
The Angiotensin-Converting Enzyme Inhibitor Lisinopril Mitigates Memory and Motor Deficits in a Drosophila Model of Alzheimer’s Disease

 and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Drosophila Genetics
2.2. Measurement of Lisinopril Concentration
2.3. Learning and Memory Assay
2.4. Climbing Assays
2.5. Measurement of Tryptophan Metabolites
2.6. Measurement of Thoracic H2O2 Levels
2.7. Statistical Analysis
3. Results
3.1. Lisinopril Improves Learning and Memory Impairment in AD Flies
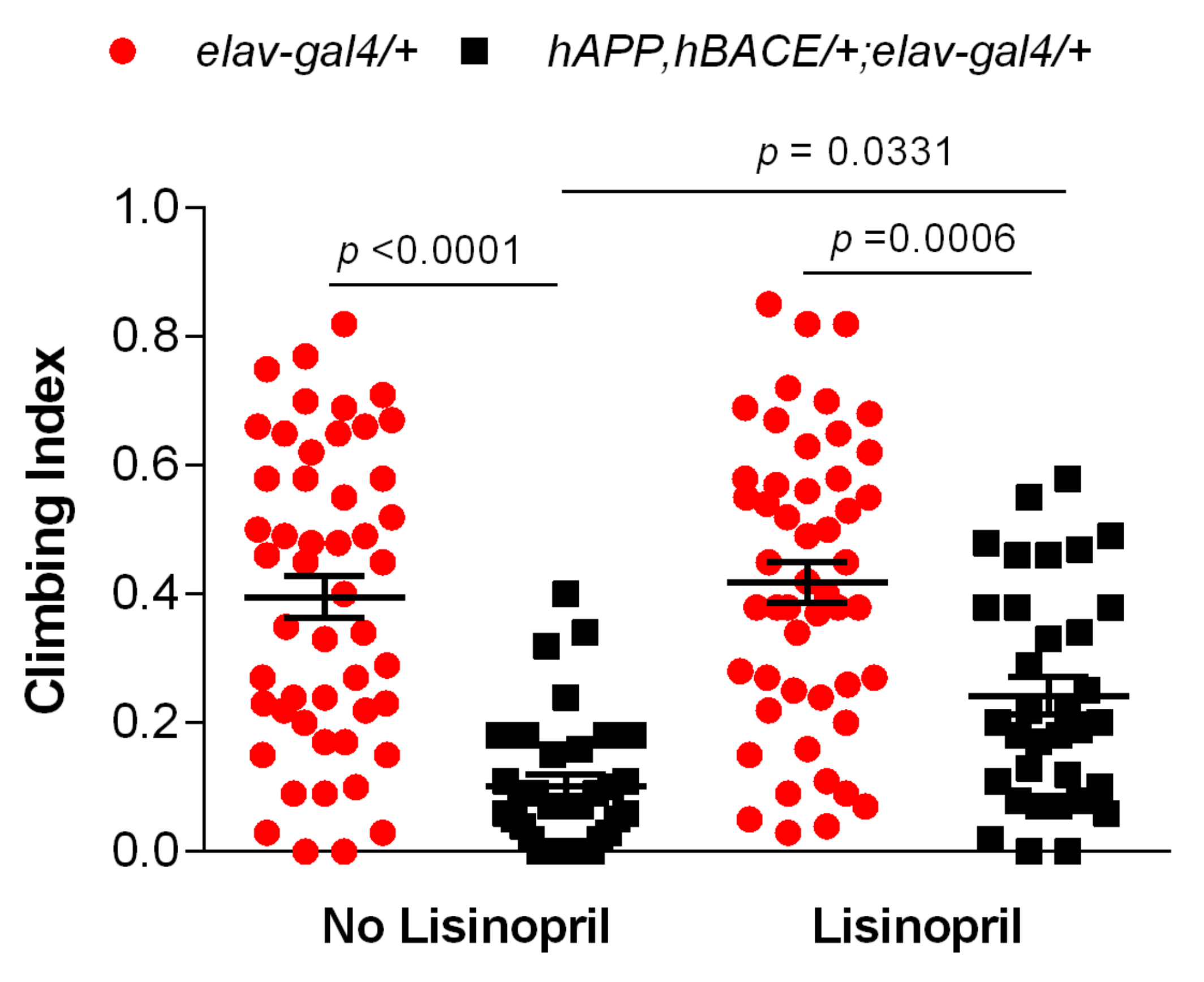
3.2. Lisinopril Improves Climbing Ability in AD Flies
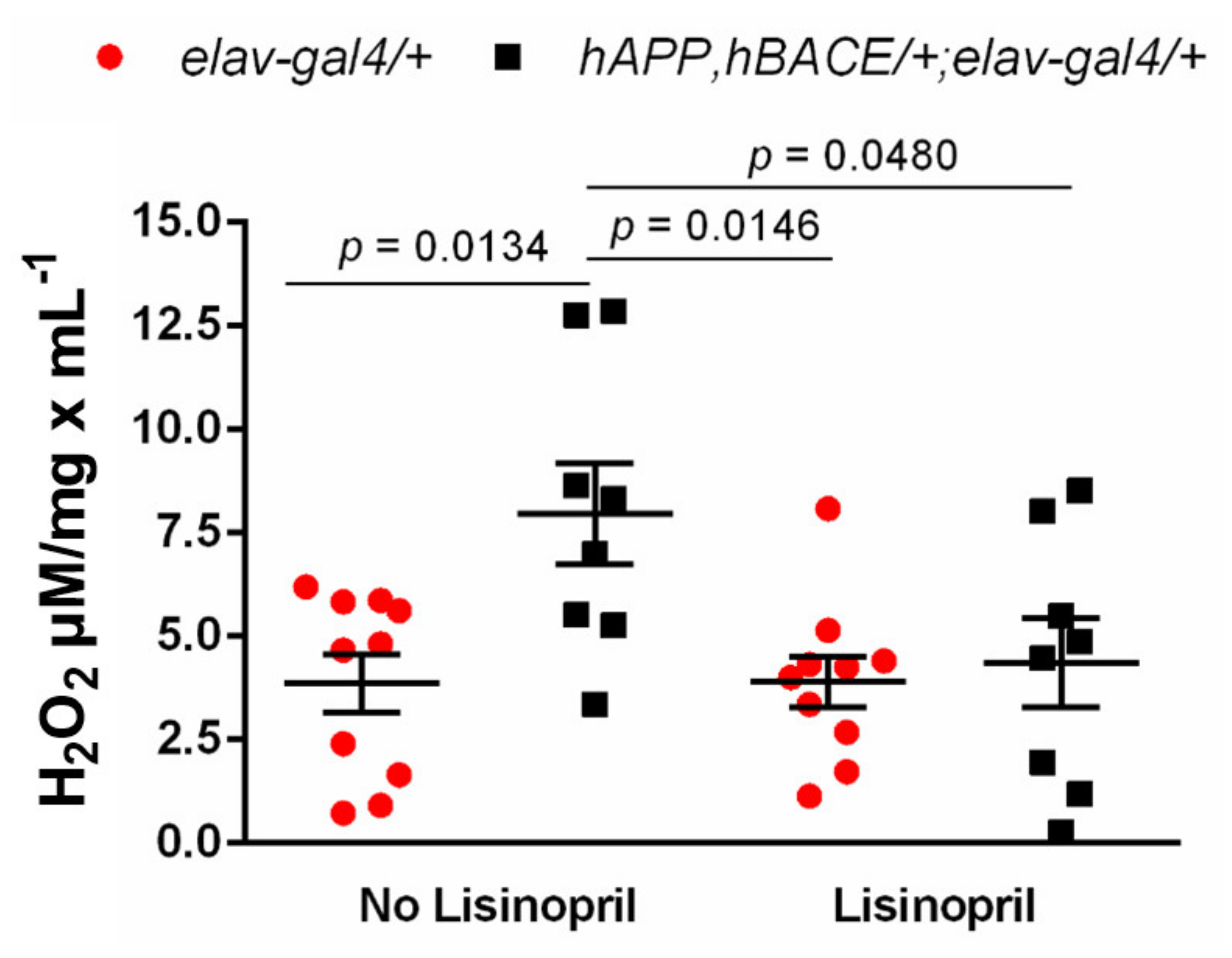
3.3. Lisinopril Lowers Thoracic H2O2 Abundance in AD Flies
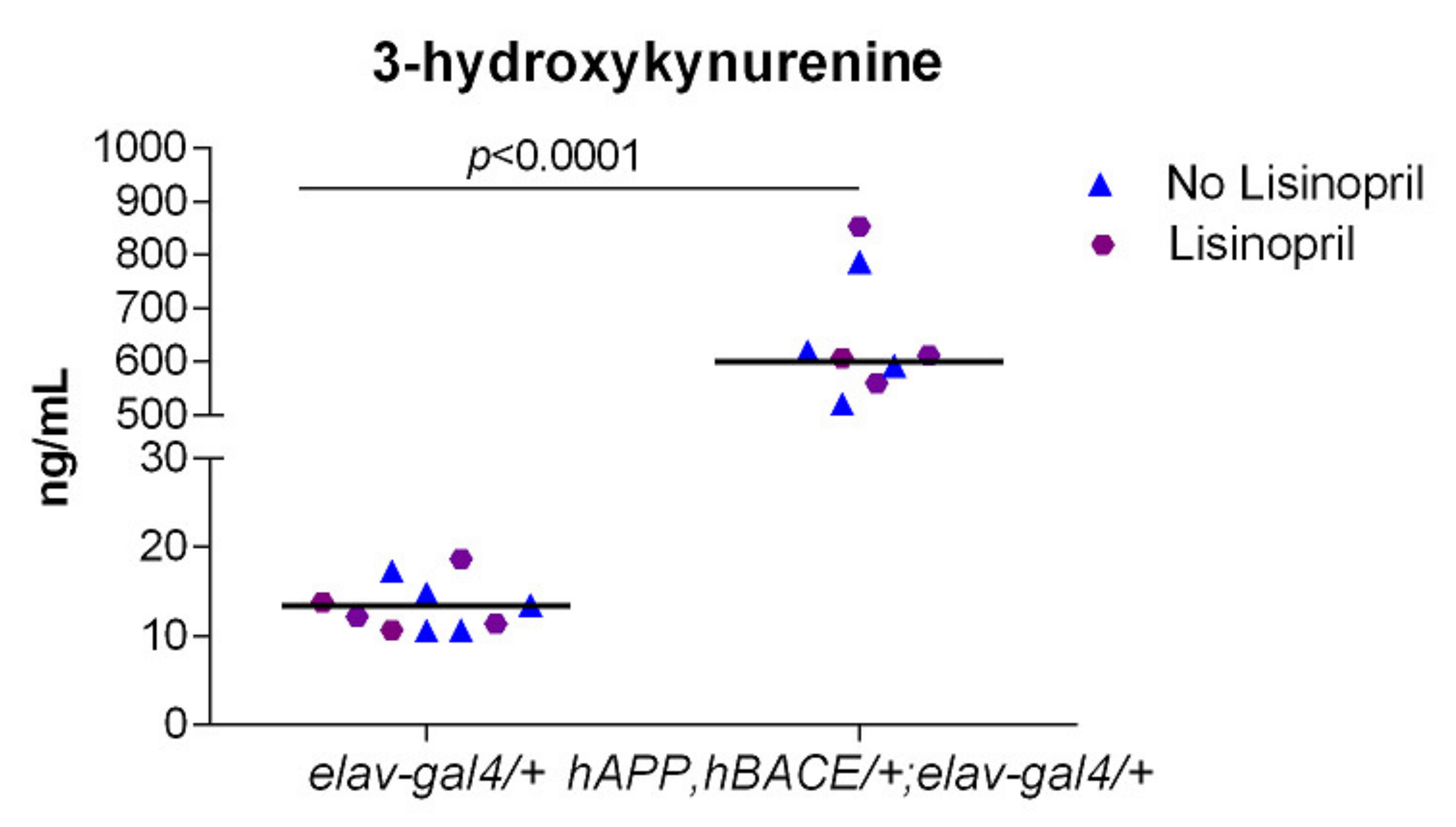
3.4. AD Flies Have Significantly Higher Levels of 3-HK in Their Heads
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kontis, V.; Bennett, J.E.; Mathers, C.D.; Li, G.; Foreman, K.; Ezzati, M. Future life expectancy in 35 industrialised countries: Projections with a Bayesian model ensemble. Lancet 2017, 389, 1323–1335. [Google Scholar] [CrossRef]
- Rocca, W.A.; Petersen, R.C.; Knopman, D.S.; Hebert, L.E.; Evans, D.A.; Hall, K.S.; Gao, S.; Unverzagt, F.W.; Langa, K.M.; Larson, E.B.; et al. Trends in the incidence and prevalence of Alzheimer’s disease, dementia, and cognitive impairment in the United States. Alzheimer’s Dement. 2011, 7, 80–93. [Google Scholar] [CrossRef]
- 2020 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2020, 16, 391–460. [CrossRef] [PubMed]
- Brookmeyer, R.; Johnson, E.; Ziegler-Graham, K.; Arrighi, H.M. Forecasting the global burden of Alzheimer’s disease. Alzheimer’s Dement. 2007, 3, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Yiannopoulou, K.G.; Papageorgiou, S.G. Current and future treatments for Alzheimer’s disease. Ther. Adv. Neurol. Disord. 2013, 6, 19–33. [Google Scholar] [CrossRef]
- Rygiel, K. Can angiotensin-converting enzyme inhibitors impact cognitive decline in early stages of Alzheimer’s disease? An overview of research evidence in the elderly patient population. J. Postgrad. Med. 2016, 62, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Quitterer, U.; AbdAlla, S. Improvements of symptoms of Alzheimer‘s disease by inhibition of the angiotensin system. Pharmacol. Res. 2020, 154, 104230. [Google Scholar] [CrossRef]
- Mogi, M.; Iwanami, J.; Horiuchi, M. Roles of brain angiotensin II in cognitive function and dementia. Int. J. Hypertens. 2012, 2012, 169649. [Google Scholar] [CrossRef]
- Paul, M.; Poyan Mehr, A.; Kreutz, R. Physiology of local renin-angiotensin systems. Physiol. Rev. 2006, 86, 747–803. [Google Scholar] [CrossRef]
- Campbell, D.J. Clinical relevance of local Renin Angiotensin systems. Front. Endocrinol. 2014, 5, 113. [Google Scholar] [CrossRef]
- Nehme, A.; Zouein, F.A.; Zayeri, Z.D.; Zibara, K. An update on the tissue renin angiotensin system and its role in physiology and pathology. J. Cardiovasc. Dev. Dis. 2019, 6, 14. [Google Scholar] [CrossRef]
- Ribeiro, V.T.; de Souza, L.C.; Simoes, E.S.A.C. Renin-angiotensin system and alzheimer’s disease pathophysiology: From the potential interactions to therapeutic perspectives. Protein Pept. Lett. 2020, 27, 484–511. [Google Scholar] [CrossRef]
- Ohrui, T.; Matsui, T.; Yamaya, M.; Arai, H.; Ebihara, S.; Maruyama, M.; Sasaki, H. Angiotensin-converting enzyme inhibitors and incidence of Alzheimer’s disease in Japan. J. Am. Geriatr. Soc. 2004, 52, 649–650. [Google Scholar] [CrossRef]
- Ho, J.K.; Nation, D.A. Memory is preserved in older adults taking AT1 receptor blockers. Alzheimer’s Res. Ther. 2017, 9, 33. [Google Scholar] [CrossRef] [PubMed]
- AbdAlla, S.; Langer, A.; Fu, X.; Quitterer, U. ACE inhibition with captopril retards the development of signs of neurodegeneration in an animal model of Alzheimer’s disease. Int. J. Mol. Sci. 2013, 14, 16917–16942. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, K.E.; Koronyo, Y.; Salumbides, B.C.; Sheyn, J.; Pelissier, L.; Lopes, D.H.; Shah, K.H.; Bernstein, E.A.; Fuchs, D.T.; Yu, J.J.; et al. Angiotensin-converting enzyme overexpression in myelomonocytes prevents Alzheimer’s-like cognitive decline. J. Clin. Investig. 2014, 124, 1000–1012. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Irvine, K.D. Drosophila as a model for understanding development and disease. Dev. Dyn. 2012, 241, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Coelho, D.S.; Schwartz, S.; Merino, M.M.; Hauert, B.; Topfel, B.; Tieche, C.; Rhiner, C.; Moreno, E. Culling less fit neurons protects against amyloid-beta-induced brain damage and cognitive and motor decline. Cell Rep. 2018, 25, 3661–3673. [Google Scholar] [CrossRef] [PubMed]
- Fournier, D.; Luft, F.C.; Bader, M.; Ganten, D.; Andrade-Navarro, M.A. Emergence and evolution of the renin-angiotensin-aldosterone system. J. Mol. Med. 2012, 90, 495–508. [Google Scholar] [CrossRef]
- Cornell, M.J.; Williams, T.A.; Lamango, N.S.; Coates, D.; Corvol, P.; Soubrier, F.; Hoheisel, J.; Lehrach, H.; Isaac, R.E. Cloning and expression of an evolutionary conserved single-domain angiotensin converting enzyme from Drosophila melanogaster. J. Biol. Chem. 1995, 270, 13613–13619. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.M.; Shin, D.R.; Yoo, O.J.; Lee, H.; Lee, J.O. Crystal structure of Drosophila angiotensin I-converting enzyme bound to captopril and lisinopril. FEBS Lett. 2003, 538, 65–70. [Google Scholar] [CrossRef]
- Gabrawy, M.M.; Campbell, S.; Carbone, M.A.; Morozova, T.V.; Arya, G.H.; Turlapati, L.B.; Walston, J.D.; Starz-Gaiano, M.; Everett, L.; Mackay, T.F.C.; et al. Lisinopril preserves physical resilience and extends life span in a genotype-specific manner in Drosophila melanogaster. J. Gerontol. A Biol. Sci. Med. Sci. 2019, 74, 1844–1852. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Gomes, S.M.; Ghalayini, J.; Iliadi, K.G.; Boulianne, G.L. Angiotensin converting enzyme inhibitors and angiotensin receptor blockers rescue memory defects in drosophila-expressing alzheimer’s disease-related transgenes independently of the canonical renin angiotensin system. eNeuro 2020, 7. [Google Scholar] [CrossRef] [PubMed]
- Fares, A.; Borrmann, D. Neurochemical aspects of alzheimer’s disease and movement disturbances: A theory of beta-amyloid and tau-protein. Am. J. Alzheimers Dis. Other Demen. 2018, 33, 535–540. [Google Scholar] [CrossRef]
- Chakraborty, R.; Vepuri, V.; Mhatre, S.D.; Paddock, B.E.; Miller, S.; Michelson, S.J.; Delvadia, R.; Desai, A.; Vinokur, M.; Melicharek, D.J.; et al. Characterization of a Drosophila Alzheimer’s disease model: Pharmacological rescue of cognitive defects. PLoS ONE 2011, 6, e20799. [Google Scholar] [CrossRef]
- Ederer, K.; Jin, K.; Bouslog, S.; Wang, L.; Gorman, G.; Rowe, G.; Abadir, P.; Raftery, D.; Moellering, D.; Promislow, D.; et al. Age- and genotype-specific effects of the angiotensin-converting enzyme inhibitor lisinopril on mitochondrial and metabolic parameters in Drosophila melanogaster. Int. J. Mol. Sci. 2018, 19, 3351. [Google Scholar] [CrossRef] [PubMed]
- Bienkowski, R.S.; Banerjee, A.; Rounds, J.C.; Rha, J.; Omotade, O.F.; Gross, C.; Morris, K.J.; Leung, S.W.; Pak, C.; Jones, S.K.; et al. The conserved, disease-associated RNA binding protein dNab2 Interacts with the fragile X protein ortholog in Drosophila neurons. Cell Rep. 2017, 20, 1372–1384. [Google Scholar] [CrossRef]
- Seugnet, L.; Suzuki, Y.; Stidd, R.; Shaw, P.J. Aversive phototaxic suppression: Evaluation of a short-term memory assay in Drosophila melanogaster. Genes Brain Behav. 2009, 8, 377–389. [Google Scholar] [CrossRef]
- Aggarwal, A.; Reichert, H.; VijayRaghavan, K. A locomotor assay reveals deficits in heterozygous Parkinson’s disease model and proprioceptive mutants in adult Drosophila. Proc. Natl. Acad. Sci. USA 2019, 116, 24830–24839. [Google Scholar] [CrossRef] [PubMed]
- Royea, J.; Hamel, E. Brain angiotensin II and angiotensin IV receptors as potential Alzheimer’s disease therapeutic targets. Geroscience 2020, 42, 1237–1256. [Google Scholar] [CrossRef]
- Ohrui, T.; Tomita, N.; Sato-Nakagawa, T.; Matsui, T.; Maruyama, M.; Niwa, K.; Arai, H.; Sasaki, H. Effects of brain-penetrating ACE inhibitors on Alzheimer disease progression. Neurology 2004, 63, 1324–1325. [Google Scholar] [CrossRef] [PubMed]
- Hajjar, I.; Okafor, M.; McDaniel, D.; Obideen, M.; Dee, E.; Shokouhi, M.; Quyyumi, A.A.; Levey, A.; Goldstein, F. Effects of candesartan vs lisinopril on neurocognitive function in older adults with executive mild cognitive impairment: A randomized clinical trial. JAMA Netw. Open 2020, 3, e2012252. [Google Scholar] [CrossRef] [PubMed]
- Dezsi, L.; Tuka, B.; Martos, D.; Vecsei, L. Alzheimer’s disease, astrocytes and kynurenines. Curr. Alzheimer Res. 2015, 12, 462–480. [Google Scholar] [CrossRef]
- Zakrocka, I.; Turski, W.A.; Kocki, T. Angiotensin-converting enzyme inhibitors modulate kynurenic acid production in rat brain cortex in vitro. Eur. J. Pharmacol. 2016, 789, 308–312. [Google Scholar] [CrossRef] [PubMed]
- Albert, M.S. Changes in cognition. Neurobiol. Aging 2011, 32, S58–S63. [Google Scholar] [CrossRef]
- Singh, B.; Sharma, B.; Jaggi, A.S.; Singh, N. Attenuating effect of lisinopril and telmisartan in intracerebroventricular streptozotocin induced experimental dementia of Alzheimer’s disease type: Possible involvement of PPAR-gamma agonistic property. J. Renin-Angiotensin-Aldosterone Syst. 2013, 14, 124–136. [Google Scholar] [CrossRef]
- Grieb, P. Intracerebroventricular Streptozotocin Injections as a Model of Alzheimer’s Disease: In Search of a Relevant Mechanism. Mol. Neurobiol. 2016, 53, 1741–1752. [Google Scholar] [CrossRef]
- Mhatre, S.D.; Satyasi, V.; Killen, M.; Paddock, B.E.; Moir, R.D.; Saunders, A.J.; Marenda, D.R. Synaptic abnormalities in a Drosophila model of Alzheimer’s disease. Dis. Models Mech. 2014, 7, 373–385. [Google Scholar] [CrossRef]
- Coelho, V.A.; Probst, V.S.; Nogari, B.M.; Teixeira, D.C.; Felcar, J.M.; Santos, D.C.; Gomes, M.V.M.; Andraus, R.A.C.; Fernandes, K.B.P. Angiotensin-II blockage, muscle strength, and exercise capacity in physically independent older adults. J. Phys. Ther. Sci. 2016, 28, 547–552. [Google Scholar] [CrossRef]
- Carter, C.S.; Onder, G.; Kritchevsky, S.B.; Pahor, M. Angiotensin-converting enzyme inhibition intervention in elderly persons: Effects on body composition and physical performance. J. Gerontol. A Biol. Sci. Med. Sci. 2005, 60, 1437–1446. [Google Scholar] [CrossRef]
- Buford, T.W.; Manini, T.M.; Hsu, F.C.; Cesari, M.; Anton, S.D.; Nayfield, S.; Stafford, R.S.; Church, T.S.; Pahor, M.; Carter, C.S. Angiotensin-converting enzyme inhibitor use by older adults is associated with greater functional responses to exercise. J. Am. Geriatr. Soc. 2012, 60, 1244–1252. [Google Scholar] [CrossRef] [PubMed]
- Flaten, H.K.; Monte, A.A. The pharmacogenomic and metabolomic predictors of ACE inhibitor and angiotensin II receptor blocker effectiveness and safety. Cardiovasc. Drugs Ther. 2017, 31, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Ábrigo, J.; Elorza, A.A.; Riedel, C.A.; Vilos, C.; Simon, F.; Cabrera, D.; Estrada, L.; Cabello-Verrugio, C. Role of oxidative stress as key regulator of muscle wasting during Cachexia. Oxidative Med. Cell. Longev. 2018, 2018, 1–17. [Google Scholar] [CrossRef]
- Kadoguchi, T.; Kinugawa, S.; Takada, S.; Fukushima, A.; Furihata, T.; Homma, T.; Masaki, Y.; Mizushima, W.; Nishikawa, M.; Takahashi, M.; et al. Angiotensin II can directly induce mitochondrial dysfunction, decrease oxidative fibre number and induce atrophy in mouse hindlimb skeletal muscle: Angiotensin II-induced skeletal muscle abnormalities. Exp. Physiol. 2015, 100, 312–322. [Google Scholar] [CrossRef]
- De Cavanagh, E.M.; Fraga, C.G.; Ferder, L.; Inserra, F. Enalapril and captopril enhance antioxidant defenses in mouse tissues. Am. J. Physiol. Regul. Integr. Comp. Physiol. 1997, 272, R514–R518. [Google Scholar] [CrossRef]
- De Cavanagh, E.M.V.; Inserra, F.; Ferder, L.; Fraga, C.G. Enalapril and captopril enhance glutathione-dependent antioxidant defenses in mouse tissues. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2000, 278, R572–R577. [Google Scholar] [CrossRef]
- Cozzoli, A.; Nico, B.; Sblendorio, V.T.; Capogrosso, R.F.; Dinardo, M.M.; Longo, V.; Gagliardi, S.; Montagnani, M.; De Luca, A. Enalapril treatment discloses an early role of angiotensin II in inflammation- and oxidative stress-related muscle damage in dystrophic mdx mice. Pharmacol. Res. 2011, 64, 482–492. [Google Scholar] [CrossRef]
- Herndon, L.A.; Schmeissner, P.J.; Dudaronek, J.M.; Brown, P.A.; Listner, K.M.; Sakano, Y.; Paupard, M.C.; Hall, D.H.; Driscoll, M. Stochastic and genetic factors influence tissue-specific decline in ageing C. elegans. Nature 2002, 419, 808–814. [Google Scholar] [CrossRef]
- Giil, L.M.; Midttun, Ø.; Refsum, H.; Ulvik, A.; Advani, R.; Smith, A.D.; Ueland, P.M. Kynurenine pathway metabolites in Alzheimer’s disease. J. Alzheimer’s Dis. 2017, 60, 495–504. [Google Scholar] [CrossRef]
- Gulaj, E.; Pawlak, K.; Bien, B.; Pawlak, D. Kynurenine and its metabolites in Alzheimer’s disease patients. Adv. Med. Sci. 2010, 55, 204–211. [Google Scholar] [CrossRef]
- Schwarz, M.J.; Guillemin, G.J.; Teipel, S.J.; Buerger, K.; Hampel, H. Increased 3-Hydroxykynurenine serum concentrations differentiate Alzheimer’s disease patients from controls. Eur. Arch. Psychiatry Clin. Neurosci. 2013, 263, 345–352. [Google Scholar] [CrossRef]
- Chiarugi, A.; Meli, E.; Moroni, F. Similarities and differences in the neuronal death processes activated by 3OH-kynurenine and quinolinic acid: Neurotoxicity of 3OH-kynurenine and QUIN. J. Neurochem. 2001, 77, 1310–1318. [Google Scholar] [CrossRef] [PubMed]
- Okuda, S.; Nishiyama, N.; Saito, H.; Katsuki, H. 3-Hydroxykynurenine, an endogenous oxidative stress generator, causes neuronal cell death with apoptotic features and region selectivity. J. Neurochem. 2002, 70, 299–307. [Google Scholar] [CrossRef]
- Braidy, N.; Grant, R. Kynurenine pathway metabolism and neuroinflammatory disease. Neural Regen. Res. 2017, 12, 39–42. [Google Scholar] [CrossRef]
- Foster, A.C.; Vezzani, A.; French, E.D.; Schwarcz, R. Kynurenic acid blocks neurotoxicity and seizures induced in rats by the related brain metabolite quinolinic acid. Neurosci. Lett. 1984, 48, 273–278. [Google Scholar] [CrossRef]
- Lugo-Huitrón, R.; Blanco-Ayala, T.; Ugalde-Muñiz, P.; Carrillo-Mora, P.; Pedraza-Chaverrí, J.; Silva-Adaya, D.; Maldonado, P.D.; Torres, I.; Pinzón, E.; Ortiz-Islas, E.; et al. On the antioxidant properties of kynurenic acid: Free radical scavenging activity and inhibition of oxidative stress. Neurotoxicol. Teratol. 2011, 33, 538–547. [Google Scholar] [CrossRef]
- Hartai, Z.; Juhasz, A.; Rimanoczy, A.; Janaky, T.; Donko, T.; Dux, L.; Penke, B.; Toth, G.K.; Janka, Z.; Kalman, J. Decreased serum and red blood cell kynurenic acid levels in Alzheimer’s disease. Neurochem. Int. 2007, 50, 308–313. [Google Scholar] [CrossRef] [PubMed]
- Breda, C.; Sathyasaikumar, K.V.; Sograte Idrissi, S.; Notarangelo, F.M.; Estranero, J.G.; Moore, G.G.L.; Green, E.W.; Kyriacou, C.P.; Schwarcz, R.; Giorgini, F. Tryptophan-2,3-dioxygenase (TDO) inhibition ameliorates neurodegeneration by modulation of kynurenine pathway metabolites. Proc. Natl. Acad. Sci. USA 2016, 113, 5435–5440. [Google Scholar] [CrossRef] [PubMed]
- Campesan, S.; Green, E.W.; Breda, C.; Sathyasaikumar, K.V.; Muchowski, P.J.; Schwarcz, R.; Kyriacou, C.P.; Giorgini, F. The kynurenine pathway modulates neurodegeneration in a drosophila model of huntington’s disease. Curr. Biol. 2011, 21, 961–966. [Google Scholar] [CrossRef] [PubMed]
- Fukui, S.; Schwarcz, R.; Rapoport, S.I.; Takada, Y.; Smith, Q.R. Blood? brain barrier transport of kynurenines: Implications for brain synthesis and metabolism. J. Neurochem. 1991, 56, 2007–2017. [Google Scholar] [CrossRef]
- Zhuravlev, A.V.; Vetrovoy, O.V.; Ivanova, P.N.; Savvateeva-Popova, E.V. 3-Hydroxykynurenine in regulation of drosophila behavior: The novel mechanisms for cardinal phenotype manifestations. Front. Physiol. 2020, 11, 971. [Google Scholar] [CrossRef] [PubMed]
- Royea, J.; Martinot, P.; Hamel, E. Memory and cerebrovascular deficits recovered following angiotensin IV intervention in a mouse model of Alzheimer’s disease. Neurobiol. Dis. 2020, 134, 104644. [Google Scholar] [CrossRef]
- Wright, J.W.; Harding, J.W. Contributions by the brain renin-angiotensin system to memory, cognition, and Alzheimer’s disease. J. Alzheimers Dis. 2019, 67, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Ferrario, C.M.; Jessup, J.; Gallagher, P.E.; Averill, D.B.; Brosnihan, K.B.; Ann Tallant, E.; Smith, R.D.; Chappell, M.C. Effects of renin-angiotensin system blockade on renal angiotensin-(1-7) forming enzymes and receptors. Kidney Int. 2005, 68, 2189–2196. [Google Scholar] [CrossRef] [PubMed]
- Gard, P.R. Cognitive-enhancing effects of angiotensin IV. BMC Neurosci. 2008, 9, S15. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolite | Source a | Df | MS | F-Value | p-Value |
|---|---|---|---|---|---|
| TRYP † | Genotype | 1 | 0.013 | 0.64 | 0.4356 |
| Treatment | 1 | 0.014 | 0.66 | 0.4269 | |
| Genotype × Treatment | 1 | 0.011 | 0.54 | 0.4728 | |
| Error | 16 | 0.020 | |||
| KYN | TRYP | 1 | 134.311 | 13.05 | 0.0026 |
| Genotype | 1 | 15.567 | 1.51 | 0.2377 | |
| Treatment | 1 | 3.374 | 0.33 | 0.5754 | |
| Genotype × Treatment | 1 | 0.740 | 0.07 | 0.7922 | |
| Error | 15 | 10.291 | |||
| KYNA† | TRYP | 1 | 0.040 | 0.59 | 0.4547 |
| Genotype | 1 | 0.017 | 0.25 | 0.6217 | |
| Treatment | 1 | 0.022 | 0.33 | 0.5761 | |
| Genotype × Treatment | 1 | 0.140 | 2.07 | 0.1706 | |
| Error | 15 | ||||
| 3-HK | TRYP | 1 | 62,309.255 | 8.69 | 0.0100 |
| Genotype | 1 | 1,549,853.363 | 216.27 | <0.0001 | |
| Treatment | 1 | 815.481 | 0.11 | 0.7405 | |
| Genotype × Treatment | 1 | 888.440 | 0.12 | 0.7297 | |
| Error | 15 | 7166.188 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thomas, J.; Smith, H.; Smith, C.A.; Coward, L.; Gorman, G.; De Luca, M.; Jumbo-Lucioni, P. The Angiotensin-Converting Enzyme Inhibitor Lisinopril Mitigates Memory and Motor Deficits in a Drosophila Model of Alzheimer’s Disease. Pathophysiology 2021, 28, 307-319. https://doi.org/10.3390/pathophysiology28020020
Thomas J, Smith H, Smith CA, Coward L, Gorman G, De Luca M, Jumbo-Lucioni P. The Angiotensin-Converting Enzyme Inhibitor Lisinopril Mitigates Memory and Motor Deficits in a Drosophila Model of Alzheimer’s Disease. Pathophysiology. 2021; 28(2):307-319. https://doi.org/10.3390/pathophysiology28020020
Chicago/Turabian StyleThomas, Jimiece, Haddon Smith, C. Aaron Smith, Lori Coward, Gregory Gorman, Maria De Luca, and Patricia Jumbo-Lucioni. 2021. "The Angiotensin-Converting Enzyme Inhibitor Lisinopril Mitigates Memory and Motor Deficits in a Drosophila Model of Alzheimer’s Disease" Pathophysiology 28, no. 2: 307-319. https://doi.org/10.3390/pathophysiology28020020
APA StyleThomas, J., Smith, H., Smith, C. A., Coward, L., Gorman, G., De Luca, M., & Jumbo-Lucioni, P. (2021). The Angiotensin-Converting Enzyme Inhibitor Lisinopril Mitigates Memory and Motor Deficits in a Drosophila Model of Alzheimer’s Disease. Pathophysiology, 28(2), 307-319. https://doi.org/10.3390/pathophysiology28020020