
Diabetic Proteinuria Revisited: Updated Physiologic Perspectives


Abstract
:1. Introduction
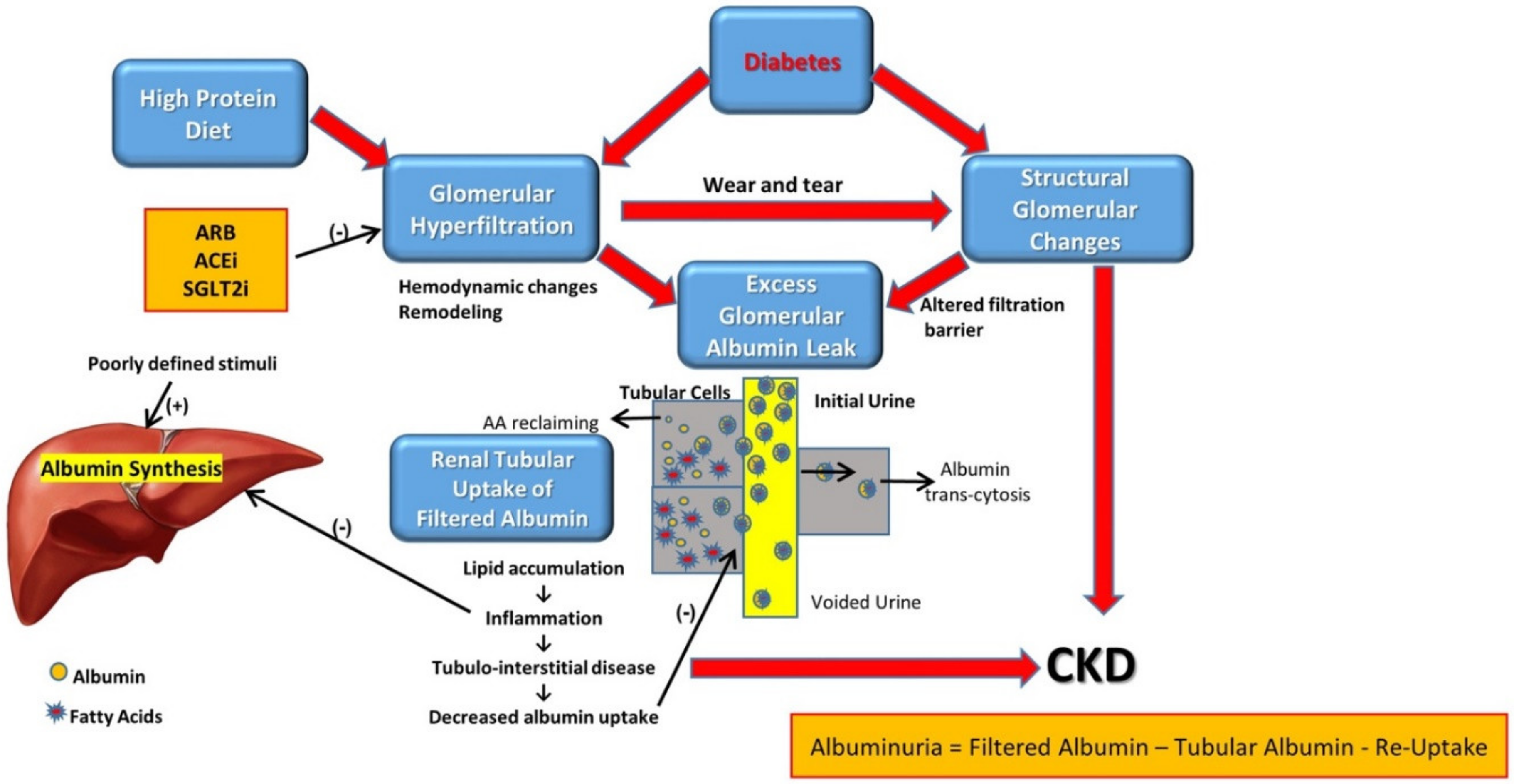
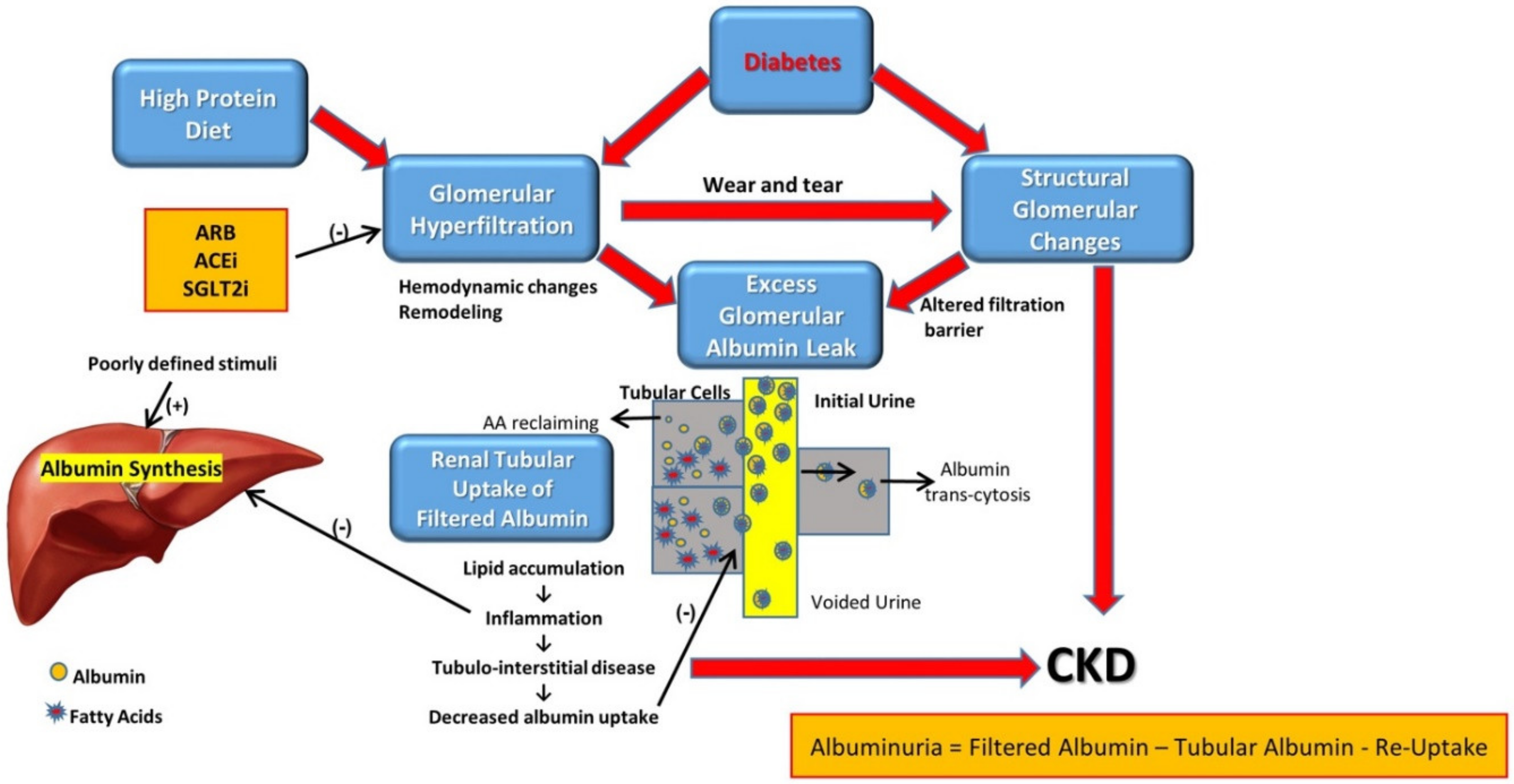
2. Pathophysiology
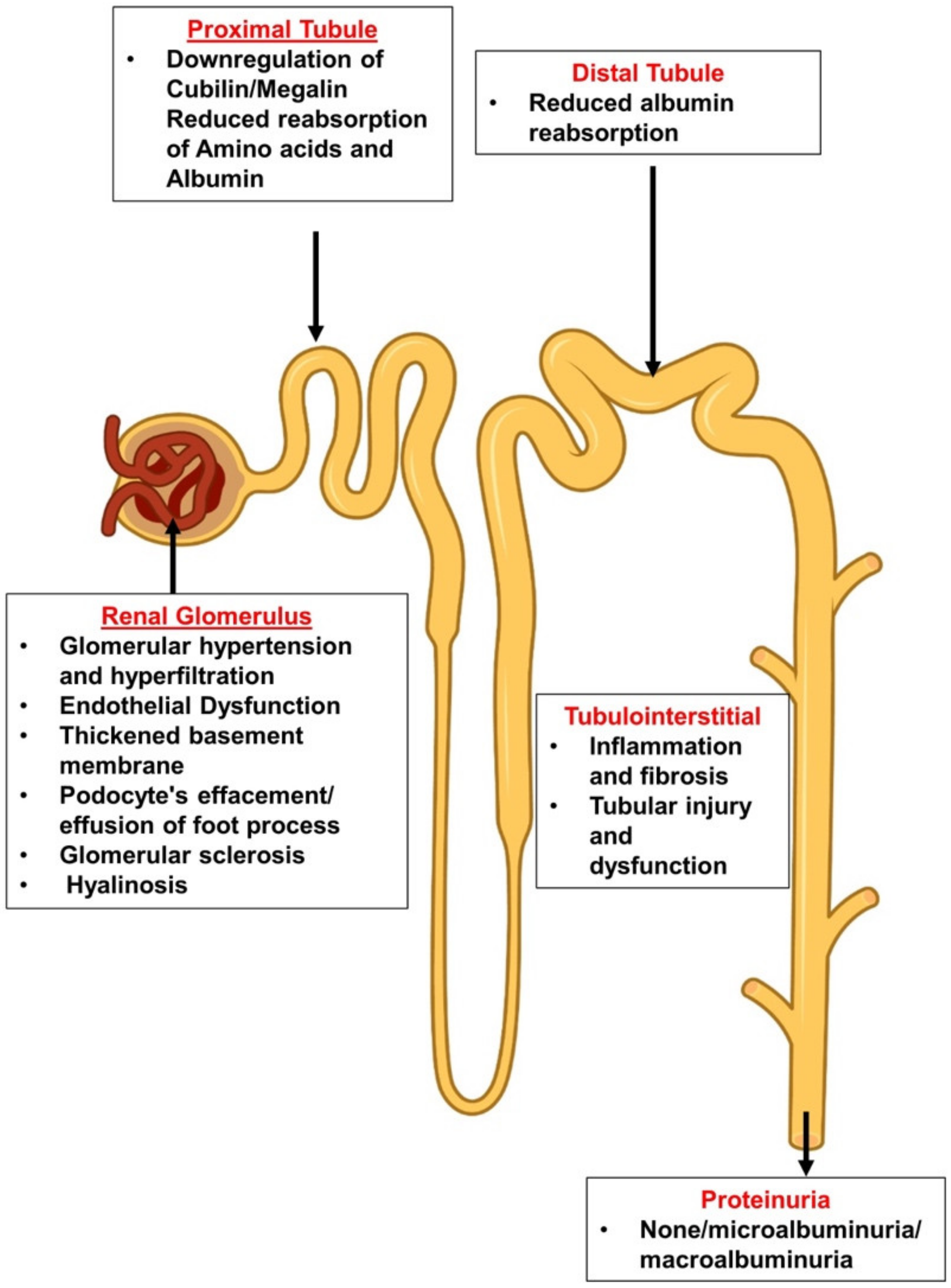
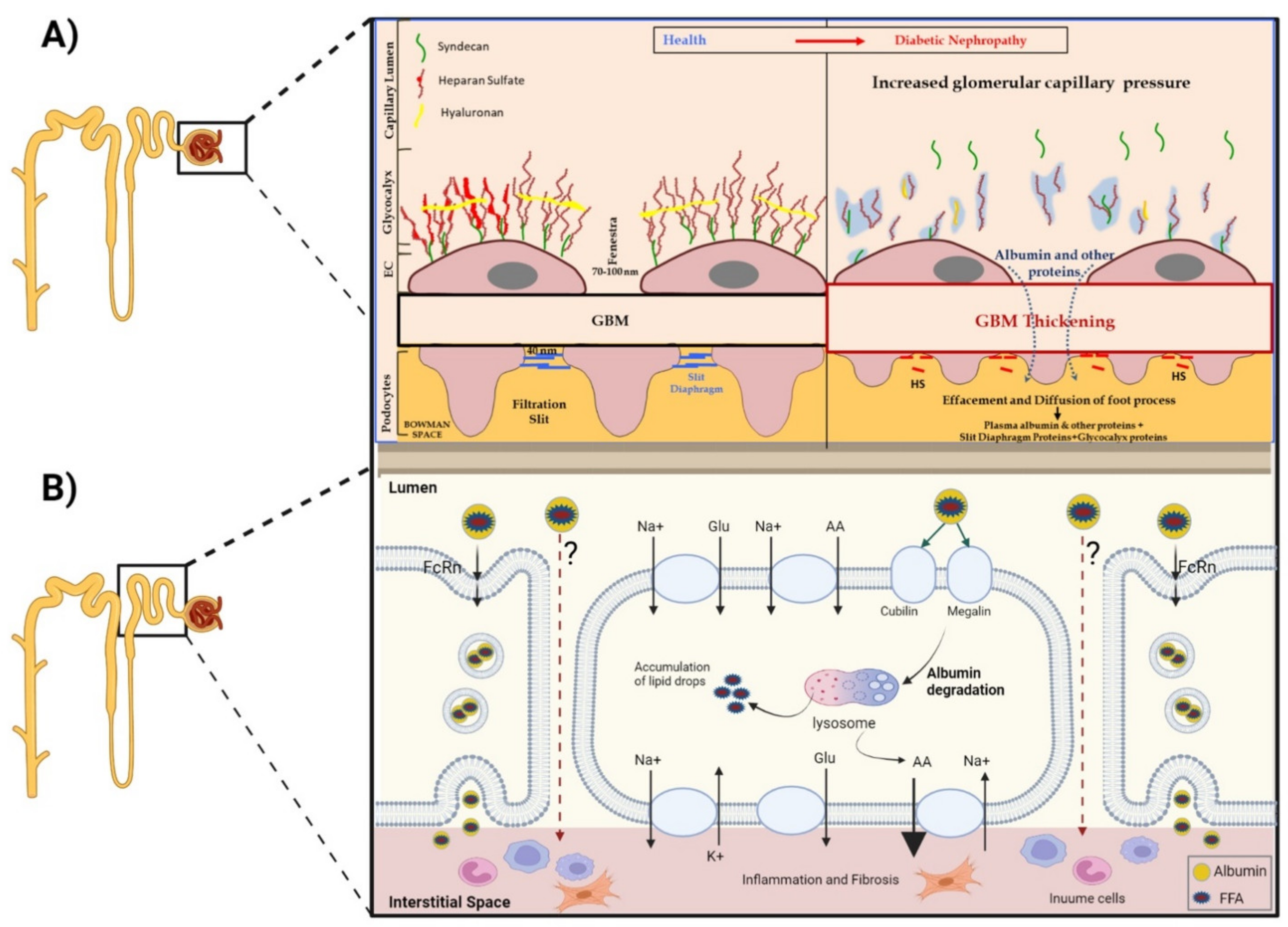
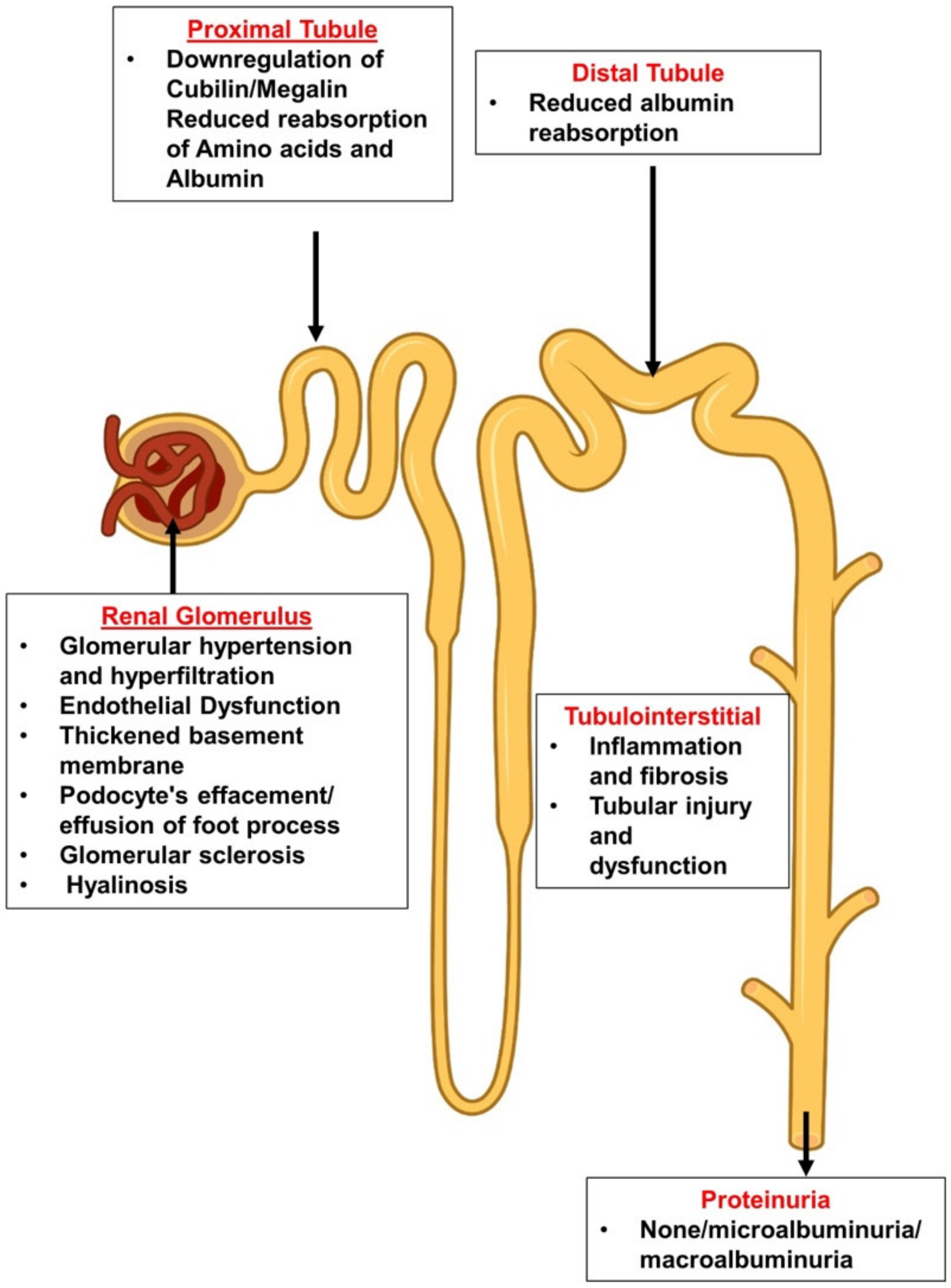
2.1. The Leaky Glomerulus—Deformed Filtration Barrier
- A.
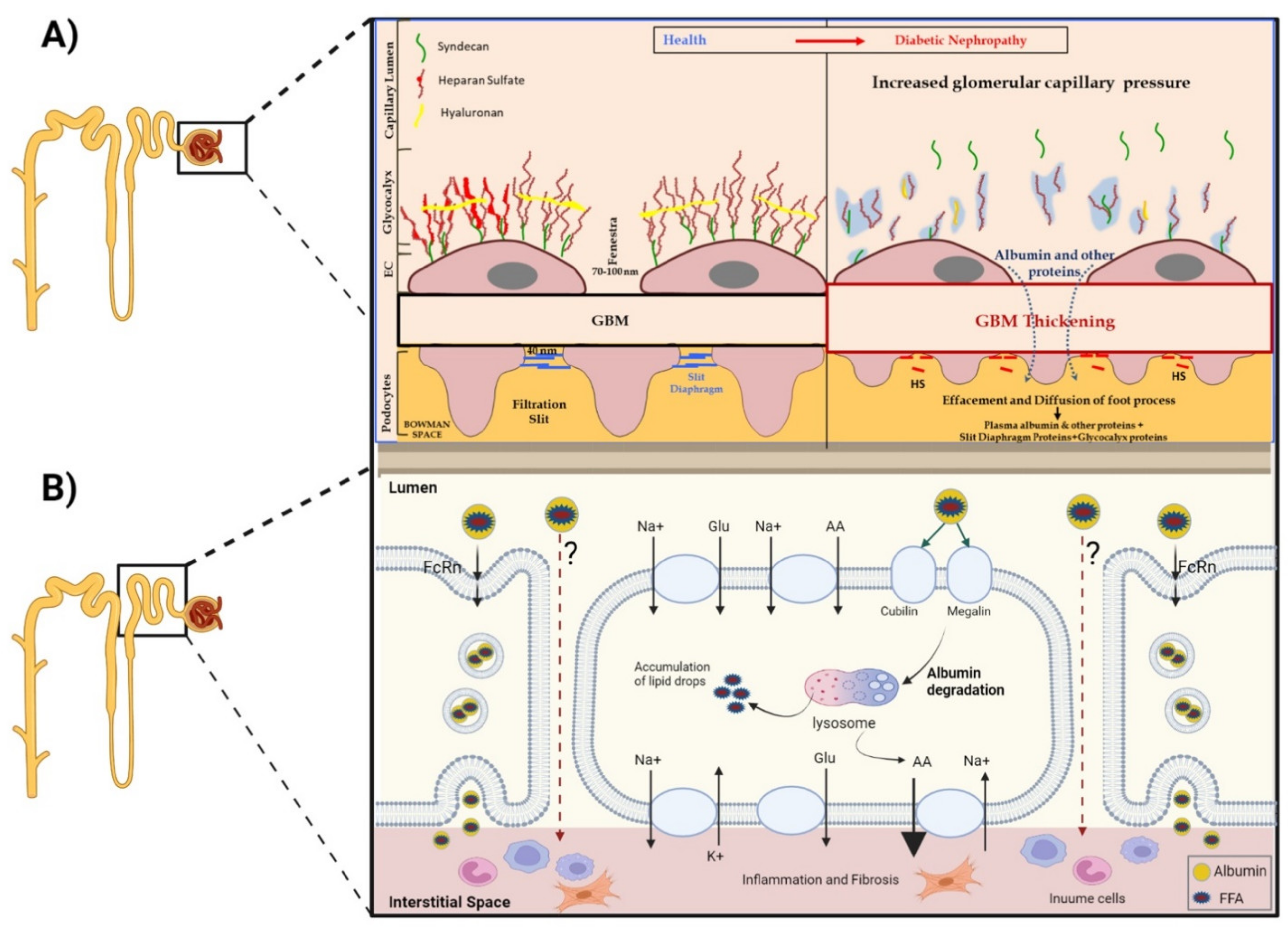
- Under normal conditions, GFB prevents leakage of high-molecular-weight proteins, including albumin, into Bowman’s space. Destruction of any of the layers of the GFB in the diabetic kidney might result in proteinuric disease. This may be initiated by promoting increased production of reactive oxygen species (ROS), the induction of AGE-induced proinflammatory signaling and increased glomerular capillary pressure and hyperfiltration. Damage to all three components of GFB in the diabetic kidney is evident by endothelial dysfunction (ED), disrupted glomerular basement membrane (GBM) with increased deposition of extracellular matrix, loss of podocyte permselectivity and progressive podocyte damage: (1) ED is characterized by structural and functional damage to the glycocalyx, and its components, especially heparan sulfate (HS), are released and appear in the urine. Indeed, derangement of the glycocalyx plays a pivotal role in the development of albuminuria secondary to enhanced vessel wall permeability with protein leak. (2) One of the milestone features of DN is thickening of GBM along with the development of proteinuria. (3) Similarly, podocyte damage is considered a crucial step in the pathogenesis of proteinuric kidney diseases, including DN. Specifically, effacement and effusion of podocyte foot processes are a hallmark feature of ultrastructural alterations characterizing proteinuric renal disease, including DN.
- B.
- Albumin is reabsorbed mainly by proximal tubular cells via two pathways: (1) a fraction of albumin binds to neonatal Fc receptor (FcRn), undergoes trans-cytosis and is reclaimed intact into the bloodstream; (2) a fraction of albumin attaches to megalin and cubilin and undergoes endocytosis and subsequent proteolysis in lysosomes, with the release of amino acids (AA) into the bloodstream. An unknown percentage of filtered albumin is likely reclaimed intact by the paracellular pathway. Free fatty acids (FFA) dissolved in albumin are up taken as well and are subjected to beta oxidation with the generation of acetyl-Co A. A fraction of FFA accumulates in tubular cells with the formation of oval fat bodies and tubular injury. This leads to the induction of inflammation that culminates in chronic and progressive tubulointerstitial disease with tubular atrophy and interstitial fibrosis.
2.2. Altered Glomerular Hemodynamics and Proteinuria
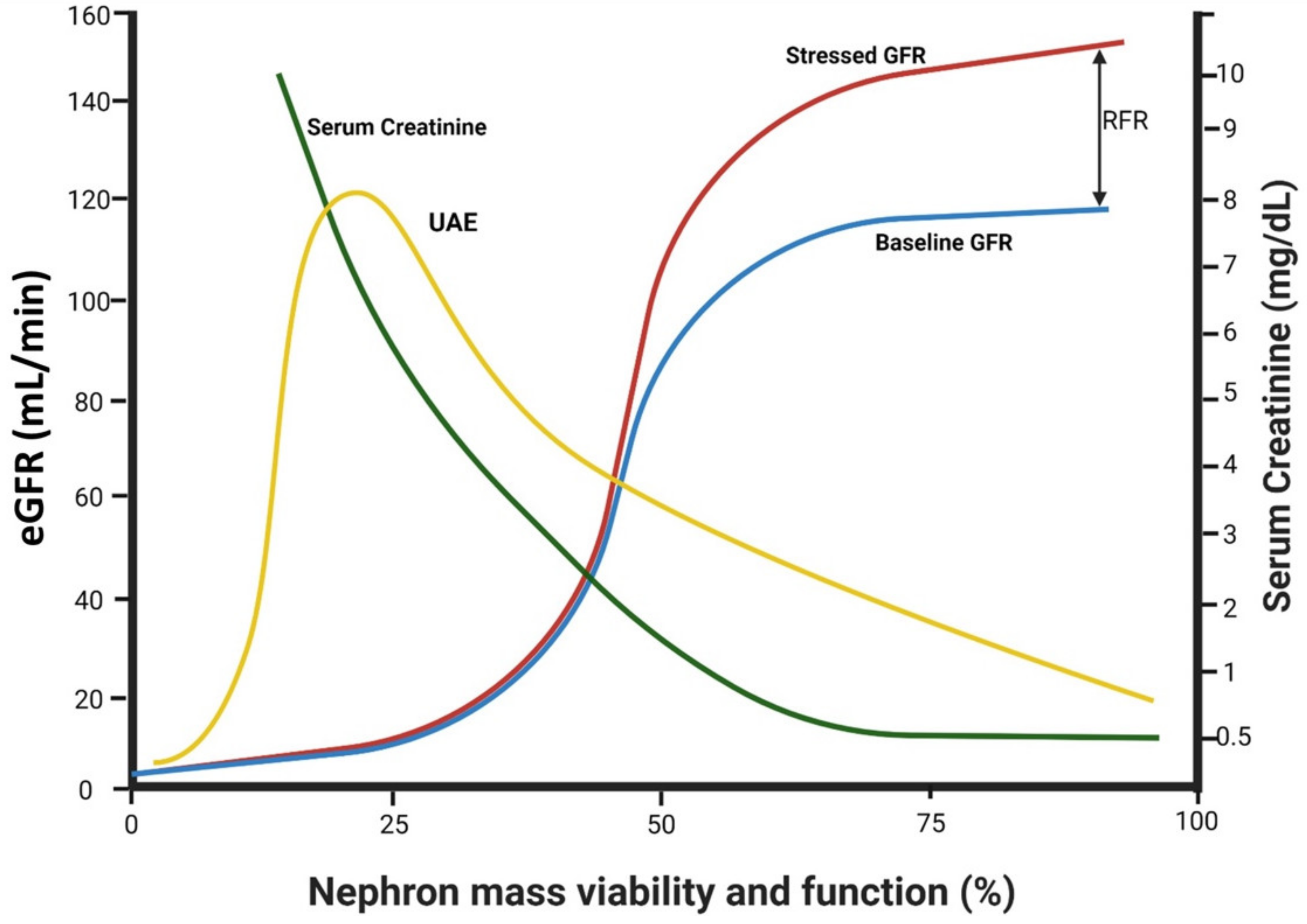
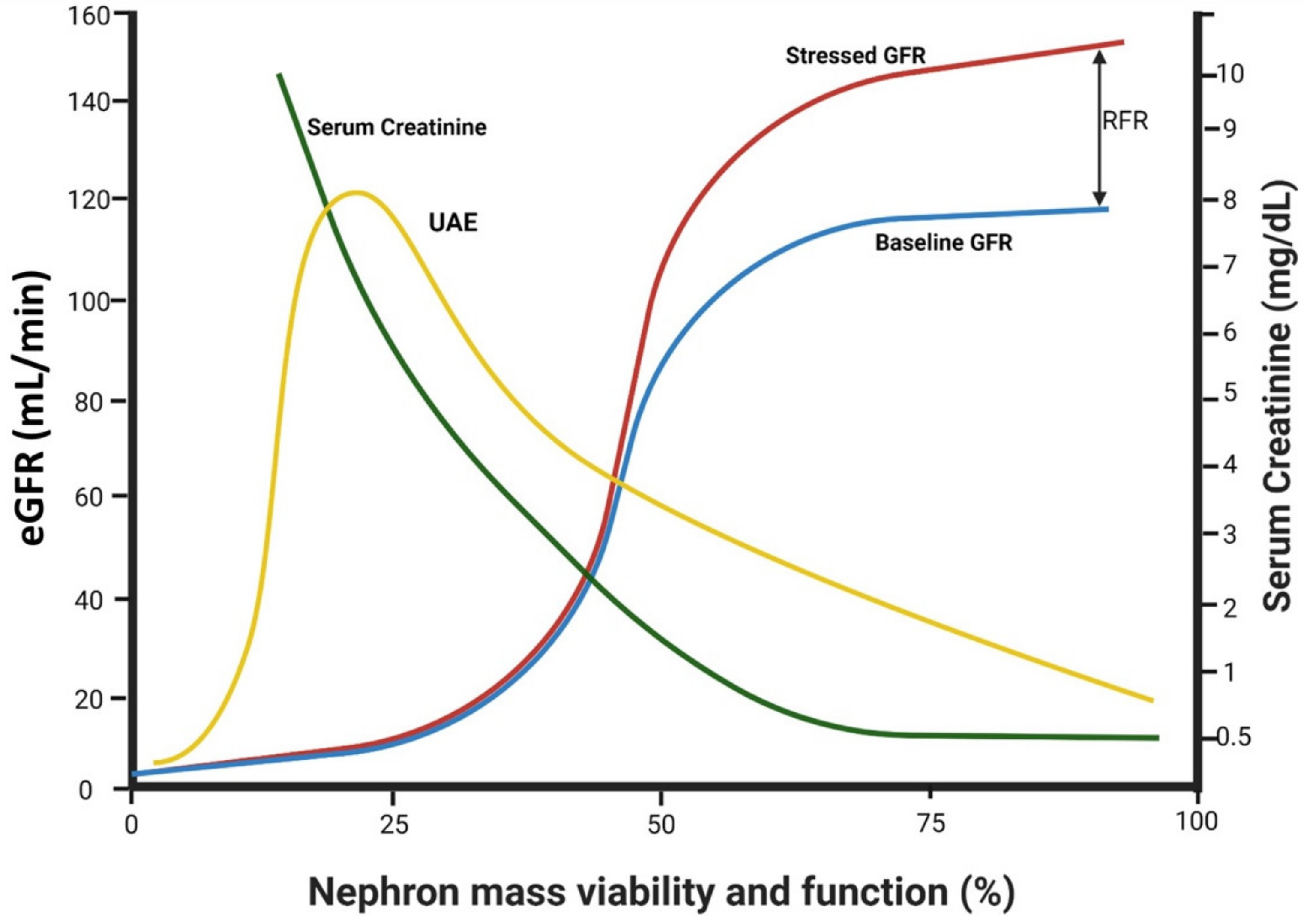
2.3. Factors Affecting Trans-Glomerular Pressure and Glomerular Protein Leak: The Role of Declining Renal Functional Reserve
2.4. Diabetic Tubulopathy—A Role in Albuminuria and Progressive DN
2.5. Albuminuria Accelerates the Progression of DN
2.6. Integrated Glomerulo-Tubular Adjustments Affecting Proteinuria
3. Clinical Perspectives
3.1. Attenuating Proteinuria Is Nephroprotective
3.2. When to Initiate Anti-Proteinuric Treatment?
3.3. The Protein Restriction Controversy
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Perkovic, V.; Agarwal, R.; Fioretto, P.; Hemmelgarn, B.R.; Levin, A.; Thomas, M.C.; Wanner, C.; Kasiske, B.L.; Wheeler, D.C.; Groop, P.H. Management of patients with diabetes and CKD: Conclusions from a “Kidney Disease: Improving Global Outcomes” (KDIGO) Controversies Conference. Kidney Int. 2016, 90, 1175–1183. [Google Scholar] [CrossRef] [PubMed]
- Gerstein, H.C.; Mann, J.F.; Yi, Q.; Zinman, B.; Dinneen, S.F.; Hoogwerf, B.; Halle, J.P.; Young, J.; Rashkow, A.; Joyce, C.; et al. Albuminuria and risk of cardiovascular events, death, and heart failure in diabetic and nondiabetic individuals. JAMA 2001, 286, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Zimmet, P.; Alberti, K.G.; Magliano, D.J.; Bennett, P.H. Diabetes mellitus statistics on prevalence and mortality: Facts and fallacies. Nat. Rev. Endocrinol. 2016, 12, 616–622. [Google Scholar] [CrossRef] [PubMed]
- Kramer, H.J.; Nguyen, Q.D.; Curhan, G.; Hsu, C.Y. Renal insufficiency in the absence of albuminuria and retinopathy among adults with type 2 diabetes mellitus. JAMA 2003, 289, 3273–3277. [Google Scholar] [CrossRef]
- Yamanouchi, M.; Furuichi, K.; Hoshino, J.; Ubara, Y.; Wada, T. Nonproteinuric diabetic kidney disease. Clin. Exp. Nephrol. 2020, 24, 573–581. [Google Scholar] [CrossRef]
- Anders, H.J.; Huber, T.B.; Isermann, B.; Schiffer, M. CKD in diabetes: Diabetic kidney disease versus nondiabetic kidney disease. Nat. Rev. Nephrol. 2018, 14, 361–377. [Google Scholar] [CrossRef]
- Chen, C.; Wang, C.; Hu, C.; Han, Y.; Zhao, L.; Zhu, X.; Xiao, L.; Sun, L. Normoalbuminuric diabetic kidney disease. Front. Med. 2017, 11, 310–318. [Google Scholar] [CrossRef]
- Bertani, T.; Gambara, V.; Remuzzi, G. Structural basis of diabetic nephropathy in microalbuminuric NIDDM patients: A light microscopy study. Diabetologia 1996, 39, 1625–1628. [Google Scholar] [CrossRef]
- Mahtal, N.; Lenoir, O.; Tharaux, P.L. Glomerular Endothelial Cell Crosstalk with Podocytes in Diabetic Kidney Disease. Front. Med. 2021, 8, 659013. [Google Scholar] [CrossRef]
- Lin, Y.C.; Chang, Y.H.; Yang, S.Y.; Wu, K.D.; Chu, T.S. Update of pathophysiology and management of diabetic kidney disease. J. Formos. Med. Assoc. 2018, 117, 662–675. [Google Scholar] [CrossRef]
- Toyoda, M.; Najafian, B.; Kim, Y.; Caramori, M.L.; Mauer, M. Podocyte detachment and reduced glomerular capillary endothelial fenestration in human type 1 diabetic nephropathy. Diabetes 2007, 56, 2155–2160. [Google Scholar] [CrossRef] [PubMed]
- Ndisang, J.F. Glomerular Endothelium and its Impact on Glomerular Filtration Barrier in Diabetes: Are the Gaps Still Illusive? Curr. Med. Chem. 2018, 25, 1525–1529. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Lee, K.; Chuang, P.Y.; Liu, Z.; He, J.C. Glomerular endothelial cell injury and cross talk in diabetic kidney disease. Am. J. Physiol.-Renal Physiol. 2015, 308, F287–F297. [Google Scholar] [CrossRef] [PubMed]
- Ceccarelli Ceccarelli, D.; Paleari, R.; Solerte, B.; Mosca, A. Re-thinking diabetic nephropathy: Microalbuminuria is just a piece of the diagnostic puzzle. Clin. Chim. Acta 2022, 524, 146–153. [Google Scholar] [CrossRef]
- Abassi, Z.; Armaly, Z.; Heyman, S.N. Glycocalyx Degradation in Ischemia-Reperfusion Injury. Am. J. Pathol. 2020, 190, 752–767. [Google Scholar] [CrossRef]
- Comper, W.D. There is little evidence that the endothelial glycocalyx has a specific role in glomerular permeability of albumin. Kidney Int. 2020, 97, 1057. [Google Scholar] [CrossRef]
- Sol, M.; Kamps, J.; van den Born, J.; van den Heuvel, M.C.; van der Vlag, J.; Krenning, G.; Hillebrands, J.L. Glomerular Endothelial Cells as Instigators of Glomerular Sclerotic Diseases. Front. Pharmacol. 2020, 11, 573557. [Google Scholar] [CrossRef]
- Kopel, J.; Pena-Hernandez, C.; Nugent, K. Evolving spectrum of diabetic nephropathy. World J. Diabetes 2019, 10, 269–279. [Google Scholar] [CrossRef]
- Siddiqi, F.S.; Advani, A. Endothelial-podocyte crosstalk: The missing link between endothelial dysfunction and albuminuria in diabetes. Diabetes 2013, 62, 3647–3655. [Google Scholar] [CrossRef]
- Daehn, I.S. Glomerular Endothelial Cell Stress and Cross-Talk with Podocytes in Early [corrected] Diabetic Kidney Disease. Front. Med. 2018, 5, 76. [Google Scholar] [CrossRef]
- Qi, H.; Casalena, G.; Shi, S.; Yu, L.; Ebefors, K.; Sun, Y.; Zhang, W.; D’Agati, V.; Schlondorff, D.; Haraldsson, B.; et al. Glomerular Endothelial Mitochondrial Dysfunction Is Essential and Characteristic of Diabetic Kidney Disease Susceptibility. Diabetes 2017, 66, 763–778. [Google Scholar] [CrossRef] [PubMed]
- Ryan, G.B.; Karnovsky, M.J. Distribution of endogenous albumin in the rat glomerulus: Role of hemodynamic factors in glomerular barrier function. Kidney Int. 1976, 9, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Naylor, R.W.; Morais, M.; Lennon, R. Complexities of the glomerular basement membrane. Nat. Rev. Nephrol. 2021, 17, 112–127. [Google Scholar] [CrossRef]
- Kriz, W.; Lowen, J.; Federico, G.; van den Born, J.; Grone, E.; Grone, H.J. Accumulation of worn-out GBM material substantially contributes to mesangial matrix expansion in diabetic nephropathy. Am. J. Physiol.-Renal Physiol. 2017, 312, F1101–F1111. [Google Scholar] [CrossRef]
- Kolset, S.O.; Reinholt, F.P.; Jenssen, T. Diabetic nephropathy and extracellular matrix. J. Histochem. Cytochem. 2012, 60, 976–986. [Google Scholar] [CrossRef]
- Goode, N.P.; Shires, M.; Crellin, D.M.; Aparicio, S.R.; Davison, A.M. Alterations of glomerular basement membrane charge and structure in diabetic nephropathy. Diabetologia 1995, 38, 1455–1465. [Google Scholar] [CrossRef]
- Sugita, E.; Hayashi, K.; Hishikawa, A.; Itoh, H. Epigenetic Alterations in Podocytes in Diabetic Nephropathy. Front. Pharmacol. 2021, 12, 759299. [Google Scholar] [CrossRef]
- Koop, K.; Eikmans, M.; Baelde, H.J.; Kawachi, H.; De Heer, E.; Paul, L.C.; Bruijn, J.A. Expression of podocyte-associated molecules in acquired human kidney diseases. J. Am. Soc. Nephrol. 2003, 14, 2063–2071. [Google Scholar] [CrossRef]
- Jefferson, J.A.; Shankland, S.J.; Pichler, R.H. Proteinuria in diabetic kidney disease: A mechanistic viewpoint. Kidney Int. 2008, 74, 22–36. [Google Scholar] [CrossRef]
- Kopp, J.B.; Anders, H.J.; Susztak, K.; Podesta, M.A.; Remuzzi, G.; Hildebrandt, F.; Romagnani, P. Podocytopathies. Nat. Rev. Dis. Primers 2020, 6, 68. [Google Scholar] [CrossRef]
- Yang, S.; Han, Y.; Liu, J.; Song, P.; Xu, X.; Zhao, L.; Hu, C.; Xiao, L.; Liu, F.; Zhang, H.; et al. Mitochondria: A Novel Therapeutic Target in Diabetic Nephropathy. Curr. Med. Chem. 2017, 24, 3185–3202. [Google Scholar] [CrossRef]
- Su, J.; Ye, D.; Gao, C.; Huang, Q.; Gui, D. Mechanism of progression of diabetic kidney disease mediated by podocyte mitochondrial injury. Mol. Biol. Rep. 2020, 47, 8023–8035. [Google Scholar] [CrossRef]
- Palmer, M.B.; Abedini, A.; Jackson, C.; Blady, S.; Chatterjee, S.; Sullivan, K.M.; Townsend, R.R.; Brodbeck, J.; Almaani, S.; Srivastava, A.; et al. The Role of Glomerular Epithelial Injury in Kidney Function Decline in Patients with Diabetic Kidney Disease in the TRIDENT Cohort. Kidney Int. Rep. 2021, 6, 1066–1080. [Google Scholar] [CrossRef]
- Carlstrom, M.; Wilcox, C.S.; Arendshorst, W.J. Renal autoregulation in health and disease. Physiol. Rev. 2015, 95, 405–511. [Google Scholar] [CrossRef]
- Tonneijck, L.; Muskiet, M.H.; Smits, M.M.; van Bommel, E.J.; Heerspink, H.J.; van Raalte, D.H.; Joles, J.A. Glomerular Hyperfiltration in Diabetes: Mechanisms, Clinical Significance, and Treatment. J. Am. Soc. Nephrol. 2017, 28, 1023–1039. [Google Scholar] [CrossRef]
- Vallon, V.; Gerasimova, M.; Rose, M.A.; Masuda, T.; Satriano, J.; Mayoux, E.; Koepsell, H.; Thomson, S.C.; Rieg, T. SGLT2 inhibitor empagliflozin reduces renal growth and albuminuria in proportion to hyperglycemia and prevents glomerular hyperfiltration in diabetic Akita mice. Am. J. Physiol.-Renal Physiol. 2014, 306, F194–F204. [Google Scholar] [CrossRef]
- Stockand, J.D.; Sansom, S.C. Glomerular mesangial cells: Electrophysiology and regulation of contraction. Physiol. Rev. 1998, 78, 723–744. [Google Scholar] [CrossRef]
- Blantz, R.C.; Singh, P. Glomerular and tubular function in the diabetic kidney. Adv. Chronic Kidney Dis. 2014, 21, 297–303. [Google Scholar] [CrossRef]
- Palm, F.; Cederberg, J.; Hansell, P.; Liss, P.; Carlsson, P.O. Reactive oxygen species cause diabetes-induced decrease in renal oxygen tension. Diabetologia 2003, 46, 1153–1160. [Google Scholar] [CrossRef]
- Heyman, S.N.; Rosenberger, C.; Rosen, S.; Khamaisi, M. Why is diabetes mellitus a risk factor for contrast-induced nephropathy? Biomed. Res. Int. 2013, 2013, 123589. [Google Scholar] [CrossRef] [Green Version]
- Mathiesen, E.R.; Hommel, E.; Olsen, U.B.; Parving, H.H. Elevated urinary prostaglandin excretion and the effect of indomethacin on renal function in incipient diabetic nephropathy. Diabet. Med. 1988, 5, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Elving, L.D.; Wetzels, J.F.; de Nobel, E.; Hoitsma, A.J.; Berden, J.H. Captopril acutely lowers albuminuria in normotensive patients with diabetic nephropathy. Am. J. Kidney Dis. 1992, 20, 559–563. [Google Scholar] [CrossRef]
- Andersen, S.; Tarnow, L.; Rossing, P.; Hansen, B.V.; Parving, H.H. Renoprotective effects of angiotensin II receptor blockade in type 1 diabetic patients with diabetic nephropathy. Kidney Int. 2000, 57, 601–606. [Google Scholar] [CrossRef] [PubMed]
- Thomson, S.C.; Vallon, V. Effects of SGLT2 inhibitor and dietary NaCl on glomerular hemodynamics assessed by micropuncture in diabetic rats. Am. J. Physiol.-Renal Physiol. 2021, 320, F761–F771. [Google Scholar] [CrossRef]
- Ogata, C.; Kamide, K.; Suzuki, Y.; Sasaki, O.; Kubota, Y.; Sato, H.; Takiuchi, S.; Horio, T.; Inenaga, T.; Kawano, Y. Evaluation of intrarenal hemodynamics by Doppler ultrasonography for renoprotective effect of angiotensin receptor blockade. Clin. Nephrol. 2005, 64, 352–357. [Google Scholar] [CrossRef]
- Ruggenenti, P.; Cortinovis, M.; Parvanova, A.; Trillini, M.; Iliev, I.P.; Bossi, A.C.; Belviso, A.; Aparicio, M.C.; Trevisan, R.; Rota, S.; et al. Preventing microalbuminuria with benazepril, valsartan, and benazepril-valsartan combination therapy in diabetic patients with high-normal albuminuria: A prospective, randomized, open-label, blinded endpoint (PROBE) study. PLoS Med. 2021, 18, e1003691. [Google Scholar] [CrossRef]
- Wanner, C.; Inzucchi, S.E.; Zinman, B. Empagliflozin and Progression of Kidney Disease in Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 1801–1802. [Google Scholar] [CrossRef]
- Nordquist, L.; Friederich-Persson, M.; Fasching, A.; Liss, P.; Shoji, K.; Nangaku, M.; Hansell, P.; Palm, F. Activation of hypoxia-inducible factors prevents diabetic nephropathy. J. Am. Soc. Nephrol. 2015, 26, 328–338. [Google Scholar] [CrossRef]
- Pullman, T.N.; Alving, A.S.; Dern, R.J.; Landowne, M. The influence of dietary protein intake on specific renal functions in normal man. J. Lab. Clin. Med. 1954, 44, 320–332. [Google Scholar]
- Sasaki, T.; Nakagawa, K.; Hata, J.; Hirakawa, Y.; Shibata, M.; Nakano, T.; Tsuboi, N.; Oda, Y.; Kitazono, T.; Yokoo, T.; et al. Pathologic Diabetic Nephropathy in Autopsied Diabetic Cases with Normoalbuminuria From a Japanese Community-Based Study. Kidney Int. Rep. 2021, 6, 3035–3044. [Google Scholar] [CrossRef]
- Jufar, A.H.; Lankadeva, Y.R.; May, C.N.; Cochrane, A.D.; Bellomo, R.; Evans, R.G. Renal functional reserve: From physiological phenomenon to clinical biomarker and beyond. Am. J. Physiol.-Regul Integr. Comp. Physiol. 2020, 319, R690–R702. [Google Scholar] [CrossRef] [PubMed]
- Gorelik, Y.; Khamaisi, M.; Abassi, Z.; Evans, R.G.; Heyman, S.N. Renal functional recovery among inpatients: A plausible marker of reduced renal functional reserve. Clin. Exp. Pharmacol. Physiol. 2021, 48, 1724–1727. [Google Scholar] [CrossRef]
- Gorelik, Y.; Bloch-Isenberg, N.; Hashoul, S.; Heyman, S.N.; Khamaisi, M. Hyperglycemia on Admission Predicts Acute Kidney Failure and Renal Functional Recovery among Inpatients. J. Clin. Med. 2021, 11, 54. [Google Scholar] [CrossRef] [PubMed]
- De Nicola, L.; Blantz, R.C.; Gabbai, F.B. Renal functional reserve in the early stage of experimental diabetes. Diabetes 1992, 41, 267–273. [Google Scholar] [CrossRef]
- Sackmann, H.; Tran-Van, T.; Tack, I.; Hanaire-Broutin, H.; Tauber, J.P.; Ader, J.L. Renal functional reserve in IDDM patients. Diabetologia 1998, 41, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Earle, K.A.; Mehrotra, S.; Dalton, R.N.; Denver, E.; Swaminathan, R. Defective nitric oxide production and functional renal reserve in patients with type 2 diabetes who have microalbuminuria of African and Asian compared with white origin. J. Am. Soc. Nephrol. 2001, 12, 2125–2130. [Google Scholar] [CrossRef]
- Zaletel, J.; Cerne, D.; Lenart, K.; Zitta, S.; Jurgens, G.; Estelberger, W.; Kocijancic, A. Renal functional reserve in patients with Type 1 diabetes mellitus. Wien. Klin. Wochenschr. 2004, 116, 246–251. [Google Scholar] [CrossRef]
- Dedov, I.I.; Mukhin, N.A.; Shestakova, M.V.; Paltzev, M.A.; Warshawskyi, V.A.; Severgina, E.S.; Nikolaev, A. Renal functional reserve in diabetic patients without clinical nephropathy: Comparisons with renal morphology. Diabet. Med. 1991, 8, S43–S47. [Google Scholar] [CrossRef]
- Nosadini, R.; Trevisan, R.; Fioretto, P.; Semplicini, A.; Sama, B.; Velussi, M.; Da Campo, G.L.; Avogaro, A.; Vizzaccaro, A.; Donadon, V. Kidney hemodynamics after ketone body and amino acid infusion in normal and IDDM subjects. Diabetes 1989, 38, 75–83. [Google Scholar] [CrossRef]
- Russo, L.M.; Bakris, G.L.; Comper, W.D. Renal handling of albumin: A critical review of basic concepts and perspective. Am. J. Kidney Dis. 2002, 39, 899–919. [Google Scholar] [CrossRef]
- Zeni, L.; Norden, A.G.W.; Cancarini, G.; Unwin, R.J. A more tubulocentric view of diabetic kidney disease. J. Nephrol. 2017, 30, 701–717. [Google Scholar] [CrossRef] [PubMed]
- Tojo, A.; Kinugasa, S. Mechanisms of glomerular albumin filtration and tubular reabsorption. Int. J. Nephrol. 2012, 2012, 481520. [Google Scholar] [CrossRef] [PubMed]
- Dickson, L.E.; Wagner, M.C.; Sandoval, R.M.; Molitoris, B.A. The proximal tubule and albuminuria: Really! J. Am. Soc. Nephrol. 2014, 25, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Castrop, H.; Schiessl, I.M. Novel routes of albumin passage across the glomerular filtration barrier. Acta Physiol. 2017, 219, 544–553. [Google Scholar] [CrossRef]
- Christensen, E.I.; Nielsen, R. Role of megalin and cubilin in renal physiology and pathophysiology. In Reviews of Physiology, Biochemistry and Pharmacology; Springer: Berlin/Heidelberg, Germany, 2006; Volume 158, pp. 1–22. [Google Scholar]
- Chaudhury, C.; Brooks, C.L.; Carter, D.C.; Robinson, J.M.; Anderson, C.L. Albumin binding to FcRn: Distinct from the FcRn-IgG interaction. Biochemistry 2006, 45, 4983–4990. [Google Scholar] [CrossRef]
- Tojo, A.; Endou, H. Intrarenal Handling of Proteins in Rats Using Fractional Micropuncture Technique. Am. J. Physiol. 1992, 263, F601–F606. [Google Scholar] [CrossRef]
- Norden, A.G.; Lapsley, M.; Lee, P.J.; Pusey, C.D.; Scheinman, S.J.; Tam, F.W.; Thakker, R.V.; Unwin, R.J.; Wrong, O. Glomerular protein sieving and implications for renal failure in Fanconi syndrome. Kidney Int. 2001, 60, 1885–1892. [Google Scholar] [CrossRef]
- Russo, L.M.; Sandoval, R.M.; McKee, M.; Osicka, T.M.; Collins, A.B.; Brown, D.; Molitoris, B.A.; Comper, W.D. The normal kidney filters nephrotic levels of albumin retrieved by proximal tubule cells: Retrieval is disrupted in nephrotic states. Kidney Int. 2007, 71, 504–513. [Google Scholar] [CrossRef] [PubMed]
- Gekle, M. Renal albumin handling: A look at the dark side of the filter. Kidney Int. 2007, 71, 479–481. [Google Scholar] [CrossRef]
- Comper, W.D.; Russo, L.M.; Vuchkova, J. Are filtered plasma proteins processed in the same way by the kidney? J. Theor. Biol. 2016, 410, 18–24. [Google Scholar] [CrossRef]
- Comper, W.D. Megalin/cubilin has a minor role in the proximal tubular cell uptake of filtered albumin. Kidney Int. 2018, 93, 1014. [Google Scholar] [CrossRef] [PubMed]
- Weyer, K.; Andersen, P.K.; Schmidt, K.; Mollet, G.; Antignac, C.; Birn, H.; Nielsen, R.; Christensen, E.I. Abolishment of proximal tubule albumin endocytosis does not affect plasma albumin during nephrotic syndrome in mice. Kidney Int. 2018, 93, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Sarav, M.; Wang, Y.; Hack, B.K.; Chang, A.; Jensen, M.; Bao, L.; Quigg, R.J. Renal FcRn reclaims albumin but facilitates elimination of IgG. J. Am. Soc. Nephrol. 2009, 20, 1941–1952. [Google Scholar] [CrossRef]
- Greive, K.A.; Balazs, N.D.H.; Comper, W.D. Protein fragments in urine have been considerably underestimated by various protein assays. Clin. Chem. 2001, 47, 1717–1719. [Google Scholar] [CrossRef] [PubMed]
- Russo, L.M.; Sandoval, R.M.; Campos, S.B.; Molitoris, B.A.; Comper, W.D.; Brown, D. Impaired tubular uptake explains albuminuria in early diabetic nephropathy. J. Am. Soc. Nephrol. 2009, 20, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Y.; Yang, J.H.; Xu, J.; Jia, J.Y.; Zhang, X.R.; Yue, X.D.; Chen, L.M.; Shan, C.Y.; Zheng, M.Y.; Han, F.; et al. Renal tubular damage may contribute more to acute hyperglycemia induced kidney injury in non-diabetic conscious rats. J. Diabetes Its Complicat. 2015, 29, 621–628. [Google Scholar] [CrossRef]
- Tian, D.; Shi, X.; Zhao, Y.; Peng, X.; Zou, L.; Xu, L.; Ma, Y.; Wen, Y.; Faulhaber-Walter, R.; Chen, L. The effect of A1 adenosine receptor in diabetic megalin loss with caspase-1/IL18 signaling. Diabetes Metab. Syndr. Obes. 2019, 12, 1583–1596. [Google Scholar] [CrossRef]
- Franzen, S.; Pihl, L.; Khan, N.; Gustafsson, H.; Palm, F. Pronounced kidney hypoxia precedes albuminuria in type 1 diabetic mice. Am. J. Physiol.-Renal Physiol. 2016, 310, F807–F809. [Google Scholar] [CrossRef]
- Rosenberger, C.; Khamaisi, M.; Abassi, Z.; Shilo, V.; Weksler-Zangen, S.; Goldfarb, M.; Shina, A.; Zibertrest, F.; Eckardt, K.U.; Rosen, S.; et al. Adaptation to hypoxia in the diabetic rat kidney. Kidney Int. 2008, 73, 34–42. [Google Scholar] [CrossRef]
- Rosenberger, C.; Khamaisi, M.; Goldfarb, M.; Shina, A.; Shilo, V.; Zilbertrest, F.; Rosen, S.; Heyman, S.N. Acute kidney injury in the diabetic rat: Studies in the isolated perfused and intact kidney. Am. J. Nephrol. 2008, 28, 831–839. [Google Scholar] [CrossRef]
- O’Neill, J.; Fasching, A.; Pihl, L.; Patinha, D.; Franzen, S.; Palm, F. Acute SGLT inhibition normalizes O2 tension in the renal cortex but causes hypoxia in the renal medulla in anaesthetized control and diabetic rats. Am. J. Physiol.-Renal Physiol. 2015, 309, F227–F234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szalat, A.; Perlman, A.; Muszkat, M.; Khamaisi, M.; Abassi, Z.; Heyman, S.N. Can SGLT2 Inhibitors Cause Acute Renal Failure? Plausible Role for Altered Glomerular Hemodynamics and Medullary Hypoxia. Drug Saf. 2018, 41, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Darawshi, S.; Yaseen, H.; Gorelik, Y.; Faor, C.; Szalat, A.; Abassi, Z.; Heyman, S.N.; Khamaisi, M. Biomarker evidence for distal tubular damage but cortical sparing in hospitalized diabetic patients with acute kidney injury (AKI) while on SGLT2 inhibitors. Renal Fail. 2020, 42, 836–844. [Google Scholar] [CrossRef] [PubMed]
- Packer, M. Role of Impaired Nutrient and Oxygen Deprivation Signaling and Deficient Autophagic Flux in Diabetic CKD Development: Implications for Understanding the Effects of Sodium-Glucose Cotransporter 2-Inhibitors. J. Am. Soc. Nephrol. 2020, 31, 907–919. [Google Scholar]
- Remuzzi, G.; Bertani, T. Pathophysiology of progressive nephropathies. N. Engl. J. Med. 1998, 339, 1448–1456. [Google Scholar] [CrossRef]
- Bakris, G.L. Slowing nephropathy progression: Focus on proteinuria reduction. Clin. J. Am. Soc. Nephrol. 2008, 3 (Suppl. 1), S3–S10. [Google Scholar] [PubMed]
- Eijkelkamp, W.B.; Zhang, Z.; Remuzzi, G.; Parving, H.H.; Cooper, M.E.; Keane, W.F.; Shahinfar, S.; Gleim, G.W.; Weir, M.R.; Brenner, B.M.; et al. Albuminuria is a target for renoprotective therapy independent from blood pressure in patients with type 2 diabetic nephropathy: Post hoc analysis from the Reduction of Endpoints in NIDDM with the Angiotensin II Antagonist Losartan (RENAAL) trial. J. Am. Soc. Nephrol. 2007, 18, 1540–1546. [Google Scholar] [CrossRef] [PubMed]
- De Zeeuw, D.; Remuzzi, G.; Parving, H.H.; Keane, W.F.; Zhang, Z.; Shahinfar, S.; Snapinn, S.; Cooper, M.E.; Mitch, W.E.; Brenner, B.M. Proteinuria, a target for renoprotection in patients with type 2 diabetic nephropathy: Lessons from RENAAL. Kidney Int. 2004, 65, 2309–2320. [Google Scholar]
- Thomas, M.E.; Schreiner, G.F. Contribution of proteinuria to progressive renal injury: Consequences of tubular uptake of fatty acid bearing albumin. Am. J. Nephrol. 1993, 13, 385–398. [Google Scholar]
- Kees-Folts, D.; Sadow, J.L.; Schreiner, G.F. Tubular catabolism of albumin is associated with the release of an inflammatory lipid. Kidney Int. 1994, 45, 1697–1709. [Google Scholar] [CrossRef]
- Thomas, M.E.; Morrison, A.R.; Schreiner, G.F. Metabolic effects of fatty acid-bearing albumin on a proximal tubule cell line. Am. J. Physiol. 1995, 268, F1177–F1184. [Google Scholar] [CrossRef] [PubMed]
- Dove, E.; Kowalewska, J. Educational Case: Nephrotic Syndrome in Older Adult. Acad Pathol. 2020, 7, 2374289520944554. [Google Scholar] [CrossRef] [PubMed]
- Markova, I.; Miklankova, D.; Huttl, M.; Kacer, P.; Skibova, J.; Kucera, J.; Sedlacek, R.; Kacerova, T.; Kazdova, L.; Malinska, H. The Effect of Lipotoxicity on Renal Dysfunction in a Nonobese Rat Model of Metabolic Syndrome: A Urinary Proteomic Approach. J. Diabetes Res. 2019, 2019, 8712979. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Gaivin, R.; Abramovich, C.; Boylan, M.; Calles, J.; Schelling, J.R. Fatty acid transport protein-2 regulates glycemic control and diabetic kidney disease progression. JCI Insight 2020, 5, e136845. [Google Scholar] [CrossRef]
- Shapiro, H.; Theilla, M.; Attal-Singer, J.; Singer, P. Effects of polyunsaturated fatty acid consumption in diabetic nephropathy. Nat. Rev. Nephrol. 2011, 7, 110–121. [Google Scholar] [CrossRef]
- Hou, Y.; Li, S.; Wu, M.; Wei, J.; Ren, Y.; Du, C.; Wu, H.; Han, C.; Duan, H.; Shi, Y. Mitochondria-targeted peptide SS-31 attenuates renal injury via an antioxidant effect in diabetic nephropathy. Am. J. Physiol.-Renal Physiol. 2016, 310, F547–F559. [Google Scholar] [CrossRef]
- Jang, H.S.; Noh, M.R.; Kim, J.; Padanilam, B.J. Defective Mitochondrial Fatty Acid Oxidation and Lipotoxicity in Kidney Diseases. Front. Med. 2020, 7, 65. [Google Scholar]
- Tang, S.C.; Lai, K.N. The pathogenic role of the renal proximal tubular cell in diabetic nephropathy. Nephrol. Dial. Transplant. 2012, 27, 3049–3056. [Google Scholar] [CrossRef]
- Opingari, E.; Verma, S.; Connelly, K.A.; Mazer, C.D.; Teoh, H.; Quan, A.; Zuo, F.; Pan, Y.; Bhatt, D.L.; Zinman, B.; et al. The impact of empagliflozin on kidney injury molecule-1: A subanalysis of the Effects of Empagliflozin on Cardiac Structure, Function, and Circulating Biomarkers in Patients with Type 2 Diabetes CardioLink-6 trial. Nephrol. Dial. Transplant. 2020, 35, 895–897. [Google Scholar] [CrossRef]
- Nishi, H.; Nangaku, M. Podocyte lipotoxicity in diabetic kidney disease. Kidney Int. 2019, 96, 809–812. [Google Scholar] [CrossRef]
- Zhang, Y.; Xiao, H.Q.; Zeng, X.T.; Zuo, H.X.; Xu, Y.C. Associations between endothelial nitric oxide synthase polymorphisms and risk of diabetic nephropathy: An updated meta-analysis. Ren. Fail. 2015, 37, 312–326. [Google Scholar] [CrossRef] [PubMed]
- Zou, H.; Wu, G.; Lv, J.; Xu, G. Relationship of angiotensin I-converting enzyme (ACE) and bradykinin B2 receptor (BDKRB2) polymorphism with diabetic nephropathy. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2017, 1863, 1264–1272. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Guan, M.; Bowden, D.W.; Ng, M.C.; Hicks, P.J.; Lea, J.P.; Ma, L.; Gao, C.; Palmer, N.D.; Freedman, B.I. Association Analysis of the Cubilin (CUBN) and Megalin (LRP2) Genes with ESRD in African Americans. Clin. J. Am. Soc. Nephrol. 2016, 11, 1034–1043. [Google Scholar] [CrossRef]
- Ihalmo, P.; Wessman, M.; Kaunisto, M.A.; Kilpikari, R.; Parkkonen, M.; Forsblom, C.; Holthofer, H.; Groop, P.H. Association analysis of podocyte slit diaphragm genes as candidates for diabetic nephropathy. Diabetologia 2008, 51, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Jotwani, V.; Scherzer, R.; Abraham, A.; Estrella, M.M.; Bennett, M.; Cohen, M.H.; Nowicki, M.; Sharma, A.; Young, M.; Tien, P.C.; et al. Association of urine alpha1-microglobulin with kidney function decline and mortality in HIV-infected women. Clin. J. Am. Soc. Nephrol. 2015, 10, 63–73. [Google Scholar] [CrossRef] [Green Version]
- Charytan, D.M. Spironolactone in dialysis: What’s old is new again. Am. J. Kidney Dis. 2016, 68, 512–514. [Google Scholar] [CrossRef]
- Ix, J.H.; Katz, R.; Bansal, N.; Foster, M.; Weiner, D.E.; Tracy, R.; Jotwani, V.; Hughes-Austin, J.; McKay, D.; Gabbai, F.; et al. Urine Fibrosis Markers and Risk of Allograft Failure in Kidney Transplant Recipients: A Case-Cohort Ancillary Study of the FAVORIT Trial. Am. J. Kidney Dis. 2017, 69, 410–419. [Google Scholar] [CrossRef]
- Anders, H.J.; Davis, J.M.; Thurau, K. Nephron Protection in Diabetic Kidney Disease. N. Engl. J. Med. 2016, 375, 2096–2098. [Google Scholar] [CrossRef]
- Cravedi, P.; Ruggenenti, P.; Remuzzi, G. Proteinuria should be used as a surrogate in CKD. Nat. Rev. Nephrol. 2012, 8, 301–306. [Google Scholar] [CrossRef]
- Brenner, B.M.; Cooper, M.E.; de Zeeuw, D.; Keane, W.F.; Mitch, W.E.; Parving, H.H.; Remuzzi, G.; Snapinn, S.M.; Zhang, Z.; Shahinfar, S. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N. Engl. J. Med. 2001, 345, 861–869. [Google Scholar] [CrossRef]
- Lewis, E.J.; Hunsicker, L.G.; Clarke, W.R.; Berl, T.; Pohl, M.A.; Lewis, J.B.; Ritz, E.; Atkins, R.C.; Rohde, R.; Raz, I.; et al. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N. Engl. J. Med. 2001, 345, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Bakris, G.L.; Agarwal, R.; Chan, J.C.; Cooper, M.E.; Gansevoort, R.T.; Haller, H.; Remuzzi, G.; Rossing, P.; Schmieder, R.E.; Nowack, C.; et al. Effect of Finerenone on Albuminuria in Patients with Diabetic Nephropathy A Randomized Clinical Trial. JAMA 2015, 314, 884–894. [Google Scholar] [CrossRef] [PubMed]
- Bakris, G.L.; Agarwal, R.; Anker, S.D.; Pitt, B.; Ruilope, L.M.; Rossing, P.; Kolkhof, P.; Nowack, C.; Schloemer, P.; Joseph, A.; et al. Effect of Finerenone on Chronic Kidney Disease Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2020, 383, 2219–2229. [Google Scholar] [CrossRef] [PubMed]
- Heerspink, H.J.L.; Stefansson, B.V.; Correa-Rotter, R.; Chertow, G.M.; Greene, T.; Hou, F.F.; Mann, J.F.E.; McMurray, J.J.V.; Lindberg, M.; Rossing, P.; et al. Dapagliflozin in Patients with Chronic Kidney Disease. N. Engl. J. Med. 2020, 383, 1436–1446. [Google Scholar] [CrossRef]
- Wiviott, S.D.; Raz, I.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Silverman, M.G.; Zelniker, T.A.; Kuder, J.F.; Murphy, S.A.; et al. Dapagliflozin and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2019, 380, 347–357. [Google Scholar] [CrossRef]
- Neal, B.; Perkovic, V.; Mahaffey, K.W.; de Zeeuw, D.; Fulcher, G.; Erondu, N.; Shaw, W.; Law, G.; Desai, M.; Matthews, D.R. Canagliflozin and Cardiovascular and Renal Events in Type 2 Diabetes. N. Engl. J. Med. 2017, 377, 644–657. [Google Scholar] [CrossRef]
- Mosenzon, O.; Wiviott, S.D.; Cahn, A.; Rozenberg, A.; Yanuv, I.; Goodrich, E.L.; Murphy, S.A.; Heerspink, H.J.L.; Zelniker, T.A.; Dwyer, J.P.; et al. Effects of dapagliflozin on development and progression of kidney disease in patients with type 2 diabetes: An analysis from the DECLARE-TIMI 58 randomised trial. Lancet Diabetes Endocrinol. 2019, 7, 606–617. [Google Scholar] [CrossRef]
- Mosenzon, O.; Leibowitz, G.; Bhatt, D.L.; Cahn, A.; Hirshberg, B.; Wei, C.; Im, K.; Rozenberg, A.; Yanuv, I.; Stahre, C.; et al. Effect of Saxagliptin on Renal Outcomes in the SAVOR-TIMI 53 Trial. Diabetes Care 2017, 40, 69–76. [Google Scholar] [CrossRef]
- Green, J.B.; Bethel, M.A.; Armstrong, P.W.; Buse, J.B.; Engel, S.S.; Garg, J.; Josse, R.; Kaufman, K.D.; Koglin, J.; Korn, S.; et al. Effect of Sitagliptin on Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 232–242. [Google Scholar] [CrossRef]
- Perkovic, V.; Toto, R.; Cooper, M.E.; Mann, J.F.E.; Rosenstock, J.; McGuire, D.K.; Kahn, S.E.; Marx, N.; Alexander, J.H.; Zinman, B.; et al. Effects of Linagliptin on Cardiovascular and Kidney Outcomes in People With Normal and Reduced Kidney Function: Secondary Analysis of the CARMELINA Randomized Trial. Diabetes Care 2020, 43, 1803–1812. [Google Scholar] [CrossRef]
- Marso, S.P.; Bain, S.C.; Consoli, A.; Eliaschewitz, F.G.; Jodar, E.; Leiter, L.A.; Lingvay, I.; Rosenstock, J.; Seufert, J.; Warren, M.L.; et al. Semaglutide and Cardiovascular Outcomes in Patients with Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 1834–1844. [Google Scholar] [CrossRef] [PubMed]
- Mann, J.F.E.; Orsted, D.D.; Brown-Frandsen, K.; Marso, S.P.; Poulter, N.R.; Rasmussen, S.; Tornoe, K.; Zinman, B.; Buse, J.B. Liraglutide and Renal Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2017, 377, 839–848. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, M.A.; Claggett, B.; Diaz, R.; Dickstein, K.; Gerstein, H.C.; Kober, L.V.; Lawson, F.C.; Ping, L.; Wei, X.; Lewis, E.F.; et al. Lixisenatide in Patients with Type 2 Diabetes and Acute Coronary Syndrome. N. Engl. J. Med. 2015, 373, 2247–2257. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://clinicaltrials.gov/ct2/show/NCT03819153 (accessed on 14 September 2022).
- Wheeler, D.C.; Jongs, N.; Stefansson, B.V.; Chertow, G.M.; Greene, T.; Hou, F.F.; Langkilde, A.M.; McMurray, J.J.V.; Rossing, P.; Nowicki, M.; et al. Safety and efficacy of dapagliflozin in patients with focal segmental glomerulosclerosis: A prespecified analysis of the dapagliflozin and prevention of adverse outcomes in chronic kidney disease (DAPA-CKD) trial. Nephrol. Dial. Transplant. 2022, 37, 1647–1656. [Google Scholar] [CrossRef]
- Mann, J.F.E.; Hansen, T.; Idorn, T.; Leiter, L.A.; Marso, S.P.; Rossing, P.; Seufert, J.; Tadayon, S.; Vilsbøll, T. Effects of once-weekly subcutaneous semaglutide on kidney function and safety in patients with type 2 diabetes: A post-hoc analysis of the SUSTAIN 1-7 randomised controlled trials. Lancet Diabetes Endocrinol. 2020, 8, 880–893. [Google Scholar] [CrossRef]
- Retnakaran, R.; Cull, C.A.; Thorne, K.I.; Adler, A.I.; Holman, R.R.; Grp, U.S. Risk factors for renal dysfunction in type 2 diabetes—UK prospective diabetes study 74. Diabetes 2006, 55, 1832–1839. [Google Scholar] [CrossRef]
- Yau, A.; Parikh, S.V.; Almaani, S. Diabetic Kidney Disease: The “Silent” Majority? Kidney Int. Rep. 2021, 6, 2939–2941. [Google Scholar] [CrossRef]
- Klemmer, P.; Grim, C.E.; Luft, F.C. Who and What Drove Walter Kempner? The Rice Diet Revisited. Hypertension 2014, 64, 684–688. [Google Scholar] [CrossRef]
- MacLaughlin, H.L.; Friedman, A.N.; Ikizler, T.A. Nutrition in Kidney Disease: Core Curriculum 2022. Am. J. Kidney Dis. 2021, 79, 437–449. [Google Scholar] [CrossRef]
- Brenner, B.M.; Meyer, T.W.; Hostetter, T.H. Dietary protein intake and the progressive nature of kidney disease: The role of hemodynamically mediated glomerular injury in the pathogenesis of progressive glomerular sclerosis in aging, renal ablation, and intrinsic renal disease. N. Engl. J. Med. 1982, 307, 652–659. [Google Scholar]
- Hostetter, T.H.; Olson, J.L.; Rennke, H.G.; Venkatachalam, M.A.; Brenner, B.M. Hyperfiltration in remnant nephrons: A potentially adverse response to renal ablation. Am. J. Physiol. 1981, 241, F85–F93. [Google Scholar] [CrossRef] [PubMed]
- Brouhard, B.H.; LaGrone, L. Effect of dietary protein restriction on functional renal reserve in diabetic nephropathy. Am. J. Med. 1990, 89, 427–431. [Google Scholar] [CrossRef]
- Juraschek, S.P.; Appel, L.J.; Anderson, C.A.; Miller, E.R., 3rd. Effect of a high-protein diet on kidney function in healthy adults: Results from the OmniHeart trial. Am. J. Kidney Dis. 2013, 61, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Farr, L.E. Assimilation of protein by young children with the nephrotic syndrome: III. Eddect of nephrotic crises on assimilation of nitrogen. Am. J. Dis. Child. 1939, 58, 939–948. [Google Scholar] [CrossRef]
- Kaysen, G.A.; Gambertoglio, J.; Jimenez, I.; Jones, H.; Hutchison, F.N. Effect of dietary protein intake on albumin homeostasis in nephrotic patients. Kidney Int. 1986, 29, 572–577. [Google Scholar] [CrossRef]
- Kaysen, G.A.; Jones, H.; Martin, V., Jr.; Hutchison, F.N. A low-protein diet restricts albumin synthesis in nephrotic rats. J. Clin. Investig. 1989, 83, 1623–1629. [Google Scholar] [CrossRef]
- Moshage, H.J.; Janssen, J.A.; Franssen, J.H.; Hafkenscheid, J.C.; Yap, S.H. Study of the molecular mechanism of decreased liver synthesis of albumin in inflammation. J. Clin. Investig. 1987, 79, 1635–1641. [Google Scholar] [CrossRef]
- Sun, L.; Kanwar, Y.S. Relevance of TNF-alpha in the context of other inflammatory cytokines in the progression of diabetic nephropathy. Kidney Int. 2015, 88, 662–665. [Google Scholar] [CrossRef]
- Feng, L.; Gao, Q.; Hu, K.; Wu, M.; Wang, Z.; Chen, F.; Mei, F.; Zhao, L.; Ma, B. Prevalence and risk factors of sarcopenia in patients with diabetes: A meta-analysis. J. Clin. Endocrinol. Metab. 2022, 107, 1470–1483. [Google Scholar] [CrossRef]
- Wong, L.; Duque, G.; McMahon, L.P. Sarcopenia and Frailty: Challenges in Mainstream Nephrology Practice. Kidney Int. Rep. 2021, 6, 2554–2564. [Google Scholar] [CrossRef]
- Oosterwijk, M.M.; Groothof, D.; Navis, G.; Bakker, S.J.L.; Laverman, G.D. High-Normal Protein Intake Is Not Associated with Faster Renal Function Deterioration in Patients with Type 2 Diabetes: A Prospective Analysis in the DIALECT Cohort. Diabetes Care 2022, 45, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Heyman, N.; Samuel, R.I.; Zaid, A. Comment on Oosterwijk et al. High-Normal Protein Intake Is Not Associated with Faster Renal Function Deterioration in Patients with Type 2 Diabetes: A Prospective Analysis in the DIALECT Cohort. Diabetes Care 2022, 45, e67–e68. [Google Scholar] [CrossRef] [PubMed]
- Kaysen, G.A.; Davies, R.W.; Hutchison, F.N. Effect of dietary protein intake and angiotensin converting enzyme inhibition in Heymann nephritis. Kidney Int. Suppl. 1989, 27, S154–S162. [Google Scholar] [PubMed]
- D’Amico, G.; Remuzzi, G.; Maschio, G.; Gentile, M.G.; Gotti, E.; Oldrizzi, L.; Manna, G.; Mecca, G.; Rugiu, C.; Fellin, G. Effect of dietary proteins and lipids in patients with membranous nephropathy and nephrotic syndrome. Clin. Nephrol. 1991, 35, 237–242. [Google Scholar] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Class | Impact on Proteinuria | Acute Effects on Kidney Function | Long-Term Impact on Kidney Function | Mechanisms Leading to Reduced Proteinuria | Mechanisms of Renal Protection |
|---|---|---|---|---|---|
| ACE inhibitors | Reduce proteinuria | Reduce GFR | Renoprotective | - Reduce glomerular hypertension via efferent arteriolar vasodilation - Improved metabolic control attenuates structural glomerular changes - Improved tubular protein re-uptake? | - Reducing proteinuria leads to attenuated tubulointerstitial disease - Blocking Ang II/aldosterone-mediated inflammation, tubular apoptosis and fibrosis |
| ARBs | Reduce proteinuria | Reduce GFR | Renoprotective | Reduce glomerular hypertension via efferent arteriolar vasodilation | - Reducing proteinuria leads to attenuated tubulointerstitial disease - Blocking Ang II/aldosterone-mediated inflammation, tubular apoptosis and fibrosis |
| SGLT2 inhibitors | Reduce proteinuria | Reduce GFR | Renoprotective | Immediate reduction in glomerular hypertension via restoration of tubulo-glomerular feedback (TGF) | - Improved metabolic control attenuates structural glomerular injury - Reducing proteinuria leads to attenuated tubulointerstitial disease - Possible additional anti-fibrotic properties |
| DPP4 antagonists | Reduce proteinuria | None | Probably no impact | Restoration of metabolic derangements leading to gradual reduction in glomerular hypertension via restoration of TGF with afferent arteriolar vasoconstriction afferent arteriolar vasoconstriction | - Improved metabolic control may attenuate structural glomerular injury - Reducing proteinuria may lead to attenuated tubulointerstitial disease |
| GLP1 analogues | Reduce proteinuria | None Mildly reduced GFR by 12 weeks | Currently unknown | Restoration of metabolic derangements leading to gradual reduction in glomerular hypertension via restoration of TGF with afferent arteriolar vasoconstriction afferent arteriolar vasoconstriction | - Improved metabolic control may attenuate structural glomerular injury - Reducing proteinuria may lead to attenuated tubulointerstitial disease |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heyman, S.N.; Raz, I.; Dwyer, J.P.; Weinberg Sibony, R.; Lewis, J.B.; Abassi, Z. Diabetic Proteinuria Revisited: Updated Physiologic Perspectives. Cells 2022, 11, 2917. https://doi.org/10.3390/cells11182917
Heyman SN, Raz I, Dwyer JP, Weinberg Sibony R, Lewis JB, Abassi Z. Diabetic Proteinuria Revisited: Updated Physiologic Perspectives. Cells. 2022; 11(18):2917. https://doi.org/10.3390/cells11182917
Chicago/Turabian StyleHeyman, Samuel N., Itamar Raz, Jamie P. Dwyer, Roni Weinberg Sibony, Julia B. Lewis, and Zaid Abassi. 2022. "Diabetic Proteinuria Revisited: Updated Physiologic Perspectives" Cells 11, no. 18: 2917. https://doi.org/10.3390/cells11182917
APA StyleHeyman, S. N., Raz, I., Dwyer, J. P., Weinberg Sibony, R., Lewis, J. B., & Abassi, Z. (2022). Diabetic Proteinuria Revisited: Updated Physiologic Perspectives. Cells, 11(18), 2917. https://doi.org/10.3390/cells11182917