–189. [Google Scholar] [CrossRef]
- Zou, J.; Chen, Z.; Wei, X.; Chen, Z.; Fu, Y.; Yang, X.; Chen, D.; Wang, R.; Jenner, P.; Lu, J.; et al. Cystatin C as a potential therapeutic mediator against Parkinson’s disease via VEGF-induced angiogenesis and enhanced neuronal autophagy in neurovascular units. Cell Death Dis. 2017, 8, e2854. [Google Scholar] [CrossRef]
- Sanz, E.; Yang, L.; Su, T.; Morris, D.R.; McKnight, G.S.; Amieux, P.S. Cell-type-specific isolation of ribosome-associated mRNA from complex tissues. Proc. Natl. Acad. Sci. USA 2009, 106, 13939–13944. [Google Scholar] [CrossRef]
- Jambusaria, A.; Hong, Z.; Zhang, L.; Srivastava, S.; Jana, A.; Toth, P.; Dai, Y.; Malik, A.B.; Rehman, J. Endothelial heterogeneity across distinct vascular beds during homeostasis and inflammation. eLife 2020, 9, e51413. [Google Scholar] [CrossRef]
- Erdő, F.; Krajcsi, P. Age-Related Functional and Expressional Changes in Efflux Pathways at the Blood-Brain Barrier. Front. Aging Neurosci. 2019, 11, 196. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, D.A.; Coelho-Santos, V.; Shih, A.Y. Pericyte Control of Blood Flow Across Microvascular Zones in the Central Nervous System. Annu. Rev. Physiol. 2022, 84, 331–354. [Google Scholar] [CrossRef] [PubMed]
- Ornelas, S.; Berthiaume, A.-A.; Bonney, S.K.; Coelho-Santos, V.; Underly, R.G.; Kremer, A.; Guérin, C.J.; Lippens, S.; Shih, A.Y. Three-dimensional ultrastructure of the brain pericyte-endothelial interface. J. Cereb. Blood Flow Metab. 2021, 41, 2185–2200. [Google Scholar] [CrossRef] [PubMed]
- Winkler, E.A.; Bell, R.D.; Zlokovic, B.V. Central nervous system pericytes in health and disease. Nat. Neurosci. 2011, 14, 1398–1405. [Google Scholar] [CrossRef] [PubMed]
- Potjewyd, G.; Kellett, K.A.; Hooper, N.M. 3D hydrogel models of the neurovascular unit to investigate blood–brain barrier dysfunction. Neuronal Signal. 2021, 5, NS20210027. [Google Scholar] [CrossRef]
- Bell, R.D.; Winkler, E.A.; Sagare, A.P.; Singh, I.; LaRue, B.; Deane, R.; Zlokovic, B.V. Pericytes Control Key Neurovascular Functions and Neuronal Phenotype in the Adult Brain and during Brain Aging. Neuron 2010, 68, 409–427. [Google Scholar] [CrossRef]
- Armulik, A.; Genové, G.; Mäe, M.; Nisancioglu, M.H.; Wallgard, E.; Niaudet, C.; He, L.; Norlin, J.; Lindblom, P.; Strittmatter, K.; et al. Pericytes regulate the blood–brain barrier. Nature 2010, 468, 557–561. [Google Scholar] [CrossRef]
- Winkler, E.A.; Sagare, A.P.; Zlokovic, B.V. The pericyte: A forgotten cell type with important implications for Alzheimer’s disease? Brain Pathol. 2014, 24, 371–386. [Google Scholar] [CrossRef]
- Wang, L.; Xiong, X.; Zhang, L.; Shen, J. Neurovascular Unit: A critical role in ischemic stroke. CNS Neurosci. Ther. 2021, 27, 7–16. [Google Scholar] [CrossRef]
- Hall, C.N.; Reynell, C.; Gesslein, B.; Hamilton, N.B.; Mishra, A.; Sutherland, B.A.; O’Farrell, F.M.; Buchan, A.M.; Lauritzen, M.; Attwell, D. Capillary pericytes regulate cerebral blood flow in health and disease. Nature 2014, 508, 55–60. [Google Scholar] [CrossRef]
- Hill, R.A.; Tong, L.; Yuan, P.; Murikinati, S.; Gupta, S.; Grutzendler, J. Regional Blood Flow in the Normal and Ischemic Brain Is Controlled by Arteriolar Smooth Muscle Cell Contractility and Not by Capillary Pericytes. Neuron 2015, 87, 95–110. [Google Scholar] [CrossRef] [Green Version]
- Cai, W.; Liu, H.; Zhao, J.; Chen, L.; Chen, J.; Lu, Z.; Hu, X. Pericytes in Brain Injury and Repair after Ischemic Stroke. Transl. Stroke Res. 2017, 8, 107–121. [Google Scholar] [CrossRef]
- Yemisci, M.; Gürsoy-Ozdemir, Y.; Vural, A.; Can, A.; Topalkara, K.; Dalkara, T. Pericyte contraction induced by oxidative-nitrative stress impairs capillary reflow despite successful opening of an occluded cerebral artery. Nat. Med. 2009, 15, 1031–1037. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, D.A.; Berthiaume, A.-A.; Grant, R.I.; Harrill, S.A.; Koski, T.; Tieu, T.; McDowell, K.P.; Faino, A.V.; Kelly, A.L.; Shih, A.Y. Brain capillary pericytes exert a substantial but slow influence on blood flow. Nat. Neurosci. 2021, 24, 633–645. [Google Scholar] [CrossRef]
- Vanlandewijck, M.; He, L.; Mäe, M.A.; Andrae, J.; Ando, K.; Del Gaudio, F.; Nahar, K.; Lebouvier, T.; Laviña, B.; Gouveia, L.; et al. A molecular atlas of cell types and zonation in the brain vasculature. Nature 2018, 554, 475–480. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.; Ayyadurai, S.; Zlokovic, B.V. Pericytes of the neurovascular unit: Key functions and signaling pathways. Nat. Neurosci. 2016, 19, 771–783. [Google Scholar] [CrossRef] [PubMed]
- Nakagomi, T.; Kubo, S.; Nakano-Doi, A.; Sakuma, R.; Lu, S.; Narita, A.; Kawahara, M.; Taguchi, A.; Matsuyama, T. Brain Vascular Pericytes Following Ischemia Have Multipotential Stem Cell Activity to Differentiate into Neural and Vascular Lineage Cells. Stem Cells 2015, 33, 1962–1974. [Google Scholar] [CrossRef]
- Berthiaume, A.A.; Schmid, F.; Stamenkovic, S.; Santos, V.; Nielson, C.; Weber, B.; Majesky, M.; Shih, A. Deficiency in pericyte remodeling as a basis for impaired capillary flow and structure during brain aging. bioRxiv 2022. [Google Scholar] [CrossRef]
- Alarcon-Martinez, L.; Villafranca-Baughman, D.; Quintero, H.; Kacerovsky, J.B.; Dotigny, F.; Murai, K.K.; Prat, A.; Drapeau, P.; Di Polo, A. Interpericyte tunnelling nanotubes regulate neurovascular coupling. Nature 2020, 585, 91–95. [Google Scholar] [CrossRef]
- VanGilder, R.L.; Rosen, C.L.; Barr, T.L.; Huber, J.D. Targeting the neurovascular unit for treatment of neurological disorders. Pharmacol. Ther. 2011, 130, 239–247. [Google Scholar] [CrossRef]
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C.; Nedergaard, M. Glial regulation of the cerebral microvasculature. Nat. Neurosci. 2007, 10, 1369–1376. [Google Scholar] [CrossRef] [PubMed]
- Attwell, D.; Buchan, A.M.; Charpak, S.; Lauritzen, M.J.; MacVicar, B.A.; Newman, E.A. Glial and neuronal control of brain blood flow. Nature 2010, 468, 232–243. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Linden, D.J.; Sapirstein, A. Astrocyte Inositol Triphosphate Receptor Type 2 and Cytosolic Phospholipase A2 Alpha Regulate Arteriole Responses in Mouse Neocortical Brain Slices. PLoS ONE 2012, 7, e42194. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-W.; Kim, W.J.; Choi, Y.K.; Song, H.S.; Son, M.J.; Gelman, I.H.; Kim, Y.-J.; Kim, K.-W. SSeCKS regulates angiogenesis and tight junction formation in blood-brain barrier. Nat. Med. 2003, 9, 900–906. [Google Scholar] [CrossRef]
- McAllister, M.S.; Krizanac-Bengez, L.; Macchia, F.; Naftalin, R.; Pedley, K.C.; Mayberg, M.; Marroni, M.; Leaman, S.; Stanness, K.A.; Janigro, D. Mechanisms of glucose transport at the blood–brain barrier: An in vitro study. Brain Res. 2001, 904, 20–30. [Google Scholar] [CrossRef]
- Mathiisen, T.M.; Lehre, K.P.; Danbolt, N.C.; Ottersen, O.P. The perivascular astroglial sheath provides a complete covering of the brain microvessels: An electron microscopic 3D reconstruction. Glia 2010, 58, 1094–1103. [Google Scholar] [CrossRef] [PubMed]
- Simard, M.; Arcuino, G.; Takano, T.; Liu, Q.S.; Nedergaard, M. Signaling at the Gliovascular Interface. J. Neurosci. 2003, 23, 9254–9262. [Google Scholar] [CrossRef] [PubMed]
- Romanos, J.; Benke, D.; Saab, A.S.; Zeilhofer, H.U.; Santello, M. Differences in glutamate uptake between cortical regions impact neuronal NMDA receptor activation. Commun. Biol. 2019, 2, 127. [Google Scholar] [CrossRef]
- Laird, D.W.; Naus, C.C.; Lampe, P.D. SnapShot: Connexins and Disease. Cell 2017, 170, 1260–1260.e1. [Google Scholar] [CrossRef]
- Freitas-Andrade, M.; Wang, N.; Bechberger, J.F.; De Bock, M.; Lampe, P.D.; Leybaert, L.; Naus, C.C. Targeting MAPK phosphorylation of Connexin43 provides neuroprotection in stroke. J. Exp. Med. 2019, 216, 916–935. [Google Scholar] [CrossRef]
- Freitas-Andrade, M.; Bechberger, J.; Wang, J.; Yeung, K.K.; Whitehead, S.N.; Hansen, R.S.; Naus, C.C. Danegaptide Enhances Astrocyte Gap Junctional Coupling and Reduces Ischemic Reperfusion Brain Injury in Mice. Biomolecules 2020, 10, 353. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Allaman, I.; Bélanger, M.; Magistretti, P.J. Astrocyte–neuron metabolic relationships: For better and for worse. Trends Neurosci. 2011, 34, 76–87. [Google Scholar] [CrossRef]
- Lan, X.; Han, X.; Li, Q.; Yang, Q.-W.; Wang, J. Modulators of microglial activation and polarization after intracerebral haemorrhage. Nat. Rev. Neurol. 2017, 13, 420–433. [Google Scholar] [CrossRef] [PubMed]
- Jayaraj, R.L.; Azimullah, S.; Beiram, R.; Jalal, F.Y.; Rosenberg, G.A. Neuroinflammation: Friend and foe for ischemic stroke. J. Neuroinflamm. 2019, 16, 142. [Google Scholar] [CrossRef] [PubMed]
- Dickson, D.W.; Mattiace, L.A.; Kure, K.; Hutchins, K.; Lyman, W.D.; Brosnan, C.F. Microglia in human disease, with an emphasis on acquired immune deficiency syndrome. Lab. Investig. 1991, 64, 135–156. [Google Scholar] [PubMed]
- Jiang, X.; Andjelkovic, A.V.; Zhu, L.; Yang, T.; Bennett, M.V.L.; Chen, J.; Keep, R.F.; Shi, Y. Blood-brain barrier dysfunction and recovery after ischemic stroke. Prog. Neurobiol. 2018, 163–164, 144–171. [Google Scholar] [CrossRef]
- Liang, Z.; Wang, X.; Hao, Y.; Qiu, L.; Lou, Y.; Zhang, Y.; Ma, D.; Feng, J. The Multifaceted Role of Astrocyte Connexin 43 in Ischemic Stroke Through Forming Hemichannels and Gap Junctions. Front. Neurol. 2020, 11, 703. [Google Scholar] [CrossRef]
- Speth, C.; Dierich, M.P.; Sopper, S. HIV-infection of the central nervous system: The tightrope walk of innate immunity. Mol. Immunol. 2005, 42, 213–228. [Google Scholar] [CrossRef]
- Aloisi, F. Immune function of microglia. Glia 2001, 36, 165–179. [Google Scholar] [CrossRef]
- Hines, D.J.; Hines, R.M.; Mulligan, S.J.; Macvicar, B.A. Microglia processes block the spread of damage in the brain and require functional chloride channels. Glia 2009, 57, 1610–1618. [Google Scholar] [CrossRef] [PubMed]
- Petersen, M.A.; Ryu, J.K.; Akassoglou, K. Fibrinogen in neurological diseases: Mechanisms, imaging and therapeutics. Nat. Rev. Neurosci. 2018, 19, 283–301. [Google Scholar] [CrossRef] [PubMed]
- Cummins, P.M. Occludin: One Protein, Many Forms. Mol. Cell. Biol. 2012, 32, 242–250. [Google Scholar] [CrossRef]
- Steinbacher, T.; Kummer, D.; Ebnet, K. Junctional adhesion molecule-A: Functional diversity through molecular promiscuity. Cell. Mol. Life Sci. 2018, 75, 1393–1409. [Google Scholar] [CrossRef] [PubMed]
- Andresen, J.; Shafi, N.I.; Bryan, R.M. Endothelial influences on cerebrovascular tone. J. Appl. Physiol. 2006, 100, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Hartsock, A.; Nelson, W.J. Adherens and tight junctions: Structure, function and connections to the actin cytoskeleton. Biochim. Biophys. Acta 2008, 1778, 660–669. [Google Scholar] [CrossRef]
- Hallmann, R.; Zhang, X.; Di Russo, J.; Li, L.; Song, J.; Hannocks, M.-J.; Sorokin, L. The regulation of immune cell trafficking by the extracellular matrix. Curr. Opin. Cell Biol. 2015, 36, 54–61. [Google Scholar] [CrossRef]
- Kalluri, R. Basement membranes: Structure, assembly and role in tumour angiogenesis. Nat. Rev. Cancer 2003, 3, 422–433. [Google Scholar] [CrossRef]
- Sorokin, L.M. The impact of the extracellular matrix on inflammation. Nat. Rev. Immunol. 2010, 10, 712–723. [Google Scholar] [CrossRef]
- Yurchenco, P.D.; Amenta, P.S.; Patton, B.L. Basement membrane assembly, stability and activities observed through a developmental lens. Matrix Biol. 2004, 22, 521–538. [Google Scholar] [CrossRef]
- Bell, A.H.; Miller, S.L.; Castillo-Melendez, M.; Malhotra, A. The Neurovascular Unit: Effects of Brain Insults During the Perinatal Period. Front. Neurosci. 2020, 13, 1452. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, F.L.; Brites, D.; Brito, M.A. Looking at the blood–brain barrier: Molecular anatomy and possible investigation approaches. Brain Res. Rev. 2010, 64, 328–363. [Google Scholar] [CrossRef] [PubMed]
- del Zoppo, G.; Milner, R.; Mabuchi, T.; Hung, S.; Wang, X.; Koziol, J. Vascular matrix adhesion and the blood–brain barrier. Biochem. Soc. Trans. 2006, 34 Pt 6, 1261–1266. [Google Scholar] [CrossRef] [PubMed]
- Gautam, J.; Zhang, X.; Yao, Y. The role of pericytic laminin in blood brain barrier integrity maintenance. Sci. Rep. 2016, 6, 36450. [Google Scholar] [CrossRef] [PubMed]
- Gautam, J.; Miner, J.H.; Yao, Y. Loss of Endothelial Laminin alpha5 Exacerbates Hemorrhagic Brain Injury. Transl. Stroke Res. 2019, 10, 705–718. [Google Scholar] [CrossRef]
- Lee, H.S.; Han, J.; Bai, H.-J.; Kim, K.-W. Brain angiogenesis in developmental and pathological processes: Regulation, molecular and cellular communication at the neurovascular interface. FEBS J. 2009, 276, 4622–4635. [Google Scholar] [CrossRef]
- Yang, Y.; Torbey, M.T. Angiogenesis and Blood-Brain Barrier Permeability in Vascular Remodeling after Stroke. Curr. Neuropharmacol. 2020, 18, 1250–1265. [Google Scholar] [CrossRef]
- Leung, D.W.; Cachianes, G.; Kuang, W.-J.; Goeddel, D.V.; Ferrara, N. Vascular Endothelial Growth Factor Is a Secreted Angiogenic Mitogen. Science 1989, 246, 1306–1309. [Google Scholar] [CrossRef] [PubMed]
- Ruhrberg, C.; Bautch, V.L. Neurovascular development and links to disease. Cell. Mol. Life Sci. 2013, 70, 1675–1684. [Google Scholar] [CrossRef]
- Abhinand, C.S.; Raju, R.; Soumya, S.J.; Arya, P.S.; Sudhakaran, P.R. VEGF-A/VEGFR2 signaling network in endothelial cells relevant to angiogenesis. J. Cell Commun. Signal. 2016, 10, 347–354. [Google Scholar] [CrossRef] [Green Version]
- Shalaby, F.; Rossant, J.; Yamaguchi, T.P.; Gertsenstein, M.; Wu, X.-F.; Breitman, M.L.; Schuh, A.C. Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature 1995, 376, 62–66. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Shaligram, S.S.; Su, H. Brain vascular biology. Handb. Clin. Neurol. 2021, 176, 49–69. [Google Scholar] [PubMed]
- Allinson, K.R.; Lee, H.S.; Fruttiger, M.; McCarty, J.; Arthur, H.M. Endothelial expression of TGFbeta type II receptor is required to maintain vascular integrity during postnatal development of the central nervous system. PLoS ONE 2012, 7, e39336. [Google Scholar] [CrossRef]
- Pandur, P.; Läsche, M.; Eisenberg, L.M.; Kühl, M. Wnt-11 activation of a non-canonical Wnt signalling pathway is required for cardiogenesis. Nature 2002, 418, 636–641. [Google Scholar] [CrossRef]
- Anderson, K.D.; Pan, L.; Yang, X.-M.; Hughes, V.C.; Walls, J.R.; Dominguez, M.G.; Simmons, M.V.; Burfeind, P.; Xue, Y.; Wei, Y.; et al. Angiogenic sprouting into neural tissue requires Gpr124, an orphan G protein-coupled receptor. Proc. Natl. Acad. Sci. USA 2011, 108, 2807–2812. [Google Scholar] [CrossRef] [PubMed]
- Kuhnert, F.; Mancuso, M.R.; Shamloo, A.; Wang, H.-T.; Choksi, V.; Florek, M.; Su, H.; Fruttiger, M.; Young, W.L.; Heilshorn, S.C.; et al. Essential Regulation of CNS Angiogenesis by the Orphan G Protein–Coupled Receptor GPR124. Science 2010, 330, 985–989. [Google Scholar] [CrossRef]
- Daneman, R.; Zhou, L.; Kebede, A.A.; Barres, B.A. Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature 2010, 468, 562–566. [Google Scholar] [CrossRef]
- Abe, M.; Oda, N.; Sato, Y.; Shibata, K.; Yamasaki, M. Augmented binding and activation of latent transforming growth factor-beta by a tryptic fragment of latency associated peptide. Endothelium 2002, 9, 25–36. [Google Scholar] [CrossRef]
- Walshe, T.E.; Saint-Geniez, M.; Maharaj, A.S.; Sekiyama, E.; Maldonado, A.E.; D’Amore, P.A. TGF-beta is required for vascular barrier function, endothelial survival and homeostasis of the adult microvasculature. PLoS ONE 2009, 4, e5149. [Google Scholar] [CrossRef]
- Tallquist, M.D.; French, W.J.; Soriano, P.; Nusse, R. Additive effects of PDGF receptor beta signaling pathways in vascular smooth muscle cell development. PLoS Biol. 2003, 1, E52. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.-L.; Yao, Y.; Norris, E.H.; Kruyer, A.; Jno-Charles, O.; Akhmerov, A.; Strickland, S. Ablation of astrocytic laminin impairs vascular smooth muscle cell function and leads to hemorrhagic stroke. J. Cell Biol. 2013, 202, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Liebner, S.; Corada, M.; Bangsow, T.; Babbage, J.; Taddei, A.; Czupalla, C.J.; Reis, M.; Felici, A.; Wolburg, H.; Fruttiger, M.; et al. Wnt/beta-catenin signaling controls development of the blood-brain barrier. J. Cell Biol. 2008, 183, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Qi, Y.; Sun, Z. The Role of Sonic Hedgehog Pathway in the Development of the Central Nervous System and Aging-Related Neurodegenerative Diseases. Front. Mol. Biosci. 2021, 8, 711710. [Google Scholar] [CrossRef]
- Farmer, W.T.; Abrahamsson, T.; Chierzi, S.; Lui, C.; Zaelzer, C.; Jones, E.V.; Bally, B.P.; Chen, G.G.; Théroux, J.-F.; Peng, J.; et al. Neurons diversify astrocytes in the adult brain through sonic hedgehog signaling. Science 2016, 351, 849–854. [Google Scholar] [CrossRef] [PubMed]
- Dohle, E.; Fuchs, S.; Kolbe, M.; Hofmann, A.; Schmidt, H.; Kirkpatrick, C.J. Sonic Hedgehog Promotes Angiogenesis and Osteogenesis in a Coculture System Consisting of Primary Osteoblasts and Outgrowth Endothelial Cells. Tissue Eng. Part A 2010, 16, 1235–1237. [Google Scholar] [CrossRef]
- Suri, C.; Jones, P.F.; Patan, S.; Bartunkova, S.; Maisonpierre, P.C.; Davis, S.; Sato, T.N.; Yancopoulos, G.D. Requisite Role of Angiopoietin-1, a Ligand for the TIE2 Receptor, during Embryonic Angiogenesis. Cell 1996, 87, 1171–1180. [Google Scholar] [CrossRef]
- Bell, R.D.; Winkler, E.A.; Singh, I.; Sagare, A.P.; Deane, R.; Wu, Z.; Holtzman, D.M.; Betsholtz, C.; Armulik, A.; Sallstrom, J.; et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature 2012, 485, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, Y.; Akamatsu, Y.; Lee, C.C.; Stetler, R.A.; Lawton, M.T.; Yang, G.-Y. Vascular remodeling after ischemic stroke: Mechanisms and therapeutic potentials. Prog. Neurobiol. 2014, 115, 138–156. [Google Scholar] [CrossRef]
- Liebeskind, D.S. Collateral Perfusion: Time for Novel Paradigms in Cerebral Ischemia. Int. J. Stroke 2012, 7, 309–310. [Google Scholar] [CrossRef]
- Kaloss, A.M.; Theus, M.H. Leptomeningeal anastomoses: Mechanisms of pial collateral remodeling in ischemic stroke. WIREs Mech. Dis. 2022, 14, e1553. [Google Scholar] [CrossRef]
- Nishijima, Y.; Akamatsu, Y.; Weinstein, P.R.; Liu, J. Collaterals: Implications in cerebral ischemic diseases and therapeutic interventions. Brain Res. 2015, 1623, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chalothorn, D.; Faber, J.E. Collateral Vessels Have Unique Endothelial and Smooth Muscle Cell Phenotypes. Int. J. Mol. Sci. 2019, 20, 3608. [Google Scholar] [CrossRef] [PubMed]
- Bang, O.Y.; Goyal, M.; Liebeskind, D.S. Collateral Circulation in Ischemic Stroke: Assessment Tools and Therapeutic Strategies. Stroke 2015, 46, 3302–3309. [Google Scholar] [CrossRef] [PubMed]
- Liebeskind, D.S. Collateral circulation. Stroke 2003, 34, 2279–2284. [Google Scholar] [CrossRef]
- Faber, J.E.; Chilian, W.M.; Deindl, E.; van Royen, N.; Simons, M. A brief etymology of the collateral circulation. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1854–1859. [Google Scholar] [CrossRef] [PubMed]
- Schaper, W. Collateral circulation: Past and present. Basic Res. Cardiol. 2009, 104, 5–21. [Google Scholar] [CrossRef] [PubMed]
- Grundmann, S.; Piek, J.J.; Pasterkamp, G.; Hoefer, I.E. Arteriogenesis: Basic mechanisms and therapeutic stimulation. Eur. J. Clin. Investig. 2007, 37, 755–766. [Google Scholar] [CrossRef]
- Heil, M.; Schaper, W. Influence of Mechanical, Cellular, and Molecular Factors on Collateral Artery Growth (Arteriogenesis). Circ. Res. 2004, 95, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Scholz, D.; Ito, W.; Fleming, I.; Deindl, E.; Sauer, A.; Wiesnet, M.; Busse, R.; Schaper, J. Ultrastructure and molecular histology of rabbit hind-limb collateral artery growth (arteriogenesis). Virchows Arch. 2000, 436, 257–270. [Google Scholar] [CrossRef] [PubMed]
- Christoforidis, G.A.; Mohammad, Y.; Kehagias, D.; Avutu, B.; Slivka, A.P. Angiographic assessment of pial collaterals as a prognostic indicator following intra-arterial thrombolysis for acute ischemic stroke. AJNR Am. J. Neuroradiol. 2005, 26, 1789–1797. [Google Scholar]
- Hayden, M.R.; Tyagi, S.C. Arteriogenesis: Angiogenesis within Unstable Atherosclerotic Plaque—Interactions with Extracellular Matrix. Curr. Interv. Cardiol. Rep. 2000, 2, 218–227. [Google Scholar] [PubMed]
- Abacı, A.; Oğuzhan, A.; Kahraman, S.; Eryol, N.K.; Ünal, S.; Arınç, H.; Ergin, A. Effect of Diabetes Mellitus on Formation of Coronary Collateral Vessels. Circulation 1999, 99, 2239–2242. [Google Scholar] [CrossRef]
- Akamatsu, Y.; Nishijima, Y.; Lee, C.C.; Yang, S.Y.; Shi, L.; An, L.; Wang, R.K.; Tominaga, T.; Liu, J. Impaired Leptomeningeal Collateral Flow Contributes to the Poor Outcome following Experimental Stroke in the Type 2 Diabetic Mice. J. Neurosci. 2015, 35, 3851–3864. [Google Scholar] [CrossRef]
- Nishijima, Y.; Akamatsu, Y.; Yang, S.Y.; Lee, C.C.; Baran, U.; Song, S.; Wang, R.; Tominaga, T.; Liu, J. Impaired Collateral Flow Compensation During Chronic Cerebral Hypoperfusion in the Type 2 Diabetic Mice. Stroke 2016, 47, 3014–3021. [Google Scholar] [CrossRef]
- Menon, B.K.; Smith, E.E.; Coutts, S.B.; Welsh, D.G.; Faber, J.E.; Goyal, M.; Hill, M.D.; Demchuk, A.M.; Damani, Z.; Cho, K.-H.; et al. Leptomeningeal collaterals are associated with modifiable metabolic risk factors. Ann. Neurol. 2013, 74, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Campbell, B.C.; Christensen, S.; Tress, B.M.; Churilov, L.; Desmond, P.M.; Parsons, M.W.; Barber, P.A.; Levi, C.; Bladin, C.; Donnan, G.; et al. Failure of Collateral Blood Flow is Associated with Infarct Growth in Ischemic Stroke. J. Cereb. Blood Flow Metab. 2013, 33, 1168–1172. [Google Scholar] [CrossRef]
- Ma, J.; Ma, Y.; Shuaib, A.; Winship, I.R. Impaired Collateral Flow in Pial Arterioles of Aged Rats During Ischemic Stroke. Transl. Stroke Res. 2020, 11, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Murtha, L.; Mcleod, D.; Pepperall, D.; McCann, S.; Beard, D.J.; Tomkins, A.J.; Holmes, W.M.; McCabe, C.; Macrae, I.M.; Spratt, N.J. Intracranial Pressure Elevation after Ischemic Stroke in Rats: Cerebral Edema is Not the Only Cause, and Short-Duration Mild Hypothermia is a Highly Effective Preventive Therapy. J. Cereb. Blood Flow Metab. 2015, 35, 592–600. [Google Scholar] [CrossRef]
- Beard, D.J.; Mcleod, D.; Logan, C.L.; Murtha, L.; Imtiaz, M.S.; Van Helden, D.F.; Spratt, N.J. Intracranial Pressure Elevation Reduces Flow through Collateral Vessels and the Penetrating Arterioles they Supply. a Possible Explanation for ‘Collateral Failure’ and Infarct Expansion after Ischemic Stroke. J. Cereb. Blood Flow Metab. 2015, 35, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Beard, D.J.; Murtha, L.A.; McLeod, D.D.; Spratt, N.J. Intracranial Pressure and Collateral Blood Flow. Stroke 2016, 47, 1695–1700. [Google Scholar] [CrossRef] [PubMed]
- Liu, J. Poststroke angiogenesis: Blood, bloom, or brood? Stroke 2015, 46, e105–e106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boengler, K.; Pipp, F.; Fernandez, B.; Ziegelhoeffer, T.; Schaper, W.; Deindl, E. Arteriogenesis is associated with an induction of the cardiac ankyrin repeat protein (carp). Cardiovasc. Res. 2003, 59, 573–581. [Google Scholar] [CrossRef]
- van Royen, N.; Hoefer, I.; Buschmann, I.; Heil, M.; Kostin, S.; Deindl, E.; Vogel, S.; Korff, T.; Augustin, H.; Bode, C.; et al. Exogenous application of transforming growth factor beta 1 stimulates arteriogenesis in the peripheral circulation. FASEB J. 2002, 16, 432–434. [Google Scholar] [CrossRef] [PubMed]
- Seiler, C.; Pohl, T.; Wustmann, K.; Hutter, D.; Nicolet, P.-A.; Windecker, S.; Eberli, F.R.; Meier, B. Promotion of collateral growth by granulocyte-macrophage colony-stimulating factor in patients with coronary artery disease: A randomized, double-blind, placebo-controlled study. Circulation 2001, 104, 2012–2017. [Google Scholar] [CrossRef]
- Buschmann, I.R.; Busch, H.J.; Mies, G.; Hossmann, K.A. Therapeutic induction of arteriogenesis in hypoperfused rat brain via granulocyte-macrophage colony-stimulating factor. Circulation 2003, 108, 610–615. [Google Scholar] [CrossRef]
- Chen, J.; Cui, X.; Zacharek, A.; Ding, G.; Shehadah, A.; Jiang, Q.; Lu, M.; Chopp, M. Niaspan treatment increases tumor necrosis factor-alpha-converting enzyme and promotes arteriogenesis after stroke. J. Cereb. Blood Flow Metab. 2009, 29, 911–920. [Google Scholar] [CrossRef]
- Jiao, H.; Wang, Z.; Liu, Y.; Wang, P.; Xue, Y. Specific role of tight junction proteins claudin-5, occludin, and ZO-1 of the blood-brain barrier in a focal cerebral ischemic insult. J. Mol. Neurosci. 2011, 44, 130–139. [Google Scholar] [CrossRef]
- Knowland, D.; Arac, A.; Sekiguchi, K.J.; Hsu, M.; Lutz, S.E.; Perrino, J.; Steinberg, G.K.; Barres, B.A.; Nimmerjahn, A.; Agalliu, D. Stepwise Recruitment of Transcellular and Paracellular Pathways Underlies Blood-Brain Barrier Breakdown in Stroke. Neuron 2014, 82, 603–617. [Google Scholar] [CrossRef]
- Ben-Zvi, A.; Lacoste, B.; Kur, E.; Andreone, B.J.; Mayshar, Y.; Yan, H.; Gu, C. Mfsd2a is critical for the formation and function of the blood–brain barrier. Nature 2014, 509, 507–511. [Google Scholar] [CrossRef]
- Andreone, B.J.; Chow, B.W.; Tata, A.; Lacoste, B.; Ben-Zvi, A.; Bullock, K.; Deik, A.A.; Ginty, D.D.; Clish, C.B.; Gu, C. Blood-Brain Barrier Permeability Is Regulated by Lipid Transport-Dependent Suppression of Caveolae-Mediated Transcytosis. Neuron 2017, 94, 581–594.e5. [Google Scholar] [CrossRef]
- Haley, M.J.; Lawrence, C.B. The blood–brain barrier after stroke: Structural studies and the role of transcytotic vesicles. J. Cereb. Blood Flow Metab. 2017, 37, 456–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nahirney, P.C.; Reeson, P.; Brown, C.E. Ultrastructural analysis of blood–brain barrier breakdown in the peri-infarct zone in young adult and aged mice. J. Cereb. Blood Flow Metab. 2016, 36, 413–425. [Google Scholar] [CrossRef] [PubMed]
- Carbonell, T.; Rama, R. Iron, oxidative stress and early neurological deterioration in ischemic stroke. Curr. Med. Chem. 2007, 14, 857–874. [Google Scholar] [CrossRef]
- Zhang, Y.; Cui, Q.; Chen, H.; Li, J. Rho kinase: A new target for treatment of cerebral ischemia/reperfusion injury. Neural Regen. Res. 2013, 8, 1180. [Google Scholar] [CrossRef]
- Gonul, E.; Duz, B.; Kahraman, S.; Kayali, H.; Kubar, A.; Timurkaynak, E. Early Pericyte Response to Brain Hypoxia in Cats: An Ultrastructural Study. Microvasc. Res. 2002, 64, 116–119. [Google Scholar] [CrossRef]
- Arimura, K.; Ago, T.; Kamouchi, M.; Nakamura, K.; Ishitsuka, K.; Kuroda, J.; Sugimori, H.; Ooboshi, H.; Sasaki, T.; Kitazono, T. PDGF receptor beta signaling in pericytes following ischemic brain injury. Curr. Neurovasc. Res. 2012, 9, 1–9. [Google Scholar] [CrossRef]
- Fernández-Klett, F.; Potas, J.; Hilpert, D.; Blazej, K.; Radke, J.; Huck, J.; Engel, O.; Stenzel, W.; Genové, G.; Priller, J. Early Loss of Pericytes and Perivascular Stromal Cell-Induced Scar Formation after Stroke. J. Cereb. Blood Flow Metab. 2013, 33, 428–439. [Google Scholar] [CrossRef]
- Underly, R.G.; Levy, M.; Hartmann, D.A.; Grant, R.I.; Watson, A.N.; Shih, A.Y. Pericytes as Inducers of Rapid, Matrix Metalloproteinase-9-Dependent Capillary Damage during Ischemia. J. Neurosci. 2017, 37, 129–140. [Google Scholar] [CrossRef]
- Nikolakopoulou, A.M.; Montagne, A.; Kisler, K.; Dai, Z.; Wang, Y.; Huuskonen, M.T.; Sagare, A.P.; Lazic, D.; Sweeney, M.D.; Kong, P.; et al. Pericyte loss leads to circulatory failure and pleiotrophin depletion causing neuron loss. Nat. Neurosci. 2019, 22, 1089–1098. [Google Scholar] [CrossRef]
- Chow, J.; Ogunshola, O.; Fan, S.Y.; Li, Y.; Ment, L.R.; Madri, J.A. Astrocyte-derived VEGF mediates survival and tube stabilization of hypoxic brain microvascular endothelial cells in vitro. Dev. Brain. Res. 2001, 130, 123–132. [Google Scholar] [CrossRef]
- Li, L.; Lundkvist, A.; Andersson, D.; Wilhelmsson, U.; Nagai, N.; Pardo, A.; Nodin, C.; Ståhlberg, A.; Aprico, K.; Larsson, K.; et al. Protective Role of Reactive Astrocytes in Brain Ischemia. J. Cereb. Blood Flow Metab. 2008, 28, 468–481. [Google Scholar] [CrossRef] [Green Version]
- Krum, J.M.; Mani, N.; Rosenstein, J.M. Roles of the endogenous VEGF receptors flt-1 and flk-1 in astroglial and vascular remodeling after brain injury. Exp. Neurol. 2008, 212, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Fordsmann, J.C.; Ko, R.W.Y.; Choi, H.B.; Thomsen, K.; Witgen, B.M.; Mathiesen, C.; Lønstrup, M.; Piilgaard, H.; MacVicar, B.A.; Lauritzen, M. Increased 20-HETE Synthesis Explains Reduced Cerebral Blood Flow but Not Impaired Neurovascular Coupling after Cortical Spreading Depression in Rat Cerebral Cortex. J. Neurosci. 2013, 33, 2562–2570. [Google Scholar] [CrossRef]
- Rovegno, M.; Saez, J.C. Role of astrocyte connexin hemichannels in cortical spreading depression. Biochim. Biophys. Acta Biomembr. 2018, 1860, 216–223. [Google Scholar] [CrossRef] [PubMed]
- Shibahara, T.; Ago, T.; Nakamura, K.; Tachibana, M.; Yoshikawa, Y.; Komori, M.; Yamanaka, K.; Wakisaka, Y.; Kitazono, T. Pericyte-Mediated Tissue Repair through PDGFRbeta Promotes Peri-Infarct Astrogliosis, Oligodendrogenesis, and Functional Recovery after Acute Ischemic Stroke. Eneuro 2020, 7, ENEURO.0474-19.2020. [Google Scholar] [CrossRef]
- Yenari, M.A.; Kauppinen, T.M.; Swanson, R.A. Microglial activation in stroke: Therapeutic targets. Neurotherapeutics 2010, 7, 378–391. [Google Scholar] [CrossRef]
- Jolivel, V.; Bicker, F.; Binamé, F.; Ploen, R.; Keller, S.; Gollan, R.; Jurek, B.; Birkenstock, J.; Poisa-Beiro, L.; Bruttger, J.; et al. Perivascular microglia promote blood vessel disintegration in the ischemic penumbra. Acta Neuropathol. 2015, 129, 279–295. [Google Scholar] [CrossRef] [PubMed]
- Lou, N.; Takano, T.; Pei, Y.; Xavier, A.L.; Goldman, S.A.; Nedergaard, M. Purinergic receptor P2RY12-dependent microglial closure of the injured blood–brain barrier. Proc. Natl. Acad. Sci. USA 2016, 113, 1074–1079. [Google Scholar] [CrossRef]
- Rodrigo, R.; Fernandez-Gajardo, R.; Gutierrez, R.; Matamala, J.M.; Carrasco, R.; Miranda-Merchak, A.; Feuerhake, W. Oxidative Stress and Pathophysiology of Ischemic Stroke: Novel Therapeutic Opportunities. CNS Neurol. Disord. Drug Targets 2013, 12, 698–714. [Google Scholar]
- Xu, C.; Tang, F.; Lu, M.; Yang, J.; Han, R.; Mei, M.; Hu, J.; Zhou, M.; Wang, H. Astragaloside IV improves the isoproterenol-induced vascular dysfunction via attenuating eNOS uncoupling-mediated oxidative stress and inhibiting ROS-NF-kappaB pathways. Int. Immunopharmacol. 2016, 33, 119–127. [Google Scholar] [CrossRef]
- van Leeuwen, E.; Hampton, M.B.; Smyth, L.C. Redox signalling and regulation of the blood-brain barrier. Int. J. Biochem. Cell Biol. 2020, 125, 105794. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, K.; Morizane, Y.; Kamami-Levy, C.; Suzuki, J.; Kayama, M.; Cai, W.; Miller, J.; Vavvas, D.G. AMP-dependent Kinase Inhibits Oxidative Stress-induced Caveolin-1 Phosphorylation and Endocytosis by Suppressing the Dissociation between c-Abl and Prdx1 Proteins in Endothelial Cells. J. Biol. Chem. 2013, 288, 20581–20591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Liao, H.-L.; Niu, X.-L.; Yuan, Y.; Lin, T.; Verna, L.; Stemerman, M.B. Low density lipoprotein induces eNOS translocation to membrane caveolae: The role of RhoA activation and stress fiber formation. Biochim. Biophys. Acta 2003, 1635, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Drab, M.; Verkade, P.; Elger, M.; Kasper, M.; Lohn, M.; Lauterbach, B.; Menne, J.; Lindschau, C.; Mende, F.; Luft, F.C.; et al. Loss of Caveolae, Vascular Dysfunction, and Pulmonary Defects in Caveolin-1 Gene-Disrupted Mice. Science 2001, 293, 2449–2452. [Google Scholar] [CrossRef] [PubMed]
- Ming, X.-F.; Viswambharan, H.; Barandier, C.; Ruffieux, J.; Kaibuchi, K.; Rusconi, S.; Yang, Z. Rho GTPase/Rho Kinase Negatively Regulates Endothelial Nitric Oxide Synthase Phosphorylation through the Inhibition of Protein Kinase B/Akt in Human Endothelial Cells. Mol. Cell. Biol. 2002, 22, 8467–8477. [Google Scholar] [CrossRef]
- Noma, K.; Kihara, Y.; Higashi, Y. Striking crosstalk of ROCK signaling with endothelial function. J. Cardiol. 2012, 60, 1–6. [Google Scholar] [CrossRef]
- Gibson, C.L.; Srivastava, K.; Sprigg, N.; Bath, P.M.W.; Bayraktutan, U. Inhibition of Rho-kinase protects cerebral barrier from ischaemia-evoked injury through modulations of endothelial cell oxidative stress and tight junctions. J. Neurochem. 2014, 129, 816–826. [Google Scholar] [CrossRef]
- Hiroi, Y.; Noma, K.; Kim, H.-H.; Sladojevic, N.; Tabit, C.E.; Li, Y.; Soydan, G.; Salomone, S.; Moskowitz, M.A.; Liao, J.K. Neuroprotection Mediated by Upregulation of Endothelial Nitric Oxide Synthase in Rho-Associated, Coiled-Coil-Containing Kinase 2 Deficient Mice. Circ. J. 2018, 82, 1195–1204. [Google Scholar] [CrossRef]
- O’Donnell, M.E. Blood-brain barrier Na transporters in ischemic stroke. Adv. Pharmacol. 2014, 71, 113–146. [Google Scholar]
- Brzica, H.; Abdullahi, W.; Ibbotson, K.; Ronaldson, P.T. Role of Transporters in Central Nervous System Drug Delivery and Blood-Brain Barrier Protection: Relevance to Treatment of Stroke. J. Central Nerv. Syst. Dis. 2017, 9, 1179573517693802. [Google Scholar] [CrossRef] [PubMed]
- Winkler, E.A.; Nishida, Y.; Sagare, A.P.; Rege, S.V.; Bell, R.D.; Perlmutter, D.; Sengillo, J.D.; Hillman, S.; Kong, P.; Nelson, A.R.; et al. GLUT1 reductions exacerbate Alzheimer’s disease vasculo-neuronal dysfunction and degeneration. Nat. Neurosci. 2015, 18, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Munji, R.N.; Soung, A.L.; Weiner, G.A.; Sohet, F.; Semple, B.D.; Trivedi, A.; Gimlin, K.; Kotoda, M.; Korai, M.; Aydin, S.; et al. Profiling the mouse brain endothelial transcriptome in health and disease models reveals a core blood–brain barrier dysfunction module. Nat. Neurosci. 2019, 22, 1892–1902. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Huang, L.; Qu, Y.; Xiao, D.; Mu, D. Pericytes in Cerebrovascular Diseases: An Emerging Therapeutic Target. Front. Cell. Neurosci. 2019, 13, 519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ElAli, A.; Thériault, P.; Rivest, S. The Role of Pericytes in Neurovascular Unit Remodeling in Brain Disorders. Int. J. Mol. Sci. 2014, 15, 6453–6474. [Google Scholar] [CrossRef] [PubMed]
- Keaney, J.; Campbell, M. The dynamic blood-brain barrier. FEBS J. 2015, 282, 4067–4079. [Google Scholar] [CrossRef]
- Zhao, C.; Ma, J.; Wang, Z.; Li, H.; Shen, H.; Li, X.; Chen, G. Mfsd2a Attenuates Blood-Brain Barrier Disruption After Sub-arachnoid Hemorrhage by Inhibiting Caveolae-Mediated Transcellular Transport in Rats. Transl. Stroke Res. 2020, 11, 1012–1027. [Google Scholar] [CrossRef]
- Chow, B.W.; Gu, C. Gradual Suppression of Transcytosis Governs Functional Blood-Retinal Barrier Formation. Neuron 2017, 93, 1325–1333.e3. [Google Scholar] [CrossRef]
- Peppiatt, C.M.; Howarth, C.; Mobbs, P.; Attwell, D. Bidirectional control of CNS capillary diameter by pericytes. Nature 2006, 443, 700–704. [Google Scholar] [CrossRef]
- Puro, D.G. Physiology and pathobiology of the pericyte-containing retinal microvasculature: New developments. Microcirculation 2007, 14, 1–10. [Google Scholar] [CrossRef]
- Freitas-Andrade, M.; Raman-Nair, J.; Lacoste, B. Structural and Functional Remodeling of the Brain Vasculature Following Stroke. Front. Physiol. 2020, 11, 948. [Google Scholar] [CrossRef]
- Molofsky, A.V.; Krenick, R.; Ullian, E.; Tsai, H.-H.; Deneen, B.; Richardson, W.D.; Barres, B.A.; Rowitch, D.H. Astrocytes and disease: A neurodevelopmental perspective. Genes Dev. 2012, 26, 891–907. [Google Scholar] [CrossRef] [PubMed]
- Clavreul, S.; Abdeladim, L.; Hernández-Garzón, E.; Niculescu, D.; Durand, J.; Ieng, S.-H.; Barry, R.; Bonvento, G.; Beaurepaire, E.; Livet, J.; et al. Cortical astrocytes develop in a plastic manner at both clonal and cellular levels. Nat. Commun. 2019, 10, 4884. [Google Scholar] [CrossRef] [PubMed]
- Batiuk, M.Y.; Martirosyan, A.; Wahis, J.; de Vin, F.; Marneffe, C.; Kusserow, C.; Koeppen, J.; Viana, J.F.; Oliveira, J.F.; Voet, T.; et al. Identification of region-specific astrocyte subtypes at single cell resolution. Nat. Commun. 2020, 11, 1220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Bornstädt, D.; Houben, T.; Seidel, J.L.; Zheng, Y.; Dilekoz, E.; Qin, T.; Sandow, N.; Kura, S.; Eikermann-Haerter, K.; Endres, M.; et al. Supply-demand mismatch transients in susceptible peri-infarct hot zones explain the origins of spreading injury depolarizations. Neuron 2015, 85, 1117–1131. [Google Scholar] [CrossRef]
- Nakamura, H.; Strong, A.J.; Dohmen, C.; Sakowitz, O.W.; Vollmar, S.; Sué, M.; Kracht, L.; Hashemi, P.; Bhatia, R.; Yoshimine, T.; et al. Spreading depolarizations cycle around and enlarge focal ischaemic brain lesions. Brain 2010, 133 Pt 7, 1994–2006. [Google Scholar] [CrossRef]
- Lauritzen, M.; Strong, A.J. ‘Spreading depression of Leao’ and its emerging relevance to acute brain injury in humans. J. Cereb. Blood Flow Metab. 2017, 37, 1553–1570. [Google Scholar] [CrossRef]
- Beck, H.; Semisch, M.; Culmsee, C.; Plesnila, N.; Hatzopoulos, A.K. Egr-1 Regulates Expression of the Glial Scar Component Phosphacan in Astrocytes after Experimental Stroke. Am. J. Pathol. 2008, 173, 77–92. [Google Scholar] [CrossRef]
- Parkhurst, C.N.; Yang, G.; Ninan, I.; Savas, J.N.; Yates, J.R., III; Lafaille, J.J.; Hempstead, B.L.; Littman, D.R.; Gan, W.-B. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 2013, 155, 1596–1609. [Google Scholar] [CrossRef]
- Haruwaka, K.; Ikegami, A.; Tachibana, Y.; Ohno, N.; Konishi, H.; Hashimoto, A.; Matsumoto, M.; Kato, D.; Ono, R.; Kiyama, H.; et al. Dual microglia effects on blood brain barrier permeability induced by systemic inflammation. Nat. Commun. 2019, 10, 5816. [Google Scholar] [CrossRef]
- Eldahshan, W.; Fagan, S.C.; Ergul, A. Inflammation within the neurovascular unit: Focus on microglia for stroke injury and recovery. Pharmacol. Res. 2019, 147, 104349. [Google Scholar] [CrossRef]
- Rawlinson, C.; Jenkins, S.; Thei, L.; Dallas, M.L.; Chen, R. Post-Ischaemic Immunological Response in the Brain: Targeting Microglia in Ischaemic Stroke Therapy. Brain Sci. 2020, 10, 159. [Google Scholar] [CrossRef] [PubMed]
- Holfelder, K.; Schittenhelm, J.; Trautmann, K.; Haybaeck, J.; Meyermann, R.; Beschorner, R. De novo expression of the hemoglobin scavenger receptor CD163 by activated microglia is not associated with hemorrhages in human brain lesions. Histol. Histopathol. 2011, 26, 1007–1017. [Google Scholar] [PubMed]
- Pedragosa, J.; Salas-Perdomo, A.; Gallizioli, M.; Cugota, R.; Miro-Mur, F.A.; Briansó, F.; Justicia, C.; Pérez-Asensio, F.; Marquez, L.; Urra, X.; et al. CNS-border associated macrophages respond to acute ischemic stroke attracting granulocytes and promoting vascular leakage. Acta Neuropathol. Commun. 2018, 6, 76. [Google Scholar] [PubMed] [Green Version]
- Koizumi, T.; Kerkhofs, D.; Mizuno, T.; Steinbusch, H.W.M.; Foulquier, S. Vessel-Associated Immune Cells in Cerebrovascular Diseases: From Perivascular Macrophages to Vessel-Associated Microglia. Front. Neurosci. 2019, 13, 1291. [Google Scholar] [CrossRef] [PubMed]
- Faraco, G.; Sugiyama, Y.; Lane, D.; Garcia-Bonilla, L.; Chang, H.; Santisteban, M.; Racchumi, G.; Murphy, M.; Van Rooijen, N.; Anrather, J.; et al. Perivascular macrophages mediate the neurovascular and cognitive dysfunction associated with hypertension. J. Clin. Investig. 2016, 126, 4674–4689. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.; Yao, Y. Basement Membrane Changes in Ischemic Stroke. Stroke 2020, 51, 1344–1352. [Google Scholar] [CrossRef]
- Thomsen, M.S.; Routhe, L.J.; Moos, T. The vascular basement membrane in the healthy and pathological brain. J. Cereb. Blood Flow Metab. 2017, 37, 3300–3317. [Google Scholar] [CrossRef]
- Hangai, M.; Kitaya, N.; Xu, J.; Chan, C.K.; Kim, J.J.; Werb, Z.; Ryan, S.J.; Brooks, P.C. Matrix Metalloproteinase-9-Dependent Exposure of a Cryptic Migratory Control Site in Collagen is Required before Retinal Angiogenesis. Am. J. Pathol. 2002, 161, 1429–1437. [Google Scholar] [CrossRef]
- Yao, Y. Basement membrane and stroke. J. Cereb. Blood Flow Metab. 2019, 39, 3–19. [Google Scholar] [CrossRef]
- Kwon, I.; Kim, E.H.; del Zoppo, G.J.; Heo, J.H. Ultrastructural and temporal changes of the microvascular basement membrane and astrocyte interface following focal cerebral ischemia. J. Neurosci. Res. 2009, 87, 668–676. [Google Scholar] [CrossRef]
- Heo, J.H.; Lucero, J.; Abumiya, T.; Koziol, J.A.; Copeland, B.R.; del Zoppo, G.J. Matrix Metalloproteinases Increase Very Early during Experimental Focal Cerebral Ischemia. J. Cereb. Blood Flow Metab. 1999, 19, 624–633. [Google Scholar] [CrossRef]
- Becker, B.F.; Jacob, M.; Leipert, S.; Salmon, A.H.J.; Chappell, D. Degradation of the endothelial glycocalyx in clinical settings: Searching for the sheddases. Br. J. Clin. Pharmacol. 2015, 80, 389–402. [Google Scholar] [CrossRef] [PubMed]
- Steiner, E.; Enzmann, G.U.; Lyck, R.; Lin, S.; Rüegg, M.A.; Kröger, S.; Engelhardt, B. The heparan sulfate proteoglycan agrin contributes to barrier properties of mouse brain endothelial cells by stabilizing adherens junctions. Cell Tissue Res. 2014, 358, 465–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwata, A.; Koike, F.; Arasaki, K.; Tamaki, M. Blood brain barrier destruction in hyperglycemic chorea in a patient with poorly controlled diabetes. J. Neurol. Sci. 1999, 163, 90–93. [Google Scholar] [CrossRef]
- Starr, J.M.; Wardlaw, J.; Ferguson, K.; MacLullich, A.; Deary, I.J.; Marshall, I. Increased blood-brain barrier permeability in type II diabetes demonstrated by gadolinium magnetic resonance imaging. J. Neurol. Neurosurg. Psychiatry 2003, 74, 70–76. [Google Scholar] [CrossRef]
- Liao, Y.J.; Ueno, M.; Nakagawa, T.; Huang, C.; Kanenishi, K.; Onodera, M.; Sakamoto, H. Oxidative damage in cerebral vessels of diabetic db/db mice. Diabetes/Metabolism Res. Rev. 2005, 21, 554–559. [Google Scholar] [CrossRef]
- Mooradian, A.D.; Haas, M.J.; Batejko, O.; Hovsepyan, M.; Feman, S.S. Statins Ameliorate Endothelial Barrier Permeability Changes in the Cerebral Tissue of Streptozotocin-Induced Diabetic Rats. Diabetes 2005, 54, 2977–2982. [Google Scholar] [CrossRef]
- Huber, J.D.; VanGilder, R.L.; Houser, K.A. Streptozotocin-induced diabetes progressively increases blood-brain barrier permeability in specific brain regions in rats. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H2660–H2668. [Google Scholar] [CrossRef]
- Hawkins, B.T.; Lundeen, T.F.; Norwood, K.M.; Brooks, H.L.; Egleton, R.D. Increased blood-brain barrier permeability and altered tight junctions in experimental diabetes in the rat: Contribution of hyperglycaemia and matrix metalloproteinases. Diabetologia 2007, 50, 202–211. [Google Scholar] [CrossRef]
- Shao, B.; Bayraktutan, U. Hyperglycaemia promotes cerebral barrier dysfunction through activation of protein kinase C-β. Diabetes, Obes. Metab. 2013, 15, 993–999. [Google Scholar] [CrossRef]
- Sun, Y.-N.; Liu, L.-B.; Xue, Y.-X.; Wang, P. Effects of insulin combined with idebenone on blood-brain barrier permeability in diabetic rats. J. Neurosci. Res. 2015, 93, 666–677. [Google Scholar] [CrossRef]
- Chao, A.; Lee, T.; Juo, S.H.; Yang, D. Hyperglycemia Increases the Production of Amyloid Beta-Peptide Leading to Decreased Endothelial Tight Junction. CNS Neurosci. Ther. 2016, 22, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Fujihara, R.; Chiba, Y.; Nakagawa, T.; Nishi, N.; Murakami, R.; Matsumoto, K.; Kawauchi, M.; Yamamoto, T.; Ueno, M. Albumin microvascular leakage in brains with diabetes mellitus. Microsc. Res. Tech. 2016, 79, 833–837. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, S.; Nakagawa, S.; Tatsumi, R.; Morofuji, Y.; Takeshita, T.; Hayashi, K.; Tanaka, K.; Matsuo, T.; Niwa, M. Glucagon-Like Peptide-1 Strengthens the Barrier Integrity in Primary Cultures of Rat Brain Endothelial Cells Under Basal and Hyperglycemia Conditions. J. Mol. Neurosci. 2016, 59, 211–219. [Google Scholar] [CrossRef]
- Yoo, D.Y.; Yim, H.S.; Jung, H.Y.; Nam, S.M.; Kim, J.W.; Choi, J.H.; Seong, J.K.; Yoon, Y.S.; Kim, D.W.; Hwang, I.K. Chronic type 2 diabetes reduces the integrity of the blood-brain barrier by reducing tight junction proteins in the hippocampus. J. Vet. Med. Sci. 2016, 78, 957–962. [Google Scholar] [CrossRef]
- Xu, Z.; Zeng, W.; Sun, J.; Chen, W.; Zhang, R.; Yang, Z.; Yao, Z.; Wang, L.; Song, L.; Chen, Y.; et al. The quantification of blood-brain barrier disruption using dynamic contrast-enhanced magnetic resonance imaging in aging rhesus monkeys with spontaneous type 2 diabetes mellitus. NeuroImage 2017, 158, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Zanotto, C.; Simão, F.; Gasparin, M.S.; Biasibetti, R.; Tortorelli, L.S.; Nardin, P.; Gonçalves, C.-A. Exendin-4 Reverses Biochemical and Functional Alterations in the Blood–Brain and Blood–CSF Barriers in Diabetic Rats. Mol. Neurobiol. 2017, 54, 2154–2166. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.R. Type 2 Diabetes Mellitus Increases the Risk of Late-Onset Alzheimer’s Disease: Ultrastructural Remodeling of the Neurovascular Unit and Diabetic Gliopathy. Brain Sci. 2019, 9, 262. [Google Scholar] [CrossRef]
- Friker, L.L.; Scheiblich, H.; Hochheiser, I.V.; Brinkschulte, R.; Riedel, D.; Latz, E.; Geyer, M.; Heneka, M.T. β-Amyloid Clustering around ASC Fibrils Boosts Its Toxicity in Microglia. Cell Rep. 2020, 30, 3743–3754.e6. [Google Scholar] [CrossRef]
- Baumbach, G.L.; Sigmund, C.D.; Faraci, F.M. Cerebral arteriolar structure in mice overexpressing human renin and angiotensinogen. Hypertension 2003, 41, 50–55. [Google Scholar] [CrossRef]
- Ueno, M.; Sakamoto, H.; Tomimoto, H.; Akiguchi, I.; Onodera, M.; Huang, C.-L.; Kanenishi, K. Blood-brain barrier is impaired in the hippocampus of young adult spontaneously hypertensive rats. Acta Neuropathol. 2004, 107, 532–538. [Google Scholar] [CrossRef] [PubMed]
- Pelisch, N.; Hosomi, N.; Mori, H.; Masaki, T.; Nishiyama, A. RAS Inhibition Attenuates Cognitive Impairment by Reducing Blood- Brain Barrier Permeability in Hypertensive Subjects. Curr. Hypertens. Rev. 2013, 9, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, M.T.; Dehghani, G.A. Acute hypertension induces brain injury and blood–brain barrier disruption through reduction of claudins mRNA expression in rat. Pathol. Res. Prac. 2014, 210, 985–990. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Yang, X.; Tao, Y.; Lan, L.; Zheng, L.; Sun, J. Tight junction disruption of blood–brain barrier in white matter lesions in chronic hypertensive rats. NeuroReport 2015, 26, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Tayebati, S.K.; Tomassoni, D.; Amenta, F. Neuroinflammatory Markers in Spontaneously Hypertensive Rat Brain: An Immunohistochemical Study. CNS Neurol. Disord. Drug Targets 2016, 15, 995–1000. [Google Scholar] [CrossRef]
- Diaz, J.R.; Kim, K.J.; Brands, M.W.; Filosa, J.A. Augmented astrocyte microdomain Ca2+ dynamics and parenchymal arteriole tone in angiotensin II-infused hypertensive mice. Glia 2019, 67, 551–565. [Google Scholar] [CrossRef]
- Methia, N.; André, P.; Hafezi-Moghadam, A.; Economopoulos, M.; Thomas, K.L.; Wagner, D.D. ApoE Deficiency Compromises the Blood Brain Barrier Especially After Injury. Mol. Med. 2001, 7, 810–815. [Google Scholar] [CrossRef]
- Nishitsuji, K.; Hosono, T.; Nakamura, T.; Bu, G.; Michikawa, M. Apolipoprotein E Regulates the Integrity of Tight Junctions in an Isoform-dependent Manner in an in Vitro Blood-Brain Barrier Model. J. Biol. Chem. 2011, 286, 17536–17542. [Google Scholar] [CrossRef]
- Ng, K.F.; Anderson, S.; Mayo, P.; Aung, H.H.; Walton, J.H.; Rutledge, J.C. Characterizing blood–brain barrier perturbations after exposure to human triglyceride-rich lipoprotein lipolysis products using MRI in a rat model. Magn. Reson. Med. 2016, 76, 1246–1251. [Google Scholar] [CrossRef]
- Mooradian, A.D. Blood-brain barrier transport of choline is reduced in the aged rat. Brain Res. 1988, 440, 328–332. [Google Scholar] [CrossRef]
- Elahy, M.; Jackaman, C.; Mamo, J.C.; Lam, V.; Dhaliwal, S.S.; Giles, C.; Nelson, D.; Takechi, R. Blood–brain barrier dysfunction developed during normal aging is associated with inflammation and loss of tight junctions but not with leukocyte recruitment. Immun. Ageing 2015, 12, 2. [Google Scholar]
- Montagne, A.; Barnes, S.R.; Sweeney, M.D.; Halliday, M.R.; Sagare, A.P.; Zhao, Z.; Toga, A.W.; Jacobs, R.E.; Liu, C.Y.; Amezcua, L.; et al. Blood-Brain Barrier Breakdown in the Aging Human Hippocampus. Neuron 2015, 85, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Bake, S.; Sohrabji, F. 17beta-estradiol differentially regulates blood-brain barrier permeability in young and aging female rats. Endocrinology 2004, 145, 5471–5475. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.C.; Clemente, L.; Liu, T.; Bowen, R.L.; Meethal, S.V.; Atwood, C.S. Reproductive hormones regulate the selective permeability of the blood-brain barrier. Biochim. Biophys. Acta 2008, 1782, 401–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burek, M.; Arias-Loza, P.A.; Roewer, N.; Förster, C.Y. Claudin-5 as a Novel Estrogen Target in Vascular Endothelium. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Maggioli, E.; McArthur, S.; Mauro, C.; Kieswich, J.; Kusters, D.H.M.; Reutelingsperger, C.P.M.; Yaqoob, M.; Solito, E. Estrogen protects the blood-brain barrier from inflammation-induced disruption and increased lymphocyte trafficking. Brain Behav. Immun. 2016, 51, 212–222. [Google Scholar] [CrossRef] [PubMed]
- Faraci, F.M.; Baumbach, G.L.; Heistad, D.D. Cerebral circulation: Humoral regulation and effects of chronic hypertension. J. Am. Soc. Nephrol. 1990, 1, 53–57. [Google Scholar] [CrossRef]
- Heistad, D.D.; Baumbach, G.L. Cerebral vascular changes during chronic hypertension: Good guys and bad guys. J. Hypertens. Suppl. 1992, 10, S71–S75. [Google Scholar] [CrossRef]
- Dorrance, A.M.; Rupp, N.C.; Nogueira, E.F. Mineralocorticoid receptor activation causes cerebral vessel remodeling and exacerbates the damage caused by cerebral ischemia. Hypertension 2006, 47, 590–595. [Google Scholar] [CrossRef]
- Mulvany, M.J.; Baumbach, G.L.; Aalkjaer, C.; Heagerty, A.M.; Korsgaard, N.; Schiffrin, E.L.; Heistad, D.D. Vascular remodeling. Hypertension 1996, 28, 505–506. [Google Scholar]
- Prado, C.M.; Ramos, S.G.; Alves-Filho, J.C.; Elias, J.; Cunha, F.Q.; Rossi, M.A. Turbulent flow/low wall shear stress and stretch differentially affect aorta remodeling in rats. J. Hypertens. 2006, 24, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Knox, C.A.; Yates, R.D.; Chen, I.L.; Klara, P.M. Effects of aging on the structural and permeability characteristics of cerebrovasculature in normotensive and hypertensive strains of rats. Acta Neuropathol. 1980, 51, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Fredriksson, K.; Kalimo, H.; Westergren, I.; Kåhrström, J.; Johansson, B.B. Blood-brain barrier leakage and brain edema in stroke-prone spontaneously hypertensive rats. Effect of chronic sympathectomy and low protein/high salt diet. Acta Neuropathol. 1987, 74, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Wu, G.; Fan, C.; Xu, J.; Jiang, S.; Yan, X.; Di, S.; Ma, Z.; Hu, W.; Yang, Y. The emerging role of signal transducer and activator of transcription 3 in cerebral ischemic and hemorrhagic stroke. Prog. Neurobiol. 2016, 137, 1–16. [Google Scholar] [CrossRef]
- Plump, A.S.; Smith, J.D.; Hayek, T.; Aalto-Setälä, K.; Walsh, A.; Verstuyft, J.G.; Rubin, E.M.; Breslow, J.L. Severe hypercholesterolemia and atherosclerosis in apolipoprotein E-deficient mice created by homologous recombination in ES cells. Cell 1992, 71, 343–353. [Google Scholar] [CrossRef]
- Dias, I.H.; Polidori, M.C.; Griffiths, H.R. Hypercholesterolaemia-induced oxidative stress at the blood–brain barrier. Biochem. Soc. Trans. 2014, 42, 1001–1005. [Google Scholar] [CrossRef]
- Farkas, E.; Luiten, P.G. Cerebral microvascular pathology in aging and Alzheimer’s disease. Prog. Neurobiol. 2001, 64, 575–611. [Google Scholar] [CrossRef]
- Farrall, A.J.; Wardlaw, J.M. Blood-brain barrier: Ageing and microvascular disease—systematic review and meta-analysis. Neurobiol. Aging 2009, 30, 337–352. [Google Scholar] [CrossRef] [PubMed]
- Mooradian, A.D. Effect of aging on the blood-brain barrier. Neurobiol. Aging 1988, 9, 31–39. [Google Scholar] [CrossRef]
- Mooradian, A.D.; Meredith, K.E. The effect of age on protein composition of rat cerebral microvessels. Neurochem. Res. 1992, 17, 665–670. [Google Scholar] [CrossRef]
- Mooradian, A.D.; Smith, T.L. The effect of experimentally induced diabetes mellitus on the lipid order and composition of rat cerebral microvessels. Neurosci. Lett. 1992, 145, 145–148. [Google Scholar] [CrossRef]
- Franceschi, C.; Campisi, J. Chronic Inflammation (Inflammaging) and Its Potential Contribution to Age-Associated Diseases. J. Gerontol. A Ser. Biol. Sci. Med. Sci. 2014, 69 (Suppl. 1), S4–S9. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, N.; Chiba, Y.; Hosono, M.; Fujii, M.; Kawamura, N.; Keino, H.; Yoshikawa, K.; Ishii, S.; Saitoh, Y.; Satoh, M.; et al. Involvement of pro-inflammatory cytokines and microglia in an age-associated neurodegeneration model, the SAMP10 mouse. Brain Res. 2007, 1185, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Donkor, E.S. Stroke in the 21(st) Century: A Snapshot of the Burden, Epidemiology, and Quality of Life. Stroke Res. Treat 2018, 2018, 3238165. [Google Scholar]
- Montano, A.; Hanley, D.F.; Hemphill, J.C., 3rd. Hemorrhagic stroke. Handb. Clin. Neurol. 2021, 176, 229–248. [Google Scholar]
- Cordonnier, C.; Demchuk, A.; Ziai, W.; Anderson, C.S. Intracerebral haemorrhage: Current approaches to acute management. Lancet 2018, 392, 1257–1268. [Google Scholar] [CrossRef]
- Godoy, D.A.; Piñero, G.R.; Koller, P.; Masotti, L.; Di Napoli, M. Steps to consider in the approach and management of critically ill patient with spontaneous intracerebral hemorrhage. World J. Crit. Care Med. 2015, 4, 213–229. [Google Scholar] [CrossRef]
- Topkoru, B.; Egemen, E.; Solaroglu, I.; Zhang, J.H.; Topkoru, E.E.B. Early Brain Injury or Vasospasm? An Overview of Common Mechanisms. Curr. Drug Targets 2017, 18, 1424–1429. [Google Scholar] [CrossRef]
- Tao, X.; Yang, W.; Zhu, S.; Que, R.; Liu, C.; Fan, T.; Wang, J.; Mo, D.; Zhang, Z.; Tan, J.; et al. Models of poststroke depression and assessments of core depressive symptoms in rodents: How to choose? Exp. Neurol. 2019, 322, 113060. [Google Scholar] [CrossRef]
- Aronowski, J.; Zhao, X. Molecular pathophysiology of cerebral hemorrhage: Secondary brain injury. Stroke 2011, 42, 1781–1786. [Google Scholar] [CrossRef]
- Jośko, J. Cerebral angiogenesis and expression of VEGF after subarachnoid hemorrhage (SAH) in rats. Brain Res. 2003, 981, 58–69. [Google Scholar] [CrossRef]
- Chu, H.; Tang, Y.; Dong, Q. Protection of Vascular Endothelial Growth Factor to Brain Edema Following Intracerebral Hemorrhage and Its Involved Mechanisms: Effect of Aquaporin-4. PLoS ONE 2013, 8, e66051. [Google Scholar]
- Tang, T.; Liu, X.-J.; Zhang, Z.-Q.; Zhou, H.-J.; Luo, J.-K.; Huang, J.-F.; Yang, Q.-D.; Li, X.-Q. Cerebral angiogenesis after collagenase-induced intracerebral hemorrhage in rats. Brain Res. 2007, 1175, 134–142. [Google Scholar] [CrossRef]
- Pan, C.; Liu, N.; Zhang, P.; Wu, Q.; Deng, H.; Xu, F.; Lian, L.; Liang, Q.; Hu, Y.; Zhu, S.; et al. EGb761 Ameliorates Neuronal Apoptosis and Promotes Angiogenesis in Experimental Intracerebral Hemorrhage via RSK1/GSK3beta Pathway. Mol. Neurobiol. 2018, 55, 1556–1567. [Google Scholar] [CrossRef] [PubMed]
- Lei, C.; Lin, S.; Zhang, C.; Tao, W.; Dong, W.; Hao, Z.; Liu, M.; Wu, B. Effects of high-mobility group box1 on cerebral angiogenesis and neurogenesis after intracerebral hemorrhage. Neuroscience 2013, 229, 12–19. [Google Scholar] [CrossRef]
- Xi, G.; Keep, R.F.; Hoff, J.T. Mechanisms of brain injury after intracerebral haemorrhage. Lancet Neurol. 2006, 5, 53–63. [Google Scholar] [CrossRef]
- Taylor, R.A.; Sansing, L.H. Microglial Responses after Ischemic Stroke and Intracerebral Hemorrhage. Clin. Dev. Immunol. 2013, 2013, 746068. [Google Scholar] [CrossRef]
- Wang, J.; Doré, S. Inflammation after intracerebral hemorrhage. J. Cereb. Blood Flow Metab. 2007, 27, 894–908. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-M.; Yin, K.-J.; Hsin, I.; Chen, S.; Fryer, J.D.; Holtzman, D.M.; Hsu, C.Y.; Xu, J. Matrix metalloproteinase-9 and spontaneous hemorrhage in an animal model of cerebral amyloid angiopathy. Ann. Neurol. 2003, 54, 379–382. [Google Scholar] [CrossRef]
- Wang, J.; Tsirka, S.E. Neuroprotection by inhibition of matrix metalloproteinases in a mouse model of intracerebral haemorrhage. Brain 2005, 128 Pt 7, 1622–1633. [Google Scholar] [CrossRef]
- Tang, J.; Liu, J.; Zhou, C.; Alexander, J.S.; Nanda, A.; Granger, D.N.; Zhang, J.H. MMP-9 Deficiency Enhances Collagenase-Induced Intracerebral Hemorrhage and Brain Injury in Mutant Mice. J. Cereb. Blood Flow Metab. 2004, 24, 1133–1145. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.Q.; Fang, Z.; Chen, X.L.; Yang, S.; Zhou, Y.F.; Mao, L.; Xia, Y.P.; Jin, H.J.; Li, Y.N.; You, M.F.; et al. Microglia-derived TNF-α mediates endothelial necroptosis aggravating blood brain–barrier disruption after ischemic stroke. Cell Death Dis. 2019, 10, 487. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.; Li, W.; Huang, S.; Yin, Z.; Xu, X.; Chen, F.; Kong, X.; Wang, H.; Zhang, J.; Lei, P. The pathological role of NLRs and AIM2 inflammasome-mediated pyroptosis in damaged blood-brain barrier after traumatic brain injury. Brain Res. 2018, 1697, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Greenwood, J.; Heasman, S.J.; Alvarez, J.I.; Prat, A.; Lyck, R.; Engelhardt, B. Review: Leucocyte-endothelial cell crosstalk at the blood-brain barrier: A prerequisite for successful immune cell entry to the brain. Neuropathol. Appl. Neurobiol. 2011, 37, 24–39. [Google Scholar] [CrossRef]
- Enzmann, D.R.; Britt, R.H.; Lyons, B.E.; Buxton, J.L.; Wilson, D.A. Natural history of experimental intracerebral hemorrhage: Sonography, computed tomography and neuropathology. AJNR Am. J. Neuroradiol. 1981, 2, 517–526. [Google Scholar]
- Gong, C.; Hoff, J.T.; Keep, R.F. Acute inflammatory reaction following experimental intracerebral hemorrhage in rat. Brain Res. 2000, 871, 57–65. [Google Scholar] [CrossRef]
- Gong, Y.; Hua, Y.; Keep, R.F.; Hoff, J.T.; Xi, G. Intracerebral hemorrhage: Effects of aging on brain edema and neurological deficits. Stroke 2004, 35, 2571–2575. [Google Scholar] [CrossRef]
- Friedrich, V.; Flores, R.; Muller, A.; Sehba, F.A. Luminal platelet aggregates in functional deficits in parenchymal vessels after subarachnoid hemorrhage. Brain Res. 2010, 1354, 179–187. [Google Scholar] [CrossRef]
- Rosengart, A.J.; Schultheiss, K.E.; Tolentino, J.; Macdonald, R.L. Prognostic Factors for Outcome in Patients with Aneurysmal Subarachnoid Hemorrhage. Stroke 2007, 38, 2315–2321. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, H.; Ayer, R.; Sugawara, T.; Chen, W.; Sozen, T.; Hasegawa, Y.; Kanamaru, K.; Zhang, J.H. Protective effects of recombinant osteopontin on early brain injury after subarachnoid hemorrhage in rats. Crit. Care Med. 2010, 38, 612–618. [Google Scholar] [CrossRef]
- Gules, I.; Satoh, M.; Nanda, A.; Zhang, J.H. Apoptosis, blood-brain barrier, and subarachnoid hemorrhage. Acta Neurochir. Suppl. 2003, 86, 483–487. [Google Scholar] [PubMed]
- Friedrich, V.; Flores, R.; Muller, A.; Sehba, F. Escape of intraluminal platelets into brain parenchyma after subarachnoid hemorrhage. Neuroscience 2010, 165, 968–975. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Gao, S.; Wang, X.; Cao, Y.; Lu, J.; Chen, S.; Lenahan, C.; Zhang, J.H.; Shao, A.; Zhang, J. Programmed Cell Deaths and Potential Crosstalk with Blood–Brain Barrier Dysfunction After Hemorrhagic Stroke. Front. Cell. Neurosci. 2020, 14, 68. [Google Scholar] [CrossRef]
- Keep, R.F.; Zhou, N.; Xiang, J.; Andjelkovic, A.V.; Hua, Y.; Xi, G. Vascular disruption and blood–brain barrier dysfunction in intracerebral hemorrhage. Fluids Barriers CNS 2014, 11, 18. [Google Scholar] [CrossRef]
- Möller, T.; Weinstein, J.; Hanisch, U.-K. Activation of Microglial Cells by Thrombin: Past, Present, and Future. Semin. Thromb. Hemost. 2006, 32 (Suppl. 1), 69–76. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Liu, X.; Ruan, H.; Chen, Y.; Feng, H. Pericyte: Potential Target for Hemorrhagic Stroke Prevention and Treatment. Curr. Drug Deliv. 2017, 14, 773–784. [Google Scholar] [CrossRef] [PubMed]
- Scimemi, A. Astrocytes and the Warning Signs of Intracerebral Hemorrhagic Stroke. Neural Plast. 2018, 2018, 7301623. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Chopp, M. Astrocytes, therapeutic targets for neuroprotection and neurorestoration in ischemic stroke. Prog. Neurobiol. 2016, 144, 103–120. [Google Scholar] [CrossRef]
- Mestriner, R.G.; Pagnussat, A.S.; Boisserand, L.S.B.; Valentim, L.; Netto, C.A. Skilled reaching training promotes astroglial changes and facilitated sensorimotor recovery after collagenase-induced intracerebral hemorrhage. Exp. Neurol. 2011, 227, 53–61. [Google Scholar] [CrossRef]
- Mestriner, R.G.; Saur, L.; Bagatini, P.B.; Baptista, P.P.A.; Vaz, S.P.; Ferreira, K.; Machado, S.A.; Xavier, L.L.; Netto, C.A. Astrocyte morphology after ischemic and hemorrhagic experimental stroke has no influence on the different recovery patterns. Behav. Brain Res. 2015, 278, 257–261. [Google Scholar] [CrossRef]
- Chen, Y.; Swanson, R.A. Astrocytes and brain injury. J. Cereb. Blood Flow Metab. 2003, 23, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhang, Z.E.; Lu, H.; Yang, Q.; Wu, H.; Wang, J. Microglial Polarization and Inflammatory Mediators After Intracerebral Hemorrhage. Mol. Neurobiol. 2017, 54, 1874–1886. [Google Scholar] [CrossRef] [PubMed]
- Zille, M.; Karuppagounder, S.S.; Chen, Y.; Gough, P.J.; Bertin, J.; Finger, J.; Milner, T.A.; Jonas, E.A.; Ratan, R.R. Neuronal Death After Hemorrhagic Stroke In Vitro and In Vivo Shares Features of Ferroptosis and Necroptosis. Stroke 2017, 48, 1033–1043. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.C.; Zhou, Y.; Fang, H.; Lin, S.; Wang, P.F.; Xiong, R.P.; Chen, J.; Xiong, X.Y.; Lv, F.L.; Liang, Q.L.; et al. Toll-like receptor 2/4 heterodimer mediates inflammatory injury in intracerebral hemorrhage. Ann. Neurol. 2014, 75, 876–889. [Google Scholar] [CrossRef]
- Huang, F.-P.; Xi, G.; Keep, R.F.; Hua, Y.; Nemoianu, A.; Hoff, J.T. Brain edema after experimental intracerebral hemorrhage: Role of hemoglobin degradation products. J. Neurosurg. 2002, 96, 287–293. [Google Scholar] [CrossRef]
- Hickenbottom, S.L.; Grotta, J.C.; Strong, R.; Denner, L.A.; Aronowski, J. Nuclear factor-kappaB and cell death after experimental intracerebral hemorrhage in rats. Stroke 1999, 30, 2472–2477; discussion 2477–2478. [Google Scholar] [CrossRef]
- Qureshi, A.I.; Ling, G.S.; Khan, J.; Suri, M.F.K.; Miskolczi, L.; Guterman, L.R.; Hopkins, L.N. Quantitative analysis of injured, necrotic, and apoptotic cells in a new experimental model of intracerebral hemorrhage. Crit. Care Med. 2001, 29, 152–157. [Google Scholar] [CrossRef]
- Wang, K.-Y.; Wu, C.-H.; Zhou, L.-Y.; Yan, X.-H.; Yang, R.-L.; Liao, L.-M.; Ge, X.-M.; Liao, Y.-S.; Li, S.-J.; Li, H.-Z.; et al. Ultrastructural Changes of Brain Tissues Surrounding Hematomas after Intracerebral Hemorrhage. Eur. Neurol. 2015, 74, 28–35. [Google Scholar] [CrossRef]
- Qureshi, A.I.; Suri, M.F.K.; Ostrow, P.T.; Kim, S.H.; Ali, Z.; Shatla, A.; Guterman, L.R.; Hopkins, L.N. Apoptosis as a form of cell death in intracerebral hemorrhage. Neurosurgery 2003, 52, 1041–1047. [Google Scholar]
- Degterev, A.; Huang, Z.; Boyce, M.; Li, Y.; Jagtap, P.; Mizushima, N.; Cuny, G.D.; Mitchison, T.J.; Moskowitz, M.A.; Yuan, J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 2005, 1, 112–119. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Lattanzi, S.; Di Napoli, M.; Ricci, S.; Divani, A.A. Matrix Metalloproteinases in Acute Intracerebral Hemorrhage. Neurotherapeutics 2020, 17, 484–496. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, G.A. Matrix metalloproteinases in neuroinflammation. Glia 2002, 39, 279–291. [Google Scholar] [CrossRef]
- Cauwe, B.; Opdenakker, G. Intracellular substrate cleavage: A novel dimension in the biochemistry, biology and pathology of matrix metalloproteinases. Crit. Rev. Biochem. Mol. Biol. 2010, 45, 351–423. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Zhang, Y.; Strong, R.; Grotta, J.C.; Aronowski, J. 15d-Prostaglandin J2 activates peroxisome proliferator-activated receptor-gamma, promotes expression of catalase, and reduces inflammation, behavioral dysfunction, and neuronal loss after intracerebral hemorrhage in rats. J. Cereb. Blood Flow Metab. 2006, 26, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Gurney, K.J.; Estrada, E.Y.; Rosenberg, G.A. Blood–brain barrier disruption by stromelysin-1 facilitates neutrophil infiltration in neuroinflammation. Neurobiol. Dis. 2006, 23, 87–96. [Google Scholar] [CrossRef]
- Hamann, G.F.; Okada, Y.; Fitridge, R.; del Zoppo, G.J. Microvascular Basal Lamina Antigens Disappear During Cerebral Ischemia and Reperfusion. Stroke 1995, 26, 2120–2126. [Google Scholar] [CrossRef]
- Black, R.A.; Rauch, C.T.; Kozlosky, C.J.; Peschon, J.J.; Slack, J.L.; Wolfson, M.F.; Castner, B.J.; Stocking, K.L.; Reddy, P.; Srinivasan, S.; et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-α from cells. Nature 1997, 385, 729–733. [Google Scholar] [CrossRef]
- Mun-Bryce, S.; Rosenberg, G.A. Matrix Metalloproteinases in Cerebrovascular Disease. J. Cereb. Blood Flow Metab. 1998, 18, 1163–1172. [Google Scholar] [CrossRef]
- Mannello, F.; Medda, V. Nuclear localization of Matrix metalloproteinases. Prog. Histochem. Cytochem. 2012, 47, 27–58. [Google Scholar] [CrossRef]
- Caplan, L.R. Caplan’s Stroke; Cambridge University Press: Cambridge, UK, 2016. [Google Scholar]
- Chobanian, A.V.; Bakris, G.L.; Black, H.R.; Cushman, W.C.; Green, L.A.; Izzo, J.L., Jr.; Jones, D.W.; Materson, B.J.; Oparil, S.; Wright, J.T., Jr.; et al. The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: The JNC 7 Report. JAMA 2003, 289, 2560–2572. [Google Scholar] [CrossRef] [PubMed]
- Lewington, S.; Clarke, R.; Qizilbash, N.; Peto, R.; Collins, R. Age-specific relevance of usual blood pressure to vascular mortality: A meta-analysis of individual data for one million adults in 61 prospective studies. Lancet 2002, 360, 1903–1913. [Google Scholar] [PubMed]
- Boehme, A.K.; Esenwa, C.; Elkind, M.S. Stroke Risk Factors, Genetics, and Prevention. Circ. Res. 2017, 120, 472–495. [Google Scholar] [CrossRef] [PubMed]
- Stansbury, J.P.; Jia, H.; Williams, L.S.; Vogel, W.B.; Duncan, P.W. Ethnic disparities in stroke: Epidemiology, acute care, and postacute outcomes. Stroke 2005, 36, 374–386. [Google Scholar] [CrossRef]
- Ergul, A.; Alhusban, A.; Fagan, S.C. Angiogenesis: A harmonized target for recovery after stroke. Stroke 2012, 43, 2270–2274. [Google Scholar] [CrossRef]
- Fisher, C.M. Pathological Observations in Hypertensive Cerebral Hemorrhage. J. Neuropathol. Exp. Neurol. 1971, 30, 536–550. [Google Scholar] [CrossRef]
- Takebayashi, S.; Kaneko, M. Electron microscopic studies of ruptured arteries in hypertensive intracerebral hemorrhage. Stroke 1983, 14, 28–36. [Google Scholar] [CrossRef]
- Ruiz-Sandoval, J.L.; Romero-Vargas, S.; Chiquete, E.; Padilla-Martίnez, J.J.; Villarreal-Careaga, J.; Cantú, C.; Arauz, A.; Barinagarrementerίa, F. Hypertensive intracerebral hemorrhage in young people: Previously unnoticed age-related clinical differences. Stroke 2006, 37, 2946–2950. [Google Scholar] [CrossRef]
- Ruίz-Sandoval, J.L.; Cantú, C.; Barinagarrementeria, F. Intracerebral hemorrhage in young people: Analysis of risk factors, location, causes, and prognosis. Stroke 1999, 30, 537–541. [Google Scholar] [CrossRef]
- Pan, P.; Weinsheimer, S.; Cooke, D.; Winkler, E.; Abla, A.; Kim, H.; Su, H. Review of treatment and therapeutic targets in brain arteriovenous malformation. J. Cereb. Blood Flow Metab. 2021, 41, 271678X211026771. [Google Scholar] [CrossRef]
- Lawton, M.T.; Rutledge, W.C.; Kim, H.; Stapf, C.; Whitehead, K.J.; Li, D.Y.; Krings, T.; Brugge, K.T.; Kondziolka, D.; Morgan, M.K.; et al. Brain arteriovenous malformations. Nat. Rev. Dis. Primers 2015, 1, 15008. [Google Scholar] [CrossRef] [PubMed]
- Shaligram, S.S.; Winkler, E.; Cooke, D.; Su, H. Risk factors for hemorrhage of brain arteriovenous malformation. CNS Neurosci. Ther. 2019, 25, 1085–1095. [Google Scholar] [CrossRef] [PubMed]
- Winkler, E.A.; Kim, C.N.; Ross, J.M.; Garcia, J.H.; Gil, E.; Oh, I.; Chen, L.Q.; Wu, D.; Catapano, J.S.; Raygor, K.; et al. A single-cell atlas of the normal and malformed human brain vasculature. Science 2022, 375, eabi7377. [Google Scholar] [CrossRef] [PubMed]
- Nikolaev, S.I.; Vetiska, S.; Bonilla, X.; Boudreau, E.; Jauhiainen, S.; Jahromi, B.R.; Khyzha, N.; DiStefano, P.V.; Suutarinen, S.; Kiehl, T.-R.; et al. Somatic Activating KRAS Mutations in Arteriovenous Malformations of the Brain. N. Engl. J. Med. 2018, 378, 250–261. [Google Scholar] [CrossRef]
- Hong, T.; Yan, Y.; Li, J.; Radovanovic, I.; Ma, X.; Shao, Y.W.; Yu, J.; Ma, Y.; Zhang, P.; Ling, F.; et al. High prevalence of KRAS/BRAF somatic mutations in brain and spinal cord arteriovenous malformations. Brain 2019, 142, 23–34. [Google Scholar] [CrossRef]
- Al-Olabi, L.; Polubothu, S.; Dowsett, K.; Andrews, K.A.; Stadnik, P.; Joseph, A.P.; Knox, R.; Pittman, A.; Clark, G.; Baird, W.; et al. Mosaic RAS/MAPK variants cause sporadic vascular malformations which respond to targeted therapy. J. Clin. Investig. 2018, 128, 1496–1508. [Google Scholar] [CrossRef]
- Wang, K.; Zhao, S.; Xie, Z.; Zhang, M.; Zhao, H.; Cheng, X.; Zhang, Y.; Niu, Y.; Liu, J.; Zhang, T.J.; et al. Exome-wide Analysis of De Novo and Rare Genetic Variants in Patients with Brain Arteriovenous Malformation. Neurology 2022, 98, e1670–e1678. [Google Scholar] [CrossRef]
- Scimone, C.; Granata, F.; Longo, M.; Mormina, E.; Turiaco, C.; Caragliano, A.A.; Donato, L.; Sidoti, A.; D’Angelo, R. Germline Mutation Enrichment in Pathways Controlling Endothelial Cell Homeostasis in Patients with Brain Arteriovenous Malformation: Implication for Molecular Diagnosis. Int. J. Mol. Sci. 2020, 21, 4321. [Google Scholar] [CrossRef]
- Kim, H.; Sidney, S.; McCulloch, C.E.; Poon, K.Y.T.; Singh, V.; Johnston, S.C.; Ko, N.U.; Achrol, A.S.; Lawton, M.T.; Higashida, R.T.; et al. Racial/Ethnic Differences in Longitudinal Risk of Intracranial Hemorrhage in Brain Arteriovenous Malformation Patients. Stroke 2007, 38, 2430–2437. [Google Scholar] [CrossRef]
- Kim, H.; Salman, R.A.-S.; McCulloch, C.E.; Stapf, C.; Young, W.L.; Mars Coinvestigators. Untreated brain arteriovenous malformation: Patient-level meta-analysis of hemorrhage predictors. Neurology 2014, 83, 590–597. [Google Scholar] [CrossRef] [Green Version]
- Mohr, J.P.; Parides, M.K.; Stapf, C.; Moquete, E.; Moy, C.S.; Overbey, J.R.; Salman, R.A.-S.; Vicaut, E.; Young, W.L.; Houdart, E.; et al. Medical management with or without interventional therapy for unruptured brain arteriovenous malformations (ARUBA): A multicentre, non-blinded, randomised trial. Lancet 2014, 383, 614–621. [Google Scholar] [CrossRef]
- Stapf, C.; Mast, H.; Sciacca, R.R.; Choi, J.H.; Khaw, A.V.; Connolly, E.S.; Pile-Spellman, J.; Mohr, J.P. Predictors of hemorrhage in patients with untreated brain arteriovenous malformation. Neurology 2006, 66, 1350–1355. [Google Scholar] [CrossRef] [PubMed]
- da Costa, L.; Wallace, M.C.; Ter Brugge, K.G.; O’Kelly, C.; Willinsky, R.A.; Tymianski, M. The natural history and predictive features of hemorrhage from brain arteriovenous malformations. Stroke 2009, 40, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Wu, Y.; Lawton, M.T.; Barbaro, N.M.; Young, W.L. Co-expression of angiogenic factors in brain arteriovenous malformations [Abstract]. J. Neurosurg. Anesthesiol. 2004, 16, 334. [Google Scholar]
- Koizumi, T.; Shiraishi, T.; Hagihara, N.; Tabuchi, K.; Hayashi, T.; Kawano, T. Expression of vascular endothelial growth factors and their receptors in and around intracranial arteriovenous malformations. Neurosurgery 2002, 50, 117–124; discussion 124–126. [Google Scholar]
- Cheng, P.; Ma, L.I.; Shaligram, S.; Walker, E.J.; Yang, S.T.; Tang, C.; Zhu, W.; Zhan, L.; Li, Q.; Zhu, X.; et al. Effect of elevation of vascular endothelial growth factor level on exacerbation of hemorrhage in mouse brain arteriovenous malformation. J. Neurosurg. 2019, 132, 1566–1573. [Google Scholar] [CrossRef]
- Chen, W.; Guo, Y.; Walker, E.J.; Shen, F.; Jun, K.; Oh, S.P.; Degos, V.; Lawton, M.T.; Tihan, T.; Davalos, D.; et al. Reduced mural cell coverage and impaired vessel integrity after angiogenic stimulation in the Alk1-deficient brain. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 305–310. [Google Scholar] [CrossRef]
- Zhu, W.; Chen, W.; Zou, D.; Wang, L.; Bao, C.; Zhan, L.; Saw, D.; Wang, S.; Winkler, E.; Li, Z.; et al. Thalidomide Reduces Hemorrhage of Brain Arteriovenous Malformations in a Mouse Model. Stroke 2018, 49, 1232–1240. [Google Scholar] [CrossRef]
- Lawton, M.T.; Jacobowitz, R.; Spetzler, R.F. Redefined role of angiogenesis in the pathogenesis of dural arteriovenous malformations. J. Neurosurg. 1997, 87, 267–274. [Google Scholar] [CrossRef]
- Zhu, Y.; Lawton, M.T.; Du, R.; Shwe, Y.; Chen, Y.; Shen, F.; Young, W.L.; Yang, G.-Y. Expression of Hypoxia-inducible Factor-1 and Vascular Endothelial Growth Factor in Response to Venous Hypertension. Neurosurgery 2006, 59, 687–696. [Google Scholar] [CrossRef]
- Gao, P.; Zhu, Y.; Ling, F.; Shen, F.; Lee, B.; Gabriel, R.A.; Hao, Q.; Yang, G.Y.; Su, H.; Young, W.L. Nonischemic cerebral venous hypertension promotes a pro-angiogenic stage through HIF-1 downstream genes and leukocyte-derived MMP-9. J. Cereb. Blood Flow Metab. 2009, 29, 1482–1490. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.Z.; Xue, Z.; Zhu, Y.; Yang, G.-Y.; Young, W.L. Matrix Metalloproteinase-9 Inhibition Attenuates Vascular Endothelial Growth Factor-Induced Intracerebral Hemorrhage. Stroke 2007, 38, 2563–2568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winkler, E.A.; Birk, H.; Burkhardt, J.-K.; Chen, X.; Yue, J.K.; Guo, D.; Rutledge, W.C.; Lasker, G.F.; Partow, C.; Tihan, T.; et al. Reductions in brain pericytes are associated with arteriovenous malformation vascular instability. J. Neurosurg. 2018, 129, 1464–1474. [Google Scholar] [CrossRef] [PubMed]
- Clatterbuck, R.; Eberhart, C.G.; Crain, B.J.; Rigamonti, D. Ultrastructural and immunocytochemical evidence that an incompetent blood-brain barrier is related to the pathophysiology of cavernous malformations. J. Neurol. Neurosurg. Psychiatry 2001, 71, 188–192. [Google Scholar] [CrossRef] [PubMed]
- Scimone, C.; Alibrandi, S.; Donato, L.; Alafaci, C.; Germanò, A.; Vinci, S.L.; D’Angelo, R.; Sidoti, A. Editome landscape of CCM-derived endothelial cells. RNA Biol. 2022, 19, 852–865. [Google Scholar] [CrossRef]
- Scimone, C.; Donato, L.; Alibrandi, S.; Esposito, T.; Alafaci, C.; D’Angelo, R.; Sidoti, A. Transcriptome analysis provides new molecular signatures in sporadic Cerebral Cavernous Malformation endothelial cells. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165956. [Google Scholar] [CrossRef]
- Labauge, P.; Laberge, S.; Brunereau, L.; Levy, C.; Tournier-Lasserve, E. Hereditary cerebral cavernous angiomas: Clinical and genetic features in 57 French families. Société Francaise de Neurochirurgie. Lancet 1998, 352, 1892–1897. [Google Scholar] [CrossRef]
- Labauge, P.; Brunereau, L.; Laberge, S.; Houtteville, J.-P. Prospective follow-up of 33 asymptomatic patients with familial cerebral cavernous malformations. Neurology 2001, 57, 1825–1828. [Google Scholar] [CrossRef]
- Denier, C.; Labauge, P.; Bergametti, F.; Marchelli, F.; Riant, F.; Arnoult, M.; Maciazek, J.; Vicaut, E.; Brunereau, L.; Tournier-Lasserve, E.; et al. Genotype-phenotype correlations in cerebral cavernous malformations patients. Ann. Neurol. 2006, 60, 550–555. [Google Scholar] [CrossRef]
- Pozzati, E.; Acciarri, N.; Tognetti, F.; Marliani, F.; Giangaspero, F. Growth, subsequent bleeding, and de novo appearance of cerebral cavernous angiomas. Neurosurgery 1996, 38, 662–669; discussion 669–670. [Google Scholar] [CrossRef]
- Maiuri, F.; Cappabianca, P.; Gangemi, M.; Caro, M.D.B.D.; Esposito, F.; Pettinato, G.; De Divitiis, O.; Mignogna, C.; Strazzullo, V.; De Divitiis, E. Clinical progression and familial occurrence of cerebral cavernous angiomas: The role of angiogenic and growth factors. Neurosurg. Focus 2006, 21, e3. [Google Scholar] [CrossRef]
- Dubovsky, J.; Zabramski, J.M.; Kurth, J.; Spetzler, R.F.; Rich, S.S.; Orr, H.T.; Weber, J.L. A gene responsible for cavernous malformations of the brain maps to chromosome 7q. Hum. Mol. Genet. 1995, 4, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Günel, M.; Awad, I.; Anson, J.; Lifton, R.P. Mapping a gene causing cerebral cavernous malformation to 7q11.2-q21. Proc. Natl. Acad. Sci. USA 1995, 92, 6620–6624. [Google Scholar] [CrossRef] [PubMed]
- Marchuk, D.; Galllone, C.J.; Morrison, L.; Clericuzio, C.L.; Hart, B.L.; Kosofsky, B.E.; Louis, D.N.; Gusella, J.F.; Davis, L.E.; Prenger, V.L. A Locus for Cerebral Cavernous Malformations Maps to Chromosome 7q in Two Families. Genomics 1995, 28, 311–314. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.W.; Iyer, L.M.; Rich, S.S.; Orr, H.T.; Gil-Nagel, A.; Kurth, J.H.; Zabramski, J.M.; Marchuk, D.; Weissenbach, J.; Clericuzio, C.L. Refined localization of the cerebral cavernous malformation gene (CCM1) to a 4-cM interval of chromosome 7q contained in a well-defined YAC contig. Genome Res. 1995, 5, 368–380. [Google Scholar] [CrossRef]
- Günel, M.; Awad, I.A.; Finberg, K.; Anson, J.A.; Steinberg, G.K.; Batjer, H.H.; Kopitnik, T.A.; Morrison, L.; Giannotta, S.L.; Nelson-Williams, C.; et al. A Founder Mutation as a Cause of Cerebral Cavernous Malformation in Hispanic Americans. N. Engl. J. Med. 1996, 334, 946–951. [Google Scholar] [CrossRef]
- Laberge, S.; Labauge, P.; Maréchal, E.; Maciazek, J.; Tournier-Lasserve, E. Genetic heterogeneity and absence of founder effect in a series of 36 French cerebral cavernous angiomas families. Eur. J. Hum. Genet. 1999, 7, 499–504. [Google Scholar] [CrossRef]
- Craig, H.D.; Günel, M.; Cepeda, O.; Johnson, E.W.; Ptacek, L.; Steinberg, G.K.; Ogilvy, C.S.; Berg, M.J.; Crawford, S.C.; Scott, R.M.; et al. Multilocus linkage identifies two new loci for a mendelian form of stroke, cerebral cavernous malformation, at 7p15-13 and 3q25.2-27. Hum. Mol. Genet. 1998, 7, 1851–1858. [Google Scholar] [CrossRef]
- Liquori, C.L.; Berg, M.J.; Siegel, A.M.; Huang, E.; Zawistowski, J.S.; Stoffer, T.; Verlaan, D.; Balogun, F.; Hughes, L.; Leedom, T.P.; et al. Mutations in a Gene Encoding a Novel Protein Containing a Phosphotyrosine-Binding Domain Cause Type 2 Cerebral Cavernous Malformations. Am. J. Hum. Genet. 2003, 73, 1459–1464. [Google Scholar] [CrossRef]
- Denier, C.; Goutagny, S.; Labauge, P.; Krivosic, V.; Arnoult, M.; Cousin, A.; Benabid, A.; Comoy, J.; Frerebeau, P.; Gilbert, B.; et al. Mutations within the MGC4607 Gene Cause Cerebral Cavernous Malformations. Am. J. Hum. Genet. 2004, 74, 326–337. [Google Scholar] [CrossRef]
- Bergametti, F.; Denier, C.; Labauge, P.; Arnoult, M.; Boetto, S.; Clanet, M.; Coubes, P.; Echenne, B.; Ibrahim, R.; Irthum, B.; et al. Mutations within the Programmed Cell Death 10 Gene Cause Cerebral Cavernous Malformations. Am. J. Hum. Genet. 2005, 76, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Porter, P.J.; Willinsky, R.A.; Harper, W.; Wallace, M.C. Cerebral cavernous malformations: Natural history and prognosis after clinical deterioration with or without hemorrhage. J. Neurosurg. 1997, 87, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Smith, W.S.; Lawton, M.T.; Halbach, V.V.; Young, W.L. Risk factors for hemorrhagic presentation in patients with dural arteriovenous fistulae. Neurosurgery 2008, 62, 628–635. [Google Scholar] [PubMed]
- Brown, R.D.; Wiebers, D.O., Jr.; Nichols, D.A. Intracranial dural arteriovenous fistulae: Angiographic predictors of intracranial hemorrhage and clinical outcome in nonsurgical patients. J. Neurosurg. 1994, 81, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Rui, Y.-N.; Hagan, J.P.; Kim, D.H. Intracranial Aneurysms: Pathology, Genetics, and Molecular Mechanisms. Neuromol. Med. 2019, 21, 325–343. [Google Scholar] [CrossRef]
- Andreasen, T.H.; Bartek, J.; Andresen, M.; Springborg, J.B.; Romner, B. Modifiable Risk Factors for Aneurysmal Subarachnoid Hemorrhage. Stroke 2013, 44, 3607–3612. [Google Scholar] [CrossRef]
- Korja, M.; Silventoinen, K.; Laatikainen, T.; Jousilahti, P.; Salomaa, V.; Hernesniemi, J.; Kaprio, J. Risk Factors and Their Combined Effects on the Incidence Rate of Subarachnoid Hemorrhage—A Population-Based Cohort Study. PLoS ONE 2013, 8, e73760. [Google Scholar] [CrossRef]
- Williams, L.N.; Brown, R.D., Jr. Management of unruptured intracranial aneurysms. Neurol. Clin. Pract. 2013, 3, 99–108. [Google Scholar] [CrossRef]
- Raymond, J.; Mohr, J.P.; Team-Aruba Collaborative Groups. The prevention of hemorrhagic stroke. A review of the rational and ethical principles of clinical trials on unruptured intracranial aneurysms and arteriovenous malformations. Interv. Neuroradiol. 2008, 14, 365–373. [Google Scholar] [CrossRef]
- Krings, T.; Mandell, D.M.; Kiehl, T.-R.; Geibprasert, S.; Tymianski, M.; Alvarez, H.; Terbrugge, K.G.; Hans, F.-J. Intracranial aneurysms: From vessel wall pathology to therapeutic approach. Nat. Rev. Neurol. 2011, 7, 547–559. [Google Scholar] [CrossRef]
- Scanarini, M.; Mingrino, S.; Zuccarello, M.; Trincia, G. Scanning electron microscopy (S.E.M.) of biopsy specimens of ruptured intracranial saccular aneurysms. Acta Neuropathol. 1978, 44, 131–134. [Google Scholar] [CrossRef] [PubMed]
- Fennell, V.S.; Kalani, M.Y.S.; Atwal, G.; Martirosyan, N.L.; Spetzler, R.F. Biology of Saccular Cerebral Aneurysms: A Review of Current Understanding and Future Directions. Front. Surg. 2016, 3, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoki, T.; Nishimura, M. The Development and the Use of Experimental Animal Models to Study the Underlying Mechanisms of CA Formation. J. Biomed. Biotechnol. 2010, 2011, 535921. [Google Scholar] [CrossRef]
- Morimoto, M.; Miyamoto, S.; Mizoguchi, A.; Kume, N.; Kita, T.; Hashimoto, N. Mouse model of cerebral aneurysm: Experimental induction by renal hypertension and local hemodynamic changes. Stroke 2002, 33, 1911–1915. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Liu, Z.; Ren, J.; Morgan, S.; Assa, C.; Liu, B. Receptor-Interacting Protein Kinase 3 Contributes to Abdominal Aortic Aneurysms via Smooth Muscle Cell Necrosis and Inflammation. Circ. Res. 2015, 116, 600–611. [Google Scholar] [CrossRef]
- Chalouhi, N.; Ali, M.S.; Jabbour, P.M.; Tjoumakaris, S.; Gonzalez, L.F.; Rosenwasser, R.H.; Koch, W.J.; Dumont, A.S. Biology of Intracranial Aneurysms: Role of Inflammation. J. Cereb. Blood Flow Metab. 2012, 32, 1659–1676. [Google Scholar] [CrossRef]
- Ahn, J.Y.; Cho, J.H.; Lee, J.W. Distal lenticulostriate artery aneurysm in deep intracerebral haemorrhage. J. Neurol. Neurosurg. Psychiatry 2007, 78, 1401–1403. [Google Scholar] [CrossRef]
- Heck, O.; Anxionnat, R.; Bracard, S.; Cai, X.; Han, S.; Feske, S.; Chou, S. Pearls & Oy-sters: Small but consequential: Intracerebral hemorrhage caused by lenticulostriate artery aneurysm. Neurology 2013, 81, 1881. [Google Scholar]
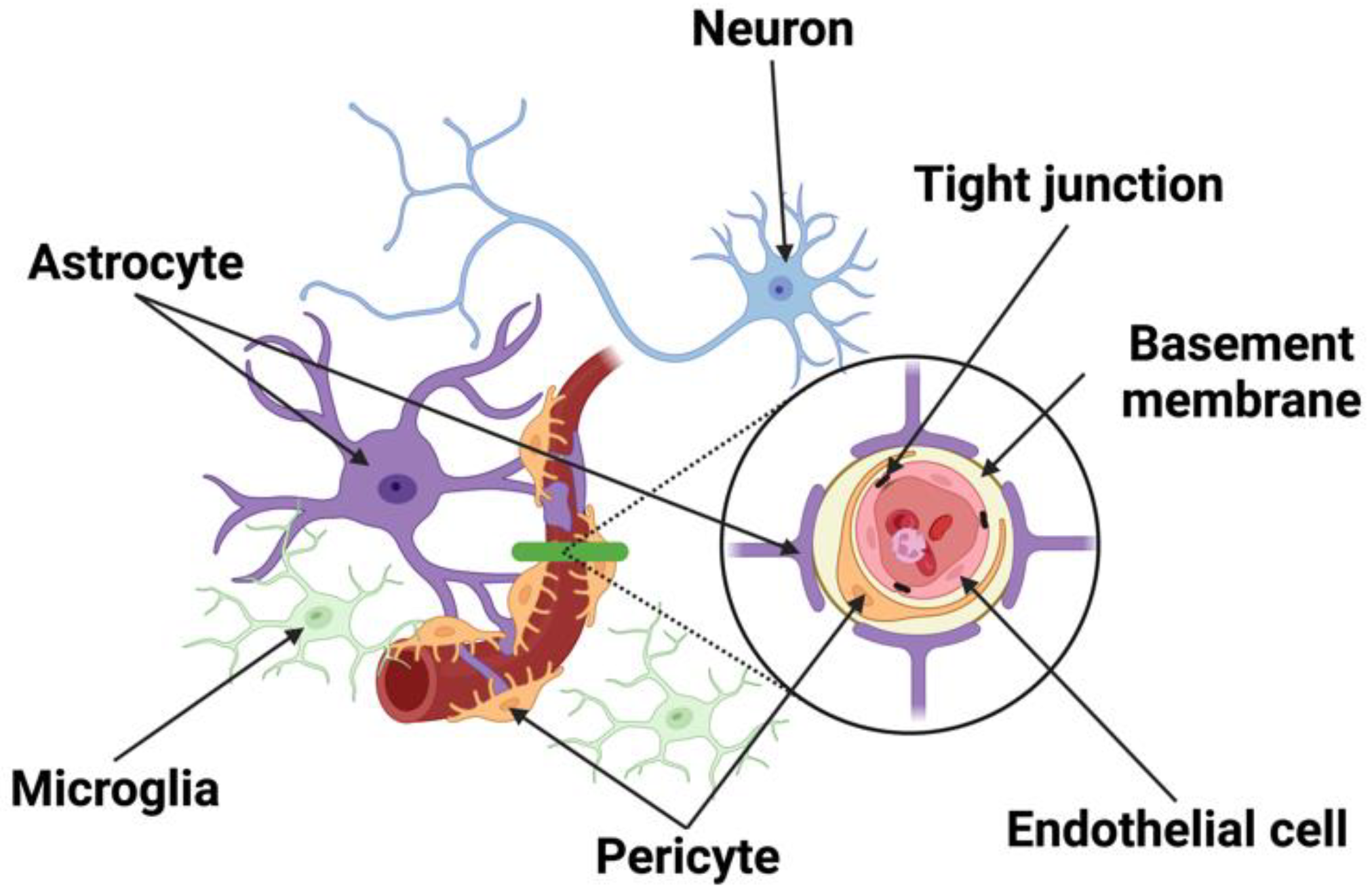
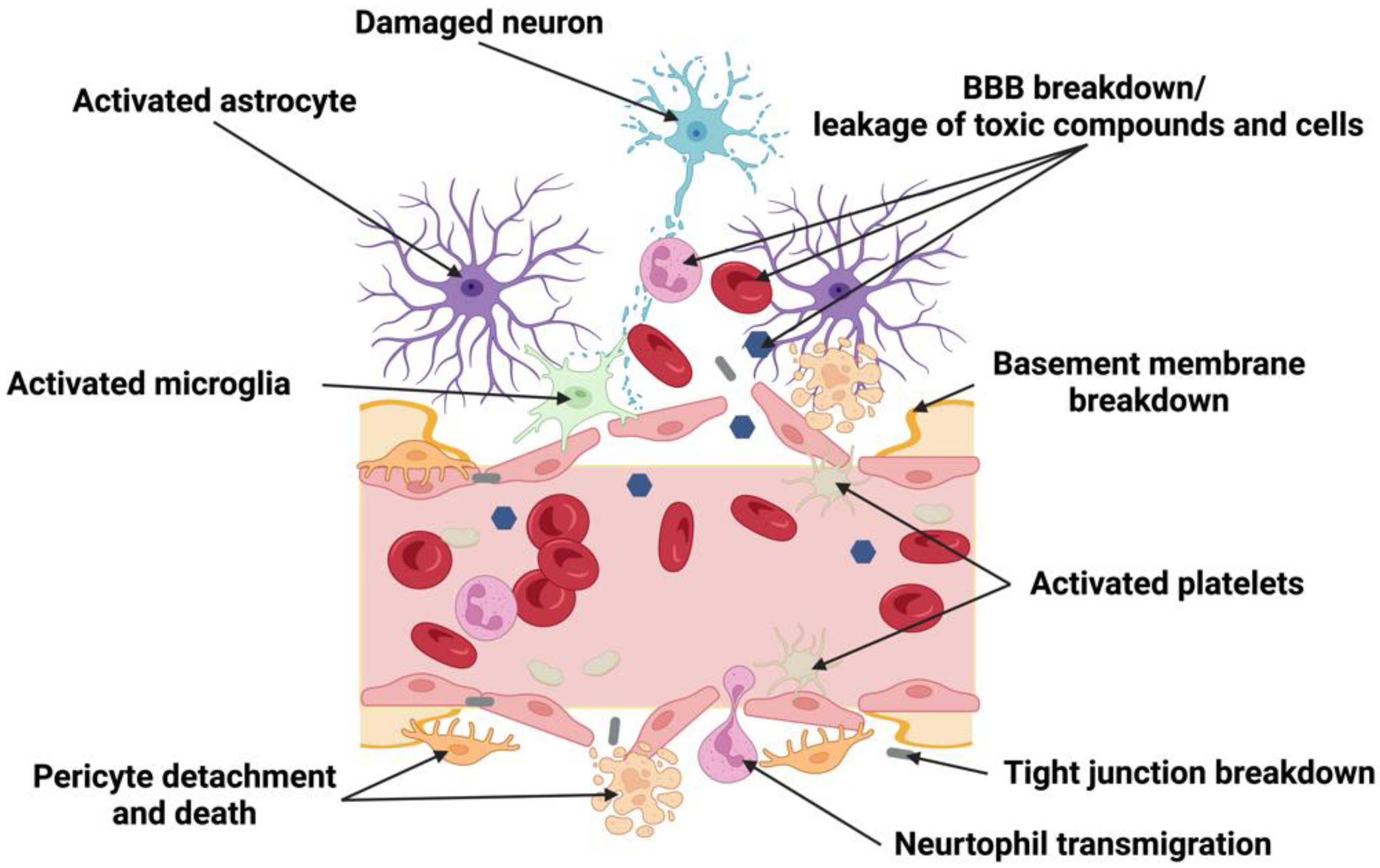

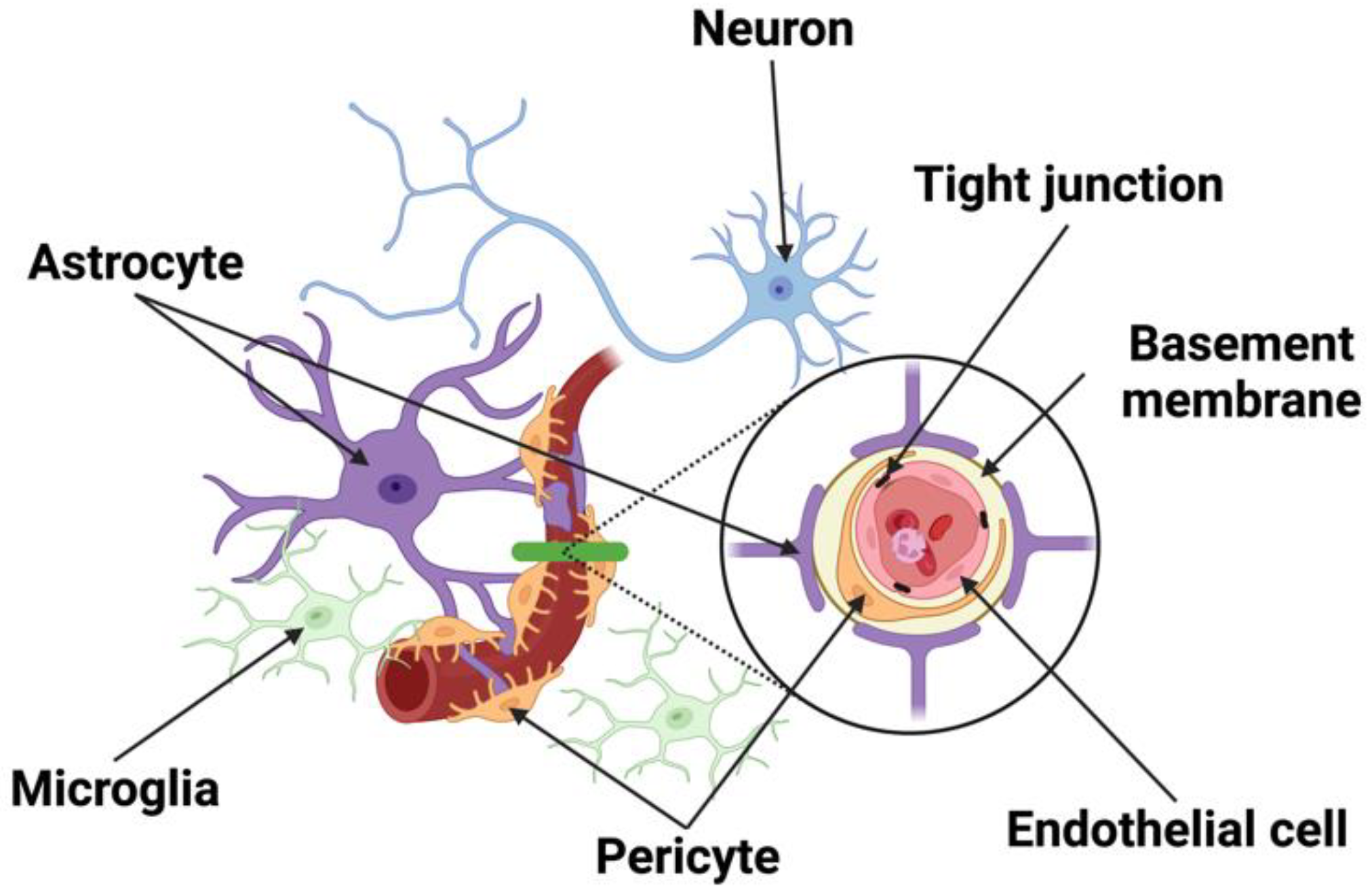{kind=link}
{kind=link}
| Endothelial cell | Following disruption of the BBB, the ferritin and the free iron accumulated in endothelial cells in brain capillaries enter the penumbra. Iron-dependent oxidative stress in the penumbra can lead to necrosis and further neurological deterioration following ischemic stroke [133]. |
| In an ischemia/reperfusion model of MCAO, BBB permeability exhibited a biphasic manner with permeability occurring at 3 and 72 h after reperfusion, and changes in claudin-5, occludin, and zonula occludens-1 protein levels [127]. | |
| Fasudil hydrochloride recovered the neurological function, improved the function of BBB inhibited RhoA protein expression, and upregulated growth-associated protein-43 and claudin-5 protein expression following cerebral ischemia/reperfusion [134]. | |
| Following a mild MCAO, tight junctions were stable during the first 24 h after reperfusion, but they underwent significant breakdown and remodeling from 48 to 58 h after reperfusion [128]. | |
| 4–24 h post-stroke, BBB breakdown was observed and vessels showed astrocyte end-foot swelling and increased endothelial vesicles [131]. | |
| Pericyte | 2 h after hypoxia, pericytes started to migrate and one of every three pericytes migrated from the original location. In the first stage of migration, spikes occurred at the abluminal surface of pericytes [135]. |
| PDGFRβ expression was induced specifically in the pericytes in peri-infarct areas and its level was gradually increased [136]. In the cultured pericytes, PDGF-B induced cell growth and anti-apoptotic responses through Akt [136]. | |
| Hypoxic-ischemic injury results in oxidative stress and pericyte constriction or eventual death [137]. | |
| In photothrombotic stroke in mice, it was postulated that rapid activation of MMP-9 secreted from pericyte somata degraded underlying tight junction complexes and resulted in plasma leakage at places where pericyte somata adjoined the capillary wall [138]. | |
| After pericyte ablation with diphtheria toxin, mice showed acute BBB breakdown, severe loss of blood flow, and a rapid neuron. Intracerebroventricular PTN infusions prevented neuron loss in pericyte-ablated mice despite persistent circulatory changes [139]. | |
| Astrocyte | Astrocytes are known to secrete pro-angiogenic factors that promote the growth of new capillaries toward the infarcted tissue [140]. |
| Reactive astrocytes have a protective role in brain ischemia, and the absence of astrocyte. IFs is linked to changes in glutamate transport, endothelin B receptor-mediated control of gap junctions, and plasminogen activator inhibitor-1 expression [141]. | |
| After CNS injury, astrocytes induce angiogenesis by endogenous VEGF and upregulation of autocrine signaling, increasing both astrocyte proliferation and facilitating the expression of growth factors [142]. | |
| CSDs induce vasoconstriction of vascular smooth muscle cells by increasing astrocytic vasoconstrictor 20-hydroxyeicosatetraenoic acid [143]. | |
| Astrocytic gap junctions were implicated in propagating CSDs and leading to exacerbated brain damage [144]. | |
| Astrocytes are activated by trophic factors produced by the pericytes within the ischemic regions, leading to peri-infarct astrogliosis [145]. | |
| Microglia and macrophage | Reactive microglia also secrete MMP-9 and MMP-3, proteases that can break down the basement membrane and exacerbate BBB leakage [146]. |
| Recruitment of microglia to blood vessels occurs within 6 h of reperfusion with significant accumulation in perilesional tissue. After 24 h of reperfusion, microglia fully enwrap small blood vessels in the peri-infarct region. Individual perivascular microglia displayed intracellular vesicles containing CD31-positive inclusions, suggesting phagocytosis of brain endothelial cells, which was correlated with BBB breakdown [147]. 72 h post-MCAO, blood vessel degradation was complete, and remaining vascular debris was cleared by microglia and immune cells | |
| P2RY12-mediated chemotaxis of microglia processes is required for the rapid closure of the BBB. Mice treated with the P2RY12 inhibitor clopidogrel, as well as those in which P2RY12 was genetically ablated, exhibited significantly diminished movement of microglial processes and failed to close laser-induced openings of the BBB. Thus, microglial cells played a previously unrecognized protective role in the maintenance of BBB integrity following cerebrovascular damage [148]. |
| Risk Factors | Findings |
|---|---|
| Diabetes and hyperglycemia | T1-weighted MRI showed a homogeneous high-intensity area in the corpus striatum in a diabetic hemichorea patient [194] |
| Increased BBB permeability with MRI was detected in patients with type II diabetes or white matter hyperintensities [195] | |
| Immunostaining for 8-OHdG, a marker of oxidative DNA damage, was seen in the vessels of the cortex of 20-week-old diabetic mice [196] | |
| BBB permeability were significantly increased in diabetic rat, 5 weeks of rosuvastatin and simvastatin therapy (10 mg/kg) improved BBB permeability [197] | |
| Progressive increase in BBB permeability to small molecules from 28 to 90 days were observed in diabetic rat Insulin treatment attenuated BBB, especially during the first few weeks; however, as diabetes progressed, it was evident that microvascular damage occurred even when hyperglycemia was controlled [198] | |
| Diabetes increases BBB permeability via a loss of tight junction proteins, but increased BBB permeability in diabetes did not result from hyperglycemia alone [199] | |
| Hyperglycemia significantly compromised the BBB integrity and enhanced total PKC activity. Elevations in NADPH oxidase and MMP-2 activities and decreases in occludin levels contributed to barrier dysfunction [200] | |
| Upregulation of TJ-associated proteins were caused by insulin and idebenone in diabetic rats. The activations of ROS, AGEs, expression levels of RAGE and NF-κB were significantly decreased after insulin treatment [201] | |
| Hyperglycemia enhances amyloid precursor protein expression with increased in human umbilical vein endothelial cells. Amyloid beta-peptide production downregulates junctional proteins causing increased BBB permeability [202] | |
| Greater immunoreactivity of albumin was observed in the vessel wall of the periventricular area of diabetic mice [203] | |
| GLP-1 inhibited the increase in production of reactive oxygen species under hyperglycemia conditions and improved the BBB integrity induced by hyperglycemia [204] | |
| Significantly increased sodium fluorescein leakage in the hippocampus was observed in diabetic rat, and tight junction markers were significantly decreased in the hippocampi [205] | |
| BBB permeability in the diabetic monkeys was significantly increased [206] | |
| EX-4 improved the permeability of the BBB and the cognitive parameters in diabetic rat [207] | |
| The astrocyte foot processes were detached, and the microglial cells played an invasive damaging role in diabetic mice. Endothelial cell deterioration was observed: loss of electron density, basement membrane thickening and rearrangement, increased transcytotic-pinocytotic vesicles, and aberrant mitochondria [208] | |
| Pericyte apoptosis was detected in vivo in hyperglycemia rats and in vitro in retinal cultures [209] | |
| Hypertension | Cerebral arterioles underwent remodeling and hypertrophy in transgenically hypertensive mice [210] |
| The staining for HRP was distributed around the vessels in the hippocampal fissure of spontaneous hypertensive rats [211] | |
| Increased BBB permeability and impaired cognitive functions was observed in angiotensin II-infused hypertensive mice; Angiotensin II receptor blocker markedly ameliorated leakage from brain microvessels and restored the cognitive decline [212] | |
| Hypertension significantly decreased the brain glutathione content and increased the brain malondialdehyde level [213]. The mRNA levels of claudins (3, 5, and 12) proteins significantly decreased in response to hypertension; | |
| Tight junction was destroyed gradually 8 weeks after hypertension, and the levels of zonula occludens-1 and occludin also decreased gradually [214] | |
| In SHR brain, an obvious glial reaction was found both for GFAP-immunoreactive astrocytes and for microglia; The pro-inflammatory IL-1β was significantly increased in CA1 sub-field of SHR hippocampus and the TNFα expression was higher in frontal cortex of SHR compared with WKY [215] | |
| Chronic hypertension significantly increased spontaneous Ca2+ events within astrocyte microdomains [216] | |
| Hyperlipidemia | 70% more spontaneous leakage of injected Evans blue dye in the brains of APOE knock-out mice [217] |
| BBB permeability was higher in APOE4 knock-in mice than in APOE3 knock-in mice, suggesting that TJ integrity in BBB is regulated by APOE in an isoform-dependent manner [218] | |
| The expression of APOE4 and lack of murine APOE led to BBB breakdown [97] | |
| Increase of mean Ki during the first 20 min after infusion of human TGRL lipolysis product was observed in mice [219] | |
| Aging | The choline carrier in old rats had reduced capacity and increased affinity [220] |
| Aged mice showed significant attenuation in the expression of BBB tight junction proteins. TNF-α in cerebral endothelial cells of aged mice was elevated and this was associated with heightened peripheral inflammation [221] | |
| Age-dependent BBB breakdown in the hippocampus was observed. The BBB breakdown in the hippocampus and its CA1 and dentate gyrus subdivisions worsened with mild cognitive impairment [222] | |
| Sex | 2- to 4-fold increase in dye extravasation in the olfactory bulb and hippocampus of reproductively senescent female mice; estrogen reduced dye extravasation in young adults compared with age-matched counterparts [223] |
| Ovariectomy induced a 2.2-fold increase in Evan’s blue dye extravasation into the brain in young female mice. The expression of the tight junction protein in microvessels was not altered [224] | |
| Treatment of endothelial cells with 17beta-estradiol led to an increase in transendothelial electric resistance and a most prominent upregulation of the tight junction protein claudin-5 expression [225]. A significant increase of claudin-5 promoter activity, mRNA, and protein levels was also detected | |
| Estradiol prevented inflammation-induced defects in barrier function in mice [226] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sato, Y.; Falcone-Juengert, J.; Tominaga, T.; Su, H.; Liu, J. Remodeling of the Neurovascular Unit Following Cerebral Ischemia and Hemorrhage. Cells 2022, 11, 2823. https://doi.org/10.3390/cells11182823
Sato Y, Falcone-Juengert J, Tominaga T, Su H, Liu J. Remodeling of the Neurovascular Unit Following Cerebral Ischemia and Hemorrhage. Cells. 2022; 11(18):2823. https://doi.org/10.3390/cells11182823
Chicago/Turabian StyleSato, Yoshimichi, Jaime Falcone-Juengert, Teiji Tominaga, Hua Su, and Jialing Liu. 2022. "Remodeling of the Neurovascular Unit Following Cerebral Ischemia and Hemorrhage" Cells 11, no. 18: 2823. https://doi.org/10.3390/cells11182823
APA StyleSato, Y., Falcone-Juengert, J., Tominaga, T., Su, H., & Liu, J. (2022). Remodeling of the Neurovascular Unit Following Cerebral Ischemia and Hemorrhage. Cells, 11(18), 2823. https://doi.org/10.3390/cells11182823