
Approaches to Measuring the Activity of Major Lipolytic and Lipogenic Enzymes In Vitro and Ex Vivo


Abstract
:1. Introduction
2. Lipolysis and Lipolytic Enzymes
2.1. Adipose Triglyceride Lipase (EC 3.1.1.3)
2.2. Hormone-Sensitive Lipase (EC 3.1.1.79)
2.3. Assays for Measurement of ATGL and HSL Activity
2.3.1. Isolation and Purification of the Lipolytic Enzymes
2.3.2. Conditions Affecting the Ex Vivo Activity of ATGL and HSL
2.3.3. Radioisotope Methods for Measurement of Lipolytic Enzyme Activity Determination
2.3.4. Spectrophotometric and Fluorescence Methods of Lipolytic Enzyme Activity Determination
2.3.5. Advantages and Disadvantages of the ATGL and HSL Activity Measurement
3. Enzymes of De Novo Lipogenesis
3.1. Acetyl-CoA Carboxylase (EC 6.4.1.2)
3.2. Fatty-Acid Synthase (EC 2.3.1.85)
3.3. Assays for Measurement of ACC and FAS Activity
3.3.1. Isolation and Purification of ACC
3.3.2. Radioisotope Methods of ACC Activity Determination
3.3.3. Spectrophotometric Methods of ACC Activity Determination
3.3.4. HPLC Measurement of ACC Activity
3.3.5. Advantages and Disadvantages of the ACC Activity Measurement
3.3.6. Isolation and Purification of FAS
3.3.7. Radioisotope Methods of FAS Activity Determination
3.3.8. The Spectrophotometric and Fluorescence Methods of FAS Activity Determination
3.3.9. Advantages and Disadvantages of the FAS Activity Measurement
4. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Nye, C.; Kim, J.; Kalhan, S.C.; Hanson, R.W. Reassessing triglyceride synthesis in adipose tissue. Trends Endocrinol. Metab. 2008, 19, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Ahmadian, M.; Duncan, R.E.; Jaworski, K.; Sarkadi-Nagy, E. Triacylglycerol metabolism in adipose tissue. Future Lipidol. 2007, 2, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Kershaw, E.E.; Hamm, J.K.; Verhagen, L.A.; Peroni, O.; Katic, M.; Flier, J.S. Adipose triglyceride lipase: Function, regulation by insulin, and comparison with adiponutrin. Diabetes 2006, 55, 148–157. [Google Scholar] [CrossRef]
- Coppack, S.W.; Jensen, M.D.; Miles, J.M. In vivo regulation of lipolysis in humans. J. Lipid Res. 1994, 35, 177–193. [Google Scholar] [CrossRef]
- Lafontan, M.; Langin, D. Lipolysis and lipid mobilization in human adipose tissue. Prog. Lipid Res. 2009, 48, 275–297. [Google Scholar] [CrossRef] [PubMed]
- Hellerstein, M.K. De novo lipogenesis in humans: Metabolic and regulatory aspects. Eur. J. Clin. Nutr. 1999, 53 (Suppl. 1), S53–S65. [Google Scholar] [CrossRef] [PubMed]
- Edens, N.K.; Leibel, R.L.; Hirsch, J. Mechanism of free fatty acid re-esterification in human adipocytes in vitro. J. Lipid Res. 1990, 31, 1423–1431. [Google Scholar] [CrossRef]
- Yen, C.-L.E.; Stone, S.J.; Koliwad, S.; Harris, C.; Farese, R.V. Thematic Review Series: Glycerolipids. DGAT enzymes and triacylglycerol biosynthesis. J. Lipid Res. 2008, 49, 2283–2301. [Google Scholar] [CrossRef]
- Ameer, F.; Scandiuzzi, L.; Hasnain, S.; Kalbacher, H.; Zaidi, N. De novo lipogenesis in health and disease. Metabolism 2014, 63, 895–902. [Google Scholar] [CrossRef]
- Morigny, P.; Boucher, J.; Arner, P.; Langin, D. Lipid and glucose metabolism in white adipocytes: Pathways, dysfunction and therapeutics. Nat. Rev. Endocrinol. 2021, 17, 276–295. [Google Scholar] [CrossRef]
- Morigny, P.; Houssier, M.; Mouisel, E.; Langin, D. Adipocyte lipolysis and insulin resistance. Biochimie 2016, 125, 259–266. [Google Scholar] [CrossRef] [PubMed]
- van Herpen, N.A.; Schrauwen-Hinderling, V.B. Lipid accumulation in non-adipose tissue and lipotoxicity. Physiol. Behav. 2008, 94, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Schaffer, J.E. Lipotoxicity: When tissues overeat. Curr. Opin. Lipidol. 2003, 14, 281–287. [Google Scholar] [CrossRef]
- Savage, D.B.; Petersen, K.F.; Shulman, G.I. Disordered lipid metabolism and the pathogenesis of insulin resistance. Physiol. Rev. 2007, 87, 507–520. [Google Scholar] [CrossRef] [PubMed]
- Balkau, B.; Valensi, P.; Eschwege, E.; Slama, G. A review of the metabolic syndrome. Diabetes Metab. 2007, 33, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, K.; Abrams, G.A. Metabolic liver disease of obesity and role of adipose tissue in the pathogenesis of nonalcoholic fatty liver disease. World J. Gastroenterol. 2007, 13, 3540–3553. [Google Scholar] [CrossRef]
- DeFronzo, R.A. Pathogenesis of type 2 diabetes mellitus. Med. Clin. N. Am. 2004, 88, 787–835. [Google Scholar] [CrossRef]
- Baracos, V.E.; Martin, L.; Korc, M.; Guttridge, D.C.; Fearon, K.C.H. Cancer-associated cachexia. Nat. Rev. Dis. Primers 2018, 4, 17105. [Google Scholar] [CrossRef]
- Smith, G.I.; Shankaran, M.; Yoshino, M.; Schweitzer, G.G.; Chondronikola, M.; Beals, J.W.; Okunade, A.L.; Patterson, B.W.; Nyangau, E.; Field, T.; et al. Insulin resistance drives hepatic de novo lipogenesis in nonalcoholic fatty liver disease. J. Clin. Investig. 2020, 130, 1453–1460. [Google Scholar] [CrossRef]
- Yilmaz, M.; Claiborn, K.C.; Hotamisligil, G.S. De Novo Lipogenesis Products and Endogenous Lipokines. Diabetes 2016, 65, 1800–1807. [Google Scholar] [CrossRef] [Green Version]
- Song, Z.; Xiaoli, A.M.; Yang, F. Regulation and Metabolic Significance of De Novo Lipogenesis in Adipose Tissues. Nutrients 2018, 10, 1383. [Google Scholar] [CrossRef] [PubMed]
- Scheja, L.; Heeren, J. The endocrine function of adipose tissues in health and cardiometabolic disease. Nat. Rev. Endocrinol. 2019, 15, 507–524. [Google Scholar] [CrossRef] [PubMed]
- Long, J.Z.; Cravatt, B.F. The metabolic serine hydrolases and their functions in mammalian physiology and disease. Chem. Rev. 2011, 111, 6022–6063. [Google Scholar] [CrossRef]
- Karlsson, M.; Contreras, J.A.; Hellman, U.; Tornqvist, H.; Holm, C. cDNA cloning, tissue distribution, and identification of the catalytic triad of monoglyceride lipase. Evolutionary relationship to esterases, lysophospholipases, and haloperoxidases. J. Biol. Chem. 1997, 272, 27218–27223. [Google Scholar] [CrossRef]
- Long, J.Z.; Nomura, D.K.; Cravatt, B.F. Characterization of monoacylglycerol lipase inhibition reveals differences in central and peripheral endocannabinoid metabolism. Chem. Biol. 2009, 16, 744–753. [Google Scholar] [CrossRef] [PubMed]
- Gamblin, C.; Rouault, C.; Lacombe, A.; Langa-Vives, F.; Farabos, D.; Lamaziere, A.; Clement, K.; Gautier, E.L.; Yvan-Charvet, L.; Dugail, I. Lysosomal Acid Lipase Drives Adipocyte Cholesterol Homeostasis and Modulates Lipid Storage in Obesity, Independent of Autophagy. Diabetes 2021, 70, 76–90. [Google Scholar] [CrossRef]
- Zechner, R.; Kienesberger, P.C.; Haemmerle, G.; Zimmermann, R.; Lass, A. Adipose triglyceride lipase and the lipolytic catabolism of cellular fat stores. J. Lipid Res. 2009, 50, 3–21. [Google Scholar] [CrossRef]
- Zimmermann, R.; Strauss, J.G.; Haemmerle, G.; Schoiswohl, G.; Birner-Gruenberger, R.; Riederer, M.; Lass, A.; Neuberger, G.; Eisenhaber, F.; Hermetter, A.; et al. Fat Mobilization in Adipose Tissue is Promoted by Adopose Triglyceride Lipase. Science 2004, 306, 1383–1386. [Google Scholar] [CrossRef]
- Zimmermann, R.; Lass, A.; Haemmerle, G.; Zechner, R. Fate of fat: The role of adipose triglyceride lipase in lipolysis. Biochim. Biophys. Acta 2009, 1791, 494–500. [Google Scholar] [CrossRef]
- Obrowsky, S.; Chandak, P.G.; Patankar, J.V.; Povoden, S.; Schlager, S.; Kershaw, E.E.; Bogner-Strauss, J.G.; Hoefler, G.; Levak-Frank, S.; Kratky, D. Adipose triglyceride lipase is a TG hydrolase of the small intestine and regulates intestinal PPARalpha signaling. J. Lipid Res. 2013, 54, 425–435. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Promes, J.A.; Harata, M.; Mishra, A.; Stephens, S.B.; Taylor, E.B.; Burand, A.J., Jr.; Sivitz, W.I.; Fink, B.D.; Ankrum, J.A.; et al. Adipose Triglyceride Lipase Is a Key Lipase for the Mobilization of Lipid Droplets in Human beta-Cells and Critical for the Maintenance of Syntaxin 1a Levels in beta-Cells. Diabetes 2020, 69, 1178–1192. [Google Scholar] [CrossRef] [PubMed]
- Cerk, I.K.; Wechselberger, L.; Oberer, M. Adipose Triglyceride Lipase Regulation: An Overview. Curr. Protein Pept. Sci. 2018, 19, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Lass, A.; Zimmermann, R.; Haemmerle, G.; Riederer, M.; Schoiswohl, G.; Schweiger, M.; Kienesberger, P.; Strauss, J.G.; Gorkiewicz, G.; Zechner, R. Adipose triglyceride lipase-mediated lipolysis of cellular fat stores is activated by CGI-58 and defective in Chanarin-Dorfman Syndrome. Cell Metab. 2006, 3, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Kimmel, A.R.; Sztalryd, C. The Perilipins: Major Cytosolic Lipid Droplet-Associated Proteins and Their Roles in Cellular Lipid Storage, Mobilization, and Systemic Homeostasis. Annu. Rev. Nutr. 2016, 36, 471–509. [Google Scholar] [CrossRef]
- Granneman, J.G.; Moore, H.P.; Mottillo, E.P.; Zhu, Z.; Zhou, L. Interactions of perilipin-5 (Plin5) with adipose triglyceride lipase. J. Biol. Chem. 2011, 286, 5126–5135. [Google Scholar] [CrossRef]
- Sztalryd, C.; Brasaemle, D.L. The perilipin family of lipid droplet proteins: Gatekeepers of intracellular lipolysis. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 1221–1232. [Google Scholar] [CrossRef]
- Yang, X.; Lu, X.; Lombes, M.; Rha, G.B.; Chi, Y.I.; Guerin, T.M.; Smart, E.J.; Liu, J. The G(0)/G(1) switch gene 2 regulates adipose lipolysis through association with adipose triglyceride lipase. Cell Metab. 2010, 11, 194–205. [Google Scholar] [CrossRef]
- Nagy, H.M.; Paar, M.; Heier, C.; Moustafa, T.; Hofer, P.; Haemmerle, G.; Lass, A.; Zechner, R.; Oberer, M.; Zimmermann, R. Adipose triglyceride lipase activity is inhibited by long-chain acyl-coenzyme A. Biochim. Biophys. Acta 2014, 1841, 588–594. [Google Scholar] [CrossRef]
- Mayer, N.; Schweiger, M.; Romauch, M.; Grabner, G.F.; Eichmann, T.O.; Fuchs, E.; Ivkovic, J.; Heier, C.; Mrak, I.; Lass, A.; et al. Development of small-molecule inhibitors targeting adipose triglyceride lipase. Nat. Chem. Biol. 2013, 9, 785–787. [Google Scholar] [CrossRef]
- Iglesias, J.; Lamontagne, J.; Erb, H.; Gezzar, S.; Zhao, S.; Joly, E.; Truong, V.L.; Skorey, K.; Crane, S.; Madiraju, S.R.; et al. Simplified assays of lipolysis enzymes for drug discovery and specificity assessment of known inhibitors. J. Lipid Res. 2016, 57, 131–141. [Google Scholar] [CrossRef] [Green Version]
- Grabner, G.F.; Guttenberger, N.; Mayer, N.; Migglautsch-Sulzer, A.K.; Lembacher-Fadum, C.; Fawzy, N.; Bulfon, D.; Hofer, P.; Zullig, T.; Hartig, L.; et al. Small-Molecule Inhibitors Targeting Lipolysis in Human Adipocytes. J. Am. Chem. Soc. 2022, 144, 6237–6250. [Google Scholar] [CrossRef] [PubMed]
- Reisenberg, M.; Singh, P.K.; Williams, G.; Doherty, P. The diacylglycerol lipases: Structure, regulation and roles in and beyond endocannabinoid signalling. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2012, 367, 3264–3275. [Google Scholar] [CrossRef] [PubMed]
- Kridel, S.J.; Axelrod, F.; Rozenkrantz, N.; Smith, J.W. Orlistat is a novel inhibitor of fatty acid synthase with antitumor activity. Cancer Res.. 2004, 64, 2070–2075. [Google Scholar] [CrossRef] [PubMed]
- McNeely, W.; Benfield, P. Orlistat. Drugs 1998, 56, 241–249, discussion 250. [Google Scholar] [CrossRef] [PubMed]
- Sjöström, L.; Rissanen, A.; Andersen, T.; Boldrin, M.; Golay, A.; Koppeschaar, H.P.F.; Krempf, M. Randomised placebo-controlled trial of orlistat for weight loss and prevention of weight regain in obese patients. Lancet 1998, 352, 167–172. [Google Scholar] [CrossRef]
- Ballinger, A.; Peikin, S.R. Orlistat: Its current status as an anti-obesity drug. Eur. J. Pharmacol. 2001, 440, 109–117. [Google Scholar] [CrossRef]
- Korbelius, M.; Vujic, N.; Sachdev, V.; Obrowsky, S.; Rainer, S.; Gottschalk, B.; Graier, W.F.; Kratky, D. ATGL/CGI-58-Dependent Hydrolysis of a Lipid Storage Pool in Murine Enterocytes. Cell Rep. 2019, 28, 1923–1934.e4. [Google Scholar] [CrossRef]
- Cornaciu, I.; Boeszoermenyi, A.; Lindermuth, H.; Nagy, H.M.; Cerk, I.K.; Ebner, C.; Salzburger, B.; Gruber, A.; Schweiger, M.; Zechner, R.; et al. The minimal domain of adipose triglyceride lipase (ATGL) ranges until leucine 254 and can be activated and inhibited by CGI-58 and G0S2, respectively. PLoS ONE 2011, 6, e26349. [Google Scholar] [CrossRef]
- Filleur, S.; Nelius, T.; de Riese, W.; Kennedy, R.C. Characterization of PEDF: A multi-functional serpin family protein. J. Cell. Biochem. 2009, 106, 769–775. [Google Scholar] [CrossRef]
- Chen, Y.; Carlessi, R.; Walz, N.; Cruzat, V.F.; Keane, K.; John, A.N.; Jiang, F.X.; Carnagarin, R.; Dass, C.R.; Newsholme, P. Pigment epithelium-derived factor (PEDF) regulates metabolism and insulin secretion from a clonal rat pancreatic beta cell line BRIN-BD11 and mouse islets. Mol. Cell. Endocrinol. 2016, 426, 50–60. [Google Scholar] [CrossRef]
- Kulminskaya, N.; Oberer, M. Protein-protein interactions regulate the activity of Adipose Triglyceride Lipase in intracellular lipolysis. Biochimie 2020, 169, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Kimmel, A.R.; Sztalryd, C. Perilipin 5, a lipid droplet protein adapted to mitochondrial energy utilization. Curr. Opin. Lipidol. 2014, 25, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Zagani, R.; El-Assaad, W.; Gamache, I.; Teodoro, J.G. Inhibition of adipose triglyceride lipase (ATGL) by the putative tumor suppressor G0S2 or a small molecule inhibitor attenuates the growth of cancer cells. Oncotarget 2015, 6, 28282–28295. [Google Scholar] [CrossRef]
- Wan, Z.; Matravadia, S.; Holloway, G.P.; Wright, D.C. FAT/CD36 regulates PEPCK expression in adipose tissue. Am. J. Physiol. Cell Physiol. 2013, 304, C478–C484. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, C.; Khatua, B.; Noel, P.; Kostenko, S.; Bag, A.; Balakrishnan, B.; Patel, K.S.; Guerra, A.A.; Martinez, M.N.; Trivedi, S.; et al. Pancreatic triglyceride lipase mediates lipotoxic systemic inflammation. J. Clin. Investig. 2020, 130, 1931–1947. [Google Scholar] [CrossRef] [PubMed]
- Fako, V.E.; Zhang, J.T.; Liu, J.Y. Mechanism of Orlistat Hydrolysis by the Thioesterase of Human Fatty Acid Synthase. ACS Catal. 2014, 4, 3444–3453. [Google Scholar] [CrossRef]
- Yeaman, S.J. Hormone-sensitive lipase--new roles for an old enzyme. Biochem. J. 2004, 379, 11–22. [Google Scholar] [CrossRef]
- Mita, T.; Furuhashi, M.; Hiramitsu, S.; Ishii, J.; Hoshina, K.; Ishimura, S.; Fuseya, T.; Watanabe, Y.; Tanaka, M.; Ohno, K.; et al. FABP4 is secreted from adipocytes by adenyl cyclase-PKA- and guanylyl cyclase-PKG-dependent lipolytic mechanisms. Obesity 2015, 23, 359–367. [Google Scholar] [CrossRef]
- Holm, C. Molecular mechanisms regulating hormone-sensitive lipase and lipolysis. Biochem. Soc. Trans. 2003, 31, 1120–1124. [Google Scholar] [CrossRef]
- Watt, M.J.; Holmes, A.G.; Pinnamaneni, S.K.; Garnham, A.P.; Steinberg, G.R.; Kemp, B.E.; Febbraio, M.A. Regulation of HSL serine phosphorylation in skeletal muscle and adipose tissue. Am. J. Physiol. Endocrinol. Metab. 2006, 290, E500–E508. [Google Scholar] [CrossRef]
- Viollet, B.; Andreelli, F.; Jorgensen, S.B.; Perrin, C.; Flamez, D.; Mu, J.; Wojtaszewski, J.F.; Schuit, F.C.; Birnbaum, M.; Richter, E.; et al. Physiological role of AMP-activated protein kinase (AMPK): Insights from knockout mouse models. Biochem. Soc. Trans. 2003, 31, 216–219. [Google Scholar] [CrossRef]
- Ueno, M.; Suzuki, J.; Hirose, M.; Sato, S.; Imagawa, M.; Zenimaru, Y.; Takahashi, S.; Ikuyama, S.; Koizumi, T.; Konoshita, T.; et al. Cardiac overexpression of perilipin 2 induces dynamic steatosis: Prevention by hormone-sensitive lipase. Am. J. Physiol. Endocrinol. Metab. 2017, 313, E699–E709. [Google Scholar] [CrossRef] [PubMed]
- Claus, T.H.; Lowe, D.B.; Liang, Y.; Salhanick, A.I.; Lubeski, C.K.; Yang, L.; Lemoine, L.; Zhu, J.; Clairmont, K.B. Specific inhibition of hormone-sensitive lipase improves lipid profile while reducing plasma glucose. J. Pharmacol. Exp. Ther. 2005, 315, 1396–1402. [Google Scholar] [CrossRef] [PubMed]
- Mottillo, E.P.; Shen, X.J.; Granneman, J.G. Role of hormone-sensitive lipase in beta-adrenergic remodeling of white adipose tissue. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E1188–E1197. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Feng, X.J.; Li, Z.M.; Li, M.; Gao, S.; He, Y.H.; Wang, J.J.; Zeng, S.Y.; Liu, X.P.; Huang, X.Y.; et al. Downregulation of adipose triglyceride lipase promotes cardiomyocyte hypertrophy by triggering the accumulation of ceramides. Arch Biochem. Biophys. 2015, 565, 76–88. [Google Scholar] [CrossRef] [PubMed]
- Ebdrup, ø.; Refsgaard, H.H.F.; Fledelius, C.; Jacobsen, P. Synthesis and Structure-Activity Relationship for a Novel Class of Potent and Selective Carbamate-Based Inhibitors of Hormone Selective Lipase with Acute In Vivo Antilipolytic Effects. J. Med. Chem. 2006, 2007, 5449–5456. [Google Scholar] [CrossRef]
- Bustanji, Y.; Issa, A.; Mohammad, M.; Hudaib, M.; Tawah, K.; Alkhatib, H.; Almasri, I.; Al-Khalidi, B. Inhibition of hormone sensitive lipase and pancreatic lipase by Rosmarinus officinalis extract and selected phenolic constituents. J. Med. Plants Res. 2010, 4, 2235–2242. [Google Scholar] [CrossRef]
- Vagelos, P.R.; Alberts, A.W.; Martin, D.B. Studies on the Mechanism of Activation of Acetyl Coenzyme A Carboxylase by Citrate. J. Biol. Chem. 1963, 238, 533–540. [Google Scholar] [CrossRef]
- Beaty, N.B.; Lane, M.D. Kinetics of activation of acetyl-CoA carboxylase by citrate. Relationship to the rate of polymerization of the enzyme. J. Biol. Chem. 1983, 258, 13043–13050. [Google Scholar] [CrossRef]
- Hardie, D.G.; Cohen, P. Purification and physicochemical properties of fatty acid synthetase and acetyl-CoA carboxylase from lactating rabbit mammary gland. Eur. J. Biochem. 1978, 92, 25–34. [Google Scholar] [CrossRef]
- Boone, A.N.; Chan, A.; Kulpa, J.E.; Brownsey, R.W. Bimodal activation of acetyl-CoA carboxylase by glutamate. J. Biol. Chem. 2000, 275, 10819–10825. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, A.; Chen, H.-Q.; Modrick, L.M.; Stefanelli, C. Activation of Acetyl-CoA Carboxylase by a Glutamateand Magnesium-Sensitive Protein Phosphatase in the Islet β-Cell. Diabetes 2001, 50, 1580–1587. [Google Scholar] [CrossRef] [PubMed]
- Brownsey, R.W.; Boone, A.N.; Elliott, J.E.; Kulpa, J.E.; Lee, W.M. Regulation of acetyl-CoA carboxylase. Biochem. Soc. Trans. 2006, 34, 223–227. [Google Scholar] [CrossRef]
- Galic, S.; Loh, K.; Murray-Segal, L.; Steinberg, G.R.; Andrews, Z.B.; Kemp, B.E. AMPK signaling to acetyl-CoA carboxylase is required for fasting- and cold-induced appetite but not thermogenesis. Elife 2018, 7, e32656. [Google Scholar] [CrossRef]
- Levert, K.L.; Waldrop, G.L.; Stephens, J.M. A biotin analog inhibits acetyl-CoA carboxylase activity and adipogenesis. J. Biol. Chem. 2002, 277, 16347–16350. [Google Scholar] [CrossRef] [PubMed]
- Munday, M.R. Regulation of mammalian acetyl-CoA carboxylase. Biochem. Soc. Trans. 2002, 30, 1059–1064. [Google Scholar] [CrossRef]
- Harwood, H.J., Jr.; Petras, S.F.; Shelly, L.D.; Zaccaro, L.M.; Perry, D.A.; Makowski, M.R.; Hargrove, D.M.; Martin, K.A.; Tracey, W.R.; Chapman, J.G.; et al. Isozyme-nonselective N-substituted bipiperidylcarboxamide acetyl-CoA carboxylase inhibitors reduce tissue malonyl-CoA concentrations, inhibit fatty acid synthesis, and increase fatty acid oxidation in cultured cells and in experimental animals. J. Biol. Chem. 2003, 278, 37099–37111. [Google Scholar] [CrossRef]
- Chen, L.; Duan, Y.; Wei, H.; Ning, H.; Bi, C.; Zhao, Y.; Qin, Y.; Li, Y. Acetyl-CoA carboxylase (ACC) as a therapeutic target for metabolic syndrome and recent developments in ACC1/2 inhibitors. Expert Opin. Investig. Drugs 2019, 28, 917–930. [Google Scholar] [CrossRef]
- Gerth, K.; Bedorf, N.; Irschik, H.; Hofle, G.; Reichenbach, H. The soraphens: A family of novel antifungal compounds from Sorangium cellulosum (Myxobacteria). I. Soraphen A1 alpha: Fermentation, isolation, biological properties. J. Antibiot. 1994, 47, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Schreurs, M.; van Dijk, T.H.; Gerding, A.; Havinga, R.; Reijngoud, D.J.; Kuipers, F. Soraphen, an inhibitor of the acetyl-CoA carboxylase system, improves peripheral insulin sensitivity in mice fed a high-fat diet. Diabetes Obes. Metab. 2009, 11, 987–991. [Google Scholar] [CrossRef]
- Jump, D.B.; Torres-Gonzalez, M.; Olson, L.K. Soraphen A, an inhibitor of acetyl CoA carboxylase activity, interferes with fatty acid elongation. Biochem. Pharmacol. 2011, 81, 649–660. [Google Scholar] [CrossRef] [PubMed]
- Harwood, H.J., Jr. Treating the metabolic syndrome: Acetyl-CoA carboxylase inhibition. Expert Opin. Ther. Targets 2005, 9, 267–281. [Google Scholar] [CrossRef] [PubMed]
- Beckers, A.; Organe, S.; Timmermans, L.; Scheys, K.; Peeters, A.; Brusselmans, K.; Verhoeven, G.; Swinnen, J.V. Chemical inhibition of acetyl-CoA carboxylase induces growth arrest and cytotoxicity selectively in cancer cells. Cancer Res. 2007, 67, 8180–8187. [Google Scholar] [CrossRef] [PubMed]
- Harriman, G.; Greenwood, J.; Bhat, S.; Huang, X.; Wang, R.; Paul, D.; Tong, L.; Saha, A.K.; Westlin, W.F.; Kapeller, R.; et al. Acetyl-CoA carboxylase inhibition by ND-630 reduces hepatic steatosis, improves insulin sensitivity, and modulates dyslipidemia in rats. Proc. Natl. Acad. Sci. USA 2016, 113, E1796–E1805. [Google Scholar] [CrossRef]
- Li, E.Q.; Zhao, W.; Zhang, C.; Qin, L.Z.; Liu, S.J.; Feng, Z.Q.; Wen, X.; Chen, C.P. Synthesis and anti-cancer activity of ND-646 and its derivatives as acetyl-CoA carboxylase 1 inhibitors. Eur. J. Pharm. Sci. 2019, 137, 105010. [Google Scholar] [CrossRef]
- Svensson, R.U.; Parker, S.J.; Eichner, L.J.; Kolar, M.J.; Wallace, M.; Brun, S.N.; Lombardo, P.S.; Van Nostrand, J.L.; Hutchins, A.; Vera, L.; et al. Inhibition of acetyl-CoA carboxylase suppresses fatty acid synthesis and tumor growth of non-small-cell lung cancer in preclinical models. Nat. Med. 2016, 22, 1108–1119. [Google Scholar] [CrossRef]
- Bourbeau, M.P.; Bartberger, M.D. Recent advances in the development of acetyl-CoA carboxylase (ACC) inhibitors for the treatment of metabolic disease. J. Med. Chem. 2015, 58, 525–536. [Google Scholar] [CrossRef]
- Esler, W.P.; Tesz, G.J.; Hellerstein, M.K.; Beysen, C.; Sivamani, R.; Turner, S.M.; Watkins, S.M.; Amor, P.A.; Carvajal-Gonzalez, S.; Geoly, F.J.; et al. Human sebum requires de novo lipogenesis, which is increased in acne vulgaris and suppressed by acetyl-CoA carboxylase inhibition. Sci. Transl. Med. 2019, 11, eaau8465. [Google Scholar] [CrossRef]
- Huard, K.; Smith, A.C.; Cappon, G.; Dow, R.L.; Edmonds, D.J.; El-Kattan, A.; Esler, W.P.; Fernando, D.P.; Griffith, D.A.; Kalgutkar, A.S.; et al. Optimizing the Benefit/Risk of Acetyl-CoA Carboxylase Inhibitors through Liver Targeting. J. Med. Chem. 2020, 63, 10879–10896. [Google Scholar] [CrossRef]
- Ryder, T.F.; Bergman, A.; King-Ahmad, A.; Amin, N.B.; Lall, M.S.; Ballard, T.E.; Kalgutkar, A.S. Pharmacokinetics, mass balance, metabolism, and excretion of the liver-targeted acetyl-CoA carboxylase inhibitor PF-05221304 (clesacostat) in humans. Xenobiotica 2022, 52, 240–253. [Google Scholar] [CrossRef]
- Huang, H.; McIntosh, A.L.; Martin, G.G.; Petrescu, A.D.; Landrock, K.K.; Landrock, D.; Kier, A.B.; Schroeder, F. Inhibitors of Fatty Acid Synthesis Induce PPAR alpha -Regulated Fatty Acid beta -Oxidative Genes: Synergistic Roles of L-FABP and Glucose. PPAR Res. 2013, 2013, 865604. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Xu, C.; Sun, M.; Luo, D.; Liao, D.F.; Cao, D. Acetyl-CoA carboxylase-alpha inhibitor TOFA induces human cancer cell apoptosis. Biochem. Biophys. Res. Commun. 2009, 385, 302–306. [Google Scholar] [CrossRef]
- Thupari, J.N.; Pinn, M.L.; Kuhajda, F.P. Fatty acid synthase inhibition in human breast cancer cells leads to malonyl-CoA-induced inhibition of fatty acid oxidation and cytotoxicity. Biochem. Biophys. Res. Commun. 2001, 285, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.W.; Yamane, H.; Zondlo, J.; Busby, J.; Wang, M. Expression, purification, and characterization of human acetyl-CoA carboxylase 2. Protein Expr. Purif. 2007, 53, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Pizer, E.S.; Jackisch, C.; Wood, F.D.; Pasternack, G.R.; Davidson, N.E.; Kuhajda, F.P. Inhibition of fatty acid synthesis induces programmed cell death in human breast cancer cells. Cancer Res. 1996, 56, 2745–2747. [Google Scholar]
- Pizer, E.S.; Chrest, F.J.; DiGiuseppe, J.A.; Han, W.F. Pharmacological inhibitors of mammalian fatty acid synthase suppress DNA replication and induce apoptosis in tumor cell lines. Cancer Res. 1998, 58, 4611–4615. [Google Scholar]
- Vance, D.; Omura, S.; Nomura, S.; Mitsuhashi, O.; Bloch, K.; Goldberg, I. Inhibition of Fatty-Acid Synthetases by Antibiotic Cerulenin. Biochem. Biophys. Res. Commun. 1972, 48, 649–656. [Google Scholar] [CrossRef]
- Vazquez-Martin, A.; Ropero, S.; Brunet, J.; Colomer, R.; Menendez, J.A. Inhibition of Fatty Acid Synthase (FASN) synergistically enhances the efficacy of 5-fluorouracil in breast carcinoma cells. Oncol. Rep. 2007, 18, 973–980. [Google Scholar] [CrossRef]
- Johansson, P.; Wiltschi, B.; Kumari, P.; Kessler, B.; Vonrhein, C.; Vonck, J.; Oesterhelt, D.; Grininger, M. Inhibition of the fungal fatty acid synthase type I multienzyme complex. Proc. Natl. Acad. Sci. USA 2008, 105, 12803–12808. [Google Scholar] [CrossRef]
- Rudolph, M.C.; Karl Maluf, N.; Wellberg, E.A.; Johnson, C.A.; Murphy, R.C.; Anderson, S.M. Mammalian fatty acid synthase activity from crude tissue lysates tracing (1)(3)C-labeled substrates using gas chromatography-mass spectrometry. Anal. Biochem. 2012, 428, 158–166. [Google Scholar] [CrossRef]
- Kuhajda, F.P.; Pizer, E.S.; Li, J.N.; Mani, N.S.; Frehywot, G.L.; Townsend, C.A. Synthesis and antitumor activity of an inhibitor of fatty acid synthase. Proc. Natl. Acad. Sci. USA 2000, 97, 3450–3454. [Google Scholar] [CrossRef] [PubMed]
- Rendina, A.R.; Cheng, D. Characterization of the inactivation of rat fatty acid synthase by C75: Inhibition of partial reactions and protection by substrates. Biochem. J. 2005, 388, 895–903. [Google Scholar] [CrossRef] [Green Version]
- Boelcke, W.P.; Teixeira, I.F.; Aquino, I.G.; Mazzaro, A.R.; Cuadra-Zelaya, F.J.M.; de Souza, A.P.; Salo, T.; Della Coletta, R.; Graner, E.; Bastos, D.C. Pharmacological fatty acid synthase inhibitors differently affect the malignant phenotype of oral cancer cells. Arch. Oral Biol. 2022, 135, 105343. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Liu, J.-Y.; Wu, X.; Zhang, J.-T. Biochemistry, molecular biology, and pharmacology of fatty acid synthase, an emerging therapeutic target and diagnosis/prognosis marke. Int. J. Biochem. Mol. Biol. 2010, 1, 69–89. [Google Scholar]
- Pemble, C.W.t.; Johnson, L.C.; Kridel, S.J.; Lowther, W.T. Crystal structure of the thioesterase domain of human fatty acid synthase inhibited by Orlistat. Nat. Struct. Mol. Biol. 2007, 14, 704–709. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.S.; Lei, J.P.; Wei, G.Q.; Chen, H.; Ma, C.Y.; Jiang, H.Z. Natural fatty acid synthase inhibitors as potent therapeutic agents for cancers: A review. Pharm. Biol. 2016, 54, 1919–1925. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Wang, Y.; Fillgrove, K.L.; Anderson, V.E. Triclosan inhibits enoyl-reductase of type I fatty acid synthase in vitro and is cytotoxic to MCF-7 and SKBr-3 breast cancer cells. Cancer Chemother. Pharmacol. 2002, 49, 187–193. [Google Scholar] [CrossRef]
- Stewart, M.J.; Parikh, S.; Xiao, G.; Tonge, P.J.; Kisker, C. Structural basis and mechanism of enoyl reductase inhibition by triclosan. J. Mol. Biol. 1999, 290, 859–865. [Google Scholar] [CrossRef]
- Sun, D.; Zhao, T.; Long, K.; Wu, M.; Zhang, Z. Triclosan down-regulates fatty acid synthase through microRNAs in HepG2 cells. Eur. J. Pharmacol. 2021, 907, 174261. [Google Scholar] [CrossRef]
- Sadowski, M.C.; Pouwer, R.H.; Gunter, J.H.; Lubik, A.A.; Quinn, R.J.; Nelson, C.C. The fatty acid synthase inhibitor triclosan: Repurposing an antimicrobial agent for targeting prostate cancer. Oncotarget 2014, 5, 9362–9381. [Google Scholar] [CrossRef]
- Recazens, E.; Mouisel, E.; Langin, D. Hormone-sensitive lipase: Sixty years later. Prog. Lipid Res. 2021, 82, 101084. [Google Scholar] [CrossRef] [PubMed]
- Bezaire, V.; Mairal, A.; Anesia, R.; Lefort, C.; Langin, D. Chronic TNFalpha and cAMP pre-treatment of human adipocytes alter HSL, ATGL and perilipin to regulate basal and stimulated lipolysis. FEBS Lett. 2009, 583, 3045–3049. [Google Scholar] [CrossRef] [PubMed]
- Langin, D.; Dicker, A.; Tavernier, G.; Hoffstedt, J.; Mairal, A.; Ryden, M.; Arner, E.; Sicard, A.; Jenkins, C.M.; Viguerie, N.; et al. Adipocyte lipases and defect of lipolysis in human obesity. Diabetes 2005, 54, 3190–3197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agustsson, T.; Ryden, M.; Hoffstedt, J.; van Harmelen, V.; Dicker, A.; Laurencikiene, J.; Isaksson, B.; Permert, J.; Arner, P. Mechanism of increased lipolysis in cancer cachexia. Cancer Res. 2007, 67, 5531–5537. [Google Scholar] [CrossRef]
- Kim, K.; Kang, J.K.; Jung, Y.H.; Lee, S.B.; Rametta, R.; Dongiovanni, P.; Valenti, L.; Pajvani, U.B. Adipocyte PHLPP2 inhibition prevents obesity-induced fatty liver. Nat. Commun. 2021, 12, 1822. [Google Scholar] [CrossRef] [PubMed]
- Heier, C.; Knittelfelder, O.; Hofbauer, H.F.; Mende, W.; Pornbacher, I.; Schiller, L.; Schoiswohl, G.; Xie, H.; Gronke, S.; Shevchenko, A.; et al. Hormone-sensitive lipase couples intergenerational sterol metabolism to reproductive success. Elife 2021, 10, e63252. [Google Scholar] [CrossRef]
- Shiau, M.Y.; Chuang, P.H.; Yang, C.P.; Hsiao, C.W.; Chang, S.W.; Chang, K.Y.; Liu, T.M.; Chen, H.W.; Chuang, C.C.; Yuan, S.Y.; et al. Mechanism of Interleukin-4 Reducing Lipid Deposit by Regulating Hormone-Sensitive Lipase. Sci. Rep. 2019, 9, 11974. [Google Scholar] [CrossRef]
- Arredondo-Amador, M.; Zambrano, C.; Kulyte, A.; Lujan, J.; Hu, K.; Sanchez de Medina, F.; Scheer, F.; Arner, P.; Ryden, M.; Martinez-Augustin, O.; et al. Circadian Rhythms in Hormone-sensitive Lipase in Human Adipose Tissue: Relationship to Meal Timing and Fasting Duration. J. Clin. Endocrinol. Metab. 2020, 105, e4407–e4416. [Google Scholar] [CrossRef]
- Zhuan, Q.; Ma, H.; Chen, J.; Luo, Y.; Luo, Y.; Gao, L.; Hou, Y.; Zhu, S.; Fu, X. Cytoplasm lipids can be modulated through hormone-sensitive lipase and are related to mitochondrial function in porcine IVM oocytes. Reprod. Fertil. Dev. 2020, 32, 667–675. [Google Scholar] [CrossRef]
- Schweiger, M.; Eichmann, T.O.; Taschler, U.; Zimmermann, R.; Zechner, R.; Lass, A. Measurement of lipolysis. Methods Enzymol. 2014, 538, 171–193. [Google Scholar] [CrossRef]
- Holm, C.; Contreras, J.A.; Verger, R.; Schotz, M.C. Large-scale purification and kinetic properties of recombinant hormone-sensitive lipase from baculovirus-insect cell systems. Methods Enzymol. 1997, 284, 272–284. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, G.R.; Kemp, B.E.; Watt, M.J. Adipocyte triglyceride lipase expression in human obesity. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E958–E964. [Google Scholar] [CrossRef] [PubMed]
- Schweiger, M.; Schreiber, R.; Haemmerle, G.; Lass, A.; Fledelius, C.; Jacobsen, P.; Tornqvist, H.; Zechner, R.; Zimmermann, R. Adipose triglyceride lipase and hormone-sensitive lipase are the major enzymes in adipose tissue triacylglycerol catabolism. J. Biol. Chem. 2006, 281, 40236–40241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haemmerle, G.; Zimmermann, R.; Hayn, M.; Theussl, C.; Waeg, G.; Wagner, E.; Sattler, W.; Magin, T.M.; Wagner, E.F.; Zechner, R. Hormone-sensitive lipase deficiency in mice causes diglyceride accumulation in adipose tissue, muscle, and testis. J. Biol. Chem. 2002, 277, 4806–4815. [Google Scholar] [CrossRef] [PubMed]
- Fredrikson, G.; Strålfors, P.; Nilsson, N.Ö.; Belfrage, P. Hormone-Sensitive Lipase from Adipose Tissue of Rat. Methods Enzymol. 1981, 71, 636–646. [Google Scholar]
- Watt, M.J.; Carey, A.L.; Wolsk-Petersen, E.; Kraemer, F.B.; Pedersen, B.K.; Febbraio, M.A. Hormone-sensitive lipase is reduced in the adipose tissue of patients with type 2 diabetes mellitus: Influence of IL-6 infusion. Diabetologia 2005, 48, 105–112. [Google Scholar] [CrossRef]
- Watt, M.J.; Heigenhauser, G.J.; Spriet, L.L. Effects of dynamic exercise intensity on the activation of hormone-sensitive lipase in human skeletal muscle. J. Physiol. 2003, 547, 301–308. [Google Scholar] [CrossRef]
- Holm, C.; Langin, D.; Manganiello, V.; Belfrage, P.; Degerman, E. Regulation of hormone-sensitive lipase activity in adipose tissue. Lipases 1997, 286, 45–67. [Google Scholar]
- Richelsen, B.; Pedersen, S.B.; Kristensen, K.; Borglum, J.D.; Norrelund, H.; Christiansen, J.S.; Jorgensen, J.O. Regulation of lipoprotein lipase and hormone-sensitive lipase activity and gene expression in adipose and muscle tissue by growth hormone treatment during weight loss in obese patients. Metabolism 2000, 49, 906–911. [Google Scholar] [CrossRef]
- Schweiger, M.; Schoiswohl, G.; Lass, A.; Radner, F.P.; Haemmerle, G.; Malli, R.; Graier, W.; Cornaciu, I.; Oberer, M.; Salvayre, R.; et al. The C-terminal region of human adipose triglyceride lipase affects enzyme activity and lipid droplet binding. J. Biol. Chem. 2008, 283, 17211–17220. [Google Scholar] [CrossRef]
- Takagi, A.; Ikeda, Y.; Kobayashi, K.; Kobayashi, K.; Ikeda, Y.; Kozawa, J.; Miyauchi, H.; Li, M.; Hashimoto, C.; Hara, Y.; et al. Newly developed selective immunoinactivation assay revealed reduction in adipose triglyceride lipase activity in peripheral leucocytes from patients with idiopathic triglyceride deposit cardiomyovasculopathy. Biochem. Biophys. Res. Commun. 2018, 495, 646–651. [Google Scholar] [CrossRef] [PubMed]
- Chandak, P.G.; Radovic, B.; Aflaki, E.; Kolb, D.; Buchebner, M.; Frohlich, E.; Magnes, C.; Sinner, F.; Haemmerle, G.; Zechner, R.; et al. Efficient phagocytosis requires triacylglycerol hydrolysis by adipose triglyceride lipase. J. Biol. Chem. 2010, 285, 20192–20201. [Google Scholar] [CrossRef]
- Reid, B.N.; Ables, G.P.; Otlivanchik, O.A.; Schoiswohl, G.; Zechner, R.; Blaner, W.S.; Goldberg, I.J.; Schwabe, R.F.; Chua, S.C., Jr.; Huang, L.S. Hepatic overexpression of hormone-sensitive lipase and adipose triglyceride lipase promotes fatty acid oxidation, stimulates direct release of free fatty acids, and ameliorates steatosis. J. Biol. Chem. 2008, 283, 13087–13099. [Google Scholar] [CrossRef]
- Mersmann, H.J. Lipoprotein and hormone-sensitive lipases in porcine adipose tissue. J. Anim. Sci. 1998, 76, 1396–1404. [Google Scholar] [CrossRef]
- Holm, C.; Olivecrona, G.; Ottosson, M. Assays of lipolytic enzymes. Methods Mol. Biol. 2001, 155, 97–119. [Google Scholar] [CrossRef]
- Sekiya, M.; Osuga, J.; Yahagi, N.; Okazaki, H.; Tamura, Y.; Igarashi, M.; Takase, S.; Harada, K.; Okazaki, S.; Iizuka, Y.; et al. Hormone-sensitive lipase is involved in hepatic cholesteryl ester hydrolysis. J. Lipid Res. 2008, 49, 1829–1838. [Google Scholar] [CrossRef] [PubMed]
- Olzmann, J.A.; Carvalho, P. Dynamics and functions of lipid droplets. Nat. Rev. Mol. Cell Biol. 2019, 20, 137–155. [Google Scholar] [CrossRef]
- Ding, Y.; Zhang, S.; Yang, L.; Na, H.; Zhang, P.; Zhang, H.; Wang, Y.; Chen, Y.; Yu, J.; Huo, C.; et al. Isolating lipid droplets from multiple species. Nat. Protoc. 2013, 8, 43–51. [Google Scholar] [CrossRef]
- Brasaemle, D.L.; Wolins, N.E. Isolation of Lipid Droplets from Cells by Density Gradient Centrifugation. Curr. Protoc. Cell Biol. 2016, 72, 3.15.1–3.15.13. [Google Scholar] [CrossRef]
- Borg, M.L.; Andrews, Z.B.; Duh, E.J.; Zechner, R.; Meikle, P.J.; Watt, M.J. Pigment epithelium-derived factor regulates lipid metabolism via adipose triglyceride lipase. Diabetes 2011, 60, 1458–1466. [Google Scholar] [CrossRef] [PubMed]
- Bradley, D.C.; Kaslow, H.R. Radiometric assays for glycerol, glucose, and glycogen. Anal. Biochem. 1989, 180, 11–16. [Google Scholar] [CrossRef]
- Slavin, B.G.; Ong, J.M.; Kern, P.A. Hormonal regulation of hormone-sensitive lipase activity and mRNA levels in isolated rat adipocytes. J. Lipid Res. 1994, 35, 1535–1541. [Google Scholar] [CrossRef]
- Amols, H.I.; Zaider, M.; Weinberger, J.; Ennis, R.; Schiff, P.B.; Reinstein, L.E. Dosimetric considerations for catheter-based beta and gamma emitters in the therapy of neointimal hyperplasia in human coronary arteries. Int. J. Radiat. Oncol. Biol. Phys. 1996, 36, 913–921. [Google Scholar] [CrossRef]
- Morak, M.; Schmidinger, H.; Riesenhuber, G.; Rechberger, G.N.; Kollroser, M.; Haemmerle, G.; Zechner, R.; Kronenberg, F.; Hermetter, A. Adipose triglyceride lipase (ATGL) and hormone-sensitive lipase (HSL) deficiencies affect expression of lipolytic activities in mouse adipose tissues. Mol. Cell. Proteom. 2012, 11, 1777–1789. [Google Scholar] [CrossRef]
- Witters, L.A.; Watts, T.D.; Daniels, D.L.; Evans, J.L. Insulin stimulates the dephosphorylation and activation of acetyl-CoA carboxylase. Proc. Natl. Acad. Sci. USA 1988, 85, 5473–5477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregolin, C.; Ryder, E.; Kleinschmidt, A.K.; Warner, R.C.; Lane, M.D. Molecular characteristics of liver acetyl CoA carboxylase. Proc. Natl. Acad. Sci. USA 1966, 56, 148–155. [Google Scholar] [CrossRef]
- Dean, D.; Daugaard, J.R.; Young, M.E.; Saha, A.; Vavvas, D.; Asp, S.; Kiens, B.; Kim, K.H.; Witters, L.; Richter, E.A.; et al. Exercise diminishes the activity of acetyl-CoA carboxylase in human muscle. Diabetes 2000, 49, 1295–1300. [Google Scholar] [CrossRef]
- Moibi, J.A.; Ekpe, E.D.; Christopherson, R.J. Acetyl-CoA carboxylase and fatty acid synthase activity and immunodetectable protein in adipose tissues of ruminants: Effect of temperature and feeding level. J. Anim. Sci. 2000, 78, 2383–2392. [Google Scholar] [CrossRef]
- Stansbie, D.; Brownsey, R.W.; Crettaz, M.; Denton, R.M. Acute effects in vivo of anti-insulin serum on rates of fatty acid synthesis and activities of acetyl-coenzyme A carboxylase and pyruvate dehydrogenase in liver and epididymal adipose tissue of fed rats. Biochem. J. 1976, 160, 413–416. [Google Scholar] [CrossRef]
- Halestrap, A.P.; Denton, R.M. Hormonal regulation of adipose-tissue acetyl-Coenzyme A carboxylase by changes in the polymeric state of the enzyme. The role of long-chain fatty acyl-Coenzyme A thioesters and citrate. Biochem. J. 1974, 142, 365–377. [Google Scholar] [CrossRef]
- Bijleveld, C.; Geelen, M.J. Measurement of acetyl-CoA carboxylase activity in isolated hepatocytes. Biochim. Biophys. Acta 1987, 918, 274–283. [Google Scholar] [CrossRef]
- Witters, L.A.; Moriarity, D.; Martin, D.B. Regulation of hepatic acetyl coenzyme A carboxylase by insulin and glucagon. J. Biol. Chem. 1979, 254, 6644–6649. [Google Scholar] [CrossRef]
- Thampy, K.G.; Wakil, S.J. Activation of acetyl-CoA carboxylase. Purification and properties of a Mn2+-dependent phosphatase. J. Biol. Chem. 1985, 260, 6318–6323. [Google Scholar] [CrossRef]
- Wong, K.; Meyers, R.; Cantley, L.C. Subcellular locations of phosphatidylinositol 4-kinase isoforms. J. Biol. Chem. 1997, 272, 13236–13241. [Google Scholar] [CrossRef]
- Zimmermann, R.; Haemmerle, G.; Wagner, E.M.; Strauss, J.G.; Kratky, D.; Zechner, R. Decreased fatty acid esterification compensates for the reduced lipolytic activity in hormone-sensitive lipase-deficient white adipose tissue. J. Lipid Res. 2003, 44, 2089–2099. [Google Scholar] [CrossRef] [Green Version]
- Chung, C.C.; Ohwaki, K.; Schneeweis, J.E.; Stec, E.; Varnerin, J.P.; Goudreau, P.N.; Chang, A.; Cassaday, J.; Yang, L.; Yamakawa, T.; et al. A fluorescence-based thiol quantification assay for ultra-high-throughput screening for inhibitors of coenzyme A production. Assay Drug Dev. Technol. 2008, 6, 361–374. [Google Scholar] [CrossRef]
- Cerne, D.; Zitnik, I.P.; Sok, M. Increased fatty acid synthase activity in non-small cell lung cancer tissue is a weaker predictor of shorter patient survival than increased lipoprotein lipase activity. Arch. Med. Res. 2010, 41, 405–409. [Google Scholar] [CrossRef]
- Topolska, M.; Martinez-Montanes, F.; Ejsing, C.S. A Simple and Direct Assay for Monitoring Fatty Acid Synthase Activity and Product-Specificity by High-Resolution Mass Spectrometry. Biomolecules 2020, 10, 118. [Google Scholar] [CrossRef]
- Jayakumar, A.; Tai, M.H.; Huang, W.Y.; al-Feel, W.; Hsu, M.; Abu-Elheiga, L.; Chirala, S.S.; Wakil, S.J. Human fatty acid synthase: Properties and molecular cloning. Proc. Natl. Acad. Sci. USA 1995, 92, 8695–8699. [Google Scholar] [CrossRef]
- Dils, R.; Carey, E.M. Fatty acid synthase from rabbit mammary gland. Methods Enzymol. 1975, 35, 74–83. [Google Scholar] [CrossRef]
- Nepokroeff, C.M.; Lakshmanan, M.R.; Porter, J.W. Fatty-acid synthase from rat liver. Methods Enzymol. 1975, 35, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Alwarawrah, Y.; Hughes, P.; Loiselle, D.; Carlson, D.A.; Darr, D.B.; Jordan, J.L.; Xiong, J.; Hunter, L.M.; Dubois, L.G.; Thompson, J.W.; et al. Fasnall, a Selective FASN Inhibitor, Shows Potent Anti-tumor Activity in the MMTV-Neu Model of HER2(+) Breast Cancer. Cell Chem. Biol. 2016, 23, 678–688. [Google Scholar] [CrossRef] [PubMed]
- Najjar, S.M.; Yang, Y.; Fernstrom, M.A.; Lee, S.J.; Deangelis, A.M.; Rjaily, G.A.; Al-Share, Q.Y.; Dai, T.; Miller, T.A.; Ratnam, S.; et al. Insulin acutely decreases hepatic fatty acid synthase activity. Cell Metab. 2005, 2, 43–53. [Google Scholar] [CrossRef]
- Roncari, D.A. Fatty acid synthase from human liver. Methods Enzymol. 1981, 71, 73–79. [Google Scholar] [CrossRef]
- Brusselmans, K.; Vrolix, R.; Verhoeven, G.; Swinnen, J.V. Induction of cancer cell apoptosis by flavonoids is associated with their ability to inhibit fatty acid synthase activity. J. Biol. Chem. 2005, 280, 5636–5645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, R.Y.; Butterworth, P.H.W.; Porter, J.W. Pigeon Liver Fatty Acid Synthase. Methods Enzymol. 1969, 14, 33–39. [Google Scholar]
- Moustaid, N.; Hainque, B.; Quignard-Boulange, A. Dexamethasone regulation of terminal differentiation in 3T3-F442A preadipocyte cell line. Cytotechnology 1988, 1, 285–293. [Google Scholar] [CrossRef]
- Puig, T.; Turrado, C.; Benhamu, B.; Aguilar, H.; Relat, J.; Ortega-Gutierrez, S.; Casals, G.; Marrero, P.F.; Urruticoechea, A.; Haro, D.; et al. Novel Inhibitors of Fatty Acid Synthase with Anticancer Activity. Clin. Cancer Res. 2009, 15, 7608–7615. [Google Scholar] [CrossRef]
- Lin, H.P.; Cheng, Z.L.; He, R.Y.; Song, L.; Tian, M.X.; Zhou, L.S.; Groh, B.S.; Liu, W.R.; Ji, M.B.; Ding, C.; et al. Destabilization of Fatty Acid Synthase by Acetylation Inhibits De Novo Lipogenesis and Tumor Cell Growth. Cancer Res. 2016, 76, 6924–6936. [Google Scholar] [CrossRef]
- Moustaid, N.; Jones, B.H.; Taylor, J.W. Insulin increases lipogenic enzyme activity in human adipocytes in primary culture. J. Nutr. 1996, 126, 865–870. [Google Scholar] [CrossRef]
- Wang, Y.; Jones Voy, B.; Urs, S.; Kim, S.; Soltani-Bejnood, M.; Quigley, N.; Heo, Y.R.; Standridge, M.; Andersen, B.; Dhar, M.; et al. The human fatty acid synthase gene and de novo lipogenesis are coordinately regulated in human adipose tissue. J. Nutr. 2004, 134, 1032–1038. [Google Scholar] [CrossRef]
- Xue, B.; Zemel, M.B. Relationship between Human Adipose Tissue Agouti and Fatty Acid Synthase (FAS). J. Nutr. 2000, 130, 2478–2481. [Google Scholar] [CrossRef]
- Chiu, S.; Mulligan, K.; Schwarz, J.M. Dietary carbohydrates and fatty liver disease: De novo lipogenesis. Curr. Opin. Clin. Nutr. Metab. Care 2018, 21, 277–282. [Google Scholar] [CrossRef]
- Jelenik, T.; Kaul, K.; Sequaris, G.; Flogel, U.; Phielix, E.; Kotzka, J.; Knebel, B.; Fahlbusch, P.; Horbelt, T.; Lehr, S.; et al. Mechanisms of Insulin Resistance in Primary and Secondary Nonalcoholic Fatty Liver. Diabetes 2017, 66, 2241–2253. [Google Scholar] [CrossRef]
- Eissing, L.; Scherer, T.; Todter, K.; Knippschild, U.; Greve, J.W.; Buurman, W.A.; Pinnschmidt, H.O.; Rensen, S.S.; Wolf, A.M.; Bartelt, A.; et al. De novo lipogenesis in human fat and liver is linked to ChREBP-beta and metabolic health. Nat. Commun. 2013, 4, 1528. [Google Scholar] [CrossRef]
- Simeone, P.; Tacconi, S.; Longo, S.; Lanuti, P.; Bravaccini, S.; Pirini, F.; Ravaioli, S.; Dini, L.; Giudetti, A.M. Expanding Roles of De Novo Lipogenesis in Breast Cancer. Int. J. Environ. Res. Public Health 2021, 18, 3575. [Google Scholar] [CrossRef]
- Hellerstein, M.K.; Schwarz, J.M.; Neese, R.A. Regulation of hepatic de novo lipogenesis in humans. Annu. Rev. Nutr. 1996, 16, 523–557. [Google Scholar] [CrossRef]
- Rios-Esteves, J.; Resh, M.D. Stearoyl CoA desaturase is required to produce active, lipid-modified Wnt proteins. Cell Rep. 2013, 4, 1072–1081. [Google Scholar] [CrossRef]
- Green, C.D.; Ozguden-Akkoc, C.G.; Wang, Y.; Jump, D.B.; Olson, L.K. Role of fatty acid elongases in determination of de novo synthesized monounsaturated fatty acid species. J. Lipid Res. 2010, 51, 1871–1877. [Google Scholar] [CrossRef]
- Lehner, R.; Kuksis, A. Biosynthesis of triacylglycerols. Prog. Lipid Res. 1996, 35, 169–201. [Google Scholar] [CrossRef]
- Izumi, A.; Hiraguchi, H.; Kodaka, M.; Ikeuchi, E.; Narita, J.; Kobayashi, R.; Matsumoto, Y.; Suzuki, T.; Yamamoto, Y.; Sato, R.; et al. MIG12 is involved in the LXR activation-mediated induction of the polymerization of mammalian acetyl-CoA carboxylase. Biochem. Biophys. Res. Commun. 2021, 567, 138–142. [Google Scholar] [CrossRef]
- Wei, J.; Tong, L. How Does Polymerization Regulate Human Acetyl-CoA Carboxylase 1? Biochemistry 2018, 57, 5495–5496. [Google Scholar] [CrossRef]
- Lawitz, E.J.; Coste, A.; Poordad, F.; Alkhouri, N.; Loo, N.; McColgan, B.J.; Tarrant, J.M.; Nguyen, T.; Han, L.; Chung, C.; et al. Acetyl-CoA Carboxylase Inhibitor GS-0976 for 12 Weeks Reduces Hepatic De Novo Lipogenesis and Steatosis in Patients With Nonalcoholic Steatohepatitis. Clin. Gastroenterol. Hepatol. 2018, 16, 1983–1991.e3. [Google Scholar] [CrossRef]
- Wang, C.; Ma, J.; Zhang, N.; Yang, Q.; Jin, Y.; Wang, Y. The acetyl-CoA carboxylase enzyme: A target for cancer therapy? Expert Rev. Anticancer Ther. 2015, 15, 667–676. [Google Scholar] [CrossRef]
- Smith, S.; Witkowski, A.; Joshi, A.K. Structural and functional organization of the animal fatty acid synthase. Prog. Lipid Res. 2003, 42, 289–317. [Google Scholar] [CrossRef]
- Jensen-Urstad, A.P.; Semenkovich, C.F. Fatty acid synthase and liver triglyceride metabolism: Housekeeper or messenger? Biochim. Biophys. Acta 2012, 1821, 747–753. [Google Scholar] [CrossRef] [Green Version]
- Rysman, E.; Brusselmans, K.; Scheys, K.; Timmermans, L.; Derua, R.; Munck, S.; Van Veldhoven, P.P.; Waltregny, D.; Daniels, V.W.; Machiels, J.; et al. De novo lipogenesis protects cancer cells from free radicals and chemotherapeutics by promoting membrane lipid saturation. Cancer Res. 2010, 70, 8117–8126. [Google Scholar] [CrossRef]
- Menendez, J.A.; Lupu, R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 2007, 7, 763–777. [Google Scholar] [CrossRef]
- Zhou, W.; Han, W.F.; Landree, L.E.; Thupari, J.N.; Pinn, M.L.; Bililign, T.; Kim, E.K.; Vadlamudi, A.; Medghalchi, S.M.; El Meskini, R.; et al. Fatty acid synthase inhibition activates AMP-activated protein kinase in SKOV3 human ovarian cancer cells. Cancer Res. 2007, 67, 2964–2971. [Google Scholar] [CrossRef] [Green Version]
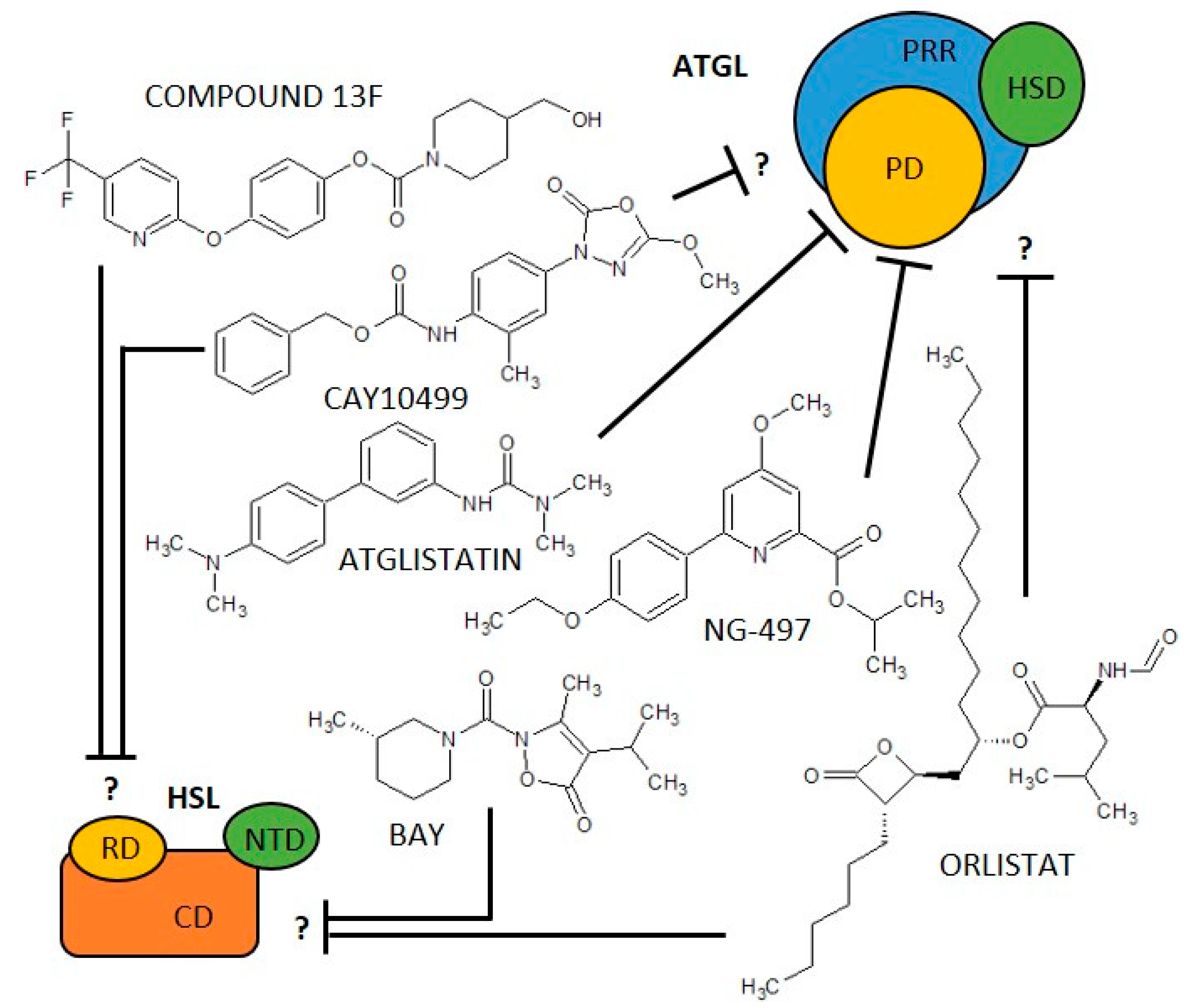
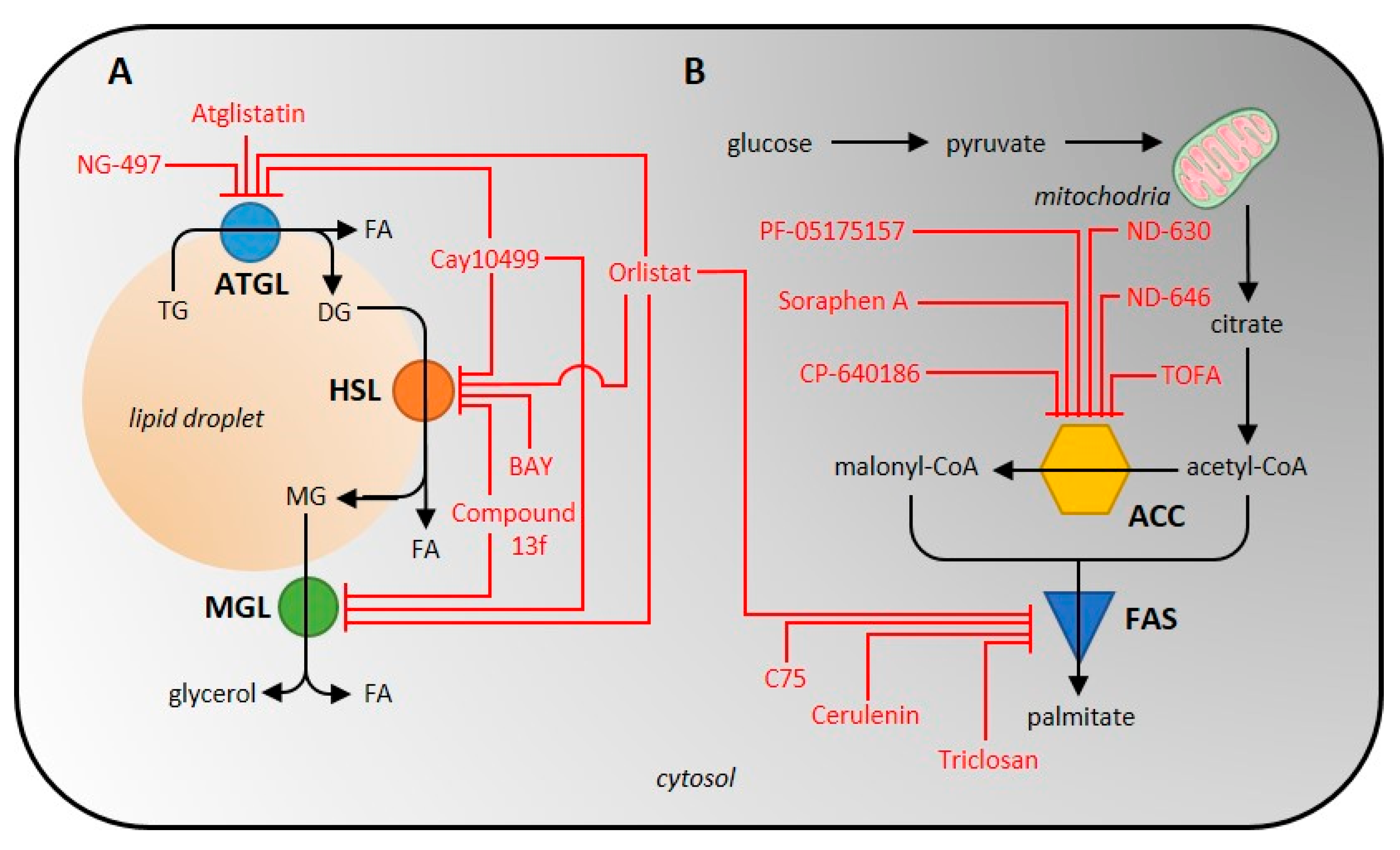
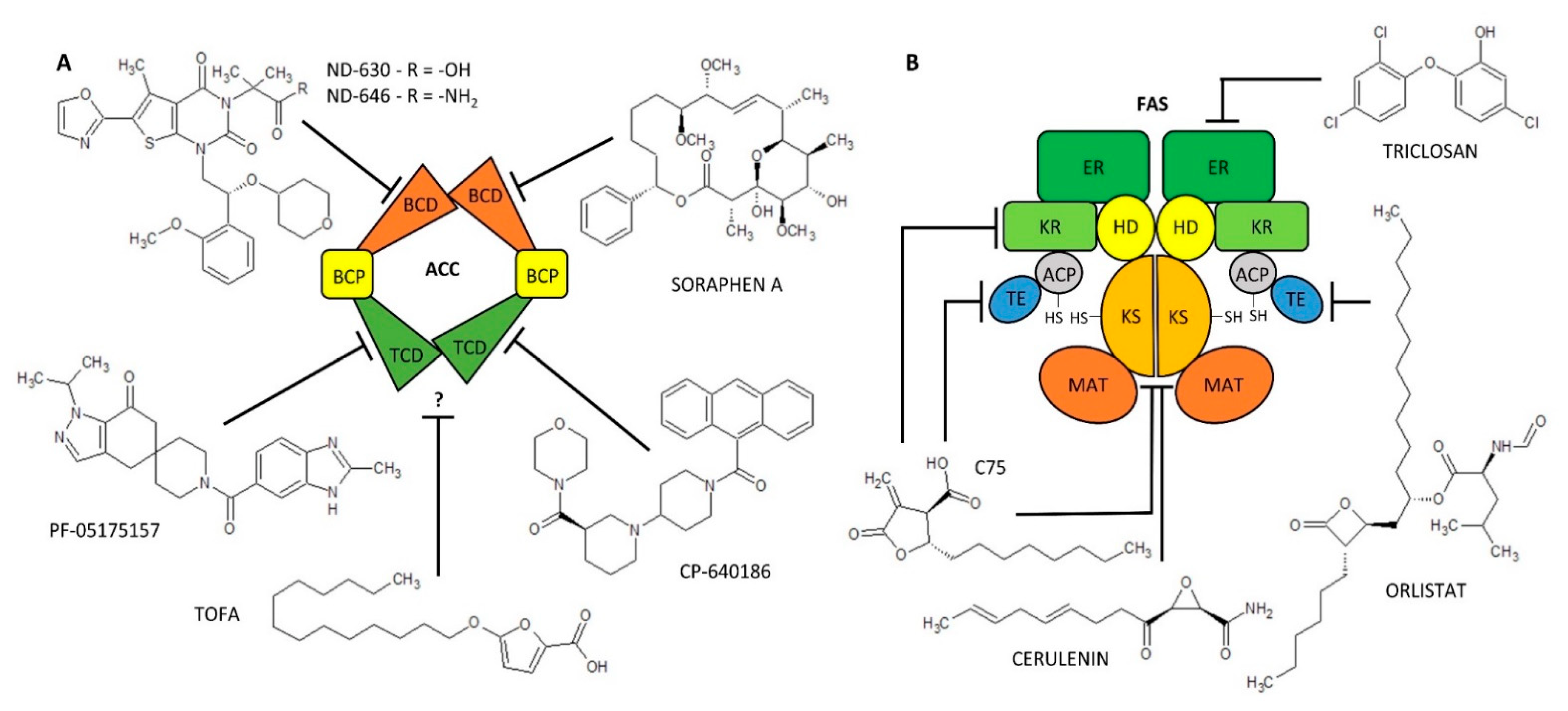

{kind=link}
{kind=link}
{kind=link}
| Enzyme (EC Code) | (Co)Activators | Negative Regulators | (Semi)Synthetic Inhibitors | ||||
|---|---|---|---|---|---|---|---|
| Name(s) | Organism or Cell Line | Enzyme Specificity 1 | Inhibition Type | IC50 [nM] | |||
| ATGL (EC 3.1.1.3) | ABHD5 [33,34,35,47,48] PEDF [34,49,50] PLIN1 [34,35,51] PLIN5 [35,52] | G0S2 [37,48,53] long acyl-CoA [38] | Atglistatin [39,40,41] | dog, goat, marmoset, mouse, rat | yes | competitive, reversible [39] | 700 [39] |
| CAY10499 [40,54] | human, mouse | no | unknown | 66 [40] | |||
| NG-497 [41] | human, rhesus monkey | yes | competitive, reversible [41] | 1300 [41] | |||
| Orlistat [40,55,56] | human, mouse | no | irreversible [56] | 1.2 [40] | |||
| HSL (EC 3.1.1.79) | FABP4 [57,58] PKA [5,59] PLIN1 [5,34] | AMPK [60,61] PLIN2 [62] | BAY 59-9435 [63,64] | 3T3-L1, human, mouse, rat | yes | non-competitive, reversible [63] | 5 [63] |
| CAY10499 [40,65] | human, mouse, rat | no | unknown | 79.8 [40] | |||
| compound 13f [40,66] | human, mouse | no | reversible [66] | 110 [66] | |||
| Orlistat [40,67] | human, mouse, rat | no | irreversible [56] | 4230 [67] | |||
| ACC (EC 6.4.1.2) | citrate [68,69] glutamate [70,71,72] | AMPK [73,74] HS-CoA [75] malonyl-CoA [75] long acyl-CoA [75,76] | CP-640186 [77,78,79,80,81,82,83] | human, monkey, mouse, rat | yes | non-competitive, reversible [78] | 53–61 [77] |
| ND-630, ND-646 [78,82,84,85,86] | human, rat | yes | reversible [84] | 1.7–6.1 [84,86,87] | |||
| PF-05175157 [88,89,90] | dog, human, rat | yes | unknown | 27–33 [88] | |||
| Soraphen A [78,79,80,81,83,87] | HepG2, LNCaP, PC-3M | yes | unknown | 1–5 [87] | |||
| TOFA [77,78,82,88,89,90,91,92,93,94] | human breast cancer cells, human colon carcinoma cells, human lung cancer cells | no | allosteric, irreversible [91] | 51 [94] | |||
| FAS (EC 2.3.1.85) | unknown | unknown | cerulenin [93,95,96,97,98,99,100] | human, mouse, MCF-7 breast cancer cells, MDA-MB-231, SKBr-3 | yes | non-competitive, irreversible [99] | 70,000–79,000 [98] |
| C75 [96,101,102,103] | HL60 cells, human, mouse, SKBr-3, rat | yes | competitive, irreversible [102] | 26,380 [103] | |||
| Orlistat [43,104,105,106] | PC-3, human, mouse | no | irreversible [56] | 900 [106] | |||
| Triclosan [107,108,109] | HepG2, MCF-7, SKBr-3 cells | no | reversible [107] | 6900–50,000 [107,110] | |||
| Enzyme | Activity Measurement | Biological Material | Reference | ||
|---|---|---|---|---|---|
| Detection Method | Substrate | Detecting Substances | |||
| ATGL | fluorescence assay | pyrene-labeled acylglycerols | pyrene | mouse AT | [144] |
| EnzChek substrate | - | human recombinant protein (HEK 293T cells) mouse recombinant protein (HEK 293T cells) | [40] | ||
| liquid scintillation counting | [9,10-3H]-triolein | [3H]-oleate | 3T3-L1 adipocytes, L6 myoblasts | [140] | |
| human AT | [122] | ||||
| human recombinant protein (COS-7 cells) | [130] | ||||
| McA-RH7777 | [133] | ||||
| mouse gonadal AT | [123] | ||||
| mouse liver | [30,133] | ||||
| mouse peritoneal macrophages | [132] | ||||
| mouse recombinant protein (COS-7 cells) | [120] | ||||
| peripheral leukocytes | [131] | ||||
| spectrophotometric assay | p-nitrophenyl esters | p-nitrophenol | mouse AT | [144] | |
| HSL | fluorescence assay | pyrene-labeled acylglycerols | pyrene | mouse AT | [144] |
| 1-S-arachidonoylthioglycerol | ThioGlo-1 aduct | human and mouse recombinant protein (HEK 293T cells) | [40] | ||
| liquid scintillation counting | cholesteryl-[1-14C]-oleate | [14C]-oleate | human AT | [129] | |
| McA-RH7777 | [133] | ||||
| mouse liver | [133,136] | ||||
| mouse peritoneal macrophages | [132] | ||||
| [3H]-oleoyl-2-O-oleylglycerol | [3H]-oleate | human recombinant protein (Sf9 cells) rat AT | [121] [125] | ||
| [32P]-ATP | [α-32P]-glycerol | rat adipocytes | [142] | ||
| [9,10-3H]-triolein | [3H]-oleate | human AT | [126,135] | ||
| mouse AT, skeletal muscle, and testis | [124] | ||||
| mouse recombinant protein (COS-7 cell) | [123] | ||||
| mouse gonadal AT | [120] | ||||
| porcine AT | [134] | ||||
| spectrophotometric assay | p-nitrophenyl esters | p-nitrophenol | mouse AT | [144] | |
| ACC | HPLC (reverse phase) | acetyl-CoA/ malonyl-CoA | acetyl-CoA/ malonyl-CoA | mouse 3T3-L1 preadipocytes | [75] |
| liquid scintillation counting | Na(K)H14CO3 | 1-[14C]-malonyl-CoA | Fao hepatoma cells | [145] | |
| chicken liver | [146] | ||||
| human skeletal muscle | [147] | ||||
| lamb AT | [148] | ||||
| rat AT | [149,150] | ||||
| rat hepatocytes | [151,152] | ||||
| rat liver | [149,153] | ||||
| rat skeletal muscle | [154] | ||||
| spectrophotometric assay | NADH | NADH | chicken liver | [146] | |
| mouse AT | [155] | ||||
| rat liver | [153] | ||||
| NADPH | NADPH | chicken liver | [68] | ||
| FAS | fluorescence assay | CoA | CPM-CoA adduct | HepG2, human epithelial SKBr-3, rat liver | [156] |
| human lung cancer | [157] | ||||
| GC-MS/LC-MS | [13C]-acetyl-CoA [13C]-malonyl-CoA | [13C]-palmitate | cow mammary gland | [158] | |
| mouse liver, mouse mammary gland | [100] | ||||
| liquid scintillation counting | 1-[14C]-acetyl-CoA | 1-[14C]-acetyl-CoA | HepG2 | [159] | |
| rabbit mammary gland | [160] | ||||
| rat liver | [161] | ||||
| 2-[14C]-malonyl-CoA | 2-[14C]-malonyl-CoA | BT474 | [162] | ||
| H35-BT, mouse liver, primary hepatocytes | [163] | ||||
| HepG2 | [159] | ||||
| human liver | [164] | ||||
| LNCaP | [165] | ||||
| pigeon liver | [166] | ||||
| rabbit mammary gland | [160] | ||||
| spectrophotometric assay | NADPH | NADPH | 3T3-F442A | [167] | |
| BT474, MCF-7, MDA-MB-231 breast cancer cells | [168] | ||||
| HCT116, HEK293T | [169] | ||||
| human adipocytes | [170] | ||||
| human AT | [171,172] | ||||
| human liver | [164] | ||||
| lamb AT | [148] | ||||
| mouse AT | [155] | ||||
| rabbit mammary gland | [70,160] | ||||
| rat liver | [101,161] | ||||
| ZR-75-30 | [169] | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wilhelm, M.; Rossmeislová, L.; Šiklová, M. Approaches to Measuring the Activity of Major Lipolytic and Lipogenic Enzymes In Vitro and Ex Vivo. Int. J. Mol. Sci. 2022, 23, 11093. https://doi.org/10.3390/ijms231911093
Wilhelm M, Rossmeislová L, Šiklová M. Approaches to Measuring the Activity of Major Lipolytic and Lipogenic Enzymes In Vitro and Ex Vivo. International Journal of Molecular Sciences. 2022; 23(19):11093. https://doi.org/10.3390/ijms231911093
Chicago/Turabian StyleWilhelm, Marek, Lenka Rossmeislová, and Michaela Šiklová. 2022. "Approaches to Measuring the Activity of Major Lipolytic and Lipogenic Enzymes In Vitro and Ex Vivo" International Journal of Molecular Sciences 23, no. 19: 11093. https://doi.org/10.3390/ijms231911093
APA StyleWilhelm, M., Rossmeislová, L., & Šiklová, M. (2022). Approaches to Measuring the Activity of Major Lipolytic and Lipogenic Enzymes In Vitro and Ex Vivo. International Journal of Molecular Sciences, 23(19), 11093. https://doi.org/10.3390/ijms231911093