
The Glyoxalase System and Methylglyoxal-Derived Carbonyl Stress in Sepsis: Glycotoxic Aspects of Sepsis Pathophysiology

 ,
, 
Abstract
:1. Introduction
1.1. Sepsis: Definitions, Incidence, Outcome, and Economic Relevance
1.2. Traditional Pathophysiology of Sepsis: PAMPs, PRRs and Reactive Metabolites
2. Different Aspects of the Stress Response in Sepsis
2.1. Neuroendocrine Reactions in Sepsis
2.2. Metabolic Changes in Sepsis: Hyperglycemia, Insulin Resistance and Respiratory Chain Uncoupling
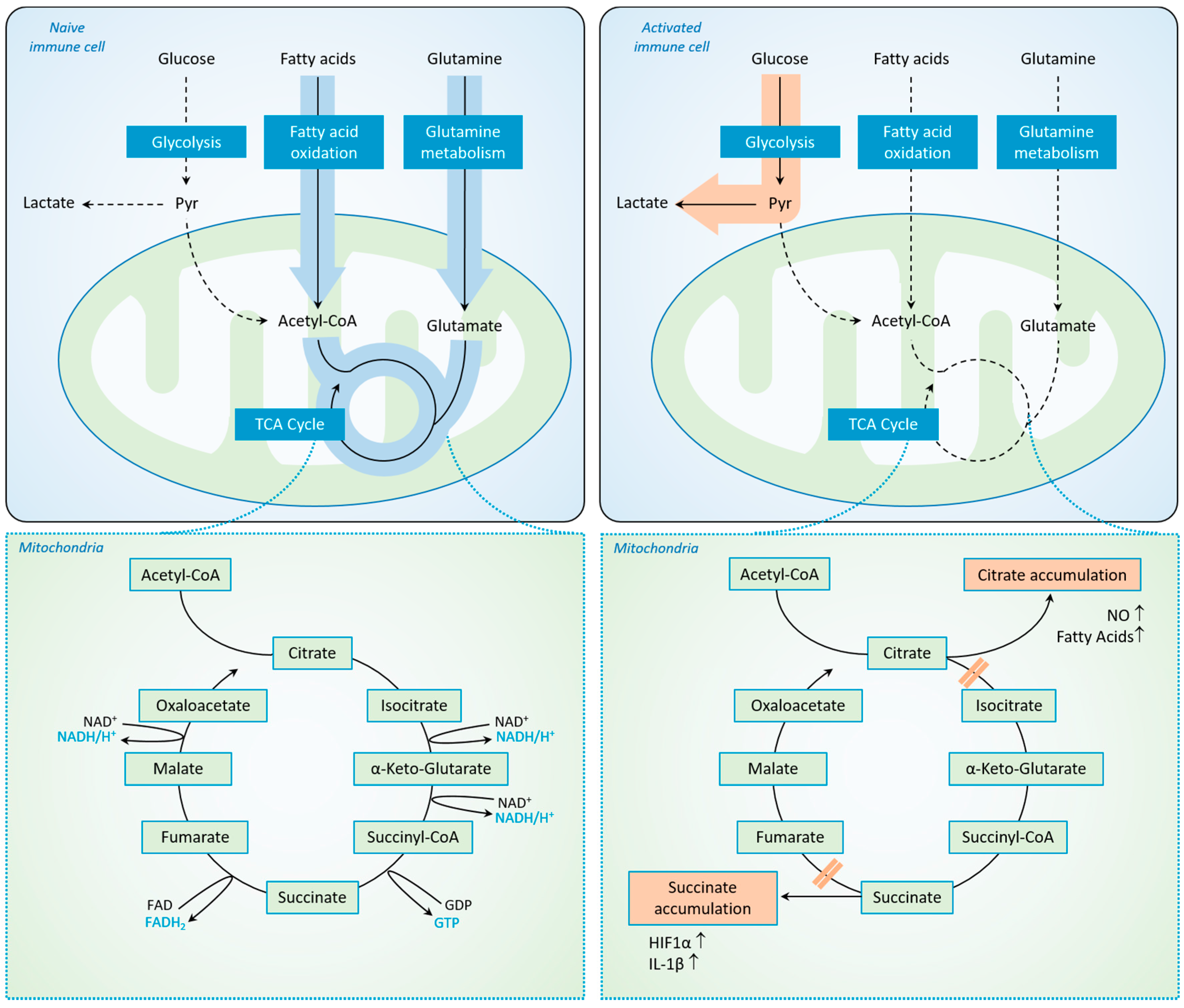
2.3. Immunometabolism in Sepsis: The “Warburg Effect” and Its Consequences for Immune Cells
2.4. Formation of ROS and RNS in Sepsis
2.5. Reactive Carbonyl Species (RCS)—An Overlooked Group of Reactive Metabolites
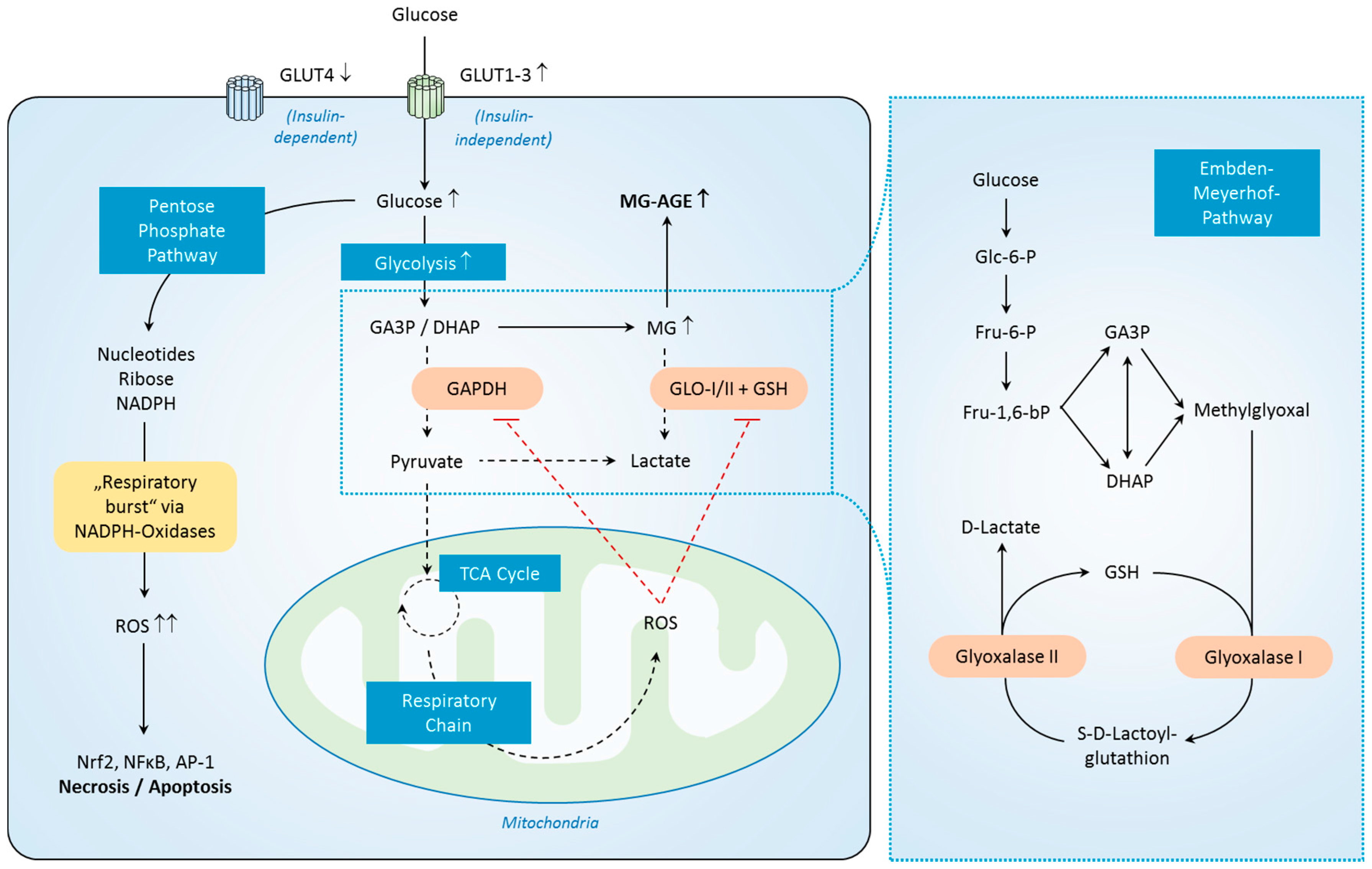
2.6. MG-Derived Carbonyl Stress in Sepsis—Diagnostic Value, Prognostic Value and Main Source of Formation
2.7. Pathophysiology of MG-Derived Carbonyl Stress in Sepsis
2.8. Regulation of MG-Derived Carbonyl Stress in Sepsis
3. Conclusive Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.-D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). J. Am. Med. Assoc. 2016, 315, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Martin, G.S.; Mannino, D.M.; Eaton, S.; Moss, M. The epidemiology of sepsis in the United States from 1979 through 2000. N. Engl. J. Med. 2003, 348, 1546–1554. [Google Scholar] [CrossRef] [PubMed]
- Bauer, M.; Brunkhorst, F.; Welte, T.; Gerlach, H.; Reinhart, K. Sepsis: Update on pathophysiology, diagnostics and therapy. Anaesthesist 2006, 55, 835–845. [Google Scholar] [CrossRef] [PubMed]
- Engel, C.; Brunkhorst, F.M.; Bone, H.-G.; Brunkhorst, R.; Gerlach, H.; Grond, S.; Gruendling, M.; Huhle, G.; Jaschinski, U.; John, S.; et al. Epidemiology of sepsis in Germany: Results from a national prospective multicenter study. Intensive Care Med. 2007, 33, 606–618. [Google Scholar] [CrossRef] [PubMed]
- Fleischmann, C.; Thomas-Rueddel, D.; Hartmann, M.; Welte, T.; Heublein, S.; Dennler, U.; Reinhart, K. Hospital Incidence and Mortality Rates of Sepsis: An Analysis of Hospital Episode (DRG) Statistics in Germany from 2007 to 2013. Dtsch. Ärztebl. 2016, 113, 159–166. [Google Scholar]
- SepNet Critical Care Trials Group. Incidence of severe sepsis and septic shock in German intensive care units: The prospective, multicentre INSEP study. Intensive Care Med. 2016. [Google Scholar] [CrossRef]
- Kaukonen, K.-M.; Bailey, M.; Suzuki, S.; Pilcher, D.; Bellomo, R. Mortality Related to Severe Sepsis and Septic Shock among Critically ill Patients in Australia and New Zealand, 2000–2012. J. Am. Med. Assoc. 2014, 311, 1308–1316. [Google Scholar] [CrossRef] [PubMed]
- Dellinger, R.P.; Carlet, J.M.; Masur, H.; Gerlach, H.; Calandra, T.; Cohen, J.; Gea-Banacloche, J.; Keh, D.; Marshall, J.C.; Parker, M.M.; et al. Surviving Sepsis Campaign guidelines for management of severe sepsis and septic shock. Crit. Care Med. 2004, 32, 858–873. [Google Scholar] [CrossRef] [PubMed]
- Dellinger, R.P.; Levy, M.M.; Rhodes, A.; Annane, D.; Gerlach, H.; Opal, S.M.; Sevransky, J.E.; Sprung, C.L.; Douglas, I.S.; Jaeschke, R.; et al. Surviving Sepsis Campaign: International guidelines for management of severe sepsis and septic shock, 2012. Intensive Care Med. 2013, 39, 165–228. [Google Scholar] [CrossRef] [PubMed]
- SSC Executive Committee. Surviving Sepsis Campaign Bundels—Revised 4/2015; SSC Executive Committee: Wakefield, MA, USA, 2015. [Google Scholar]
- Ritthaler, U.; Deng, Y.; Zhang, Y.; Greten, J.; Abel, M.; Sido, B.; Allenberg, J.; Otto, G.; Roth, H.; Bierhaus, A. Expression of receptors for advanced glycation end products in peripheral occlusive vascular disease. Am. J. Pathol. 1995, 146, 688–694. [Google Scholar] [PubMed]
- Weigand, M.A.; Hörner, C.; Bardenheuer, H.J.; Bouchon, A. The systemic inflammatory response syndrome. Best Pract. Res. Clin. Anaesthesiol. 2004, 18, 455–475. [Google Scholar] [CrossRef] [PubMed]
- Carré, J.E.; Singer, M. Cellular energetic metabolism in sepsis: The need for a systems approach. Biochim. Biophys. Acta 2008, 1777, 763–771. [Google Scholar] [CrossRef] [PubMed]
- Hotchkiss, R.S.; Karl, I.E. The pathophysiology and treatment of sepsis. N. Engl. J. Med. 2003, 348, 138–150. [Google Scholar] [CrossRef] [PubMed]
- Abraham, E.; Singer, M. Mechanisms of sepsis-induced organ dysfunction. Crit. Care Med. 2007, 35, 2408–2416. [Google Scholar] [CrossRef] [PubMed]
- Lide, D.R. CRC Handbook of Chemistry and Physics, 85th ed.; CRC Press: Boca Raton, FL, USA, 2004. [Google Scholar]
- Gutteridge, J.M.; Mitchell, J. Redox imbalance in the critically ill. Br. Med. Bull. 1999, 55, 49–75. [Google Scholar] [CrossRef] [PubMed]
- Bone, R.C.; Balk, R.A.; Cerra, F.B.; Dellinger, R.P.; Fein, A.M.; Knaus, W.A.; Schein, R.M.; Sibbald, W.J. DEfinitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The accp/sccm consensus conference committee. American college of chest physicians/society of critical care medicine. Chest 1992, 101, 1644–1655. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.M.; Fink, M.P.; Marshall, J.C.; Abraham, E.; Angus, D.; Cook, D.; Cohen, J.; Opal, S.M.; Vincent, J.-L.; Ramsay, G.; et al. International Sepsis Definitions Conference. 2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference. Intensive Care Med. 2003, 29, 530–538. [Google Scholar] [CrossRef] [PubMed]
- Hotchkiss, R.S.; Monneret, G.; Payen, D. Sepsis-induced immunosuppression: From cellular dysfunctions to immunotherapy. Nat. Rev. Immunol. 2013, 13, 862–874. [Google Scholar] [CrossRef] [PubMed]
- Preiser, J.-C.; Ichai, C.; Orban, J.-C.; Groeneveld, A.B.J. Metabolic response to the stress of critical illness. Br. J. Anaesth. 2014, 113, 945–954. [Google Scholar] [CrossRef] [PubMed]
- Dungan, K.M.; Braithwaite, S.S.; Preiser, J.-C. Stress hyperglycaemia. Lancet 2009, 373, 1798–1807. [Google Scholar] [CrossRef]
- Marik, P.E.; Bellomo, R. Stress hyperglycemia: An essential survival response! Crit. Care 2013, 17, 305. [Google Scholar] [CrossRef] [PubMed]
- Marik, P.E.; Raghavan, M. Stress-hyperglycemia, insulin and immunomodulation in sepsis. Intensive Care Med. 2004, 30, 748–756. [Google Scholar] [CrossRef] [PubMed]
- Krinsley, J.S. Association between hyperglycemia and increased hospital mortality in a heterogeneous population of critically ill patients. Mayo Clin. Proc. 2003, 78, 1471–1478. [Google Scholar] [CrossRef] [PubMed]
- Capes, S.E.; Hunt, D.; Malmberg, K.; Gerstein, H.C. Stress hyperglycaemia and increased risk of death after myocardial infarction in patients with and without diabetes: A systematic overview. Lancet 2000, 355, 773–778. [Google Scholar] [CrossRef]
- Gale, S.C.; Sicoutris, C.; Reilly, P.M.; Schwab, C.W.; Gracias, V.H. Poor glycemic control is associated with increased mortality in critically ill trauma patients. Am. Surg. 2007, 73, 454–460. [Google Scholar] [PubMed]
- Langley, J.; Adams, G. Insulin-based regimens decrease mortality rates in critically ill patients: A systematic review. Diabetes Metab. Res. Rev. 2007, 23, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Wiener, R.S.; Wiener, D.C.; Larson, R.J. Benefits and risks of tight glucose control in critically ill adults: A meta-analysis. J. Am. Med. Assoc. 2008, 300, 933–944. [Google Scholar] [CrossRef] [PubMed]
- NICE-SUGAR Study Investigators; Finfer, S.; Chittock, D.R.; Su, S.Y.-S.; Blair, D.; Foster, D.; Dhingra, V.; Bellomo, R.; Cook, D.; Dodek, P.; et al. Intensive versus conventional glucose control in critically ill patients. N. Engl. J. Med. 2009, 360, 1283–1297. [Google Scholar] [PubMed]
- Fink, M.P. Cytopathic hypoxia. Mitochondrial dysfunction as mechanism contributing to organ dysfunction in sepsis. Crit. Care Clin. 2001, 17, 219–237. [Google Scholar] [CrossRef]
- Fink, M.P. Cytopathic hypoxia and sepsis: Is mitochondrial dysfunction pathophysiologically important or just an epiphenomenon. Pediatr. Crit. Care Med. 2015, 16, 89–91. [Google Scholar] [CrossRef] [PubMed]
- Losser, M.-R.; Damoisel, C.; Payen, D. Bench-to-bedside review: Glucose and stress conditions in the intensive care unit. Crit. Care 2010, 14, 231. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, P.C.; Müller, M.; Graeve, L. Löffler/Petrides Biochemie und Pathobiochemie; Vollständig Überarbeitete Auflage; Springer: Berlin/Heidelberg, Germany, 2014; Volume 9. [Google Scholar]
- Lang, C.H.; Dobrescu, C.; Mészáros, K. Insulin-mediated glucose uptake by individual tissues during sepsis. Metabolism 1990, 39, 1096–1107. [Google Scholar] [CrossRef]
- Fan, J.; Li, Y.H.; Wojnar, M.M.; Lang, C.H. Endotoxin-induced alterations in insulin-stimulated phosphorylation of insulin receptor, IRS-1, and MAP kinase in skeletal muscle. Shock Augusta Ga 1996, 6, 164–170. [Google Scholar] [CrossRef]
- Gamelli, R.L.; Liu, H.; He, L.K.; Hofmann, C.A. Alterations of glucose transporter mRNA and protein levels in brain following thermal injury and sepsis in mice. Shock Augusta Ga 1994, 1, 395–400. [Google Scholar] [CrossRef]
- Maratou, E.; Dimitriadis, G.; Kollias, A.; Boutati, E.; Lambadiari, V.; Mitrou, P.; Raptis, S.A. Glucose transporter expression on the plasma membrane of resting and activated white blood cells. Eur. J. Clin. Investig. 2007, 37, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.A.; Stallons, L.J.; Schnellmann, R.G. Renal cortical hexokinase and pentose phosphate pathway activation through the EGFR/Akt signaling pathway in endotoxin-induced acute kidney injury. Am. J. Physiol. Ren. Physiol. 2014, 307, F435–F444. [Google Scholar] [CrossRef] [PubMed]
- Huynh, A.; DuPage, M.; Priyadharshini, B.; Sage, P.T.; Quiros, J.; Borges, C.M.; Townamchai, N.; Gerriets, V.A.; Rathmell, J.C.; Sharpe, A.H.; et al. Control of PI(3) kinase in Treg cells maintains homeostasis and lineage stability. Nat. Immunol. 2015, 16, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Roberts, D.J.; Miyamoto, S. Hexokinase II integrates energy metabolism and cellular protection: Akting on mitochondria and TORCing to autophagy. Cell Death Differ. 2015, 22, 248–257. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.J.; Kishton, R.J.; Rathmell, J. A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 2016, 16, 553–565. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.P.; Sabatini, D.M. Cancer Cell Metabolism: Warburg and Beyond. Cell 2008, 134, 703–707. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Prados, J.-C.; Través, P.G.; Cuenca, J.; Rico, D.; Aragonés, J.; Martín-Sanz, P.; Cascante, M.; Boscá, L. Substrate Fate in Activated Macrophages: A Comparison between Innate, Classic, and Alternative Activation. J. Immunol. 2010, 185, 605–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krawczyk, C.M.; Holowka, T.; Sun, J.; Blagih, J.; Amiel, E.; DeBerardinis, R.J.; Cross, J.R.; Jung, E.; Thompson, C.B.; Jones, R.G.; et al. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood 2010, 115, 4742–4749. [Google Scholar] [CrossRef] [PubMed]
- Doughty, C.A.; Bleiman, B.F.; Wagner, D.J.; Dufort, F.J.; Mataraza, J.M.; Roberts, M.F.; Chiles, T.C. Antigen receptor-mediated changes in glucose metabolism in B lymphocytes: Role of phosphatidylinositol 3-kinase signaling in the glycolytic control of growth. Blood 2006, 107, 4458–4465. [Google Scholar] [CrossRef] [PubMed]
- Michalek, R.D.; Gerriets, V.A.; Jacobs, S.R.; Macintyre, A.N.; MacIver, N.J.; Mason, E.F.; Sullivan, S.A.; Nichols, A.G.; Rathmell, J.C. Cutting edge: Distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J. Immunol. Baltim. Md 1950 2011, 186, 3299–3303. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, R.P.; Loftus, R.M.; Keating, S.E.; Liou, K.T.; Biron, C.A.; Gardiner, C.M.; Finlay, D.K. mTORC1-dependent metabolic reprogramming is a prerequisite for NK cell effector function. J. Immunol. Baltim. Md 1950 2014, 193, 4477–4484. [Google Scholar] [CrossRef] [PubMed]
- Palsson-McDermott, E.M.; O’Neill, L.A.J. The Warburg effect then and now: From cancer to inflammatory diseases. BioEssays News Rev. Mol. Cell. Dev. Biol. 2013, 35, 965–973. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Hu, H.; Chang, R.; Zhong, J.; Knabel, M.; O’Meally, R.; Cole, R.N.; Pandey, A.; Semenza, G.L. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell 2011, 145, 732–744. [Google Scholar] [CrossRef] [PubMed]
- Palsson-McDermott, E.M.; Curtis, A.M.; Goel, G.; Lauterbach, M.A.R.; Sheedy, F.J.; Gleeson, L.E.; van den Bosch, M.W.M.; Quinn, S.R.; Domingo-Fernandez, R.; Johnston, D.G.W.; et al. Pyruvate kinase M2 regulates Hif-1α activity and IL-1β induction and is a critical determinant of the warburg effect in LPS-activated macrophages. Cell Metab. 2015, 21, 65–80. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.-S.; Hisata, S.; Park, M.-A.; DeNicola, G.M.; Ryter, S.W.; Nakahira, K.; Choi, A.M.K. mTORC1-Induced HK1-Dependent Glycolysis Regulates NLRP3 Inflammasome Activation. Cell Rep. 2015, 12, 102–115. [Google Scholar] [CrossRef] [PubMed]
- Jha, A.K.; Huang, S.C.-C.; Sergushichev, A.; Lampropoulou, V.; Ivanova, Y.; Loginicheva, E.; Chmielewski, K.; Stewart, K.M.; Ashall, J.; Everts, B.; et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 2015, 42, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Gerriets, V.A.; Kishton, R.J.; Nichols, A.G.; Macintyre, A.N.; Inoue, M.; Ilkayeva, O.; Winter, P.S.; Liu, X.; Priyadharshini, B.; Slawinska, M.E.; et al. Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation. J. Clin. Investig. 2015, 125, 194–207. [Google Scholar] [CrossRef] [PubMed]
- Beier, U.H.; Angelin, A.; Akimova, T.; Wang, L.; Liu, Y.; Xiao, H.; Koike, M.A.; Hancock, S.A.; Bhatti, T.R.; Han, R.; et al. Essential role of mitochondrial energy metabolism in Foxp3+ T-regulatory cell function and allograft survival. FASEB J. 2015, 29, 2315–2326. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.Z.; Wang, R.; Huang, G.; Vogel, P.; Neale, G.; Green, D.R.; Chi, H. HIF1α–dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J. Exp. Med. 2011, 208, 1367–1376. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, S.; Yang, K.; Guy, C.; Vogel, P.; Neale, G.; Chi, H. Treg cells require the phosphatase PTEN to restrain TH1 and TFH cell responses. Nat. Immunol. 2015, 16, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-H.; Curtis, J.D.; Maggi, L.B.; Faubert, B.; Villarino, A.V.; O’Sullivan, D.; Huang, S.C.-C.; van der Windt, G.J.W.; Blagih, J.; Qiu, J.; et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 2013, 153, 1239–1251. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, R.; Jia, J.; Arif, A.; Ray, P.S.; Fox, P.L. The GAIT system: A gatekeeper of inflammatory gene expression. Trends Biochem. Sci. 2009, 34, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Hertzog, P.J.; Ravasi, T.; Hume, D.A. Interferon-gamma: An overview of signals, mechanisms and functions. J. Leukoc. Biol. 2004, 75, 163–189. [Google Scholar] [CrossRef] [PubMed]
- Dahlgren, C.; Karlsson, A. Respiratory burst in human neutrophils. J. Immunol. Methods 1999, 232, 3–14. [Google Scholar] [CrossRef]
- Baldridge, C.W.; Gerard, R.W. The Extra Respiration of Phagocytosis. Am. J. Physiol. Leg. Content 1932, 103, 235–236. [Google Scholar]
- Cepinskas, G.; Wilson, J.X. Inflammatory Response in Microvascular Endothelium in Sepsis: Role of Oxidants. J. Clin. Biochem. Nutr. 2008, 42, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Ince, C.; Mayeux, P.R.; Nguyen, T.; Gomez, H.; Kellum, J.A.; Ospina-Tascón, G.A.; Hernandez, G.; Murray, P.; de Backer, D.; ADQI XIV Workgroup. THE ENDOTHELIUM IN SEPSIS. Shock Augusta Ga 2016, 45, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Santos, S.S.; Brunialti, M.K.C.; Rigato, O.; Machado, F.R.; Silva, E.; Salomao, R. Generation of nitric oxide and reactive oxygen species by neutrophils and monocytes from septic patients and association with outcomes. Shock Augusta Ga 2012, 38, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Cobb, J.P. Nitric Oxide Synthase Inhibition as Therapy for Sepsis: A Decade of Promise. Surg. Infect. 2001, 2, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Poderoso, J.J.; Carreras, M.C.; Lisdero, C.; Riobó, N.; Schöpfer, F.; Boveris, A. Nitric Oxide Inhibits Electron Transfer and Increases Superoxide Radical Production in Rat Heart Mitochondria and Submitochondrial Particles. Arch. Biochem. Biophys. 1996, 328, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.-J.; Kwon, Y.-G.; Chung, H.-T.; Lee, S.-K.; Simmons, R.L.; Billiar, T.R.; Kim, Y.-M. Antioxidant Enzymes Suppress Nitric Oxide Production through the Inhibition of NF-κB Activation: Role of H2O2 and Nitric Oxide in Inducible Nitric Oxide Synthase Expression in Macrophages. Nitric Oxide 2001, 5, 504–513. [Google Scholar] [CrossRef] [PubMed]
- Cuzzocrea, S.; Mazzon, E.; Paola, R.D.; Esposito, E.; Macarthur, H.; Matuschak, G.M.; Salvemini, D. A Role for Nitric Oxide-Mediated Peroxynitrite Formation in a Model of Endotoxin-Induced Shock. J. Pharmacol. Exp. Ther. 2006, 319, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Beltrán, B.; Orsi, A.; Clementi, E.; Moncada, S. Oxidative stress and S-nitrosylation of proteins in cells. Br. J. Pharmacol. 2000, 129, 953–960. [Google Scholar] [CrossRef] [PubMed]
- Arulkumaran, N.; Deutschman, C.S.; Pinsky, M.R.; Zuckerbraun, B.; Schumacker, P.T.; Gomez, H.; Gomez, A.; Murray, P.; Kellum, J.A. Mitochondrial Function in Sepsis. Shock 2016, 45, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Boulos, M.; Astiz, M.E.; Barua, R.S.; Osman, M. Impaired mitochondrial function induced by serum from septic shock patients is attenuated by inhibition of nitric oxide synthase and poly(ADP-ribose) synthase. Crit. Care Med. 2003, 31, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Vanasco, V.; Saez, T.; Magnani, N.D.; Pereyra, L.; Marchini, T.; Corach, A.; Inés Vaccaro, M.; Corach, D.; Evelson, P.; Alvarez, S. Cardiac mitochondrial biogenesis in endotoxemia is not accompanied by mitochondrial function recovery. Free Radic. Biol. Med. 2014, 77, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Levy, R.J.; Vijayasarathy, C.; Raj, N.R.; Avadhani, N.G.; Deutschman, C.S. Competitive and noncompetitive inhibition of myocardial cytochrome C oxidase in sepsis. Shock Augusta Ga 2004, 21, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Lowes, D.A.; Webster, N.R.; Murphy, M.P.; Galley, H.F. Antioxidants that protect mitochondria reduce interleukin-6 and oxidative stress, improve mitochondrial function, and reduce biochemical markers of organ dysfunction in a rat model of acute sepsis. Br. J. Anaesth. 2013, 110, 472–480. [Google Scholar] [CrossRef] [PubMed]
- Brealey, D.; Karyampudi, S.; Jacques, T.S.; Novelli, M.; Stidwill, R.; Taylor, V.; Smolenski, R.T.; Singer, M. Mitochondrial dysfunction in a long-term rodent model of sepsis and organ failure. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 286, R491–R497. [Google Scholar] [CrossRef] [PubMed]
- Crouser, E.D.; Julian, M.W.; Dorinsky, P.M. Ileal VO2-O2 alterations induced by endotoxin correlate with severity of mitochondrial injury. Am. J. Respir. Crit. Care Med. 1999, 160, 1347–1353. [Google Scholar] [CrossRef] [PubMed]
- King, C.J.; Tytgat, S.; Delude, R.L.; Fink, M.P. Ileal mucosal oxygen consumption is decreased in endotoxemic rats but is restored toward normal by treatment with aminoguanidine. Crit. Care Med. 1999, 27, 2518–2524. [Google Scholar] [CrossRef] [PubMed]
- Brealey, D.; Brand, M.; Hargreaves, I.; Heales, S.; Land, J.; Smolenski, R.; Davies, N.A.; Cooper, C.E.; Singer, M. Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet 2002, 360, 219–223. [Google Scholar] [CrossRef]
- Sjövall, F.; Morota, S.; Frostner, E.Å.; Hansson, M.J.; Elmér, E. Cytokine and nitric oxide levels in patients with sepsis—Temporal evolvement and relation to platelet mitochondrial respiratory function. PLoS ONE 2014, 9, e103756. [Google Scholar] [CrossRef] [PubMed]
- Sjövall, F.; Morota, S.; Hansson, M.J.; Friberg, H.; Gnaiger, E.; Elmér, E. Temporal increase of platelet mitochondrial respiration is negatively associated with clinical outcome in patients with sepsis. Crit. Care Lond. Engl. 2010, 14, R214. [Google Scholar] [CrossRef] [PubMed]
- Babior, B.M. Phagocytes and oxidative stress. Am. J. Med. 2000, 109, 33–44. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Mao, K.; Chen, S.; Chen, M.; Ma, Y.; Wang, Y.; Huang, B.; He, Z.; Zeng, Y.; Hu, Y.; Sun, S.; et al. Nitric oxide suppresses NLRP3 inflammasome activation and protects against LPS-induced septic shock. Cell Res. 2013, 23, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Emre, Y.; Hurtaud, C.; Nübel, T.; Criscuolo, F.; Ricquier, D.; Cassard-Doulcier, A.-M. Mitochondria contribute to LPS-induced MAPK activation via uncoupling protein UCP2 in macrophages. Biochem. J. 2007, 402, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Rousset, S.; Emre, Y.; Join-Lambert, O.; Hurtaud, C.; Ricquier, D.; Cassard-Doulcier, A.-M. The uncoupling protein 2 modulates the cytokine balance in innate immunity. Cytokine 2006, 35, 135–142. [Google Scholar] [CrossRef] [PubMed]
- West, A.P.; Shadel, G.S.; Ghosh, S. Mitochondria in innate immune responses. Nat. Rev. Immunol. 2011, 11, 389–402. [Google Scholar] [CrossRef] [PubMed]
- West, A.P.; Brodsky, I.E.; Rahner, C.; Woo, D.K.; Erdjument-Bromage, H.; Tempst, P.; Walsh, M.C.; Choi, Y.; Shadel, G.S.; Ghosh, S. TLR signaling augments macrophage bactericidal activity through mitochondrial ROS. Nature 2011, 472, 476–480. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S.; Schumacker, P.T.; Arch, R.H. Reactive Oxygen Species Are Downstream Products of TRAF-mediated Signal Transduction. J. Biol. Chem. 2001, 276, 42728–42736. [Google Scholar] [CrossRef] [PubMed]
- Hensley, K.; Robinson, K.A.; Gabbita, S.P.; Salsman, S.; Floyd, R.A. Reactive oxygen species, cell signaling, and cell injury. Free Radic. Biol. Med. 2000, 28, 1456–1462. [Google Scholar] [CrossRef]
- Lander, H.M. An essential role for free radicals and derived species in signal transduction. FASEB J. 1997, 11, 118–124. [Google Scholar] [PubMed]
- Genestra, M. Oxyl radicals, redox-sensitive signalling cascades and antioxidants. Cell. Signal. 2007, 19, 1807–1819. [Google Scholar] [CrossRef] [PubMed]
- Tonks, N.K. Redox redux: Revisiting PTPs and the control of cell signaling. Cell 2005, 121, 667–670. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B.; Gutteridge, J.M.C. Free Radicals in Biology and Medicine; Oxford University Press: Oxford, UK, 2015. [Google Scholar]
- Aslan, M.; Ozben, T. Oxidants in receptor tyrosine kinase signal transduction pathways. Antioxid. Redox Signal. 2003, 5, 781–788. [Google Scholar] [CrossRef] [PubMed]
- Schreck, R.; Rieber, P.; Baeuerle, P.A. Reactive oxygen intermediates as apparently widely used messengers in the activation of the NF-κ B transcription factor and HIV-1. EMBO J. 1991, 10, 2247–2258. [Google Scholar] [PubMed]
- Ahmed, N.; Dobler, D.; Dean, M.; Thornalley, P.J. Peptide mapping identifies hotspot site of modification in human serum albumin by methylglyoxal involved in ligand binding and esterase activity. J. Biol. Chem. 2005, 280, 5724–5732. [Google Scholar] [CrossRef] [PubMed]
- Dobler, D.; Ahmed, N.; Song, L.; Eboigbodin, K.E.; Thornalley, P.J. Increased dicarbonyl metabolism in endothelial cells in hyperglycemia induces anoikis and impairs angiogenesis by RGD and GFOGER motif modification. Diabetes 2006, 55, 1961–1969. [Google Scholar] [CrossRef] [PubMed]
- Kalapos, M.P. The tandem of free radicals and methylglyoxal. Chem. Biol. Interact. 2008, 171, 251–271. [Google Scholar] [CrossRef] [PubMed]
- Thornalley, P.J.; Langborg, A.; Minhas, H.S. Formation of glyoxal, methylglyoxal and 3-deoxyglucosone in the glycation of proteins by glucose. Biochem. J. 1999, 344 Pt 1, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Veech, R.L.; Raijman, L.; Dalziel, K.; Krebs, H.A. Disequilibrium in the triose phosphate isomerase system in rat liver. Biochem. J. 1969, 115, 837–842. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, N.; Thornalley, P.J. Measurement of methylglyoxal by stable isotopic dilution analysis LC-MS/MS with corroborative prediction in physiological samples. Nat. Protoc. 2014, 9, 1969–1979. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, N.; Xue, M.; Thornalley, P.J. Dicarbonyls and glyoxalase in disease mechanisms and clinical therapeutics. Glycoconj. J. 2016, 33, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Vander Jagt, D.L. Methylglyoxal, diabetes mellitus and diabetic complications. Drug Metabol. Drug Interact. 2008, 23, 93–124. [Google Scholar] [PubMed]
- Beisswenger, P.J.; Howell, S.K.; Nelson, R.G.; Mauer, M.; Szwergold, B.S. α-Oxoaldehyde metabolism and diabetic complications. Biochem. Soc. Trans. 2003, 31, 1358–1363. [Google Scholar] [CrossRef] [PubMed]
- Bierhaus, A.; Fleming, T.; Stoyanov, S.; Leffler, A.; Babes, A.; Neacsu, C.; Sauer, S.K.; Eberhardt, M.; Schnölzer, M.; Lasitschka, F.; et al. Methylglyoxal modification of Nav1.8 facilitates nociceptive neuron firing and causes hyperalgesia in diabetic neuropathy. Nat. Med. 2012, 18, 926–933. [Google Scholar] [CrossRef] [PubMed]
- Brenner, T.; Fleming, T.; Uhle, F.; Silaff, S.; Schmitt, F.; Salgado, E.; Ulrich, A.; Zimmermann, S.; Bruckner, T.; Martin, E.; et al. Methylglyoxal as a new biomarker in patients with septic shock: An observational clinical study. Crit. Care Lond. Engl. 2014, 18, 683. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.M.; Ong, C.Y.; Walker, M.J.; McEwan, A.G. Defence against methylglyoxal in Group A Streptococcus: A role for Glyoxylase I in bacterial virulence and survival in neutrophils? Pathog. Dis. 2016, 74, ftv122. [Google Scholar] [CrossRef] [PubMed]
- Baskaran, S.; Rajan, D.P.; Balasubramanian, K.A. Formation of methylglyoxal by bacteria isolated from human faeces. J. Med. Microbiol. 1989, 28, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Booth, I.R.; Ferguson, G.P.; Miller, S.; Li, C.; Gunasekera, B.; Kinghorn, S. Bacterial production of methylglyoxal: A survival strategy or death by misadventure? Biochem. Soc. Trans. 2003, 31, 1406–1408. [Google Scholar] [CrossRef] [PubMed]
- Cooper, R.A. Metabolism of methylglyoxal in microorganisms. Annu. Rev. Microbiol. 1984, 38, 49–68. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, G.P.; Tötemeyer, S.; MacLean, M.J.; Booth, I.R. Methylglyoxal production in bacteria: Suicide or survival? Arch. Microbiol. 1998, 170, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Brenner, T.; Fleming, T.H.; Spranz, D.; Schemmer, P.; Bruckner, T.; Uhle, F.; Martin, E.O.; Weigand, M.A.; Hofer, S. Reactive Metabolites and AGE-RAGE-Mediated Inflammation in Patients following Liver Transplantation. Mediat. Inflamm. 2013, 2013, 501430. [Google Scholar] [CrossRef] [PubMed]
- Uhle, F.; Lichtenstern, C.; Brenner, T.; Fleming, T.; Koch, C.; Hecker, A.; Heiss, C.; Nawroth, P.P.; Hofer, S.; Weigand, M.A.; et al. Role of the RAGE Axis during the Immune Response after Severe Trauma: A Prospective Pilot Study. Mediat. Inflamm. 2015, 2015, e691491. [Google Scholar] [CrossRef] [PubMed]
- Lo, T.W.; Westwood, M.E.; McLellan, A.C.; Selwood, T.; Thornalley, P.J. Binding and modification of proteins by methylglyoxal under physiological conditions. A kinetic and mechanistic study with N α-acetylarginine, N α-acetylcysteine, and N α-acetyllysine, and bovine serum albumin. J. Biol. Chem. 1994, 269, 32299–32305. [Google Scholar] [PubMed]
- Westwood, M.E.; Thornalley, P.J. Induction of synthesis and secretion of interleukin 1 β in the human monocytic THP-1 cells by human serum albumins modified with methylglyoxal and advanced glycation endproducts. Immunol. Lett. 1996, 50, 17–21. [Google Scholar] [CrossRef]
- Abordo, E.A.; Westwood, M.E.; Thornalley, P.J. Synthesis and secretion of macrophage colony stimulating factor by mature human monocytes and human monocytic THP-1 cells induced by human serum albumin derivatives modified with methylglyoxal and glucose-derived advanced glycation endproducts. Immunol. Lett. 1996, 53, 7–13. [Google Scholar] [CrossRef]
- Westwood, M.E.; Argirov, O.K.; Abordo, E.A.; Thornalley, P.J. Methylglyoxal-modified arginine residues—A signal for receptor-mediated endocytosis and degradation of proteins by monocytic THP-1 cells. Biochim. Biophys. Acta 1997, 1356, 84–94. [Google Scholar] [CrossRef]
- Du, J.; Zeng, J.; Ou, X.; Ren, X.; Cai, S. Methylglyoxal downregulates Raf-1 protein through a ubiquitination-mediated mechanism. Int. J. Biochem. Cell Biol. 2006, 38, 1084–1091. [Google Scholar] [CrossRef] [PubMed]
- Hoon, S.; Gebbia, M.; Costanzo, M.; Davis, R.W.; Giaever, G.; Nislow, C. A global perspective of the genetic basis for carbonyl stress resistance. G3 Bethesda Md 2011, 1, 219–231. [Google Scholar] [CrossRef] [PubMed]
- Berner, A.K.; Brouwers, O.; Pringle, R.; Klaassen, I.; Colhoun, L.; McVicar, C.; Brockbank, S.; Curry, J.W.; Miyata, T.; Brownlee, M.; et al. Protection against methylglyoxal-derived AGEs by regulation of glyoxalase 1 prevents retinal neuroglial and vasodegenerative pathology. Diabetologia 2012, 55, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Fleming, T.H.; Humpert, P.M.; Nawroth, P.P.; Bierhaus, A. Reactive metabolites and AGE/RAGE-mediated cellular dysfunction affect the aging process: A mini-review. Gerontology 2011, 57, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Dhar, A.; Dhar, I.; Desai, K.M.; Wu, L. Methylglyoxal scavengers attenuate endothelial dysfunction induced by methylglyoxal and high concentrations of glucose. Br. J. Pharmacol. 2010, 161, 1843–1856. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, N.; Thornalley, P.J. Dicarbonyl stress in cell and tissue dysfunction contributing to ageing and disease. Biochem. Biophys. Res. Commun. 2015, 458, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Pedchenko, V.K.; Chetyrkin, S.V.; Chuang, P.; Ham, A.-J.L.; Saleem, M.A.; Mathieson, P.W.; Hudson, B.G.; Voziyan, P.A. Mechanism of perturbation of integrin-mediated cell-matrix interactions by reactive carbonyl compounds and its implication for pathogenesis of diabetic nephropathy. Diabetes 2005, 54, 2952–2960. [Google Scholar] [CrossRef] [PubMed]
- Duran-Jimenez, B.; Dobler, D.; Moffatt, S.; Rabbani, N.; Streuli, C.H.; Thornalley, P.J.; Tomlinson, D.R.; Gardiner, N.J. Advanced glycation end products in extracellular matrix proteins contribute to the failure of sensory nerve regeneration in diabetes. Diabetes 2009, 58, 2893–2903. [Google Scholar] [CrossRef] [PubMed]
- Morcos, M.; Du, X.; Pfisterer, F.; Hutter, H.; Sayed, A.A.R.; Thornalley, P.; Ahmed, N.; Baynes, J.; Thorpe, S.; Kukudov, G.; et al. Glyoxalase-1 prevents mitochondrial protein modification and enhances lifespan in Caenorhabditis elegans. Aging Cell 2008, 7, 260–269. [Google Scholar] [CrossRef] [PubMed]
- Kalapos, M.P.; Littauer, A.; de Groot, H. Has reactive oxygen a role in methylglyoxal toxicity? A study on cultured rat hepatocytes. Arch. Toxicol. 1993, 67, 369–372. [Google Scholar] [CrossRef] [PubMed]
- Wu, L. The pro-oxidant role of methylglyoxal in mesenteric artery smooth muscle cells. Can. J. Physiol. Pharmacol. 2005, 83, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Abordo, E.A.; Minhas, H.S.; Thornalley, P.J. Accumulation of α-oxoaldehydes during oxidative stress: A role in cytotoxicity. Biochem. Pharmacol. 1999, 58, 641–648. [Google Scholar] [CrossRef]
- Hyslop, P.A.; Hinshaw, D.B.; Halsey, W.A.; Schraufstätter, I.U.; Sauerheber, R.D.; Spragg, R.G.; Jackson, J.H.; Cochrane, C.G. Mechanisms of oxidant-mediated cell injury. The glycolytic and mitochondrial pathways of ADP phosphorylation are major intracellular targets inactivated by hydrogen peroxide. J. Biol. Chem. 1988, 263, 1665–1675. [Google Scholar] [PubMed]
- Yao, D.; Brownlee, M. Hyperglycemia-induced reactive oxygen species increase expression of the receptor for advanced glycation end products (RAGE) and RAGE ligands. Diabetes 2010, 59, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.-H.; Wu, H.-J.; Shiao, N.-H. Apoptotic signaling in methylglyoxal-treated human osteoblasts involves oxidative stress, c-Jun N-terminal kinase, caspase-3, and p21-activated kinase 2. J. Cell. Biochem. 2007, 100, 1056–1069. [Google Scholar] [CrossRef] [PubMed]
- Seo, K.; Ki, S.H.; Shin, S.M. Methylglyoxal induces mitochondrial dysfunction and cell death in liver. Toxicol. Res. 2014, 30, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Thornalley, P.J. Glyoxalase I—Structure, function and a critical role in the enzymatic defence against glycation. Biochem. Soc. Trans. 2003, 31, 1343–1348. [Google Scholar] [CrossRef] [PubMed]
- Lohmann, K. Beitrag zur enzymatischen Umwandlung von synthetischem Methylglyoxal in Milchsäure. Biochemie 1932, 254, 332–354. [Google Scholar]
- Dakin, H.D.; Dudley, H.W. On Glyoxalase. J. Biol. Chem. 1913, 14, 423–431. [Google Scholar]
- Neubauer, C. Über die Zerstorung von Milchsäurealdehyd und Methylglyoxal durch tierische Organe. Biochemie 1912, 49, 502–506. [Google Scholar]
- Rabbani, N.; Thornalley, P.J. Glyoxalase Centennial conference: Introduction, history of research on the glyoxalase system and future prospects. Biochem. Soc. Trans. 2014, 42, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, N.; Thornalley, P.J. Dicarbonyls linked to damage in the powerhouse: Glycation of mitochondrial proteins and oxidative stress. Biochem. Soc. Trans. 2008, 36, 1045–1050. [Google Scholar] [CrossRef] [PubMed]
- Kanwar, M.; Kowluru, R.A. Role of Glyceraldehyde 3-Phosphate Dehydrogenase in the Development and Progression of Diabetic Retinopathy. Diabetes 2009, 58, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Schlotterer, A.; Kukudov, G.; Bozorgmehr, F.; Hutter, H.; Du, X.; Oikonomou, D.; Ibrahim, Y.; Pfisterer, F.; Rabbani, N.; Thornalley, P.; et al. C. elegans as Model for the Study of High Glucose—Mediated Life Span Reduction. Diabetes 2009, 58, 2450–2456. [Google Scholar] [CrossRef] [PubMed]
- Bierhaus, A.; Chevion, S.; Chevion, M.; Hofmann, M.; Quehenberger, P.; Illmer, T.; Luther, T.; Berentshtein, E.; Tritschler, H.; Müller, M.; et al. Advanced glycation end product-induced activation of NF-κB is suppressed by α-lipoic acid in cultured endothelial cells. Diabetes 1997, 46, 1481–1490. [Google Scholar] [CrossRef] [PubMed]
- Bierhaus, A.; Humpert, P.M.; Morcos, M.; Wendt, T.; Chavakis, T.; Arnold, B.; Stern, D.M.; Nawroth, P.P. Understanding RAGE, the receptor for advanced glycation end products. J. Mol. Med. Berl. Ger. 2005, 83, 876–886. [Google Scholar] [CrossRef] [PubMed]
- Lander, H.M.; Tauras, J.M.; Ogiste, J.S.; Hori, O.; Moss, R.A.; Schmidt, A.M. Activation of the receptor for advanced glycation end products triggers a p21(ras)-dependent mitogen-activated protein kinase pathway regulated by oxidant stress. J. Biol. Chem. 1997, 272, 17810–17814. [Google Scholar] [CrossRef] [PubMed]
- Thornalley, P.J. Glutathione-dependent detoxification of alpha-oxoaldehydes by the glyoxalase system: Involvement in disease mechanisms and antiproliferative activity of glyoxalase I inhibitors. Chem. Biol. Interact. 1998, 111–112, 137–151. [Google Scholar] [CrossRef]
- Bierhaus, A.; Schiekofer, S.; Schwaninger, M.; Andrassy, M.; Humpert, P.M.; Chen, J.; Hong, M.; Luther, T.; Henle, T.; Klöting, I.; et al. Diabetes-associated sustained activation of the transcription factor nuclear factor-kappaB. Diabetes 2001, 50, 2792–2808. [Google Scholar] [CrossRef] [PubMed]
- Bierhaus, A.; Stoyanov, S.; Haag, G.; Konrade, I. RAGE-deficiency reduces diabetes-associated impairment of glyoxalase-1 in neuronal cells. Diabetes 2006, 55, A511. [Google Scholar]
- Liliensiek, B.; Weigand, M.A.; Bierhaus, A.; Nicklas, W.; Kasper, M.; Hofer, S.; Plachky, J.; Gröne, H.-J.; Kurschus, F.C.; Schmidt, A.M.; et al. Receptor for advanced glycation end products (RAGE) regulates sepsis but not the adaptive immune response. J. Clin. Investig. 2004, 113, 1641–1650. [Google Scholar] [CrossRef] [PubMed]
- Hofer, S.; Uhle, F.; Fleming, T.; Hell, C.; Schmoch, T.; Bruckner, T.; Weigand, M.A.; Brenner, T. RAGE-mediated inflammation in patients with septic shock. J. Surg. Res. 2016, 202, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Van Herreweghe, F.; Mao, J.; Chaplen, F.W.R.; Grooten, J.; Gevaert, K.; Vandekerckhove, J.; Vancompernolle, K. Tumor necrosis factor-induced modulation of glyoxalase I activities through phosphorylation by PKA results in cell death and is accompanied by the formation of a specific methylglyoxal-derived AGE. Proc. Natl. Acad. Sci. USA 2002, 99, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Bourne, H.; Lichtenstein, L.; Melmon, K.; Henney, C.; Weinstein, Y.; Shearer, G. Modulation of inflammation and immunity by cyclic AMP. Science 1974, 184, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Janssens, P.A.; Lowrey, P. Hormonal regulation of hepatic glycogenolysis in the carp, Cyprinus carpio. Am. J. Physiol. Regul. Integr. Comp. Physiol. 1987, 252, R653–R660. [Google Scholar]
- Beavo, J.A.; Brunton, L.L. Cyclic nucleotide research—Still expanding after half a century. Nat. Rev. Mol. Cell Biol. 2002, 3, 710–718. [Google Scholar] [CrossRef] [PubMed]
- Zuo, L.; Shi, L.; Yan, F. The reciprocal interaction of sympathetic nervous system and cAMP-PKA-NF-κB pathway in immune suppression after experimental stroke. Neurosci. Lett. 2016, 627, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Park, T.; Chen, H.; Kevala, K.; Lee, J.-W.; Kim, H.-Y. N-Docosahexaenoylethanolamine ameliorates LPS-induced neuroinflammation via cAMP/PKA-dependent signaling. J. Neuroinflamm. 2016, 13, 284. [Google Scholar] [CrossRef] [PubMed]
- Rivkin, I.; Neutze, J.A. Influence of cyclic nucleotides and a phosphodiesterase inhibitor on in vitro human blood neutrophil chemotaxis. Arch. Int. Pharmacodyn. Ther. 1977, 228, 196–204. [Google Scholar] [PubMed]
- Marone, G.; Columbo, M.; Triggiani, M.; Cirillo, R.; Genovese, A.; Formisano, S. Inhibition of IgE-mediated release of histamine and peptide leukotriene from human basophils and mast cells by forskolin. Biochem. Pharmacol. 1987, 36, 13–20. [Google Scholar] [CrossRef]
- Nielson, C.P. Beta-adrenergic modulation of the polymorphonuclear leukocyte respiratory burst is dependent upon the mechanism of cell activation. J. Immunol. 1987, 139, 2392–2397. [Google Scholar] [PubMed]
- Dent, G.; Giembycz, M.A.; Evans, P.M.; Rabe, K.F.; Barnes, P.J. Suppression of human eosinophil respiratory burst and cyclic AMP hydrolysis by inhibitors of type IV phosphodiesterase: Interaction with the beta adrenoceptor agonist albuterol. J. Pharmacol. Exp. Ther. 1994, 271, 1167–1174. [Google Scholar] [PubMed]
- Lorenowicz, M.J.; Fernandez-Borja, M.; Hordijk, P.L. cAMP signaling in leukocyte transendothelial migration. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1014–1022. [Google Scholar] [CrossRef] [PubMed]
- Skalhegg, B.S.; Tasken, K. Specificity in the cAMP/PKA signaling pathway. Differential expression, regulation, and subcellular localization of subunits of PKA. Front. Biosci. 2000, 5, D678–D693. [Google Scholar] [CrossRef] [PubMed]
- Colledge, M.; Scott, J.D. AKAPs: From structure to function. Trends Cell Biol. 1999, 9, 216–221. [Google Scholar] [CrossRef]
- Gerlo, S.; Kooijman, R.; Beck, I.M.; Kolmus, K.; Spooren, A.; Haegeman, G. Cyclic AMP: A selective modulator of NF-κB action. Cell. Mol. Life Sci. 2011, 68, 3823–3841. [Google Scholar] [CrossRef] [PubMed]
- Morgenstern, J.; Fleming, T.; Schumacher, D.; Eckstein, V.; Freichel, M.; Herzig, S.; Nawroth, P. Loss of glyoxalase 1 induces compensatory mechanism to achieve dicarbonyl detoxification in mammalian Schwann cells. J. Biol. Chem. 2017, 292, 3224–3238. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Hormone | Pathway | Metabolic Changes |
|---|---|---|
| Increased | ||
| Cortisol |
|
|
| Nor-/Epinephrine |
|
|
| Vasopressin |
|
|
| Insulin (although its effects are impaired by peripheral insulin resistance) |
| In hepatocytes and skeletal muscle
|
| Glucagon |
|
|
| Reduced | ||
| Thyroid-stimulating hormone (TSH) ↓, triiodothyronine (T3) ↓, thyroxine (T4) ↓ |
|
|
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schmoch, T.; Uhle, F.; Siegler, B.H.; Fleming, T.; Morgenstern, J.; Nawroth, P.P.; Weigand, M.A.; Brenner, T. The Glyoxalase System and Methylglyoxal-Derived Carbonyl Stress in Sepsis: Glycotoxic Aspects of Sepsis Pathophysiology. Int. J. Mol. Sci. 2017, 18, 657. https://doi.org/10.3390/ijms18030657
Schmoch T, Uhle F, Siegler BH, Fleming T, Morgenstern J, Nawroth PP, Weigand MA, Brenner T. The Glyoxalase System and Methylglyoxal-Derived Carbonyl Stress in Sepsis: Glycotoxic Aspects of Sepsis Pathophysiology. International Journal of Molecular Sciences. 2017; 18(3):657. https://doi.org/10.3390/ijms18030657
Chicago/Turabian StyleSchmoch, Thomas, Florian Uhle, Benedikt H. Siegler, Thomas Fleming, Jakob Morgenstern, Peter P. Nawroth, Markus A. Weigand, and Thorsten Brenner. 2017. "The Glyoxalase System and Methylglyoxal-Derived Carbonyl Stress in Sepsis: Glycotoxic Aspects of Sepsis Pathophysiology" International Journal of Molecular Sciences 18, no. 3: 657. https://doi.org/10.3390/ijms18030657
APA StyleSchmoch, T., Uhle, F., Siegler, B. H., Fleming, T., Morgenstern, J., Nawroth, P. P., Weigand, M. A., & Brenner, T. (2017). The Glyoxalase System and Methylglyoxal-Derived Carbonyl Stress in Sepsis: Glycotoxic Aspects of Sepsis Pathophysiology. International Journal of Molecular Sciences, 18(3), 657. https://doi.org/10.3390/ijms18030657