Pathophysiology of Lung Disease and Wound Repair in Cystic Fibrosis



Abstract
1. Introduction
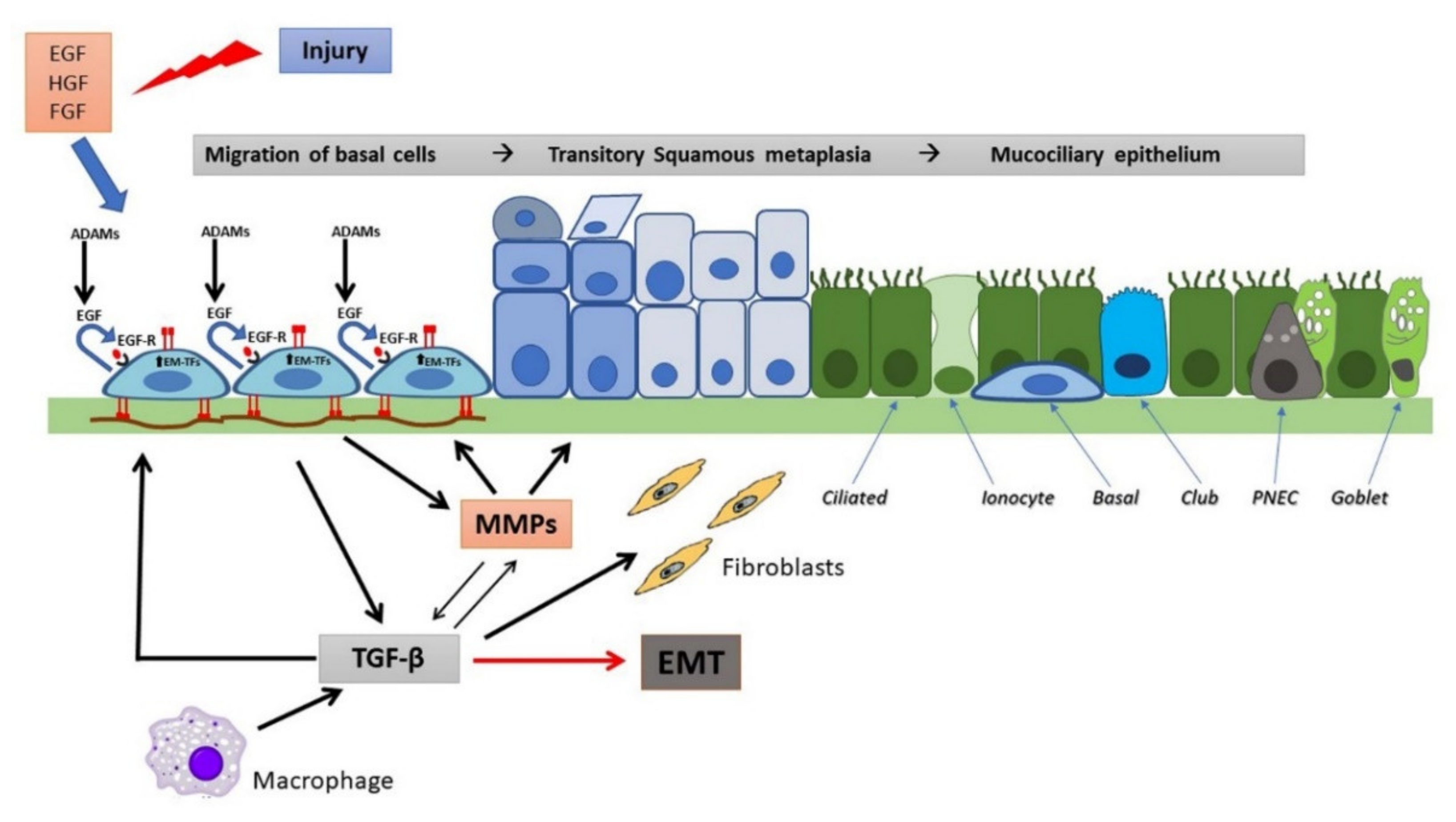
Events Involved in Epithelial Repair and EMT
2. Pathological Processes in the CF Airways
3. Airway Epithelial Regeneration, Wound Repair and CF
3.1. Airway Epithelial Regeneration in Xenograft Models
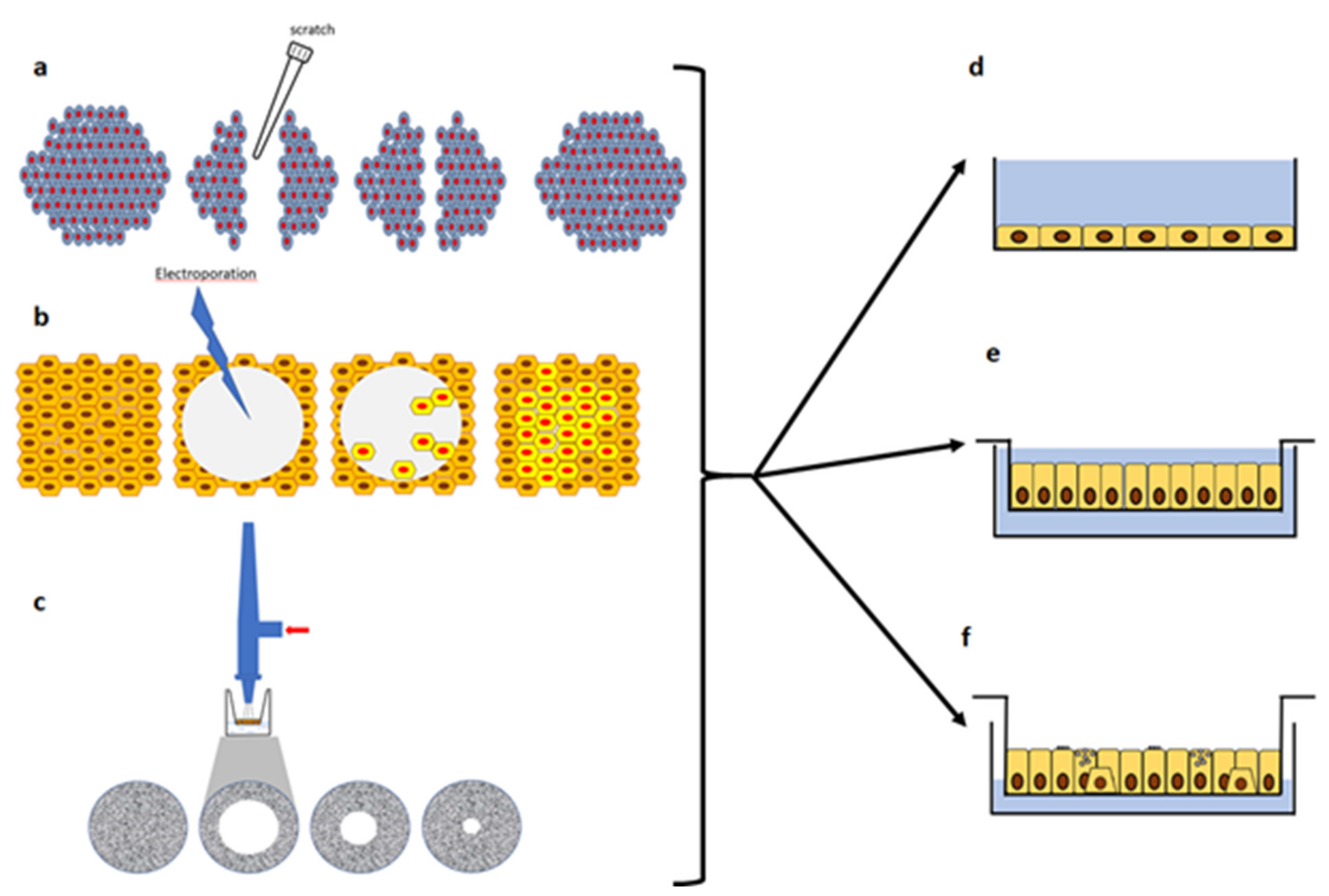
3.2. In-Vitro Models of Injury and Repair with Submerged Cultures
3.3. Wound Repair in Polarized and ALI Cultures
3.4. Cellular and Molecular Events in CF Wound Repair
4. Modulation of Wound Repair in CF
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Elborn, J.S. Cystic fibrosis. Lancet 2016, 388, 2519–2531. [Google Scholar] [CrossRef]
- Amaral, M.D. Novel personalized therapies for cystic fibrosis: Treating the basic defect in all patients. J. Intern. Med. 2015, 277, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Marson, F.A.L.; Bertuzzo, C.S.; Ribeiro, J.D. Classification of CFTR mutation classes. Lancet Respir. Med. 2016, 4, e37–e38. [Google Scholar] [CrossRef]
- De Boeck, K.; Amaral, M.D. Progress in therapies for cystic fibrosis. Lancet Respir. Med. 2016, 4, 662–674. [Google Scholar] [CrossRef]
- Boucher, R.C. Cystic fibrosis: A disease of vulnerability to airway surface dehydration. Trends Mol. Med. 2007, 13, 231–240. [Google Scholar] [CrossRef]
- Haq, I.J.; Gray, M.A.; Garnett, J.P.; Ward, C.; Brodlie, M. Airway surface liquid homeostasis in cystic fibrosis: Pathophysiology and therapeutic targets. Thorax 2016, 71, 284–287. [Google Scholar] [CrossRef]
- Stutts, M.J.; Canessa, C.M.; Olsen, J.C.; Hamrick, M.; Cohn, J.A.; Rossier, B.C.; Boucher, R.C. CFTR as a cAMP-dependent regulator of sodium channel. Science 1995, 269, 847–850. [Google Scholar] [CrossRef] [PubMed]
- Mall, M.; Hipper, A.; Greger, R.; Kunzelmann, K. Wild type but not deltaF508 CFTR inhibits Na+ conductance when coexpressed in Xenopus oocytes. Febs Lett. 1996, 381, 47–52. [Google Scholar] [CrossRef]
- Donaldson, S.H.; Boucher, R.C. Sodium channels and cystic fibrosis. Chest 2007, 132, 1631–1636. [Google Scholar] [CrossRef]
- Pezzulo, A.A.; Tang, X.X.; Hoegger, M.J.; Alaiwa, M.H.A.; Ramachandran, S.; Moninger, T.O.; Karp, P.H.; Wohlford-Lenane, C.L.; Haagsman, H.P.; van Eijk, M.; et al. Reduced airway surface pH impairs bacterial killing in the porcine cystic fibrosis lung. Nature 2012, 487, 109–113. [Google Scholar] [CrossRef]
- Gustafsson, J.K.; Ermund, A.; Ambort, D.; Johansson, M.E.; Nilsson, H.E.; Thorell, K.; Hebert, H.; Sjovall, H.; Hansson, G.C. Bicarbonate and functional CFTR channel are required for proper mucin secretion and link cystic fibrosis with its mucus phenotype. J. Exp. Med. 2012, 209, 1263–1272. [Google Scholar] [CrossRef] [PubMed]
- Conese, M.; Tirelli, A.S.; Alicandro, G.; Di Gioia, S.; Carbone, A.; Castellani, S.; Colombo, C. Biomarkers of Inflammation and Remodelling in Cystic Fibrosis. Clin. Immunol. Endocr. Metab. Drugs 2016, 3, 92–108. [Google Scholar] [CrossRef]
- Larson, J.E.; Cohen, J.C. Developmental paradigm for early features of cystic fibrosis. Pediatr. Pulmonol. 2005, 40, 371–377. [Google Scholar] [CrossRef] [PubMed]
- LeSimple, P.; Liao, J.; Robert, R.; Gruenert, D.C.; Hanrahan, J.W. Cystic fibrosis transmembrane conductance regulator trafficking modulates the barrier function of airway epithelial cell monolayers. J. Physiol. 2010, 588, 1195–1209. [Google Scholar] [CrossRef] [PubMed]
- Hajj, R.; Lesimple, P.; Nawrocki-Raby, B.; Birembaut, P.; Puchelle, E.; Coraux, C. Human airway surface epithelial regeneration is delayed and abnormal in cystic fibrosis. J. Pathol. 2007, 211, 340–350. [Google Scholar] [CrossRef]
- Castellani, S.; Guerra, L.; Favia, M.; Di Gioia, S.; Casavola, V.; Conese, M. NHERF1 and CFTR restore tight junction organisation and function in cystic fibrosis airway epithelial cells: Role of ezrin and the RhoA/ROCK pathway. Lab. Investig. 2012, 92, 1527–1540. [Google Scholar] [CrossRef]
- Rout-Pitt, N.; Farrow, N.; Parsons, D.; Donnelley, M. Epithelial mesenchymal transition (EMT): A universal process in lung diseases with implications for cystic fibrosis pathophysiology. Respir. Res. 2018, 19, 136. [Google Scholar] [CrossRef]
- Amaral, M.D.; Quaresma, M.C.; Pankonien, I. What Role Does CFTR Play in Development, Differentiation, Regeneration and Cancer? Int. J. Mol. Sci. 2020, 21, 3133. [Google Scholar] [CrossRef] [PubMed]
- Byun, J.S.; Gardner, K. Wounds that will not heal: Pervasive cellular reprogramming in cancer. Am. J. Pathol. 2013, 182, 1055–1064. [Google Scholar] [CrossRef]
- Favia, M.; Guerra, L.; Fanelli, T.; Cardone, R.A.; Monterisi, S.; Di Sole, F.; Castellani, S.; Chen, M.; Seidler, U.; Reshkin, S.J.; et al. Na+/H+ exchanger regulatory factor 1 overexpression-dependent increase of cytoskeleton organization is fundamental in the rescue of F508del cystic fibrosis transmembrane conductance regulator in human airway CFBE41o-cells. Mol. Biol. Cell 2010, 21, 73–86. [Google Scholar] [CrossRef]
- Nilsson, H.E.; Dragomir, A.; Lazorova, L.; Johannesson, M.; Roomans, G.M. CFTR and tight junctions in cultured bronchial epithelial cells. Exp. Mol. Pathol. 2010, 88, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Lasalvia, M.; Castellani, S.; D’Antonio, P.; Perna, G.; Carbone, A.; Colia, A.L.; Maffione, A.B.; Capozzi, V.; Conese, M. Human airway epithelial cells investigated by atomic force microscopy: A hint to cystic fibrosis epithelial pathology. Exp. Cell Res. 2016, 348, 46–55. [Google Scholar] [CrossRef]
- Monterisi, S.; Favia, M.; Guerra, L.; Cardone, R.A.; Marzulli, D.; Reshkin, S.J.; Casavola, V.; Zaccolo, M. CFTR regulation in human airway epithelial cells requires integrity of the actin cytoskeleton and compartmentalized cAMP and PKA activity. J. Cell Sci. 2012, 125, 1106–1117. [Google Scholar] [CrossRef] [PubMed]
- Guggino, W.B.; Stanton, B.A. New insights into cystic fibrosis: Molecular switches that regulate CFTR. Nat. Rev. Mol. Cell Biol. 2006, 7, 426–436. [Google Scholar] [CrossRef]
- Castellani, S.; Favia, M.; Guerra, L.; Carbone, A.; Abbattiscianni, A.C.; Di Gioia, S.; Casavola, V.; Conese, M. Emerging relationship between CFTR, actin and tight junction organization in cystic fibrosis airway epithelium. Histol. Histopathol. 2017, 32, 445–459. [Google Scholar] [PubMed]
- Short, D.B.; Trotter, K.W.; Reczek, D.; Kreda, S.M.; Bretscher, A.; Boucher, R.C.; Stutts, M.J.; Milgram, S.L. An apical PDZ protein anchors the cystic fibrosis transmembrane conductance regulator to the cytoskeleton. J. Biol. Chem. 1998, 273, 19797–19801. [Google Scholar] [CrossRef]
- Moyer, B.D.; Duhaime, M.; Shaw, C.; Denton, J.; Reynolds, D.; Karlson, K.H.; Pfeiffer, J.; Wang, S.; Mickle, J.E.; Milewski, M.; et al. The PDZ-interacting domain of cystic fibrosis transmembrane conductance regulator is required for functional expression in the apical plasma membrane. J. Biol. Chem. 2000, 275, 27069–27074. [Google Scholar] [CrossRef]
- Sun, F.; Hug, M.J.; Bradbury, N.A.; Frizzell, R.A. Protein kinase A associates with cystic fibrosis transmembrane conductance regulator via an interaction with ezrin. J. Biol. Chem. 2000, 275, 14360–14366. [Google Scholar] [CrossRef]
- Guerra, L.; Fanelli, T.; Favia, M.; Riccardi, S.M.; Busco, G.; Cardone, R.A.; Carrabino, S.; Weinman, E.J.; Reshkin, S.J.; Conese, M.; et al. Na+/H+ exchanger regulatory factor isoform 1 overexpression modulates cystic fibrosis transmembrane conductance regulator (CFTR) expression and activity in human airway 16HBE14o- cells and rescues DeltaF508 CFTR functional expression in cystic fibrosis cells. J. Biol. Chem. 2005, 280, 40925–40933. [Google Scholar]
- Weiser, N.; Molenda, N.; Urbanova, K.; Bahler, M.; Pieper, U.; Oberleithner, H.; Schillers, H. Paracellular permeability of bronchial epithelium is controlled by CFTR. Cell Physiol. Biochem. 2011, 28, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Molina, S.A.; Stauffer, B.; Moriarty, H.K.; Kim, A.H.; McCarty, N.A.; Koval, M. Junctional abnormalities in human airway epithelial cells expressing F508del CFTR. Am. J. Physiol. Cell Mol. Physiol. 2015, 309, L475–L487. [Google Scholar] [CrossRef]
- Ruan, Y.C.; Wang, Y.; Da Silva, N.; Kim, B.; Diao, R.Y.; Hill, E.; Brown, D.; Chan, H.C.; Breton, S. CFTR interacts with ZO-1 to regulate tight junction assembly and epithelial differentiation through the ZONAB pathway. J. Cell Sci. 2014, 127, 4396–4408. [Google Scholar] [CrossRef]
- Aasen, T. Connexins: Junctional and non-junctional modulators of proliferation. Cell Tissue Res. 2015, 360, 685–699. [Google Scholar] [CrossRef] [PubMed]
- Vinken, M. Introduction: Connexins, pannexins and their channels as gatekeepers of organ physiology. Cell Mol. Life Sci. 2015, 72, 2775–2778. [Google Scholar] [CrossRef]
- Huang, S.; Dudez, T.; Scerri, I.; Thomas, M.A.; Giepmans, B.N.; Suter, S.; Chanson, M. Defective activation of c-Src in cystic fibrosis airway epithelial cells results in loss of tumor necrosis factor-alpha-induced gap junction regulation. J. Biol. Chem. 2003, 278, 8326–8332. [Google Scholar] [CrossRef] [PubMed]
- Losa, D.; Kohler, T.; Bellec, J.; Dudez, T.; Crespin, S.; Bacchetta, M.; Boulanger, P.; Hong, S.S.; Morel, S.; Nguyen, T.H.; et al. Pseudomonas aeruginosa-induced apoptosis in airway epithelial cells is mediated by gap junctional communication in a JNK-dependent manner. J. Immunol. 2014, 192, 4804–4812. [Google Scholar] [CrossRef]
- Chanson, M.; Kotsias, B.A.; Peracchia, C.; O’Grady, S.M. Interactions of connexins with other membrane channels and transporters. Prog. Biophys. Mol. Biol. 2007, 94, 233–244. [Google Scholar] [CrossRef][Green Version]
- Rejman, J.; Colombo, C.; Conese, M. Engraftment of bone marrow-derived stem cells to the lung in a model of acute respiratory infection by Pseudomonas aeruginosa. Mol. Ther. 2009, 17, 1257–1265. [Google Scholar] [CrossRef] [PubMed]
- Adam, D.; Roux-Delrieu, J.; Luczka, E.; Bonnomet, A.; Lesage, J.; Merol, J.C.; Polette, M.; Abely, M.; Coraux, C. Cystic fibrosis airway epithelium remodelling: Involvement of inflammation. J. Pathol. 2015, 235, 408–419. [Google Scholar] [CrossRef]
- de Bentzmann, S.; Polette, M.; Zahm, J.-M.; Hinnrasky, J.K.C.; Bajolet, O.; Klossek, J.-M.; Filloux, A.; Lazdunski, A.; Puchelle, E. Pseudomonas aeruginosa virulence factors delay airway epithelial wound repair by altering the actin cytoskeleton and inducing overactivation of epithelial matrix metalloproteinase-2. Lab. Investig. 2000, 80, 209–219. [Google Scholar] [CrossRef]
- Ruffin, M.; Bilodeau, C.; Maille, E.; LaFayette, S.L.; McKay, G.A.; Trinh, N.T.; Beaudoin, T.; Desrosiers, M.Y.; Rousseau, S.; Nguyen, D.; et al. Quorum-sensing inhibition abrogates the deleterious impact of Pseudomonas aeruginosa on airway epithelial repair. FASEB J. 2016, 30, 3011–3025. [Google Scholar] [CrossRef]
- Saint-Criq, V.; Villeret, B.; Bastaert, F.; Kheir, S.; Hatton, A.; Cazes, A.; Xing, Z.; Sermet-Gaudelus, I.; Garcia-Verdugo, I.; Edelman, A.; et al. Pseudomonas aeruginosa LasB protease impairs innate immunity in mice and humans by targeting a lung epithelial cystic fibrosis transmembrane regulator-IL-6-antimicrobial-repair pathway. Thorax 2018, 73, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Shute, J.; Marshall, L.; Bodey, K.; Bush, A. Growth factors in cystic fibrosis-when more is not enough. Paediatr. Respir. Rev. 2003, 4, 120–127. [Google Scholar] [CrossRef]
- Courtney, J.M.; Ennis, M.; Elborn, J.S. Cytokines and inflammatory mediators in cystic fibrosis. J. Cyst. Fibros. 2004, 3, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Virella-Lowell, I.; Herlihy, J.D.; Liu, B.; Lopez, C.; Cruz, P.; Muller, C.; Baker, H.V.; Flotte, T.R. Effects of CFTR, interleukin-10, and Pseudomonas aeruginosa on gene expression profiles in a CF bronchial epithelial cell Line. Mol. Ther. 2004, 10, 562–573. [Google Scholar] [CrossRef] [PubMed]
- Crosby, L.M.; Waters, C.M. Epithelial repair mechanisms in the lung. Am. J. Physiol. Cell Mol. Physiol. 2010, 298, L715–L731. [Google Scholar] [CrossRef] [PubMed]
- Stone, R.C.; Pastar, I.; Ojeh, N.; Chen, V.; Liu, S.; Garzon, K.I.; Tomic-Canic, M. Epithelial-mesenchymal transition in tissue repair and fibrosis. Cell Tissue Res. 2016, 365, 495–506. [Google Scholar] [CrossRef]
- Pain, M.; Bermudez, O.; Lacoste, P.; Royer, P.J.; Botturi, K.; Tissot, A.; Brouard, S.; Eickelberg, O.; Magnan, A. Tissue remodelling in chronic bronchial diseases: From the epithelial to mesenchymal phenotype. Eur. Respir. Rev. 2014, 23, 118–130. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.Y.; Guilford, P.; Thiery, J.P. Early events in cell adhesion and polarity during epithelial-mesenchymal transition. J. Cell Sci. 2012, 125, 4417–4422. [Google Scholar] [CrossRef]
- Yilmaz, M.; Christofori, G. EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev. 2009, 28, 15–33. [Google Scholar] [CrossRef] [PubMed]
- Niehrs, C. The complex world of WNT receptor signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 767–779. [Google Scholar] [CrossRef]
- Kourtidis, A.; Ngok, S.P.; Anastasiadis, P.Z. p120 catenin: An essential regulator of cadherin stability, adhesion-induced signaling, and cancer progression. Prog. Mol. Biol. Transl. Sci. 2013, 116, 409–432. [Google Scholar]
- Bax, N.A.; Pijnappels, D.A.; van Oorschot, A.A.; Winter, E.M.; de Vries, A.A.; van Tuyn, J.; Braun, J.; Maas, S.; Schalij, M.J.; Atsma, D.E.; et al. Epithelial-to-mesenchymal transformation alters electrical conductivity of human epicardial cells. J. Cell Mol. Med. 2011, 15, 2675–2683. [Google Scholar] [CrossRef]
- Peinado, H.; Olmeda, D.; Cano, A. Snail, Zeb and bHLH factors in tumour progression: An alliance against the epithelial phenotype? Nat. Rev. Cancer 2007, 7, 415–428. [Google Scholar] [CrossRef]
- De Craene, B.; Berx, G. Regulatory networks defining EMT during cancer initiation and progression. Nat. Rev. Cancer 2013, 13, 97–110. [Google Scholar] [CrossRef]
- Thiery, J.P.; Sleeman, J.P. Complex networks orchestrate epithelial-mesenchymal transitions. Nat. Rev. Mol. Cell Biol. 2006, 7, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, M.; Christofori, G. Mechanisms of motility in metastasizing cells. Mol. Cancer Res. 2010, 8, 629–642. [Google Scholar] [CrossRef]
- McNiven, M.A. Breaking away: Matrix remodeling from the leading edge. Trends Cell Biol. 2013, 23, 16–21. [Google Scholar] [CrossRef]
- Ridley, A.J. Life at the leading edge. Cell 2011, 145, 1012–1022. [Google Scholar] [CrossRef]
- Nelson, W.J. Remodeling epithelial cell organization: Transitions between front-rear and apical-basal polarity. Cold Spring Harb. Perspect. Biol. 2009, 1, a000513. [Google Scholar] [CrossRef] [PubMed]
- Godde, N.J.; Galea, R.C.; Elsum, I.A.; Humbert, P.O. Cell polarity in motion: Redefining mammary tissue organization through EMT and cell polarity transitions. J. Mammary Gland. Biol. Neoplasia 2010, 15, 149–168. [Google Scholar] [CrossRef]
- Mendez, M.G.; Kojima, S.; Goldman, R.D. Vimentin induces changes in cell shape, motility, and adhesion during the epithelial to mesenchymal transition. FASEB J. 2010, 24, 1838–1851. [Google Scholar] [CrossRef]
- Yang, X.; Pursell, B.; Lu, S.; Chang, T.K.; Mercurio, A.M. Regulation of beta 4-integrin expression by epigenetic modifications in the mammary gland and during the epithelial-to-mesenchymal transition. J. Cell Sci. 2009, 122, 2473–2480. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Kugler, M.C.; Wei, Y.; Kim, K.K.; Li, X.; Brumwell, A.N.; Chapman, H.A. Integrin alpha3beta1-dependent beta-catenin phosphorylation links epithelial Smad signaling to cell contacts. J. Cell Biol. 2009, 184, 309–322. [Google Scholar] [CrossRef]
- Maschler, S.; Wirl, G.; Spring, H.; Bredow, D.V.; Sordat, I.; Beug, H.; Reichmann, E. Tumor cell invasiveness correlates with changes in integrin expression and localization. Oncogene 2005, 24, 2032–2041. [Google Scholar] [CrossRef]
- Mise, N.; Savai, R.; Yu, H.; Schwarz, J.; Kaminski, N.; Eickelberg, O. Zyxin is a transforming growth factor-beta (TGF-beta)/Smad3 target gene that regulates lung cancer cell motility via integrin alpha5beta1. J. Biol. Chem. 2012, 287, 31393–31405. [Google Scholar] [CrossRef] [PubMed]
- Koenig, A.; Mueller, C.; Hasel, C.; Adler, G.; Menke, A. Collagen type I induces disruption of E-cadherin-mediated cell-cell contacts and promotes proliferation of pancreatic carcinoma cells. Cancer Res. 2006, 66, 4662–4671. [Google Scholar] [CrossRef]
- Herard, A.L.; Pierrot, D.; Hinnrasky, J.; Kaplan, H.; Sheppard, D.; Puchelle, E.; Zahm, J.M. Fibronectin and its alpha 5 beta 1-integrin receptor are involved in the wound-repair process of airway epithelium. Am. J. Physiol. 1996, 271, L726–L733. [Google Scholar] [CrossRef]
- Nistico, P.; Bissell, M.J.; Radisky, D.C. Epithelial-mesenchymal transition: General principles and pathological relevance with special emphasis on the role of matrix metalloproteinases. Cold Spring Harb. Perspect. Biol. 2012, 4, a011908. [Google Scholar] [CrossRef]
- Munshi, H.G.; Stack, M.S. Reciprocal interactions between adhesion receptor signaling and MMP regulation. Cancer Metastasis Rev. 2006, 25, 45–56. [Google Scholar] [CrossRef]
- Shah, P.P.; Fong, M.Y.; Kakar, S.S. PTTG induces EMT through integrin alphaVbeta3-focal adhesion kinase signaling in lung cancer cells. Oncogene 2012, 31, 3124–3135. [Google Scholar] [CrossRef]
- Radisky, D.C.; Levy, D.D.; Littlepage, L.E.; Liu, H.; Nelson, C.M.; Fata, J.E.; Leake, D.; Godden, E.L.; Albertson, D.G.; Nieto, M.A.; et al. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature 2005, 436, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, D. Integrin-mediated activation of latent transforming growth factor beta. Cancer Metastasis Rev. 2005, 24, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.E.; Weinberg, S.H.; Lemmon, C.A. Mechanochemical Signaling of the Extracellular Matrix in Epithelial-Mesenchymal Transition. Front. Cell Dev. Biol. 2019, 7, 135. [Google Scholar] [CrossRef] [PubMed]
- Aiello, N.M.; Brabletz, T.; Kang, Y.; Nieto, M.A.; Weinberg, R.A.; Stanger, B.Z. Upholding a role for EMT in pancreatic cancer metastasis. Nature 2017, 547, E7–E8. [Google Scholar] [CrossRef]
- Valles, A.M.; Boyer, B.; Badet, J.; Tucker, G.C.; Barritault, D.; Thiery, J.P. Acidic fibroblast growth factor is a modulator of epithelial plasticity in a rat bladder carcinoma cell line. Proc. Natl. Acad. Sci. USA 1990, 87, 1124–1128. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, P.J.; Ebner, R.; Lopez, A.R.; Derynck, R. TGF-beta induced transdifferentiation of mammary epithelial cells to mesenchymal cells: Involvement of type I receptors. J. Cell Biol. 1994, 127, 2021–2036. [Google Scholar] [CrossRef]
- Pardo-Saganta, A.; Law, B.M.; Tata, P.R.; Villoria, J.; Saez, B.; Mou, H.; Zhao, R.; Rajagopal, J. Injury induces direct lineage segregation of functionally distinct airway basal stem/progenitor cell subpopulations. Cell Stem Cell 2015, 16, 184–197. [Google Scholar] [CrossRef]
- Watson, J.K.; Rulands, S.; Wilkinson, A.C.; Wuidart, A.; Ousset, M.; Van Keymeulen, A.; Gottgens, B.; Blanpain, C.; Simons, B.D.; Rawlins, E.L. Clonal Dynamics Reveal Two Distinct Populations of Basal Cells in Slow-Turnover Airway Epithelium. Cell Rep. 2015, 12, 90–101. [Google Scholar] [CrossRef]
- Barbry, P.; Cavard, A.; Chanson, M.; Jaffe, A.B.; Plasschaert, L.W. Regeneration of airway epithelial cells to study rare cell states in cystic fibrosis. J. Cyst. Fibros. 2020, 19 (Suppl 1), S42–S46. [Google Scholar] [CrossRef]
- Zaragosi, L.E.; Deprez, M.; Barbry, P. Using single-cell RNA sequencing to unravel cell lineage relationships in the respiratory tract. Biochem. Soc. Trans. 2020, 48, 327–336. [Google Scholar] [CrossRef]
- Ingram, J.L.; Bonner, J.C. EGF and PDGF receptor tyrosine kinases as therapeutic targets for chronic lung diseases. Curr. Mol. Med. 2006, 6, 409–421. [Google Scholar] [CrossRef]
- Knight, D. Epithelium-fibroblast interactions in response to airway inflammation. Immunol. Cell Biol. 2001, 79, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Gee, J.M.; Knowlden, J.M. ADAM metalloproteases and EGFR signalling. Breast Cancer Res. 2003, 5, 223–224. [Google Scholar] [CrossRef] [PubMed]
- Branchett, W.J.; Lloyd, C.M. Regulatory cytokine function in the respiratory tract. Mucosal Immunol. 2019, 12, 589–600. [Google Scholar] [CrossRef]
- Bonner, J.C. Mesenchymal cell survival in airway and interstitial pulmonary fibrosis. Fibrogenesis Tissue Repair 2010, 3, 15. [Google Scholar] [CrossRef] [PubMed]
- Puchelle, E.; Zahm, J.M.; Tournier, J.M.; Coraux, C. Airway epithelial repair, regeneration, and remodeling after injury in chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc. 2006, 3, 726–733. [Google Scholar] [CrossRef]
- Coraux, C.; Martinella-Catusse, C.; Nawrocki-Raby, B.; Hajj, R.; Burlet, H.; Escotte, S.; Laplace, V.; Birembaut, P.; Puchelle, E. Differential expression of matrix metalloproteinases and interleukin-8 during regeneration of human airway epithelium in vivo. J. Pathol. 2005, 206, 160–169. [Google Scholar] [CrossRef]
- Lechapt-Zalcman, E.; Pruliere-Escabasse, V.; Advenier, D.; Galiacy, S.; Charriere-Bertrand, C.; Coste, A.; Harf, A.; d’Ortho, M.P.; Escudier, E. Transforming growth factor-beta1 increases airway wound repair via MMP-2 upregulation: A new pathway for epithelial wound repair? Am. J. Physiol. Cell Mol. Physiol. 2006, 290, L1277–L1282. [Google Scholar] [CrossRef]
- Spurzem, J.R.; Gupta, J.; Veys, T.; Kneifl, K.R.; Rennard, S.I.; Wyatt, T.A. Activation of protein kinase A accelerates bovine bronchial epithelial cell migration. Am. J. Physiol. Cell Mol. Physiol. 2002, 282, L1108–L1116. [Google Scholar] [CrossRef] [PubMed]
- Waters, C.M.; Savla, U. Keratinocyte growth factor accelerates wound closure in airway epithelium during cyclic mechanical strain. J. Cell Physiol. 1999, 181, 424–432. [Google Scholar] [CrossRef]
- Andersen, D.H. Cystic fibrosis of the pancreas and its relation to celiac disease: A clinical and pathological study. Am. J. Dis. Child. 1938, 56, 344–399. [Google Scholar] [CrossRef]
- Andersen, D.H. Cystic fibrosis of the pancreas, vitamin A deficiency and bronchiectasis. J. Pediatr. 1939, 15, 763–771. [Google Scholar] [CrossRef]
- Blackfan, K.D.; Wolbach, S.B. Vitamin A deficiency in infants. J. Pediatr. 1933, 3, 679–706. [Google Scholar] [CrossRef]
- Gibson, R.L.; Burns, J.L.; Ramsey, B.W. Pathophysiology and management of pulmonary infections in cystic fibrosis. Am. J. Respir. Crit. Care Med. 2003, 168, 918–951. [Google Scholar] [CrossRef]
- Hubeau, C.; Lorenzato, M.; Couetil, J.P.; Hubert, D.; Dusser, D.; Puchelle, E.; Gaillard, D. Quantitative analysis of inflammatory cells infiltrating the cystic fibrosis airway mucosa. Clin. Exp. Immunol. 2001, 124, 69–76. [Google Scholar] [CrossRef]
- Dovey, M.; Wisseman, C.L.; Roggli, V.L.; Roomans, G.M.; Shelburne, J.D.; Spock, A. Ultrastructural morphology of the lung in cystic fibrosis. J. Submicrosc. Cytol. Pathol. 1989, 21, 521–534. [Google Scholar]
- Carson, J.L.; Collier, A.M.; Gambling, T.M.; Knowles, M.R.; Boucher, R.C. Ultrastructure of airway epithelial cell membranes among patients with cystic fibrosis. Hum. Pathol. 1990, 21, 640–647. [Google Scholar] [CrossRef]
- Conese, M.; Castellani, S.; D’Oria, S.; di Gioia, S.; Montemurro, P. Role of Neutrophils in Cystic Fibrosis Lung Disease. In Role of Neutrophils in Disease Pathogenesis; Khajah, M.A., Ed.; IntechOpen Limited: London, UK, 2017; pp. 119–141. [Google Scholar]
- Suzuki, T.; Yamashita, C.; Zemans, R.L.; Briones, N.; Van Linden, A.; Downey, G.P. Leukocyte elastase induces lung epithelial apoptosis via a PAR-1-, NF-kappaB-, and p53-dependent pathway. Am. J. Respir. Cell Mol. Biol. 2009, 41, 742–755. [Google Scholar] [CrossRef] [PubMed]
- Fischer, B.M.; Wong, J.K.; Degan, S.; Kummarapurugu, A.B.; Zheng, S.; Haridass, P.; Voynow, J.A. Increased expression of senescence markers in cystic fibrosis airways. Am. J. Physiol. Cell Mol. Physiol. 2013, 304, L394–L400. [Google Scholar] [CrossRef]
- Voynow, J.A.; Fischer, B.M.; Malarkey, D.E.; Burch, L.H.; Wong, T.; Longphre, M.; Ho, S.B.; Foster, W.M. Neutrophil elastase induces mucus cell metaplasia in mouse lung. Am. J. Physiol. Cell Mol. Physiol. 2004, 287, L1293–L1302. [Google Scholar] [CrossRef]
- Park, J.A.; Sharif, A.S.; Shiomi, T.; Kobzik, L.; Kasahara, D.I.; Tschumperlin, D.J.; Voynow, J.; Drazen, J.M. Human neutrophil elastase-mediated goblet cell metaplasia is attenuated in TACE-deficient mice. Am. J. Physiol. Cell Mol. Physiol. 2013, 304, L701–L707. [Google Scholar] [CrossRef] [PubMed]
- De Rose, V.; Molloy, K.; Gohy, S.; Pilette, C.; Greene, C.M. Airway Epithelium Dysfunction in Cystic Fibrosis and COPD. Mediat. Inflamm. 2018, 2018, 1309746. [Google Scholar] [CrossRef] [PubMed]
- Decraene, A.; Willems-Widyastuti, A.; Kasran, A.; De Boeck, K.; Bullens, D.M.; Dupont, L.J. Elevated expression of both mRNA and protein levels of IL-17A in sputum of stable Cystic Fibrosis patients. Respir. Res. 2010, 11, 177. [Google Scholar] [CrossRef] [PubMed]
- Roussel, L.; Rousseau, S. IL-17 primes airway epithelial cells lacking functional Cystic Fibrosis Transmembrane conductance Regulator (CFTR) to increase NOD1 responses. Biochem. Biophys. Res. Commun. 2010, 391, 505–509. [Google Scholar] [CrossRef]
- Voynow, J.A.; Fischer, B.M.; Roberts, B.C.; Proia, A.D. Basal-like cells constitute the proliferating cell population in cystic fibrosis airways. Am. J. Respir. Crit. Care Med. 2005, 172, 1013–1018. [Google Scholar] [CrossRef] [PubMed]
- Leigh, M.W.; Kylander, J.E.; Yankaskas, J.R.; Boucher, R.C. Cell proliferation in bronchial epithelium and submucosal glands of cystic fibrosis patients. Am. J. Respir. Cell Mol. Biol. 1995, 12, 605–612. [Google Scholar] [CrossRef] [PubMed]
- Piorunek, T.; Marszalek, A.; Biczysko, W.; Gozdzik, J.; Cofta, S.; Seget, M. Correlation between the stage of cystic fibrosis and the level of morphological changes in adult patients. J. Physiol. Pharm. 2008, 59 (Suppl 6), 565–572. [Google Scholar]
- Tiddens, H.A.; Koopman, L.P.; Lambert, R.K.; Elliott, W.M.; Hop, W.C.; van der Mark, T.W.; de Boer, W.J.; de Jongste, J.C. Cartilaginous airway wall dimensions and airway resistance in cystic fibrosis lungs. Eur. Respir. J. 2000, 15, 735–742. [Google Scholar] [CrossRef]
- Durieu, I.; Peyrol, S.; Gindre, D.; Bellon, G.; Durand, D.V.; Pacheco, Y. Subepithelial fibrosis and degradation of the bronchial extracellular matrix in cystic fibrosis. Am. J. Respir. Crit. Care Med. 1998, 158, 580–588. [Google Scholar] [CrossRef] [PubMed]
- Hilliard, T.N.; Regamey, N.; Shute, J.K.; Nicholson, A.G.; Alton, E.W.; Bush, A.; Davies, J.C. Airway remodelling in children with cystic fibrosis. Thorax 2007, 62, 1074–1080. [Google Scholar] [CrossRef]
- Bruce, M.C.; Poncz, L.; Klinger, J.D.; Stern, R.C.; Tomashefski, J.F., Jr.; Dearborn, D.G. Biochemical and pathologic evidence for proteolytic destruction of lung connective tissue in cystic fibrosis. Am. Rev. Respir. Dis. 1985, 132, 529–535. [Google Scholar] [PubMed]
- Regamey, N.; Jeffery, P.K.; Alton, E.W.; Bush, A.; Davies, J.C. Airway remodelling and its relationship to inflammation in cystic fibrosis. Thorax 2011, 66, 624–629. [Google Scholar] [CrossRef] [PubMed]
- Iosifidis, T.; Garratt, L.W.; Coombe, D.R.; Knight, D.A.; Stick, S.M.; Kicic, A. Airway epithelial repair in health and disease: Orchestrator or simply a player? Respirology 2016, 21, 438–448. [Google Scholar] [CrossRef] [PubMed]
- Gaggar, A.; Hector, A.; Bratcher, P.E.; Mall, M.A.; Griese, M.; Hartl, D. The role of matrix metalloproteinases in cystic fibrosis lung disease. Eur. Respir. J. 2011, 38, 721–727. [Google Scholar] [CrossRef] [PubMed]
- Sagel, S.D.; Kapsner, R.K.; Osberg, I. Induced sputum matrix metalloproteinase-9 correlates with lung function and airway inflammation in children with cystic fibrosis. Pediatr. Pulmonol. 2005, 39, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Garratt, L.W.; Sutanto, E.N.; Ling, K.M.; Looi, K.; Iosifidis, T.; Martinovich, K.M.; Shaw, N.C.; Kicic-Starcevich, E.; Knight, D.A.; Ranganathan, S.; et al. Matrix metalloproteinase activation by free neutrophil elastase contributes to bronchiectasis progression in early cystic fibrosis. Eur. Respir. J. 2015, 46, 384–394. [Google Scholar] [CrossRef]
- McKelvey, M.C.; Weldon, S.; McAuley, D.F.; Mall, M.A.; Taggart, C.C. Targeting Proteases in Cystic Fibrosis Lung Disease. Paradigms, Progress, and Potential. Am. J. Respir. Crit. Care Med. 2020, 201, 141–147. [Google Scholar] [CrossRef]
- Harris, W.T.; Muhlebach, M.S.; Oster, R.A.; Knowles, M.R.; Clancy, J.P.; Noah, T.L. Plasma TGF-beta(1) in pediatric cystic fibrosis: Potential biomarker of lung disease and response to therapy. Pediatr. Pulmonol. 2011, 46, 688–695. [Google Scholar] [CrossRef]
- Harris, W.T.; Muhlebach, M.S.; Oster, R.A.; Knowles, M.R.; Noah, T.L. Transforming growth factor-beta(1) in bronchoalveolar lavage fluid from children with cystic fibrosis. Pediatr. Pulmonol. 2009, 44, 1057–1064. [Google Scholar] [CrossRef]
- Kramer, E.L.; Clancy, J.P. TGFbeta as a therapeutic target in cystic fibrosis. Expert Opin. Targets 2018, 22, 177–189. [Google Scholar] [CrossRef]
- Sun, H.; Harris, W.T.; Kortyka, S.; Kotha, K.; Ostmann, A.J.; Rezayat, A.; Sridharan, A.; Sanders, Y.; Naren, A.P.; Clancy, J.P. Tgf-beta downregulation of distinct chloride channels in cystic fibrosis-affected epithelia. PLoS ONE 2014, 9, e106842. [Google Scholar] [CrossRef]
- Snodgrass, S.M.; Cihil, K.M.; Cornuet, P.K.; Myerburg, M.M.; Swiatecka-Urban, A. Tgf-beta1 inhibits Cftr biogenesis and prevents functional rescue of DeltaF508-Cftr in primary differentiated human bronchial epithelial cells. PLoS ONE 2013, 8, e63167. [Google Scholar]
- Howe, K.L.; Wang, A.; Hunter, M.M.; Stanton, B.A.; McKay, D.M. TGFbeta down-regulation of the CFTR: A means to limit epithelial chloride secretion. Exp. Cell Res. 2004, 298, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, Y.; Miyata, M.; Hatsushika, K.; Ohnuma, Y.; Katoh, R.; Ogawa, H.; Okumura, K.; Masuyama, K.; Nakao, A. TGF-beta signaling may play a role in the development of goblet cell hyperplasia in a mouse model of allergic rhinitis. Allergol. Int. 2010, 59, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Le, A.V.; Cho, J.Y.; Miller, M.; McElwain, S.; Golgotiu, K.; Broide, D.H. Inhibition of allergen-induced airway remodeling in Smad 3-deficient mice. J. Immunol. 2007, 178, 7310–7316. [Google Scholar] [CrossRef] [PubMed]
- Harris, W.T.; Kelly, D.R.; Zhou, Y.; Wang, D.; MacEwen, M.; Hagood, J.S.; Clancy, J.P.; Ambalavanan, N.; Sorscher, E.J. Myofibroblast differentiation and enhanced TGF-B signaling in cystic fibrosis lung disease. PLoS ONE 2013, 8, e70196. [Google Scholar] [CrossRef]
- Nicola, T.; Kabir, F.L.; Coric, T.; Wall, S.B.; Zhang, W.; James, M.; MacEwen, M.; Ren, C.; Halloran, B.; Ambalavanan, N.; et al. CFTR dysfunction increases endoglin and TGF-beta signaling in airway epithelia. Physiol. Rep. 2019, 7, e13977. [Google Scholar] [CrossRef]
- Denney, L.; Byrne, A.J.; Shea, T.J.; Buckley, J.S.; Pease, J.E.; Herledan, G.M.; Walker, S.A.; Gregory, L.G.; Lloyd, C.M. Pulmonary Epithelial Cell-Derived Cytokine TGF-beta1 Is a Critical Cofactor for Enhanced Innate Lymphoid Cell Function. Immunity 2015, 43, 945–958. [Google Scholar] [CrossRef]
- Aschner, Y.; Downey, G.P. Transforming Growth Factor-beta: Master Regulator of the Respiratory System in Health and Disease. Am. J. Respir. Cell Mol. Biol. 2016, 54, 647–655. [Google Scholar] [CrossRef]
- Harris, W.T.; Boyd, J.T.; McPhail, G.L.; Brody, A.S.; Szczesniak, R.D.; Korbee, L.L.; Baker, M.L.; Clancy, J.P. Constrictive Bronchiolitis in Cystic Fibrosis Adolescents with Refractory Pulmonary Decline. Ann. Am. Thorac. Soc. 2016, 13, 2174–2183. [Google Scholar] [CrossRef]
- Kreda, S.M.; Davis, C.W.; Rose, M.C. CFTR, mucins, and mucus obstruction in cystic fibrosis. Cold Spring Harb. Perspect. Med. 2012, 2, a009589. [Google Scholar] [CrossRef]
- Argast, G.M.; Campbell, J.S.; Brooling, J.T.; Fausto, N. Epidermal growth factor receptor transactivation mediates tumor necrosis factor-induced hepatocyte replication. J. Biol. Chem. 2004, 279, 34530–34536. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.N.; Woodbury, R.L.; Kathmann, L.E.; Opresko, L.K.; Zangar, R.C.; Wiley, H.S.; Thrall, B.D. Induced autocrine signaling through the epidermal growth factor receptor contributes to the response of mammary epithelial cells to tumor necrosis factor alpha. J. Biol. Chem. 2004, 279, 18488–18496. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Yan, C.; Gieling, R.G.; Kida, Y.; Garner, W.; Li, W.; Han, Y.P. Tumor necrosis factor-alpha induced expression of matrix metalloproteinase-9 through p21-activated kinase-1. BMC Immunol. 2009, 10, 15. [Google Scholar] [CrossRef]
- Maille, E.; Trinh, N.T.; Prive, A.; Bilodeau, C.; Bissonnette, E.; Grandvaux, N.; Brochiero, E. Regulation of normal and cystic fibrosis airway epithelial repair processes by TNF-alpha after injury. Am. J. Physiol. Cell Mol. Physiol. 2011, 301, L945–L955. [Google Scholar] [CrossRef] [PubMed]
- Casalino-Matsuda, S.M.; Monzon, M.E.; Forteza, R.M. Epidermal growth factor receptor activation by epidermal growth factor mediates oxidant-induced goblet cell metaplasia in human airway epithelium. Am. J. Respir. Cell Mol. Biol. 2006, 34, 581–591. [Google Scholar] [CrossRef] [PubMed]
- Takeyama, K.; Jung, B.; Shim, J.J.; Burgel, P.R.; Dao-Pick, T.; Ueki, I.F.; Protin, U.; Kroschel, P.; Nadel, J.A. Activation of epidermal growth factor receptors is responsible for mucin synthesis induced by cigarette smoke. Am. J. Physiol. Cell Mol. Physiol. 2001, 280, L165–L172. [Google Scholar] [CrossRef] [PubMed]
- Atherton, H.C.; Jones, G.; Danahay, H. IL-13-induced changes in the goblet cell density of human bronchial epithelial cell cultures: MAP kinase and phosphatidylinositol 3-kinase regulation. Am. J. Physiol. Cell Mol. Physiol. 2003, 285, L730–L739. [Google Scholar] [CrossRef] [PubMed]
- Stolarczyk, M.; Scholte, B.J. The EGFR-ADAM17 Axis in Chronic Obstructive Pulmonary Disease and Cystic Fibrosis Lung Pathology. Mediat. Inflamm. 2018, 2018, 1067134. [Google Scholar] [CrossRef] [PubMed]
- Val, S.; Belade, E.; George, I.; Boczkowski, J.; Baeza-Squiban, A. Fine PM induce airway MUC5AC expression through the autocrine effect of amphiregulin. Arch. Toxicol. 2012, 86, 1851–1859. [Google Scholar] [CrossRef] [PubMed]
- Chokki, M.; Mitsuhashi, H.; Kamimura, T. Metalloprotease-dependent amphiregulin release mediates tumor necrosis factor-alpha-induced IL-8 secretion in the human airway epithelial cell line NCI-H292. Life Sci. 2006, 78, 3051–3057. [Google Scholar] [CrossRef]
- Zhou, Y.; Lee, J.Y.; Lee, C.M.; Cho, W.K.; Kang, M.J.; Koff, J.L.; Yoon, P.O.; Chae, J.; Park, H.O.; Elias, J.A.; et al. Amphiregulin, an epidermal growth factor receptor ligand, plays an essential role in the pathogenesis of transforming growth factor-beta-induced pulmonary fibrosis. J. Biol. Chem. 2012, 287, 41991–42000. [Google Scholar] [CrossRef] [PubMed]
- Stolarczyk, M.; Veit, G.; Schnur, A.; Veltman, M.; Lukacs, G.L.; Scholte, B.J. Extracellular oxidation in cystic fibrosis airway epithelium causes enhanced EGFR/ADAM17 activity. Am. J. Physiol. Cell Mol. Physiol. 2018, 314, L555–L568. [Google Scholar] [CrossRef]
- Coraux, C.; Roux, J.; Jolly, T.; Birembaut, P. Epithelial cell-extracellular matrix interactions and stem cells in airway epithelial regeneration. Proc. Am. Thorac. Soc. 2008, 5, 689–694. [Google Scholar] [CrossRef] [PubMed]
- Zahm, J.M.; Kaplan, H.; Herard, A.L.; Doriot, F.; Pierrot, D.; Somelette, P.; Puchelle, E. Cell migration and proliferation during the in vitro wound repair of the respiratory epithelium. Cell Motil. Cytoskelet. 1997, 37, 33–43. [Google Scholar] [CrossRef]
- Herard, A.L.; Zahm, J.M.; Pierrot, D.; Hinnrasky, J.; Fuchey, C.; Puchelle, E. Epithelial barrier integrity during in vitro wound repair of the airway epithelium. Am. J. Respir. Cell Mol. Biol. 1996, 15, 624–632. [Google Scholar] [CrossRef]
- McDowell, E.M.; Becci, P.J.; Schurch, W.; Trump, B.F. The respiratory epithelium. VII. Epidermoid metaplasia of hamster tracheal epithelium during regeneration following mechanical injury. J. Natl. Cancer Instig. 1979, 62, 995–1008. [Google Scholar]
- Sacco, O.; Silvestri, M.; Sabatini, F.; Sale, R.; Defilippi, A.C.; Rossi, G.A. Epithelial cells and fibroblasts: Structural repair and remodelling in the airways. Paediatr. Respir. Rev. 2004, 5 (Suppl A), S35–S40. [Google Scholar] [CrossRef]
- White, S.R.; Dorscheid, D.R.; Rabe, K.F.; Wojcik, K.R.; Hamann, K.J. Role of very late adhesion integrins in mediating repair of human airway epithelial cell monolayers after mechanical injury. Am. J. Respir. Cell Mol. Biol. 1999, 20, 787–796. [Google Scholar] [CrossRef]
- Pilewski, J.M.; Latoche, J.D.; Arcasoy, S.M.; Albelda, S.M. Expression of integrin cell adhesion receptors during human airway epithelial repair in vivo. Am. J. Physiol. 1997, 273, L256–L263. [Google Scholar] [CrossRef] [PubMed]
- Buisson, A.C.; Zahm, J.M.; Polette, M.; Pierrot, D.; Bellon, G.; Puchelle, E.; Birembaut, P.; Tournier, J.M. Gelatinase B is involved in the in vitro wound repair of human respiratory epithelium. J. Cell Physiol. 1996, 166, 413–426. [Google Scholar] [CrossRef]
- Legrand, C.; Gilles, C.; Zahm, J.M.; Polette, M.; Buisson, A.C.; Kaplan, H.; Birembaut, P.; Tournier, J.M. Airway epithelial cell migration dynamics. MMP-9 role in cell-extracellular matrix remodeling. J. Cell Biol. 1999, 146, 517–529. [Google Scholar] [CrossRef]
- Buisson, A.C.; Gilles, C.; Polette, M.; Zahm, J.M.; Birembaut, P.; Tournier, J.M. Wound repair-induced expression of a stromelysins is associated with the acquisition of a mesenchymal phenotype in human respiratory epithelial cells. Lab. Investig. 1996, 74, 658–669. [Google Scholar]
- Dunsmore, S.E.; Saarialho-Kere, U.K.; Roby, J.D.; Wilson, C.L.; Matrisian, L.M.; Welgus, H.G.; Parks, W.C. Matrilysin expression and function in airway epithelium. J. Clin. Investig. 1998, 102, 1321–1331. [Google Scholar] [CrossRef] [PubMed]
- Romberger, D.J.; Beckmann, J.D.; Claassen, L.; Ertl, R.F.; Rennard, S.I. Modulation of fibronectin production of bovine bronchial epithelial cells by transforming growth factor-beta. Am. J. Respir. Cell Mol. Biol. 1992, 7, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Spurzem, J.R.; Sacco, O.; Rickard, K.A.; Rennard, S.I. Transforming growth factor-beta increases adhesion but not migration of bovine bronchial epithelial cells to matrix proteins. J. Lab. Clin. Med. 1993, 122, 92–102. [Google Scholar]
- Neurohr, C.; Nishimura, S.L.; Sheppard, D. Activation of transforming growth factor-beta by the integrin alphavbeta8 delays epithelial wound closure. Am. J. Respir. Cell Mol. Biol. 2006, 35, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Puchelle, E.; Peault, B. Human airway xenograft models of epithelial cell regeneration. Respir. Res. 2000, 1, 125–128. [Google Scholar] [CrossRef]
- Trinh, N.T.; Prive, A.; Maille, E.; Noel, J.; Brochiero, E. EGF and K+ channel activity control normal and cystic fibrosis bronchial epithelia repair. Am. J. Physiol. Cell Mol. Physiol. 2008, 295, L866–L880. [Google Scholar] [CrossRef] [PubMed]
- Schiller, K.R.; Maniak, P.J.; O’Grady, S.M. Cystic fibrosis transmembrane conductance regulator is involved in airway epithelial wound repair. Am. J. Physiol. Cell Physiol. 2010, 299, C912–C921. [Google Scholar] [CrossRef]
- Trinh, N.T.; Bardou, O.; Prive, A.; Maille, E.; Adam, D.; Lingee, S.; Ferraro, P.; Desrosiers, M.Y.; Coraux, C.; Brochiero, E. Improvement of defective cystic fibrosis airway epithelial wound repair after CFTR rescue. Eur. Respir. J. 2012, 40, 1390–1400. [Google Scholar] [CrossRef]
- Hussain, R.; Umer, H.M.; Björkqvist, M.; Roomans, G.M. ENaC, iNOS, mucins expression and wound healing in cystic fibrosis airway epithelial and submucosal cells. Cell Biol. Int. Rep. 2014, 21, 25–38. [Google Scholar]
- Itokazu, Y.; Pagano, R.E.; Schroeder, A.S.; O’Grady, S.M.; Limper, A.H.; Marks, D.L. Reduced GM1 ganglioside in CFTR-deficient human airway cells results in decreased beta1-integrin signaling and delayed wound repair. Am. J. Physiol. Cell Physiol. 2014, 306, C819–C830. [Google Scholar] [CrossRef]
- Castellani, S.; Di Gioia, S.; di Toma, L.; Conese, M. Human Cellular Models for the Investigation of Lung Inflammation and Mucus Production in Cystic Fibrosis. Anal. Cell Pathol. 2018, 2018, 3839803. [Google Scholar] [CrossRef] [PubMed]
- Wiszniewski, L.; Jornot, L.; Dudez, T.; Pagano, A.; Rochat, T.; Lacroix, J.S.; Suter, S.; Chanson, M. Long-term cultures of polarized airway epithelial cells from patients with cystic fibrosis. Am. J. Respir. Cell Mol. Biol. 2006, 34, 39–48. [Google Scholar] [CrossRef]
- Fulcher, M.L.; Gabriel, S.; Burns, K.A.; Yankaskas, J.R.; Randell, S.H. Well-differentiated human airway epithelial cell cultures. Methods Mol. Med. 2005, 107, 183–206. [Google Scholar] [PubMed]
- Dupuit, F.; Gaillard, D.; Hinnransky, J.; Mongodin, E.; De Bentzmann, S.; Copreni, E.; Puchelle, E. Differentiation and functional human airway epithelium regeneration in tracheal xenografts. Am. J. Physiol. 2000, 278, L165–L176. [Google Scholar]
- Escotte, S.; Catusse, C.; Coraux, C.; Puchelle, E. Reconstitution of human airway tissue in the humanized xenograft model. J. Cyst. Fibros. 2004, 3 (Suppl 2), 63–65. [Google Scholar] [CrossRef]
- Castillon, N.; Avril-Delplanque, A.; Coraux, C.; Delenda, C.; Peault, B.; Danos, O.; Puchelle, E. Regeneration of a well-differentiated human airway surface epithelium by spheroid and lentivirus vector-transduced airway cells. J. Gene Med. 2004, 6, 846–856. [Google Scholar] [CrossRef] [PubMed]
- Zepeda, M.L.; Chinoy, M.R.; Wilson, J.M. Characterization of stem cells in human airway capable of reconstituting a fully differentiated bronchial epithelium. Somat Cell Mol. Genet. 1995, 21, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, J.F.; Schlossberg, H.; Yankaskas, J.; Dudus, L. Progenitor cells of the adult human airway involved in submucosal gland development. Development 1995, 121, 2031–2046. [Google Scholar]
- Coraux, C.; Hajj, R.; Lesimple, P.; Puchelle, E. In vivo models of human airway epithelium repair and regeneration. Eur. Resp. Rev. 2005, 14, 131–136. [Google Scholar] [CrossRef]
- Péault, B.; Tirouvanziam, R.; Sombardier, M.-N.; Chen, S.; Perricaudet, M.; Gaillard, D. Gene transfer to human fetal pulmonary tissue developed in immunodeficient SCID mice. Hum. Gene Ther. 1994, 5, 1131–1137. [Google Scholar] [CrossRef]
- Delplanque, A.; Coraux, C.; Tirouvanziam, R.; Khazaal, I.; Puchelle, E.; Ambros, P.; Gaillard, G.; Péault, B. Epithelial stem cell-mediated development of the human respiratory mucosa in SCID mice. J. Cell Sci. 2000, 113, 767–778. [Google Scholar]
- Avril-Delplanque, A.; Casal, I.; Castillon, N.; Hinnrasky, J.; Puchelle, E.; Peault, B. Aquaporin-3 expression in human fetal airway epithelial progenitor cells. Stem Cells 2005, 23, 992–1001. [Google Scholar] [CrossRef]
- Trinh, N.T.; Prive, A.; Kheir, L.; Bourret, J.C.; Hijazi, T.; Amraei, M.G.; Noel, J.; Brochiero, E. Involvement of KATP and KvLQT1 K+ channels in EGF-stimulated alveolar epithelial cell repair processes. Am. J. Physiol. Cell Mol. Physiol. 2007, 293, L870–L882. [Google Scholar] [CrossRef]
- Huang, W.; Jin, A.; Zhang, J.; Wang, C.; Tsang, L.L.; Cai, Z.; Zhou, X.; Chen, H.; Chan, H.C. Upregulation of CFTR in patients with endometriosis and its involvement in NFkappaB-uPAR dependent cell migration. Oncotarget 2017, 8, 66951–66959. [Google Scholar] [CrossRef]
- Chen, J.; Chen, Y.; Chen, Y.; Yang, Z.; You, B.; Ruan, Y.C.; Peng, Y. Epidermal CFTR Suppresses MAPK/NF-kappaB to Promote Cutaneous Wound Healing. Cell Physiol. Biochem. 2016, 39, 2262–2274. [Google Scholar] [CrossRef]
- Dong, J.; Jiang, X.; Zhang, X.; Liu, K.S.; Zhang, J.; Chen, J.; Yu, M.K.; Tsang, L.L.; Chung, Y.W.; Wang, Y.; et al. Dynamically Regulated CFTR Expression and Its Functional Role in Cutaneous Wound Healing. J. Cell Physiol. 2015, 230, 2049–2058. [Google Scholar] [CrossRef]
- Chiu, W.T.; Tran, T.V.; Pan, S.C.; Huang, H.K.; Chen, Y.C.; Wong, T.W. Cystic Fibrosis Transmembrane Conductance Regulator: A Possible New Target for Photodynamic Therapy Enhances Wound Healing. Adv. Wound Care 2019, 8, 476–486. [Google Scholar] [CrossRef] [PubMed]
- Adam, D.; Bilodeau, C.; Sognigbe, L.; Maille, E.; Ruffin, M.; Brochiero, E. CFTR rescue with VX-809 and VX-770 favors the repair of primary airway epithelial cell cultures from patients with class II mutations in the presence of Pseudomonas aeruginosa exoproducts. J. Cyst. Fibros. 2018, 17, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Kelly, M.; Trudel, S.; Brouillard, F.; Bouillaud, F.; Colas, J.; Nguyen-Khoa, T.; Ollero, M.; Edelman, A.; Fritsch, J. Cystic fibrosis transmembrane regulator inhibitors CFTR(inh)-172 and GlyH-101 target mitochondrial functions, independently of chloride channel inhibition. J. Pharm. Exp. Ther. 2010, 333, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Rejman, J.; Di Gioia, S.; Bragonzi, A.; Conese, M. Pseudomonas aeruginosa infection destroys the barrier function of lung epithelium and enhances polyplex-mediated transfection. Hum. Gene Ther. 2007, 18, 642–652. [Google Scholar] [CrossRef] [PubMed]
- Nomura, K.; Obata, K.; Keira, T.; Miyata, R.; Hirakawa, S.; Takano, K.; Kohno, T.; Sawada, N.; Himi, T.; Kojima, T. Pseudomonas aeruginosa elastase causes transient disruption of tight junctions and downregulation of PAR-2 in human nasal epithelial cells. Respir. Res. 2014, 15, 21. [Google Scholar] [CrossRef]
- Geiser, T.K.; Kazmierczak, B.I.; Garrity-Ryan, L.K.; Matthay, M.A.; Engel, J.N. Pseudomonas aeruginosa ExoT inhibits in vitro lung epithelial wound repair. Cell Microbiol. 2001, 3, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Maille, E.; Ruffin, M.; Adam, D.; Messaoud, H.; Lafayette, S.L.; McKay, G.; Nguyen, D.; Brochiero, E. Quorum Sensing Down-Regulation Counteracts the Negative Impact of Pseudomonas aeruginosa on CFTR Channel Expression, Function and Rescue in Human Airway Epithelial Cells. Front. Cell Infect. Microbiol. 2017, 7, 470. [Google Scholar] [CrossRef] [PubMed]
- Sagel, S.D.; Wagner, B.D.; Anthony, M.M.; Emmett, P.; Zemanick, E.T. Sputum biomarkers of inflammation and lung function decline in children with cystic fibrosis. Am. J. Respir. Crit. Care Med. 2012, 186, 857–865. [Google Scholar] [CrossRef]
- Sly, P.D.; Gangell, C.L.; Chen, L.; Ware, R.S.; Ranganathan, S.; Mott, L.S.; Murray, C.P.; Stick, S.M.; Investigators, A.C. Risk factors for bronchiectasis in children with cystic fibrosis. N. Engl. J. Med. 2013, 368, 1963–1970. [Google Scholar] [CrossRef]
- Garratt, L.W.; Sutanto, E.N.; Ling, K.M.; Looi, K.; Iosifidis, T.; Martinovich, K.M.; Shaw, N.C.; Buckley, A.G.; Kicic-Starcevich, E.; Lannigan, F.J.; et al. Alpha-1 Antitrypsin Mitigates the Inhibition of Airway Epithelial Cell Repair by Neutrophil Elastase. Am. J. Respir. Cell Mol. Biol. 2016, 54, 341–349. [Google Scholar] [CrossRef]
- Zoso, A.; Sofoluwe, A.; Bacchetta, M.; Chanson, M. Transcriptomic profile of cystic fibrosis airway epithelial cells undergoing repair. Sci. Data 2019, 6, 240. [Google Scholar] [CrossRef]
- Crespin, S.; Bacchetta, M.; Huang, S.; Dudez, T.; Wiszniewski, L.; Chanson, M. Approaches to study differentiation and repair of human airway epithelial cells. Methods Mol. Biol. 2011, 742, 173–185. [Google Scholar] [PubMed]
- Quaresma, M.C.; Pankonien, I.; Clarke, L.A.; Sousa, L.S.; Silva, I.A.L.; Railean, V.; Dousova, T.; Fuxe, J.; Amaral, M.D. Mutant CFTR Drives TWIST1 mediated epithelial-mesenchymal transition. Cell Death Dis. 2020, 11, 920. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Whitney, E.M.; Gao, S.Y.; Yang, V.W. Transcriptional profiling of Kruppel-like factor 4 reveals a function in cell cycle regulation and epithelial differentiation. J. Mol. Biol. 2003, 326, 665–677. [Google Scholar] [CrossRef]
- McConnell, B.B.; Yang, V.W. Mammalian Kruppel-like factors in health and diseases. Physiol. Rev. 2010, 90, 1337–1381. [Google Scholar] [CrossRef] [PubMed]
- Crespin, S.; Bacchetta, M.; Bou Saab, J.; Tantilipikorn, P.; Bellec, J.; Dudez, T.; Nguyen, T.H.; Kwak, B.R.; Lacroix, J.S.; Huang, S.; et al. Cx26 regulates proliferation of repairing basal airway epithelial cells. Int. J. Biochem. Cell Biol. 2014, 52, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Kardami, E.; Dang, X.; Iacobas, D.A.; Nickel, B.E.; Jeyaraman, M.; Srisakuldee, W.; Makazan, J.; Tanguy, S.; Spray, D.C. The role of connexins in controlling cell growth and gene expression. Prog. Biophys. Mol. Biol. 2007, 94, 245–264. [Google Scholar] [CrossRef]
- Sousa, L.; Pankonien, I.; Simoes, F.B.; Chanson, M.; Amaral, M.D. Impact of KLF4 on Cell Proliferation and Epithelial Differentiation in the Context of Cystic Fibrosis. Int. J. Mol. Sci. 2020, 21, 6717. [Google Scholar] [CrossRef]
- Sousa, L.; Pankonien, I.; Clarke, L.A.; Silva, I.; Kunzelmann, K.; Amaral, M.D. KLF4 Acts as a wt-CFTR Suppressor through an AKT-Mediated Pathway. Cells 2020, 9, 1607. [Google Scholar] [CrossRef]
- Kirk, K.L. CFTR channels and wound healing. Focus on “Cystic fibrosis transmembrane conductance regulator is involved in airway epithelial wound repair”. Am. J. Physiol. Cell Physiol. 2010, 299, C888–C890. [Google Scholar] [CrossRef]
- Schwab, A. Ion channels and transporters on the move. News Physiol. Sci. 2001, 16, 29–33. [Google Scholar] [CrossRef][Green Version]
- Schwab, A.; Nechyporuk-Zloy, V.; Fabian, A.; Stock, C. Cells move when ions and water flow. Pflug. Arch. 2007, 453, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.H.; Reid, B.; Fontaine, J.H.; Miller, L.A.; Hyde, D.M.; Mogilner, A.; Zhao, M. Airway epithelial wounds in rhesus monkey generate ionic currents that guide cell migration to promote healing. J. Appl. Physiol. 2011, 111, 1031–1041. [Google Scholar] [CrossRef][Green Version]
- Poulsen, J.H.; Fischer, H.; Illek, B.; Machen, T.E. Bicarbonate conductance and pH regulatory capability of cystic fibrosis transmembrane conductance regulator. Proc. Natl. Acad. Sci. USA 1994, 91, 5340–5344. [Google Scholar] [CrossRef] [PubMed]
- Reddy, M.M.; Quinton, P.M. Control of dynamic CFTR selectivity by glutamate and ATP in epithelial cells. Nature 2003, 423, 756–760. [Google Scholar] [CrossRef]
- Krahling, H.; Mally, S.; Eble, J.A.; Noel, J.; Schwab, A.; Stock, C. The glycocalyx maintains a cell surface pH nanoenvironment crucial for integrin-mediated migration of human melanoma cells. Pflug. Arch. 2009, 458, 1069–1083. [Google Scholar] [CrossRef] [PubMed]
- Stock, C.; Cardone, R.A.; Busco, G.; Krahling, H.; Schwab, A.; Reshkin, S.J. Protons extruded by NHE1: Digestive or glue? Eur. J. Cell Biol. 2008, 87, 591–599. [Google Scholar] [CrossRef]
- Todeschini, A.R.; Hakomori, S.I. Functional role of glycosphingolipids and gangliosides in control of cell adhesion, motility, and growth, through glycosynaptic microdomains. Biochim. Biophys. Acta 2008, 1780, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Grassme, H.; Henry, B.; Ziobro, R.; Becker, K.A.; Riethmuller, J.; Gardner, A.; Seitz, A.P.; Steinmann, J.; Lang, S.; Ward, C.; et al. beta1-Integrin Accumulates in Cystic Fibrosis Luminal Airway Epithelial Membranes and Decreases Sphingosine, Promoting Bacterial Infections. Cell Host Microbe 2017, 21, 707–718.e8. [Google Scholar] [CrossRef]
- Badaoui, M.; Zoso, A.; Idris, T.; Bacchetta, M.; Simonin, J.; Lemeille, S.; Wehrle-Haller, B.; Chanson, M. Vav3 Mediates Pseudomonas aeruginosa Adhesion to the Cystic Fibrosis Airway Epithelium. Cell Rep. 2020, 32, 107842. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Sohal, S.S.; Reid, D.; Soltani, A.; Ward, C.; Weston, S.; Muller, H.K.; Wood-Baker, R.; Walters, E.H. Evaluation of epithelial mesenchymal transition in patients with chronic obstructive pulmonary disease. Respir. Res. 2011, 12, 130. [Google Scholar] [CrossRef]
- Jonsdottir, H.R.; Arason, A.J.; Palsson, R.; Franzdottir, S.R.; Gudbjartsson, T.; Isaksson, H.J.; Gudmundsson, G.; Gudjonsson, T.; Magnusson, M.K. Basal cells of the human airways acquire mesenchymal traits in idiopathic pulmonary fibrosis and in culture. Lab. Investig. 2015, 95, 1418–1428. [Google Scholar] [CrossRef] [PubMed]
- Jolly, M.K.; Ward, C.; Eapen, M.S.; Myers, S.; Hallgren, O.; Levine, H.; Sohal, S.S. Epithelial-mesenchymal transition, a spectrum of states: Role in lung development, homeostasis, and disease. Dev. Dyn. 2018, 247, 346–358. [Google Scholar] [CrossRef] [PubMed]
- Clarke, L.A.; Botelho, H.M.; Sousa, L.; Falcao, A.O.; Amaral, M.D. Transcriptome meta-analysis reveals common differential and global gene expression profiles in cystic fibrosis and other respiratory disorders and identifies CFTR regulators. Genomics 2015, 106, 268–277. [Google Scholar] [CrossRef]
- Hackett, T.L.; Warner, S.M.; Stefanowicz, D.; Shaheen, F.; Pechkovsky, D.V.; Murray, L.A.; Argentieri, R.; Kicic, A.; Stick, S.M.; Bai, T.R.; et al. Induction of epithelial-mesenchymal transition in primary airway epithelial cells from patients with asthma by transforming growth factor-beta1. Am. J. Respir. Crit. Care Med. 2009, 180, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- Grande, M.T.; Sanchez-Laorden, B.; Lopez-Blau, C.; De Frutos, C.A.; Boutet, A.; Arevalo, M.; Rowe, R.G.; Weiss, S.J.; Lopez-Novoa, J.M.; Nieto, M.A. Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat. Med. 2015, 21, 989–997. [Google Scholar] [CrossRef]
- Schaeffer, D.; Somarelli, J.A.; Hanna, G.; Palmer, G.M.; Garcia-Blanco, M.A. Cellular migration and invasion uncoupled: Increased migration is not an inexorable consequence of epithelial-to-mesenchymal transition. Mol. Cell Biol. 2014, 34, 3486–3499. [Google Scholar] [CrossRef][Green Version]
- De Boeck, K.; Davies, J.C. Where are we with transformational therapies for patients with cystic fibrosis? Curr. Opin. Pharm. 2017, 34, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Keown, K.; Brown, R.; Doherty, D.F.; Houston, C.; McKelvey, M.C.; Creane, S.; Linden, D.; McAuley, D.F.; Kidney, J.C.; Weldon, S.; et al. Airway Inflammation and Host Responses in the Era of CFTR Modulators. Int. J. Mol. Sci. 2020, 21, 6379. [Google Scholar] [CrossRef] [PubMed]
- Van Goor, F.; Straley, K.S.; Cao, D.; Gonzalez, J.; Hadida, S.; Hazlewood, A.; Joubran, J.; Knapp, T.; Makings, L.R.; Miller, M.; et al. Rescue of DeltaF508-CFTR trafficking and gating in human cystic fibrosis airway primary cultures by small molecules. Am. J. Physiol. Cell Mol. Physiol. 2006, 290, L1117–L1130. [Google Scholar] [CrossRef] [PubMed]
- Wainwright, C.E.; Elborn, J.S.; Ramsey, B.W.; Marigowda, G.; Huang, X.; Cipolli, M.; Colombo, C.; Davies, J.C.; De Boeck, K.; Flume, P.A.; et al. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N. Engl. J. Med. 2015, 373, 220–231. [Google Scholar] [CrossRef]
- Van Goor, F.; Hadida, S.; Grootenhuis, P.D.; Burton, B.; Stack, J.H.; Straley, K.S.; Decker, C.J.; Miller, M.; McCartney, J.; Olson, E.R.; et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc. Natl. Acad. Sci. USA 2011, 108, 18843–18848. [Google Scholar] [CrossRef] [PubMed]
- Melis, N.; Tauc, M.; Cougnon, M.; Bendahhou, S.; Giuliano, S.; Rubera, I.; Duranton, C. Revisiting CFTR inhibition: A comparative study of CFTRinh -172 and GlyH-101 inhibitors. Br. J. Pharm. 2014, 171, 3716–3727. [Google Scholar] [CrossRef]
- Cruz, F.F.; Rocco, P.R.M. The potential of mesenchymal stem cell therapy for chronic lung disease. Expert Rev. Respir. Med. 2020, 14, 31–39. [Google Scholar] [CrossRef]
- Caretti, A.; Peli, V.; Colombo, M.; Zulueta, A. Lights and Shadows in the Use of Mesenchymal Stem Cells in Lung Inflammation, a Poorly Investigated Topic in Cystic Fibrosis. Cells 2019, 9, 20. [Google Scholar] [CrossRef]
- Conese, M.; Beccia, E.; Castellani, S.; Di Gioia, S.; Colombo, C.; Angiolillo, A.; Carbone, A. The long and winding road: Stem cells for cystic fibrosis. Expert Opin. Biol. Ther. 2018, 18, 281–292. [Google Scholar] [CrossRef]
- Cruz, F.F.; Rocco, P.R.M. Stem-cell extracellular vesicles and lung repair. Stem Cell Investig. 2017, 4, 78. [Google Scholar] [CrossRef] [PubMed]
- Paracchini, V.; Carbone, A.; Colombo, F.; Castellani, S.; Mazzucchelli, S.; Gioia, S.D.; Degiorgio, D.; Seia, M.; Porretti, L.; Colombo, C.; et al. Amniotic mesenchymal stem cells: A new source for hepatocyte-like cells and induction of CFTR expression by coculture with cystic fibrosis airway epithelial cells. J. Biomed. Biotechnol. 2012, 2012, 575471. [Google Scholar] [CrossRef] [PubMed]
- Carbone, A.; Castellani, S.; Favia, M.; Diana, A.; Paracchini, V.; Di Gioia, S.; Seia, M.; Casavola, V.; Colombo, C.; Conese, M. Correction of defective CFTR/ENaC function and tightness of cystic fibrosis airway epithelium by amniotic mesenchymal stromal (stem) cells. J. Cell Mol. Med. 2014, 18, 1631–1643. [Google Scholar] [CrossRef] [PubMed]
- Carbone, A.; Zefferino, R.; Beccia, E.; Casavola, V.; Castellani, S.; Di Gioia, S.; Giannone, V.; Seia, M.; Angiolillo, A.; Colombo, C.; et al. Gap Junctions Are Involved in the Rescue of CFTR-Dependent Chloride Efflux by Amniotic Mesenchymal Stem Cells in Coculture with Cystic Fibrosis CFBE41o- Cells. Stem Cells Int. 2018, 2018, 1203717. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutation Class | Class I | Class II | Class III | Class IV | Class V | Class VI | |
|---|---|---|---|---|---|---|---|
| IA | IB | ||||||
| CFTR defect | No mRNA | No protein | No traffic | Impaired gating | Decreased conductance | Less protein | Less stable |
| Mutation example | Dele2,3(21 kb), 1717-1G→A | G542X, W1282X, 1609delCA | Phe508del, N1303K, M1101K | G551D, S549R, G1349D | R117H, R334W, A455E | 3272-26A→G, 3849+10 kg C→T | c. 120del123, rPhe580del |
| Phenotype severity | More-severe disease | Less-severe disease | |||||
| Cellular Models | Type of Culture | Type of Wound | Effects on Wound Closure | Effects on Proliferation/Migration | Modulation | Reference |
|---|---|---|---|---|---|---|
| Immortalized cell lines: normal (NuLi) and CF (CuFi-1, F508del homozygous) bronchial cells | Submerged on plastic | Mechanical injury of monolayers (pipette tip scratch assay). Wound closure was evaluated by light microscopy. | CuFi-1 monolayers showed a slower wound repair up to 33% as compared with NuLi in the absence or presence of exogenous EGF. | CuFi-1 cells exhibited slower migration (by 25%) than NuLi cells. CuFi-1 cells showed a proliferation rate similar to NuLi cells. | Inhibition of K+ channels decreased EGF-stimulated wound repair in both cell lines. CFTRinh-172 did not affect significantly wound closure in both cell phenotypes. | Trinh et al., 2008 [162] |
| Immortalized cell lines: Calu-3 (normal human lung adenocarcinoma cell line); UNCCF1T (CF human bronchial epithelial cells; F508del homozygous). Primary cell lines: NHBE (normal human bronchial epithelial cells). | Submerged on plastic | Circular lesion produced by lethal electroporation. Wound closure was measured by continuous impedance sensing (CIS) combined with phase-contrast imaging. | UNCCF1T showed delayed wound closure as compared with NHBE cells | UNCCF1T showed slower cell migration (by 1.7-fold) as compared with NHBE cells | CFTRinh-172 delayed wound closure of both Calu-3 and NHBE cells. A CFTR-specific shRNA (shCFTR) delayed wound closure in Calu-3 cells. | Schiller et al., 2010 [163] |
| Immortalized cell lines: NuLi and CuFi-1 cells. Primary cell lines: non-CF and CF human airway epithelial cells (from nasal polyps) | Submerged on plastic | Mechanical injury of monolayers (pipette tip scratch assay). Wound closure was evaluated by light microscopy. | CuFi-1 monolayers showed a slower wound-repair rate as compared with NuLi in the absence or presence of exogenous EGF. CF primary airway monolayers showed a reduced wound-repair rate as compared with non-CF monolayers. | CuFi-1 cells showed a proliferation rate similar to NuLi cells. | TNF-α chronic exposure (24–48 h) enhanced CuFi-1 and NuLi wound-repair rate in the absence and presence of exogenous EGF. TNF-α increased cell migration in wounded NuLi and CuFi-1 monolayers, despite inhibition of cell growth. TNF-α stimulated wound repair in non-CF and CF primary monolayers. | Maille et al., 2011 [138] |
| Immortalized cell lines: NuLi and CuFi-1 cells; IB3 (F508del/W1282X) and S9 (wt-CFTR genetically repaired IB3 cells); CFBE41o- transduced with wt-CFTR (CFBE-wt) or F508del-CFTR (CFBE-F508del). Primary cell lines: non-CF and CF bronchial and nasal epithelial cells. | Submerged on plastic | Mechanical injury of monolayers (pipette tip scratch assay). Wound closure was evaluated by light microscopy | CuFi-1 monolayers showed a slower wound-repair rate as compared with NuLi. The wound closure rate of CF human bronchial cell monolayers was 63% slower than that of non-CF bronchial monolayers. The wound closure in CF human nasal monolayers was delayed when compared with non-CF (53% slower wound-repair rate). S9 and CFBE-wt showed a higher wound-repair rate than IB3 and CFBE-F508del, resepctively. | CuFi-1 cells exhibited slower migration (by 25%) than NuLi cells. CuFi-1 cells showed a proliferation rate similar to NuLi cells. | CFTR silencing by siRNA and the CFTR inhibitor GlyH101 elicited a significant decrease in wound repair in non-CF primary nasal epithelial cell monolayers. CFTR inhibition by GlyH101 reduced significanlty both cell migration and proliferation in non-CF primary nasal epithelial cell monolayers. The CFTR corrector VRT-325 enhanced the wound repair rate of CFBE-F508del and CF primary bronchial epithelial cell monolayers. | Trinh et al., 2012 [164] |
| Immortalized cell lines: 16HBE14o-(wt CFTR) (normal human bronchial epithelial cells; CFBE41o-(F508del homozygous), and its corresponding plasmid-corrected CFBE41o-pCep4, overexpressing wtCFTR) cells; Calu-3 and CFSMEo- (CF submucosal gland epithelial cells, F508del/2QX) cells | Submerged on plastic | Mechanical injury of monolayers (pipette tip scratch assay). Wound closure was evaluated by light microscopy | CFBE cells showed a slower wond closure compared to corrected CFBE cells but not in resepct to 16HBE cells. CFSME showed a significant delay in wound repair time compared to Calu-3 cells | CFBE, corrected CFBE, and 16HBE showed the same proliferation and migration rates in non-wounded conditions. | CFTRinh-172 and forskolin induced a delay in wound repair in 16HBE, CFBE and corrected CFBE cells. | Hussain et al., 2014 [165] |
| Immortalized cell lines: CFTR-silenced (shRNA) and control Calu-3 cells. | Submerged on plastic | Circular lesion produced by lethal electroporation. Wound closure was measured by continuous impedance sensing (CIS) combined with phase-contrast imaging. | CFTR-silenced cells showed a reduced rate of electrode coverage as compared with control cells. | Cell migration was slower in CFTR-silenced cells than in control cells. | Ganglioside GM1 partially restored the wound-repair defect in CFTR-silenced cells. | Itokazu et al., 2014 [166] |
| Primary cell lines: non-CF and CF airway epithelial cells (MucilAir™ and MucilAir™-CF) | ALI cultures | Circular wounds obtained by an airbrush linked to a pressure regulator. Wound closure was evaluated by light microscopy. | CF cultures showed a higher wound repair rate than non-CF cultures at early time pints (12 and 24 h). | In non-CF cultures, the Ki-67-labeling index reached its maximum at 48 h post-wounding and was higher in the front area as compared with the front area. In CF cultures, the maximum of the Ki-67-labeling index was reached at 36 h, with no significant difference between front and back areas. | In non-CF cultures, CX26 mRNA expression paralleled the behavior of Ki-67 labeling index. Also KLF4 showed a transient increase during the early timepoints after injury. In CF cutlures, the increase in Cx26 mRNA expression peaked at 60 h post-wounding, with no significant difference between front and back areas. KLF4 mRNA levels remained unchanged during wound repair. | Crespin et al., 2014 [198] |
| Primary cell lines: non-CF and CF nasal and bronchial epithelial cells. | Submerged on plastic and ALI cultures | Mechanical injury of monolayers (pipette tip scratch assay). Wound closure was evaluated by light microscopy. | P. aeruginosa exoproducts inhibited wound-repair rates in non-CF cells, affected cell trajectories and impaired their directional migration ability toward the opposite side of the wounds. A dose-dependent inhibition of wound-repair rates in CF cells was observed in the presence of increasing concentrations of P. aeruginosa exoproducts. | P. aeruginosa exoproducts decreased the percentage of proliferative primary non-CF cells at 6 h of repair. | Quorum sensing inihibitor HDMF can reduce the exoproducts-induced wound-repair defect of non-CF and CF monolayers and as well as of highly differentiated cell cultures. | Ruffin et al., 2106 [41] |
| Primary cell lines: non-CF and CF bronchial epithelial cells | Submerged on plastic | Mechanical injury of monolayers (WoundMaker device). Wound closure was evaluated by IncuCyte live-cell imaging system. | NE exposure determined a dose-dependent delay/inhibition of wound repair at 30–72 h. | NE exposure caused increased cell detachment of viable cells, reduction in cell viability, apoptosis, and reduction in cell proliferation. | Wound closure by CF cells initially exposed to 100 nM NE and then treated with 1 mM α1AT was significantly increased over 100 nM NE alone. | Garratt et al., 2016 [192] |
| Primary cell lines: non-CF and CF bronchial and nasal epithelial cells. CF cells were obtained from 4 homozygous F508del patients and 4 patients carrying F508del and another class II mutation (N1303 K or I507del). | Submerged (monolayers) and ALI cultures | Monolayers: mechanical injury (pipette tip scratch assay). Wound closure was evaluated by light microscopy. ALI cultures: mechanical wound by a glass Pasteur pipette connected to the vacuum. Wound closure was evaluated by time-lapse microscopy. | CF ALI cultures showed a delay in wound repair as compared with non-CF cultures. | Proliferation and migration were not assessed. | In all patients, the improvement in the wounding repair rate over a 6h-period of monolayers was higher after CFTR rescue with Orkambi® (VX-809 + VX-770), compared to VX-809 alone. Orkambi® treatment sighltly improved the repair rates of CF monolayers in the presence of P. aeruginosa exoproducts. Similar results were obtained with CF differentiated cultures. | Adam et al., 2018 [184] |
| Primary cell lines: non-CF and CF bronchial epithelial cells. | ALI cultures | Circular wounds obtained by an airbrush linked to a pressure regulator. | Significant differences in gene expression of different cell types between CF and non-CF cultures at post-wound, wound closure and post-wound closure were not observed. Comparison of gene expression by RNA sequencing determined that CF cultures had up- and down-regulated genes as compard with non-CF cultures at all the conditions (non-wounded, post-wounding, wound closure, and post-wound closure). | Both cultures showed high proliferation during wound as assessed by the expression of MKI67 gene. | P. aeruginosa flagellin determined up- and down-regulation of genes when CF and non-CF cultures were compared pre-wounding and at wound closure. | Zoso et al., 2019 [193] |
| Immortalized cell lines: CFBE41o- cells stably overexpressing wt- or F508del-CFTR. Primary cell lines: non-CF and CF bronchial cells. epithelial cells | Polarized on filter (CFBE cell lines) or ALI cultures (primary bronchial cells). | Mechanical injury of monolayers (pipette tip scratch assay). Wound closure was evaluated by light microscopy. | wt-CFTR CFBE and fully differentiated bronchial epithelial cells closed the wounds 1.5–2 times faster than corresponding CF cells. | Primary CF bronchial cells (three different CFTR genotypes) exhibiting 3-fold higher cell proliferation rates vs control cells. | The triple drug combo VX-445/VX-661/VX-770 restored the epithelial phenotype in F508del-CFTR CFBE reducing mesenchymal cell markers, but no effect on wound repair rate was assessed. | Quaresma et al., 2020 [195] |
| Immortalized cell lines: CFBE41o- cells stably overexpressing wt- or F508del-CFTR. | Polarized on filter. | Mechanical injury of monolayers (pipette tip scratch assay). Wound closure was evaluated by light microscopy. | wt-CFTR CFBE closed the wounds faster than corresponding CF cells, although significance was not assessed. | CF cells showed higher proliferation than non-CF cells | KLF4 KO had no major impact on cell proliferation in the CF context. KLF4 KO significantly decreased TEER of wt-CFTR cells whilst increasing TEER of F508del-CFTR cells. KLF4 KO in F508del-CFTR cells determined a delay in wound closure, while having no effect on wt cells. | Sousa et al., 2020 [200] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Conese, M.; Di Gioia, S. Pathophysiology of Lung Disease and Wound Repair in Cystic Fibrosis. Pathophysiology 2021, 28, 155-188. https://doi.org/10.3390/pathophysiology28010011
Conese M, Di Gioia S. Pathophysiology of Lung Disease and Wound Repair in Cystic Fibrosis. Pathophysiology. 2021; 28(1):155-188. https://doi.org/10.3390/pathophysiology28010011
Chicago/Turabian StyleConese, Massimo, and Sante Di Gioia. 2021. "Pathophysiology of Lung Disease and Wound Repair in Cystic Fibrosis" Pathophysiology 28, no. 1: 155-188. https://doi.org/10.3390/pathophysiology28010011
APA StyleConese, M., & Di Gioia, S. (2021). Pathophysiology of Lung Disease and Wound Repair in Cystic Fibrosis. Pathophysiology, 28(1), 155-188. https://doi.org/10.3390/pathophysiology28010011