Abstract
The transcription factor MYC plays a pivotal role in regulating various cellular processes and has been implicated in tumorigenesis across multiple cancer types. MYC has emerged as a master regulator governing tumor intrinsic and tumor microenvironment interactions, supporting tumor progression and driving drug resistance. This review paper aims to provide an overview and discussion of the intricate mechanisms through which MYC influences tumorigenesis and therapeutic resistance in cancer. We delve into the signaling pathways and molecular networks orchestrated by MYC in the context of tumor intrinsic characteristics, such as proliferation, replication stress and DNA repair. Furthermore, we explore the impact of MYC on the tumor microenvironment, including immune evasion, angiogenesis and cancer-associated fibroblast remodeling. Understanding MYC’s multifaceted role in driving drug resistance and tumor progression is crucial for developing targeted therapies and combination treatments that may effectively combat this devastating disease. Through an analysis of the current literature, this review’s goal is to shed light on the complexities of MYC-driven oncogenesis and its potential as a promising therapeutic target.
Keywords:
MYC; tumorigenesis; therapeutic resistance; replication stress; DNA repair; immune evasion 1. Objectives
This review paper has two primary objectives: firstly, to provide a comprehensive overview of how MYC influences tumorigenesis and therapeutic resistance, dissecting its role in cell proliferation, survival, DNA repair, immune evasion, angiogenesis and fibroblast remodeling. Secondly, this review aims to highlight MYC’s potential as a therapeutic target for innovative cancer treatment approaches by exploring strategies such as inhibiting MYC expression, destabilizing its protein, disrupting MYC/MAX dimerization and combining MYC inhibitors with DNA-damaging agents.
In summary, this review seeks to comprehensively explore MYC’s intricate role in tumorigenesis and therapeutic resistance and to highlight the promise of MYC as a therapeutic target. By addressing these objectives, we aim to contribute to the growing body of knowledge surrounding MYC-driven oncogenesis and its implications for cancer treatment.
2. The Physiological Function of MYC
The MYC gene encodes a multifunctional nuclear phosphoprotein that controls a variety of cellular functions. MYC proteins largely function as an essential global transcription factor, regulating genes involved in several different cellular processes, including cell growth, cell cycles, differentiation, apoptosis, angiogenesis, metabolism, DNA repair, protein translation, mitochondrial biogenesis, immune response and stem cell formation [1,2]. Because of its ability to regulate widespread gene expression, MYC expression is tightly controlled in normal cells. MYC activation in normal cells is prevented from causing tumorigenesis through multiple genetic and epigenetically controlled checkpoint mechanisms, including proliferative arrest, apoptosis and cellular senescence [3].
The MYC gene family consists of three members, c-MYC, L-MYC and N-MYC, with c-MYC being the most widely expressed member and all containing essentially the same conserved regions that are functionally important [2,4,5]. All contain three domain-type structures, an N-terminal region containing the transactivation domain with conserved regions known as MYC boxes (MB); a central region, also containing conserved MBs, implicated in nuclear localization as well as stability control; and a C-terminal region involved in binding to DNA and comprising the basic helix–loop–helix leucine zipper (bHLHZ) domain [6]. MYC dimerizes with its partner MAX through bHLHZ domains resulting in a stable DNA-binding heterodimer, which is essential for MYC to regulate gene transcription [2,6]. The primary mechanism by which MYC regulates gene expression is through the binding of MYC/MAX heterodimers to E-Box sequences in the regulatory regions of target genes [2,6].
3. MYC Is Often Activated in Human Cancers
MYC alterations have been reported to occur in approximately 70% of human malignancies [2,7]. MYC can be genetically activated directly through genomic amplification, chromosomal translocation, retroviral integration, the activation of super enhancers and mutations [2,8,9,10]. Additionally, MYC can be activated through downstream growth signaling from other oncogenes, including RAS, SRC and NOTCH [9,11], or the inactivation of tumor suppressor genes, such as Adenomatous polyposis coli (APC), Phosphatase and tensin homolog (PTEN) and Protein phosphatase 2A PP2A [12,13,14,15], leading to increased MYC gene expression, translation and/or protein stability.
It has been demonstrated that even relatively small constitutive changes in the MYC expression level >2-fold -fold relative to normal) have biological consequences and impact tumorigenesis [8,16]. Although MYC is one of the most activated oncogenes implicated in the pathogenesis of human cancers, its activation alone generally cannot induce tumorigenesis; rather, it results in the activation of checkpoints, including those through p53, ARF, BIM and PTEN which can cause cell growth arrest or death [17,18,19]. Thus, MYC cooperates with many other oncogenic or tumor suppressor genes to initiate tumorigenesis [3]. Its activation is also generally essential for tumorigenesis as shown in several animal models of cancer in which MYC alteration is required for tumor initiation, progression or maintenance [19,20,21,22,23]. Therefore, tumors with dysregulated MYC have been considered as “MYC-driven” and/or “MYC-addicted” tumors.
A recent report analyzing somatic copy-number alterations (SCNAs) across human cancers has identified 76 amplification regions that are altered at a significant frequency across multiple cancer types, and the most frequent of these focal SCNAs is MYC amplifications [24]. The MYC oncogene is a central driver in multiple cancers, such as breast cancer [25], liver cancer [26], colorectal carcinoma [27], prostatic neoplasia [28], ovarian cancer and lung cancer [8]. Moreover, high levels of MYC deregulation are associated with aggressive conditions and a poor prognosis. For example, in the triple-negative form of breast cancer (TNBC), the most aggressive subtype of breast cancer and the most difficult to treat, MYC is amplified in approximately 57% of cases in contrast to only 7–13% in luminal A-type (ER/PR-positive) cancers (a breast cancer subtype with a more favorable outcome) [23,29].
4. Mechanisms of MYC Activation/Phosphorylation
In physiologic conditions, MYC is under extraordinarily tight regulation by cells [30,31,32,33]. Different factors act to control MYC mRNA expression, stability, export and translation [30,31,32,33]. In addition to transcriptional and mRNA regulation, MYC protein stability and activity are regulated by several post-translational modifications as well as multiple ubiquitin ligases [31,32,33]. The conserved MYC Box 1 (MB1) region of MYC’s transactivation domain is influenced by two sequential and interdependent phosphorylation events on Ser62 (pS62) and Thr58 (pT58). The phosphorylation of MYC enhances its recruitment to target genes [34,35]. MYC is stabilized when it is phosphorylated on Ser62 by extracellular receptor kinase (ERK) or cyclin-dependent protein kinase 2 (CDK2), but it is targeted for degradation when it is phosphorylated on Thr58 by glycogen synthase kinase (GSK-3) via the ubiquitin–proteasome pathway [14,34,35].
The best characterized arm of MYC-induced tumorigeneses relies on the RAS pathway [14]. MYC is activated and stabilized downstream of RAS-induced growth stimuli, which phosphorylate MYC at Ser62. There are at least two effector pathways through which RAS promotes the stability of MYC: the RAF–MEK–ERK kinase cascade and the phosphatidylinositol 3-kinase (PI3K)–protein kinase B (AKT) pathway which inhibits GSK-3β [14,15]. The phosphorylation of MYC on Ser62, after a growth stimulatory signal, results in its stabilization, but also its subsequent phosphorylation at Thr58 by the GSK3 kinase. The phosphorylation of Thr58 then facilitates the dephosphorylation of Ser62 by the protein phosphatase 2A (PP2A), leading to the degradation of MYC through the ubiquitin pathway [36,37]. PI3K–AKT pathway activation by RAS leads to the phosphorylation and inhibition of GSK-3β, facilitating the stabilization of MYC [38]. In contrast, PP2A, which acts as a negative regulator of the PI3K–AKT pathway [39], directly dephosphorylates Ser62 and stimulates the degradation of MYC [15].
PP2A is a major serine/threonine phosphatase with specificity for its substrates. A scaffolding A component, a catalytic C subunit and a third, highly changeable regulatory B subunit make up the heterotrimeric phosphatase PP2A [40]. Structural A and catalytic C subunits have two isoforms, α and β. More than 23 isoforms of the regulatory B subunit exist, and they are divided into four distinct families called B/B55, B′/B56, B″ and B‴. The B56α subunit is the only known B subunit capable of directly inhibiting the stability and activity of MYC [40,41,42]. PP2A-B56α dephosphorylates the Ser62 residue, targeting the phosphorylated MYC protein at Thr58 for ubiquitin-mediated proteasomal degradation [43]. Additionally, PP2A containing the B56α subunit can activate GSK-3β by dephosphorylating it [44]. Conversely, PP2A-B55α can be targeted to MYC in a complex with EYA3 to dephosphorylate Thr58, and this is associated with increased MYC stabiility [42]. Together, increasing our knowledge of key post-translational regulatory events of MYC may provide therapeutic approaches aimed at destabilizing MYC protein [6,45,46].
5. The Interplay between RAS and MYC
The intricate interplay between the oncogenes RAS and MYC has been a subject of intense investigation since the groundbreaking discovery in 1983 by Land and colleagues, which unveiled the concept of oncogenic cooperation between these two entities [47]. This discovery revealed the strong reliance that exists among individual oncogenic mutations. Over the past three decades, researchers have sought to unravel the complex mechanisms that underlie the cooperative effects of RAS and MYC in cancer, yet many aspects of this interaction remain shrouded in mystery.
Early studies primarily focused on the individual cell-intrinsic outcomes driven by RAS and MYC. These studies highlighted RAS and MYC synergistic induction and the stabilization of key cell cycle proteins, which played pivotal roles in cellular proliferation and progression [48,49,50], with even MYC stabilization being recognized as a part of this intricate interplay [14]. Studies also delved into the mutual disruption of RAS-induced senescence by MYC [51,52] and the RAS-mediated inhibition of MYC-induced apoptosis [51,53]. Furthermore, researchers delved into the capacity of MYC to overcome barriers to self-renewal in cells driven by RAS mutations [54].
Conversely, critical insights into RAS and MYC’s cooperation emerged from co-transgenic in vivo experiments conducted in mice. These studies unveiled that the synergistic oncogenic partnership between RAS and MYC extends beyond their isolated effects on cells and involves intricate interactions within the complex tumor microenvironment [55,56,57,58]. Such interactions necessitate a comprehensive examination of the dynamic interplay that these oncogenes establish with the tumor stroma. Recent research has taken a focused approach to investigate the collaborative impact of Myc dysregulation on the development and advancement of KRas-driven lung tumors within an in vivo context [55]. This study adeptly delineates the sequential events through which Myc orchestrates the transformation of adenomas into aggressive, inflammatory and immune-suppressed adenocarcinomas. Collectively, these findings offer deeper insights into the complex role of MYC in the progression of tumors.
To comprehensively understand the full scope of RAS–MYC cooperation, future research endeavors should focus on unraveling the complex network of interactions within the tumor microenvironment and how these interactions influence cancer progression. By delving into the intricate details of this interplay, we may uncover novel therapeutic avenues that target the RAS–MYC axis, potentially leading to transformative strategies for cancer treatment.
6. Cell Intrinsic Role of MYC in Tumorigenesis
The precise mechanisms by which the deregulation of the MYC oncoprotein contributes to cancer formation, maintenance and progression are still unclear. MYC deregulation likely induces tumorigenesis via multiple mechanisms, mostly related to its broad ability to regulate the expression of a large number of different genes [1,2,59]. In general, we can state that the cell-intrinsic mechanisms by which MYC induces tumorigenesis include enhancing two fundamental cellular functions: proliferation and survival. MYC enhances cell proliferation via stimulating metabolism, protein synthesis, cell cycle progression and DNA replication; however, this can lead to genomic instability [59]. Then, to ensure cell survival against this stress and for the maintenance of genomic integrity during DNA replication, MYC enhances cell survival via stimulating DNA repair and suppressing cell death [60,61,62]. Taken together, data suggest that MYC plays dual roles in inducing and surviving replication stress.
6.1. The Impact of MYC Overexpression on Replication Stress, Genomic Instability and Oncogenic Transformation
MYC overexpression activates downstream genes, stimulating the cell cycle and DNA synthesis and causing genomic instability [59,63,64]. It directly triggers cell cycle progression by activating cyclin D, CDK4 and E2F transcription factors [65,66,67]. MYC affects gene stability, microRNAs and noncoding RNAs, leading to chromosomal alterations [59,68,69,70,71]. MYC amplification is associated with various chromosomal changes, including breaks, translocations, deletions, inversions, aneuploidy and extrachromosomal elements [59,70,71].
MYC increases metabolism to fuel tumorigenesis, promoting ATP production and cellular building blocks for cell division and DNA replication, providing a transformation advantage [72,73]. MYC-overexpressing cells exhibit increased glucose and glutamine utilization, stimulating fatty acid and cholesterol synthesis [74,75,76]. This metabolic increase helps sustain high rates of DNA replication in MYC-transformed cells, but the deregulated replication and loss of checkpoints lead to genomic instability and double-stranded DNA breaks (DSBs) [72].
Thus, deregulated MYC induces DSBs by increasing replication and causing replication stress and the accumulation of reactive oxygen species (ROS) [77,78,79]. MYC-overexpression-induced DSBs in normal cells upregulate the formation of γH2AX foci, a biomarker of DSBs, stimulating cellular checkpoint mechanisms such as cell cycle arrest, apoptosis and premature senescence as part of the DNA damage response (DDR), delaying tumorigenesis [73,80]. MYC overexpression in normal cells can have no impact or varying effects, including proliferative arrest, senescence and apoptosis [17]. However, in MYC-transformed cells, activated MYC promotes DSB repair in response to DNA damage, enabling cancer cell survival [60,61] (Figure 1). The specific mechanisms that oncogenically activate MYC and alter its functional output in cancer cells remain poorly understood. Evidence suggests that modified post-translational modifications, changes in binding partners and multimerization play important roles in this context [34,81,82,83,84,85,86]. Recent studies have demonstrated that MYC forms multimeric structures in response to perturbations, affecting its interactome and enabling tumor cells to proliferate under stressful conditions, limiting DNA double-strand break formation during S-phase [84]. Further research is needed to unravel these intricate molecular mechanisms to advance our understanding of MYC-driven tumorigenesis and identify potential therapeutic targets.
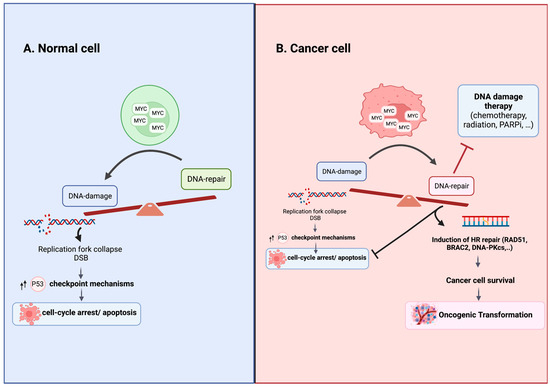
Figure 1.
Consequences of MYC Overexpression in Normal and Cancer Cells. (A) In normal cells with intact cell cycle checkpoints, MYC overexpression tips the balance in favor of damage over repair by driving replication stress and stimulating checkpoint mechanisms that lead to cell cycle arrest, senescence or apoptosis. (B) In cancer cells with altered cellular checkpoints, MYC overexpression upregulates homologous recombination DNA repair, promoting cell survival and resistance to DNA damage agent treatment. The specific mechanisms responsible for oncogenically activating MYC and modulating its functional output in cancer cells are not fully elucidated. The altered post-translational modifications, shifts in binding partners and the formation of multimeric structures are believed to play roles in regulating MYC’s function in cancer cells [81,82,83,84]. Figure was created with BioRender.com, accessed on 2 August 2023.
6.2. MYC-Induced DSB Repair in Chemoresistance
The accumulation of genomic instability renders the cell genome vulnerable to DSB-induced lethality. The p53 tumor suppressor, also known as the “guardian of the genome”, accumulates in response to DSBs and subsequently induces the transcription of its downstream target genes, which are required for the induction of senescence or apoptosis [77,87]. However, MYC-driven cancer cells survive the accumulation of DNA-damage and maintain DNA replication. In normal fibroblasts, MYC activates the ARF–Mdm2–p53 tumor suppressor pathway, enhancing p53-dependent apoptosis (Figure 1). In contrast, the overexpression of MYC in cancer broadly represses anticancer proteins that promote apoptosis (Figure 1). It has been demonstrated that MYC transgenic mice with MYC-driven tumors also overexpress the anti-apoptosis protein Mdm2, which acts as a negative regulator of p53 [88]. Additionally, MYC has been shown to protect cancer cells from radiation-induced DNA damage and apoptosis, while inducing DNA repair in response to radiation [89]. Thus, it is not surprising that the MYC oncoprotein is closely linked to chemoresistance in different tumor types [60,90,91,92]. It promotes cell survival by increasing DNA repair machinery and suppressing pro-apoptotic processes [60,90,91,92].
Consistent with this, MYC was found to be associated with the promoter region of various DSB repair-related genes, such as NBS1, Ku70, Rad51, BRCA2, Rad50 and the DNA-dependent protein kinase catalytic subunit (DNA-PKcs), and activates their transcription [60,93]. DSBs are repaired through two pathways: homologous recombination (HR) and nonhomologous end joining (NHEJ), which differ in terms of their fidelity and template requirements [77]. HR is predominant during S- and G2-phases, relying on RAD51, RAD51B, RAD51C, RAD51D, XRCC2, XRCC3, BRCA1 and BRCA2 for repair using the sister chromatid as a template [77]. NHEJ functions throughout the cell cycle and depends on the DNA-protein kinase complex and Ku for DSB repair [77]. Silencing MYC in HeLa cells significantly reduces DSB’s repair capability, and MYC regulates RAD51 expression, with MYC induction leading to upregulation and MYC knockdown, resulting in decreased RAD51 expression [60,61,93,94]. MYC’s ability to protect cancer cell genomes from DNA damage can prevent catastrophic damage and promote survival in the face of genomic instability, which can support oncogenic transformation (Figure 1). Targeting MYC may enhance cancer cell sensitivity to DNA damage and serve as a potential strategy for anticancer therapy.
The suppression of MYC expression or function can reverse tumorigenesis by reversing abnormal DNA replication and DNA repair processes, leading to cell cycle arrest, apoptosis, senescence and the accumulation of DNA damage [22,60,90,91,92,95]. Targeting MYC in cancer cells with high genomic instability can induce cell death and enhance vulnerability to DNA-damaging agents (Figure 2). The inhibition of MYC-mediated DSB repair following treatment with DNA-damaging agents holds promise for overcoming MYC-induced drug resistance and chemoresistance.
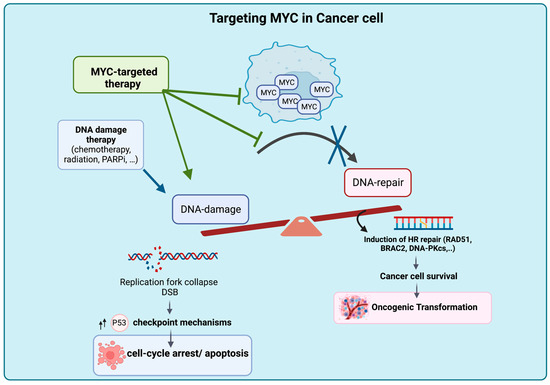
Figure 2.
Proposed Mechanism of Tumor Intrinsic Cell Death Upon MYC Deactivation. Targeting MYC results in accumulation of DNA damage due to the blockage of DNA DSB repair, leading to cell death and rendering the cancer cell genome vulnerable to the effects of DNA-damaging agents. Figure was created with BioRender.com, accessed on 2 August 2023.
7. MYC as a Regulator of the Tumor Microenvironment Leading to Drug Resistance
Tumorigenesis is a complex process that not only affects malignant cells’ genetic events but also influences the surrounding tumor microenvironment (TME). The TME includes endothelial cells, fibroblasts and immune cells among many others, in addition to the extracellular matrix produced by these cells. The crosstalk between tumor cells and mesenchymal stromal cells is thought to be critical for both cancer development and drug resistance [96,97]. More recently, novel studies have shown that MYC plays a role in tumorigenesis in cell intrinsic signaling and has a broader spectrum of functions in the tumor microenvironment [55,98,99,100] (Figure 3). It is acknowledged that stromal cells alter tumor cell drug responses, with cancer-associated fibroblasts (CAFs) and inflammatory cells comprising most stromal cells in the tumor microenvironment. Variations in fibroblast cells and immune profiles have been linked to tumorigenesis and therapeutic responses [101,102].
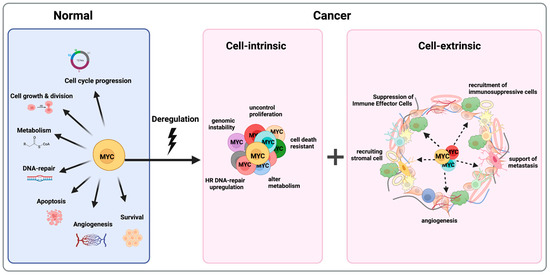
Figure 3.
MYC is a Master Regulator of Almost All the Hallmarks of Cancer. MYC controls a variety of cellular functions (on the left); deregulation of MYC leads to tumors “addicted” to MYC because of both tumor cell-intrinsic (in the middle) and cell-extrinsic mechanisms (on the right). Figure was created with BioRender.com, accessed on 2 August 2023.
Apart from its cell-intrinsic role in tumorigenesis, MYC plays a crucial role in shaping the tumor microenvironment and establishing a nurturing niche for cancer cells (Figure 3). Through its transcriptional activity, MYC orchestrates a variety of molecular changes that contribute to the development of a tumor-permissive microenvironment and educating tumor-infiltrating cells [103,104]. This microenvironment not only promotes cancer cell survival and growth but also fosters drug resistance. MYC contributes to angiogenesis, CAFs’ metabolic changes, immune evasion, invasion and migration, which all lead to distant drug resistance [55,105,106,107].
One key aspect of MYC’s programming of the tumor microenvironment is its ability to stimulate angiogenesis, the formation of new blood vessels. MYC promotes the secretion of angiogenic factors such as vascular endothelial growth factor (VEGF), which stimulates the growth of blood vessels, ensuring an adequate oxygen and nutrient supply to the tumor [98,107,108]. This enhanced vascular network facilitates cancer cell survival and enables the cells’ rapid proliferation. Emerging evidence indicates that MYC upregulation is not limited to cancer cells but also extends to cancer-associated fibroblasts (CAFs) [109]. CAFs have been shown to play a crucial role in promoting tumor angiogenesis [110,111]. CAFs achieve this by secreting cytokines that attract endothelial cells, facilitating the formation of tumor-associated blood vessels and nodal metastases. This finding aligns with previous research by Baudino et al., who demonstrated in mouse models that MYC plays a crucial role in regulating cytokines involved in lymphangiogenesis, including VEGF-C and VEGF-D [112]. Consequently, the upregulation of MYC expression may contribute to the development of nodal metastases. The presence of MYC-expressing fibroblasts in the local metastatic environment assists in promoting colonization by creating a microenvironment conducive to lymphangiogenesis, thereby supporting the survival of cancer cells [109,112]. In addition, MYC expression in CAFs directly regulates the expression of genes involved in glucose metabolism, including lactate dehydrogenase A [109,113]. It has been reported that CAFs exhibit an elevated expression of glycolytic enzymes [114]. This implies that cancer cells can take advantage of altered CAF metabolism to facilitate tumor growth and vascularization [109,113,114]. These observations strongly suggest that MYC plays an essential role in regulating CAFs within the tumor microenvironment, further emphasizing its multifaceted involvement in cancer progression and the shaping of the tumor microenvironment.
7.1. Immune Evasion and MYC
MYC promotes immunosuppression and contributes to the recruitment of immunosuppressive cells within the tumor microenvironment [55,115]. This section discusses the mechanisms by which MYC supports immune evasion in cancer.
7.1.1. MYC Induces the Recruitment of Immunosuppressive Cells
MYC plays a significant role in promoting the recruitment of immunosuppressive cells within the tumor microenvironment, including regulatory T cells (Treg) [23,55,103,116,117]. MYC in tumor cells influences Treg accumulation, activation and metabolic programming [118]. MYC overactivation promotes glycolysis in tumor cells, creating a low-glucose environment that favors Treg generation [119,120,121]. MYC also regulates the secretion of chemokines and cytokines, such as CCL22, CCL17, TGF-β and IL-10, which attract and activate Tregs [55,107,122]. Additionally, MYC influences the recruitment of myeloid-derived suppressor cells (MDSCs) and tumor-associated macrophages (TAMs) [103,117,123,124,125,126,127,128]. These immunosuppressive cells establish an immune-evading microenvironment that hampers effector immune cell function and promotes tumor growth.
7.1.2. Suppression of Immune Effector Cells and Escape from Immune Recognition
Effector immune cells, including T cells and NK cells, play a crucial role in recognizing and eliminating cancer cells. MYC can modulate the function of these cells to suppress their cytotoxic activity [55,129]. For instance, MYC activation in cancer cells can downregulate the expression of major histocompatibility complex (MHC) molecules, impairing antigen presentation and the subsequent activation of T cells [129]. Additionally, MYC-induced metabolic reprogramming creates a nutrient-deprived microenvironment that impairs multiple immune cell types [55,119,120,121]. Furthermore, MYC contributes to immune evasion in tumor cells by inducing the expression of PD-L1, which suppresses the attack from immune cells against the cancer cells [55,130,131,132]. These mechanisms contribute to the establishment of an immunosuppressive tumor microenvironment, allowing cancer cells to evade immune surveillance, which suggests that targeting MYC could be a therapeutic strategy to counteract immune evasion and enhance cancer treatment outcomes.
MYC inhibition reverses the immunosuppressive tumor microenvironment, restores immune cell activation and enhances the production of immune-stimulatory molecules [133,134,135,136]. Preclinical studies demonstrate the potential of MYC inhibition to improve immune response and synergize with immunotherapies [137,138,139]. Targeting MYC is promising for restoring immune response, modulating the tumor microenvironment and improving cancer patient outcomes.
8. MYC as a Therapeutic Target for Cancer
Experimental evidence has demonstrated that inhibiting MYC expression can reverse tumorigenesis, providing a proof of concept for the pharmacological targeting of this oncoprotein to impede tumor cell growth [2,99,140]. However, developing specific pharmacological agents to target MYC is challenging due to its unique characteristics. MYC’s disordered structure, lack of hydrophobic pockets and absence of catalytic activity make it difficult to bind with small molecules or conventional enzyme inhibitors. Moreover, MYC’s nuclear localization presents challenges for targeting with large molecules, such as monoclonal antibodies.
Despite these obstacles, recent pharmaceutical-based strategies have emerged to target MYC and hinder tumor growth [2,14,46,140,141,142]. These approaches include inhibiting MYC expression at the transcriptional level, blocking MYC translation through the PI3K/AKT/mTOR pathway, destabilizing MYC using inhibitors of USP7, AURKA, and PLK1 or activators of PP2A at the posttranslational level, and utilizing Omomyc to disrupt the MYC/MAX dimeric complex binding to DNA. These strategies offer potential avenues to inhibit MYC’s function and suppress tumor growth (Figure 4). Further research and development in this field are essential for advancing our understanding and therapeutic targeting of MYC in cancer treatment.
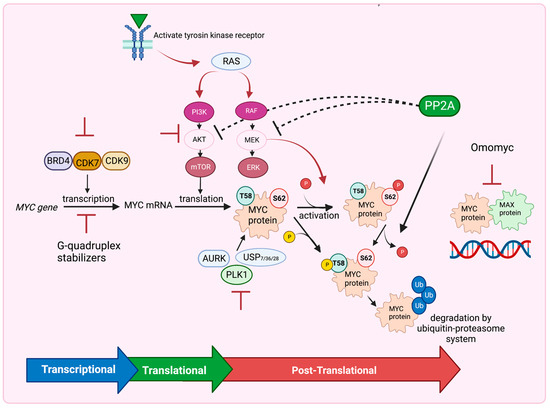
Figure 4.
Several Strategies to Target MYC. Inhibitors of BRD4, CDK7 and CDK9 or G-quadruplex stabilizers inhibit MYC expression at the transcriptional level. Inhibition of the PI3K/AKT/mTOR pathway blocks MYC translation, whereas inhibition of RAF/MEK/ERK pathway, USP7, 28, 36, AURK and PLK1 inhibitors destabilize MYC at the posttranslational level. PP2A also dephosphorylates MYC at Ser62 and destabilizes MYC leading to MYC degradation by the ubiquitin–proteasome system. PP2A also inhibits the PI3K/AKT/mTOR and RAF/MEK/ERK pathways. Omomyc drug functions to interrupt the MYC–MAX dimeric complex binding to DNA. Figure was created with BioRender.com, accessed on 5 August 2023 and adapted from [14,141,142].
8.1. Targeting MYC Gene Transcription
Targeting MYC transcriptional regulation is a promising strategy for cancer treatment [46,140,143]. The BRD4 inhibitor (JQ1) competes with BRD4 for binding to acetylated lysines, displacing BRD4 from super-enhancers within the MYC oncogene and reducing MYC expression, resulting in anti-cancer effects in hematopoietic cancers, PDAC and MYCN-driven cancers [144,145]. The inhibition of CDK7 and/or CDK9, which is critical for MYC transcription, reduces MYC expression and downregulates MYC target genes. Inhibitors against CDK7 and CDK9 demonstrate potent anti-tumor effects in MYC-driven cancers, including T cell acute lymphoblastic leukemia, mixed-lineage leukemia, neuroblastomas and small cell lung cancers [146]. Additionally, the MYC promoter possesses a structurally actionable element [143,147]. Several studies have demonstrated the potential of specific small-molecule ligands, such as cationic porphyrins and quindolines (e.g., CX-33543 or quarfloxin), to stabilize G-quadruplexes within the MYC promoter, leading to the downregulation of MYC expression [143,148,149]. Further research should focus on optimizing these therapies and exploring combination treatments for enhanced efficacy.
8.2. Targeting MYC mRNA Translation
Targeting MYC mRNA translation provides an alternative approach to combating MYC-driven cancers [143,150,151]. The PI3K/AKT/mTOR pathway, frequently dysregulated in various cancers, plays a crucial role in protein synthesis through mTOR complexes 1 and 2 (mTORC1 and mTORC2). The mTORC1-mediated phosphorylation of eukaryotic translation initiation factor 4E-binding protein 1 (4EBP1) releases its inhibition on eIF4E, leading to the enhanced translation of mRNAs with long 5′-untranslated regions (5′-UTRs) and complex RNA secondary structures, including MYC mRNA. The pharmacological inhibition of the PI3K/AKT/mTOR pathway significantly reduces MYC levels and demonstrates therapeutic efficacy in MYC-driven cancers [150,151]. Additionally, cytoplasmic polyadenylation element-binding protein (CPEB) controls polyadenylation-induced translation and can recognize cytoplasmic polyadenylation elements (CPEs) in the 3-UTRs of MYC mRNA. CPEB inhibits c-MYC expression by promoting the deadenylation and decay of its mRNA [152]. CPEB family proteins are often downregulated in human cancers, so restoring their expression could lead to MYC inhibition in MYC-driven cancers [153].
8.3. Targeting MYC Stability
Targeting MYC stability offers a potential approach for suppressing MYC-dependent cancers [32,33]. The ubiquitin–proteasome system tightly regulates MYC stability, with phosphorylation at Thr58 triggering polyubiquitination by the E3 ligase FBW7 and subsequent proteasomal degradation [154,155]. Deubiquitinating enzymes, such as USP28, USP36 and USP7, counteract MYC degradation mediated by FBW7, leading to MYC stabilization and tumor cell proliferation [156,157]. Additionally, AURKA and PLK1 play crucial roles in maintaining MYC expression. AURKA forms a complex with N-MYC, protecting it from FBW7-mediated degradation, and inhibitors of AURKA disrupt the MYC–AURKA complex, promoting N-MYC degradation and tumor regression [158]. PLK1 and MYC create a positive feedforward activation loop that sustains a high expression, and PLK1 inhibitors induce apoptosis in MYC-overexpressing tumor cells, highlighting their potential as therapeutics for MYC-dependent cancers [159]. Targeting these mechanisms could destabilize MYC and offer therapeutic benefits for MYC-driven cancers.
The PP2A serine/threonine phosphatase is frequently inhibited in most human cancers, and it primarily functions as a tumor suppressor by diminishing the activation of key oncogenic regulators, including MYC, ERK and AKT [160]. PP2A directly dephosphorylates MYC, resulting in its degradation, making MYC one of the well-characterized substrates of PP2A [160,161,162].
Recent studies have demonstrated the effectiveness of re-engineering FDA-approved tricyclic neuroleptics, particularly small-molecule activators of PP2A (SMAPs), in inducing apoptosis, promoting the dephosphorylation of PP2A targets such as MYC and AKT and suppressing tumor growth in mouse models [20,163].
A recent study by Leonard et al. provides insight into the direct activation of PP2A by an SMAP molecule called DT-061, one of the lead-engineered compounds based on tricyclic neuroleptics [164]. The researchers describe the 3D structure of the DT-061-bound PP2A trimeric complex and reveal that DT-061 occupies a unique intersubunit pocket, which directly binds and selectively stabilizes the PP2A-B56α holoenzyme [164]. This binding and stabilization mechanism of DT-061 on PP2A-B56α enhances its antitumor function [164]. The dephosphorylation of the most-studied MYC-activating phosphorylation event on Serine 62 by PP2A-B56α suggests that DT-061 has potential as an approach for targeting active MYC through the stabilization of PP2A-B56α [161,164]. The in vivo efficacy of DT-061 in murine tumor models further supports its therapeutic potential, although further investigation is needed to assess its effects on immune function in disease [20].
8.4. Targeting the MYC–MAX Complex
The formation of the MYC/MAX complex is essential for MYC to bind to DNA and activate the transcription of target genes [165]. This complex adopts a parallel, left-handed, four-helix bundle structure, in which each monomer consists of two R-helices separated by a loop [166]. Although this structure does not exhibit obvious binding sites for small-molecule inhibitors, researchers have conducted screenings to identify molecules that can block the interaction. Among them, the peptide mimetic IIA6B17 has been identified as a small-molecule inhibitor of MYC/MAX dimerization [167]. Another compound known as 10058-F4 has demonstrated the ability to disrupt the MYC/MAX complex specifically in HL60 cells [166]. Omomyc, a well-known inhibitor, is a mutant mini-peptide with a basic helix–loop–helix structure that sequesters MYC in an inactive complex, preventing MYC-induced tumorigenesis in multiple mouse tumor models [168].
8.5. Enhancing Therapeutic Efficacy: Combining MYC Targeting with DNA Damage Agents, including PARP Inhibitors
PARP inhibitors (PARPi) are a novel class of anticancer therapies that competitively bind to the catalytically active site of PARP molecules, interfering with their DNA repair function by competing with NAD+ [169]. The PARP inhibitors exhibit varying degrees of potency in terms of enzymatic inhibition and PARP trapping effects [170,171,172]. For example, olaparib and talazoparib have similar levels of catalytic inhibition, but talazoparib shows approximately a 100-fold higher potency than olaparib in trapping PARP–DNA complexes [173,174]; however, the clinical significance of these modes of action are still under study. These inhibitors have shown effectiveness in treating tumors with defects in homologous recombination repair (HR). Specifically, PARP inhibitors have been utilized to target tumors harboring mutations in the key HR genes, Breast Cancer Associated 1 and 2 (BRCA1 and BRCA2) [175]. Several PARP inhibitors have received approval for the treatment of BRCA-mutated ovarian, breast and pancreatic cancers. There are currently 269 clinical trials registered to investigate the potential of PARP inhibitors as an anticancer therapy in chemo-resistant germline or somatic BRCA1/2 mutated breast, ovarian, lung and pancreatic cancers [169].
Acquired resistance to PARPi is a significant challenge in cancer treatment, and one of the mechanisms behind this resistance is the restoration of homologous recombination (HR) capacity [175]. This can occur through reversion mutations, in which BRCA1/2 function is restored by secondary mutations [176,177]. Studies have shown that reversion mutations in BRCA1/2 are observed in patients with breast, ovarian and pancreatic carcinomas and are associated with the development of PARPi-resistant tumors [176,177,178]. Additionally, high levels of MYC have been found to enhance the expression of RAD51, a key protein involved in homologous recombination repair (HRR) [93,94]. Elevated levels of c-MYC and RAD51 in TNBC patients have been associated with resistance to PARP inhibitors [179,180].
The prevalence of BRCA1/2 mutations among patient cases is relatively low, and a substantial number of these patients develop resistance to PARP inhibitors, resulting in treatment failure [181,182,183]. Consequently, there is a considerable focus on exploring combination therapies to enhance the effectiveness of treatments and broaden the scope of patients who can derive benefits from PARP inhibitors. One approach is to combine PARPi with agents that induce HR defects in tumors with intact HR function, thereby rendering them sensitive to PARP inhibition [184]. Additionally, strategies that block DNA repair pathways by inducing hypoxia or interfering with DNA damage cell cycle checkpoints have been explored to enhance the effectiveness of PARPi. Examples include inhibitors targeting signaling through the PI3K pathway and cell cycle checkpoints [184]. The upregulation of MYC has been linked to the activation of the homologous recombination DNA repair pathway, including the increased expression of RAD51 [60,61,93,94,185]. Indeed, it has been demonstrated that these molecular alterations, specifically upregulated RAD51 expression, contribute to the development of resistance to PARP inhibitors in cells with defective BRCA1 [179]. Recent evidence has shown that inhibiting the downstream signaling of MYC through cyclin-dependent kinase (CDK) inhibitors can enhance cancer cell sensitivity to PARP inhibitors, regardless of the BRCA status, in triple-negative breast cancer (TNBC) [180]. Despite these observations, the specific role of MYC in regulating DNA repair mechanisms and its impact on therapy response have often been overlooked. This intriguing association between MYC overexpression, enhanced HR DNA repair and resistance to DNA-damaging agents, such as PARPi, has prompted the need for further investigation.
9. Discussion and Future Directions
In conclusion, MYC is a multifunctional nuclear phosphoprotein that plays a critical role in regulating various cellular processes, including cell growth, the cell cycle, differentiation, metabolism and DNA repair. MYC alterations are prevalent in human cancers, contributing to tumorigenesis and drug resistance. Even though oncogenic MYC leads to the production of rapidly dividing cancer cells and an increase in genomic instability, one would expect these cells to be more vulnerable to DNA-damaging therapy. However, the opposite happens; high-MYC cancer cells exhibit prolonged survival even after DNA-damaging chemotherapy [21,23,186]. This evidence strongly indicates that oncogenic MYC in cancer cells triggers the development of a highly efficient DNA repair system, effectively countering the genomic damage induced by rapid proliferation, as well as chemotherapy and radiation therapy [89]. Understanding MYC-induced DNA repair mechanisms may offer opportunities to enhance cancer cell sensitivity to DNA-damaging agents and overcome drug resistance.
Moreover, MYC’s impact extends beyond cell-intrinsic signaling, influencing the tumor microenvironment by promoting angiogenesis, immune evasion and the recruitment of immunosuppressive cells. Targeting MYC in cancer cells and the tumor microenvironment may provide a promising strategy for anticancer therapy and improving treatment outcomes. Despite the challenges in targeting MYC directly, emerging pharmaceutical-based approaches offer hope for inhibiting MYC expression and function as well as impeding tumor growth. Numerous studies have consistently demonstrated that targeting MYC activity or expression results in a notable reduction in tumor growth across diverse preclinical tumor models [20,21,46,187,188]. Notably, emerging evidence highlights the impact of MYC deregulation in suppressing the immune response to tumors, whereas inhibiting MYC activity shows promise in stimulating an anti-tumor response [55,99,100,133,134,135]. Future research should focus on establishing the broader potential of MYC inhibitors in enhancing anti-tumor immunity. Continued research in this area is vital for advancing our understanding of MYC’s role in cancer and developing effective therapeutic interventions targeting this master oncoprotein.
Future directions in MYC-related research encompass various aspects aimed at deepening our understanding of its role in cancer and harnessing its potential for therapeutic applications:
- Unraveling DNA Repair Mechanisms: Investigating the precise mechanisms underlying MYC-induced DNA repair can unveil novel vulnerabilities in cancer cells. This knowledge could lead to the development of strategies that sensitize high-MYC cancer cells to DNA-damaging agents, ultimately overcoming drug resistance.
- Microenvironment Modulation: Further exploring how MYC impacts the tumor microenvironment, especially its influence on immune evasion and angiogenesis, can provide insights for designing therapies that not only target cancer cells but also disrupt the supportive network around them. This could potentially enhance the effectiveness of anticancer treatments.
- Refining MYC Inhibitors: Despite challenges, refining pharmaceutical-based approaches to inhibit MYC expression and function remains a promising avenue. Future research could focus on designing more potent and selective MYC inhibitors that effectively halt its oncogenic effects, leading to improved outcomes in cancer treatment.
- Immunomodulation Strategies: Understanding the interplay between MYC deregulation, immune suppression and anti-tumor immunity is critical. Exploring the potential of MYC inhibitors to enhance anti-tumor immune responses could open up new avenues for immunomodulatory therapies.
- Patient-Derived Models: Utilizing patient-derived models, such as organoids and xenografts, can offer more clinically relevant insights into MYC-targeted therapies and help bridge the gap between laboratory research and clinical application.
- Clinical Translations: Transitioning findings from preclinical models to clinical settings is vital. The rigorous testing of MYC inhibitors in clinical trials across different cancer types can help us evaluate their safety, efficacy and potential to improve patient outcomes.
- Combination Therapies: Exploring combination therapies that integrate MYC inhibition with existing treatments, such as DNA-damaging agents or immunotherapies, might offer synergistic effects and enhance therapeutic responses. Identifying optimal combinations is a promising avenue for future investigations.
In summary, future research endeavors should focus on gaining a deeper understanding of MYC’s multifaceted roles in cancer biology and translating these insights into innovative therapeutic strategies. This includes refining existing approaches, exploring combinatorial treatments and uncovering novel facets of MYC’s influence on tumor progression and the immune response. Such efforts are promising for advancing cancer treatment and improving patient outcomes.
Author Contributions
Z.O.D. wrote the manuscript and R.C.S. contributed to writing, and provided valuable feedback. Both authors read and approved the final manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
Rosalie C. Sears was funded by R01 CA186241, U01 CA224012, National Cancer Institute U54CA209988 and Brenden-Colson Center foundation. Zinab O. Doha was funded by the Saudi Arabian Cultural Mission (SACM) to the United States and Taibah University in Medina, Saudi Arabia.
Conflicts of Interest
Zinab O. Doha declares no conflict of interest. Rosalie C. Sears: Consultant: Larkspur Biosciences. Scientific Advisory Board: RAPPTA Therapeutics. Sponsored Research Support: Cardiff Oncology, Astra Zeneca Partner of Choice grant award.
References
- Beaulieu, M.-E.; Castillo, F.; Soucek, L. Structural and Biophysical Insights into the Function of the Intrinsically Disordered Myc Oncoprotein. Cells 2020, 9, 1038. [Google Scholar] [CrossRef]
- Duffy, M.J.; O’Grady, S.; Tang, M.; Crown, J. MYC as a target for cancer treatment. Cancer Treat. Rev. 2021, 94, 102154. [Google Scholar] [CrossRef]
- Gabay, M.; Li, Y.; Felsher, D.W. MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harb. Perspect. Med. 2014, 4, a014241. [Google Scholar] [CrossRef]
- Meyer, N.; Penn, L.Z. Reflecting on 25 years with MYC. Nat. Rev. Cancer 2008, 8, 976–990. [Google Scholar] [CrossRef]
- Dang, C.V. A time for MYC: Metabolism and therapy. In Cold Spring Harbor Symposia on Quantitative Biology; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2016. [Google Scholar]
- Conacci-Sorrell, M.; McFerrin, L.; Eisenman, R.N. An overview of MYC and its interactome. Cold Spring Harb. Perspect. Med. 2014, 4, a014357. [Google Scholar] [CrossRef]
- Carroll, P.A.; Freie, B.W.; Mathsyaraja, H.; Eisenman, R.N. The MYC transcription factor network: Balancing metabolism, proliferation and oncogenesis. Front. Med. 2018, 12, 412–425. [Google Scholar] [CrossRef]
- Schaub, F.X.; Dhankani, V.; Berger, A.C.; Trivedi, M.; Richardson, A.B.; Shaw, R.; Zhao, W.; Zhang, X.; Ventura, A.; Liu, Y. Pan-cancer alterations of the MYC oncogene and its proximal network across the cancer genome atlas. Cell Syst. 2018, 6, 282–300.e282. [Google Scholar] [CrossRef]
- Casey, S.C.; Baylot, V.; Felsher, D.W. The MYC oncogene is a global regulator of the immune response. Blood 2018, 131, 2007–2015. [Google Scholar] [CrossRef]
- Mariani-Costantini, R.; Escot, C.; Theillet, C.; Gentile, A.; Merlo, G.; Lidereau, R.; Callahan, R. In situ c-myc expression and genomic status of the c-myc locus in infiltrating ductal carcinomas of the breast. Cancer Res. 1988, 48, 199–205. [Google Scholar]
- Sears, R.; Leone, G.; DeGregori, J.; Nevins, J.R. Ras enhances Myc protein stability. Mol. Cell 1999, 3, 169–179. [Google Scholar] [CrossRef]
- Nussinov, R.; Tsai, C.-J.; Jang, H.; Korcsmáros, T.; Csermely, P. Oncogenic KRAS signaling and YAP1/β-catenin: Similar cell cycle control in tumor initiation. In Seminars in Cell & Developmental Biology; Elsevier: Amsterdam, The Netherlands, 2016. [Google Scholar]
- Pippa, R.; Odero, M.D. The Role of MYC and PP2A in the Initiation and Progression of Myeloid Leukemias. Cells 2020, 9, 544. [Google Scholar] [CrossRef] [PubMed]
- Sears, R.; Nuckolls, F.; Haura, E.; Taya, Y.; Tamai, K.; Nevins, J.R. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev. 2000, 14, 2501–2514. [Google Scholar] [CrossRef]
- Yeh, E.; Cunningham, M.; Arnold, H.; Chasse, D.; Monteith, T.; Ivaldi, G.; Hahn, W.C.; Stukenberg, P.T.; Shenolikar, S.; Uchida, T. A signalling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nat. Cell Biol. 2004, 6, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, J.W.; Zhao, X.; De Cecco, M.; Peterson, A.L.; Pagliaroli, L.; Manivannan, J.; Hubbard, G.B.; Ikeno, Y.; Zhang, Y.; Feng, B. Reduced expression of MYC increases longevity and enhances healthspan. Cell 2015, 160, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Evan, G.I.; Wyllie, A.H.; Gilbert, C.S.; Littlewood, T.D.; Land, H.; Brooks, M.; Waters, C.M.; Penn, L.Z.; Hancock, D.C. Induction of apoptosis in fibroblasts by c-myc protein. Cell 1992, 69, 119–128. [Google Scholar] [CrossRef]
- Nilsson, J.A.; Cleveland, J.L. Myc pathways provoking cell suicide and cancer. Oncogene 2003, 22, 9007–9021. [Google Scholar] [CrossRef]
- Murphy, D.J.; Junttila, M.R.; Pouyet, L.; Karnezis, A.; Shchors, K.; Bui, D.A.; Brown-Swigart, L.; Johnson, L.; Evan, G.I. Distinct thresholds govern Myc’s biological output in vivo. Cancer Cell 2008, 14, 447–457. [Google Scholar] [CrossRef]
- Risom, T.; Wang, X.; Liang, J.; Zhang, X.; Pelz, C.; Campbell, L.G.; Eng, J.; Chin, K.; Farrington, C.; Narla, G.; et al. Deregulating MYC in a model of HER2+ breast cancer mimics human intertumoral heterogeneity. J. Clin. Investig. 2020, 130, 231–246. [Google Scholar] [CrossRef]
- Farrell, A.S.; Joly, M.M.; Allen-Petersen, B.L.; Worth, P.J.; Lanciault, C.; Sauer, D.; Link, J.; Pelz, C.; Heiser, L.M.; Morton, J.P.; et al. MYC regulates ductal-neuroendocrine lineage plasticity in pancreatic ductal adenocarcinoma associated with poor outcome and chemoresistance. Nat. Commun. 2017, 8, 1728. [Google Scholar] [CrossRef]
- Mahauad-Fernandez, W.D.; Rakhra, K.; Felsher, D.W. Generation of a Tetracycline Regulated Mouse Model of MYC-Induced T-Cell Acute Lymphoblastic Leukemia. Methods Mol. Biol. 2021, 2318, 297–312. [Google Scholar]
- Doha, Z.O.; Wang, X.; Calistri, N.; Eng, J.; Daniel, C.J.; Ternes, L.; Kim, E.N.; Pelz, C.; Munks, M.; Betts, C.; et al. A Novel Mouse Model that Recapitulates the Heterogeneity of Human Triple Negative Breast Cancer. bioRxiv 2022. [Google Scholar] [CrossRef]
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010, 463, 899–905. [Google Scholar] [CrossRef] [PubMed]
- Korangath, P.; Teo, W.W.; Sadik, H.; Han, L.; Mori, N.; Huijts, C.M.; Wildes, F.; Bharti, S.; Zhang, Z.; Santa-Maria, C.A. Targeting Glutamine Metabolism in Breast Cancer with AminooxyacetateTargeting Glutamine Metabolism in Breast Cancer. Clin. Cancer Res. 2015, 21, 3263–3273. [Google Scholar] [CrossRef] [PubMed]
- Shachaf, C.M.; Kopelman, A.M.; Arvanitis, C.; Karlsson, Å.; Beer, S.; Mandl, S.; Bachmann, M.H.; Borowsky, A.D.; Ruebner, B.; Cardiff, R.D. MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature 2004, 431, 1112–1117. [Google Scholar] [CrossRef]
- Ben-David, E.; Bester, A.C.; Shifman, S.; Kerem, B. Transcriptional Dynamics in Colorectal Carcinogenesis: New Insights into the Role of c-Myc and miR17 in Benign to Cancer TransformationTranscriptional Dynamics in Colorectal Carcinogenesis. Cancer Res. 2014, 74, 5532–5540. [Google Scholar] [CrossRef]
- Koh, C.M.; Gurel, B.; Sutcliffe, S.; Aryee, M.J.; Schultz, D.; Iwata, T.; Uemura, M.; Zeller, K.I.; Anele, U.; Zheng, Q. Alterations in nucleolar structure and gene expression programs in prostatic neoplasia are driven by the MYC oncogene. Am. J. Pathol. 2011, 178, 1824–1834. [Google Scholar] [CrossRef]
- AlSultan, D.; Kavanagh, E.; O’Grady, S.; Eustace, A.J.; Castell, A.; Larsson, L.-G.; Crown, J.; Madden, S.F.; Duffy, M.J. The novel low molecular weight MYC antagonist MYCMI-6 inhibits proliferation and induces apoptosis in breast cancer cells. Investig. New Drugs 2021, 39, 587–594. [Google Scholar] [CrossRef]
- Levens, D. You don’t muck with MYC. Genes Cancer 2010, 1, 547–554. [Google Scholar] [CrossRef]
- Farrell, A.S.; Sears, R.C. MYC degradation. Cold Spring Harb. Perspect. Med. 2014, 4, a014365. [Google Scholar] [CrossRef]
- Hann, S.R. Role of post-translational modifications in regulating c-Myc proteolysis, transcriptional activity and biological function. In Seminars in Cancer Biology; Elsevier: Amsterdam, The Netherlands, 2006. [Google Scholar]
- Sun, X.-X.; Li, Y.; Sears, R.C.; Dai, M.-S. Targeting the MYC ubiquitination-proteasome degradation pathway for cancer therapy. Front. Oncol. 2021, 11, 679445. [Google Scholar] [CrossRef]
- Farrell, A.S.; Pelz, C.; Wang, X.; Daniel, C.J.; Wang, Z.; Su, Y.; Janghorban, M.; Zhang, X.; Morgan, C.; Impey, S. Pin1 regulates the dynamics of c-Myc DNA binding to facilitate target gene regulation and oncogenesis. Mol. Cell. Biol. 2013, 33, 2930–2949. [Google Scholar] [CrossRef]
- Benassi, B.; Fanciulli, M.; Fiorentino, F.; Porrello, A.; Chiorino, G.; Loda, M.; Zupi, G.; Biroccio, A. c-Myc phosphorylation is required for cellular response to oxidative stress. Mol. Cell 2006, 21, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Arnold, H.K.; Zhang, X.; Daniel, C.J.; Tibbitts, D.; Escamilla-Powers, J.; Farrell, A.; Tokarz, S.; Morgan, C.; Sears, R.C. The Axin1 scaffold protein promotes formation of a degradation complex for c-Myc. EMBO J. 2009, 28, 500–512. [Google Scholar] [CrossRef] [PubMed]
- Gregory, M.A.; Hann, S.R. c-Myc proteolysis by the ubiquitin-proteasome pathway: Stabilization of c-Myc in Burkitt’s lymphoma cells. Mol. Cell. Biol. 2000, 20, 2423–2435. [Google Scholar] [CrossRef] [PubMed]
- Cross, D.A.; Alessi, D.R.; Cohen, P.; Andjelkovich, M.; Hemmings, B.A. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 1995, 378, 785–789. [Google Scholar] [CrossRef]
- Hemmings, B.A.; Restuccia, D.F. PI3K-PKB/Akt pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011189. [Google Scholar] [CrossRef]
- Eichhorn, P.J.; Creyghton, M.P.; Bernards, R. Protein phosphatase 2A regulatory subunits and cancer. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2009, 1795, 1–15. [Google Scholar] [CrossRef]
- Ruvolo, P.P. The broken “Off” switch in cancer signaling: PP2A as a regulator of tumorigenesis, drug resistance, and immune surveillance. BBA Clin. 2016, 6, 87–99. [Google Scholar] [CrossRef]
- Zhang, L.; Zhou, H.; Li, X.; Vartuli, R.L.; Rowse, M.; Xing, Y.; Rudra, P.; Ghosh, D.; Zhao, R.; Ford, H.L. Eya3 partners with PP2A to induce c-Myc stabilization and tumor progression. Nat. Commun. 2018, 9, 1047. [Google Scholar] [CrossRef]
- Arnold, H.K.; Sears, R.C. Protein phosphatase 2A regulatory subunit B56α associates with c-Myc and negatively regulates c-Myc accumulation. Mol. Cell. Biol. 2006, 26, 2832–2844. [Google Scholar] [CrossRef]
- Lin, C.-F.; Chen, C.-L.; Chiang, C.-W.; Jan, M.-S.; Huang, W.-C.; Lin, Y.-S. GSK-3β acts downstream of PP2A and the PI 3-kinase-Akt pathway, and upstream of caspase-2 in ceramide-induced mitochondrial apoptosis. J. Cell Sci. 2007, 120, 2935–2943. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, W.; Weiss, W. Myc proteins as therapeutic targets. Oncogene 2010, 29, 1249–1259. [Google Scholar] [CrossRef] [PubMed]
- Allen-Petersen, B.L.; Sears, R.C. Mission possible: Advances in MYC therapeutic targeting in cancer. BioDrugs 2019, 33, 539–553. [Google Scholar] [CrossRef] [PubMed]
- Land, H.; Parada, L.F.; Weinberg, R.A. Tumorigenic conversion of primary embryo fibroblasts requires at least two cooperating oncogenes. Nature 1983, 304, 596–602. [Google Scholar] [CrossRef] [PubMed]
- Born, T.L.; Frost, J.A.; Schönthal, A.; Prendergast, G.C.; Feramisco, J.R. c-Myc cooperates with activated Ras to induce the cdc2 promoter. Mol. Cell. Biol. 1994, 14, 5710–5718. [Google Scholar]
- Leone, G.; DeGregori, J.; Sears, R.; Jakoi, L.; Nevins, J.R. Myc and Ras collaborate in inducing accumulation of active cyclin E/Cdk2 and E2F. Nature 1997, 387, 422–426. [Google Scholar] [CrossRef]
- Wang, C.; Lisanti, M.P.; Liao, D.J. Reviewing once more the c-myc and Ras collaboration: Converging at the cyclin D1-CDK4 complex and challenging basic concepts of cancer biology. Cell Cycle 2011, 10, 57–67. [Google Scholar] [CrossRef]
- Evan, G.; Littlewood, T. A matter of life and cell death. Science 1998, 281, 1317–1322. [Google Scholar] [CrossRef]
- Vaqué, J.P.; Navascues, J.; Shiio, Y.; Laiho, M.; Ajenjo, N.; Mauleon, I.; Matallanas, D.; Crespo, P.; León, J. Myc antagonizes Ras-mediated growth arrest in leukemia cells through the inhibition of the Ras-ERK-p21Cip1 pathway. J. Biol. Chem. 2005, 280, 1112–1122. [Google Scholar] [CrossRef]
- Tsuneoka, M.; Mekada, E. Ras/MEK signaling suppresses Myc-dependent apoptosis in cells transformed by c-myc and activated ras. Oncogene 2000, 19, 115–123. [Google Scholar] [CrossRef]
- Dong, J.; Sutor, S.; Jiang, G.; Cao, Y.; Asmann, Y.W.; Wigle, D.A. c-Myc regulates self-renewal in bronchoalveolar stem cells. PLoS ONE 2011, 6, e23707. [Google Scholar] [CrossRef]
- Kortlever, R.M.; Sodir, N.M.; Wilson, C.H.; Burkhart, D.L.; Pellegrinet, L.; Brown Swigart, L.; Littlewood, T.D.; Evan, G.I. Myc Cooperates with Ras by Programming Inflammation and Immune Suppression. Cell 2017, 171, 1301–1315.e1314. [Google Scholar] [CrossRef] [PubMed]
- Alexander, W.S.; Adams, J.M.; Cory, S. Oncogene cooperation in lymphocyte transformation: Malignant conversion of E mu-myc transgenic pre-B cells in vitro is enhanced by vH-ras or v-raf but not v-abl. Mol. Cell. Biol. 1989, 9, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Podsypanina, K.; Politi, K.; Beverly, L.J.; Varmus, H.E. Oncogene cooperation in tumor maintenance and tumor recurrence in mouse mammary tumors induced by Myc and mutant Kras. Proc. Natl. Acad. Sci. USA 2008, 105, 5242–5247. [Google Scholar] [CrossRef]
- Andres, A.-C.; van der Valk, M.A.; Schönenberger, C.; Flückiger, F.; LeMeur, M.; Gerlinger, P.; Groner, B. Ha-ras and c-myc oncogene expression interferes with morphological and functional differentiation of mammary epithelial cells in single and double transgenic mice. Genes Dev. 1988, 2, 1486–1495. [Google Scholar] [CrossRef] [PubMed]
- Kuzyk, A.; Mai, S. c-MYC-induced genomic instability. Cold Spring Harb. Perspect. Med. 2014, 4, a014373. [Google Scholar] [CrossRef] [PubMed]
- Kumari, A.; Folk, W.P.; Sakamuro, D. The Dual Roles of MYC in Genomic Instability and Cancer Chemoresistance. Genes 2017, 8, 158. [Google Scholar] [CrossRef]
- McMahon, S.B. MYC and the control of apoptosis. Cold Spring Harb. Perspect. Med. 2014, 4, a014407. [Google Scholar] [CrossRef]
- Curti, L.; Campaner, S. MYC-induced replicative stress: A double-edged sword for cancer development and treatment. Int. J. Mol. Sci. 2021, 22, 6168. [Google Scholar] [CrossRef]
- Cerni, C.; Mougneau, E.; Zerlin, M.; Julius, M.; Marcu, K.; Cuzin, F. c-myc and functionally related oncogenes induce both high rates of sister chromatid exchange and abnormal karyotypes in rat fibroblasts. In Mechanisms in B-Cell Neoplasia, Proceedings of the National Cancer Institute, National Institutes of Health, Bethesda, MD, USA, 24–26 March 1986; Springer: Cham, Switzerland, 1986. [Google Scholar]
- Dominguez-Sola, D.; Ying, C.Y.; Grandori, C.; Ruggiero, L.; Chen, B.; Li, M.; Galloway, D.A.; Gu, W.; Gautier, J.; Dalla-Favera, R. Non-transcriptional control of DNA replication by c-Myc. Nature 2007, 448, 445–451. [Google Scholar] [CrossRef]
- Bretones, G.; Delgado, M.D.; León, J. Myc and cell cycle control. Biochim. Biophys. Acta (BBA)-Gene Regul. Mech. 2015, 1849, 506–516. [Google Scholar] [CrossRef] [PubMed]
- Dong, P.; Maddali, M.V.; Srimani, J.K.; Thélot, F.; Nevins, J.R.; Mathey-Prevot, B.; You, L. Division of labour between Myc and G1 cyclins in cell cycle commitment and pace control. Nat. Commun. 2014, 5, 4750. [Google Scholar] [CrossRef] [PubMed]
- Sears, R.; Ohtani, K.; Nevins, J.R. Identification of positively and negatively acting elements regulating expression of the E2F2 gene in response to cell growth signals. Mol. Cell. Biol. 1997, 17, 5227–5235. [Google Scholar] [CrossRef] [PubMed]
- Huppi, K.; Volfovsky, N.; Runfola, T.; Jones, T.L.; Mackiewicz, M.; Martin, S.E.; Mushinski, J.F.; Stephens, R.; Caplen, N.J. The identification of microRNAs in a genomically unstable region of human chromosome 8q24. Mol. Cancer Res. 2008, 6, 212–221. [Google Scholar] [CrossRef]
- Huppi, K.; Pitt, J.; Wahlberg, B.; Caplen, N.J. Genomic instability and mouse microRNAs. Toxicol. Mech. Methods 2011, 21, 325–333. [Google Scholar] [CrossRef]
- Mai, S.; Fluri, M.; Siwarski, D.; Huppi, K. Genomic instability in MycER-activated Rat1A-MycER cells. Chromosome Res. 1996, 4, 365–371. [Google Scholar] [CrossRef]
- Kuttler, F.; Mai, S. Formation of non-random extrachromosomal elements during development, differentiation and oncogenesis. In Seminars in Cancer Biology; Elsevier: Amsterdam, The Netherlands, 2007. [Google Scholar]
- Stine, Z.E.; Walton, Z.E.; Altman, B.J.; Hsieh, A.L.; Dang, C.V. MYC, Metabolism, and Cancer. Cancer Discov. 2015, 5, 1024–1039. [Google Scholar] [CrossRef]
- Campaner, S.; Amati, B. Two sides of the Myc-induced DNA damage response: From tumor suppression to tumor maintenance. Cell Div. 2012, 7, 6. [Google Scholar] [CrossRef]
- Kim, J.-w.; Zeller, K.I.; Wang, Y.; Jegga, A.G.; Aronow, B.J.; O’Donnell, K.A.; Dang, C.V. Evaluation of myc E-box phylogenetic footprints in glycolytic genes by chromatin immunoprecipitation assays. Mol. Cell. Biol. 2004, 24, 5923–5936. [Google Scholar] [CrossRef]
- Gao, P.; Tchernyshyov, I.; Chang, T.-C.; Lee, Y.-S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458, 762–765. [Google Scholar] [CrossRef]
- Edmunds, L.R.; Sharma, L.; Kang, A.; Lu, J.; Vockley, J.; Basu, S.; Uppala, R.; Goetzman, E.S.; Beck, M.E.; Scott, D. c-Myc programs fatty acid metabolism and dictates acetyl-CoA abundance and fate. J. Biol. Chem. 2014, 289, 25382–25392. [Google Scholar] [CrossRef] [PubMed]
- De Zio, D.; Cianfanelli, V.; Cecconi, F. New insights into the link between DNA damage and apoptosis. Antioxid. Redox Signal 2013, 19, 559–571. [Google Scholar] [CrossRef] [PubMed]
- Vafa, O.; Wade, M.; Kern, S.; Beeche, M.; Pandita, T.K.; Hampton, G.M.; Wahl, G.M. c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: A mechanism for oncogene-induced genetic instability. Mol. cell 2002, 9, 1031–1044. [Google Scholar] [CrossRef] [PubMed]
- Khanna, K.K.; Jackson, S.P. DNA double-strand breaks: Signaling, repair and the cancer connection. Nat. Genet. 2001, 27, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Bartkova, J.; Rezaei, N.; Liontos, M.; Karakaidos, P.; Kletsas, D.; Issaeva, N.; Vassiliou, L.-V.F.; Kolettas, E.; Niforou, K.; Zoumpourlis, V.C. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 2006, 444, 633–637. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Cunningham, M.; Zhang, X.; Tokarz, S.; Laraway, B.; Troxell, M.; Sears, R.C. Phosphorylation regulates c-Myc’s oncogenic activity in the mammary gland. Cancer Res. 2011, 71, 925–936. [Google Scholar] [CrossRef]
- Dingar, D.; Tu, W.B.; Resetca, D.; Lourenco, C.; Tamachi, A.; De Melo, J.; Houlahan, K.E.; Kalkat, M.; Chan, P.K.; Boutros, P.C.; et al. MYC dephosphorylation by the PP1/PNUTS phosphatase complex regulates chromatin binding and protein stability. Nat. Commun. 2018, 9, 3502. [Google Scholar] [CrossRef]
- Amati, B.; Brooks, M.W.; Levy, N.; Littlewood, T.D.; Evan, G.I.; Land, H. Oncogenic activity of the c-Myc protein requires dimerization with Max. Cell 1993, 72, 233–245. [Google Scholar] [CrossRef]
- Solvie, D.; Baluapuri, A.; Uhl, L.; Fleischhauer, D.; Endres, T.; Papadopoulos, D.; Aziba, A.; Gaballa, A.; Mikicic, I.; Isaakova, E.; et al. MYC multimers shield stalled replication forks from RNA polymerase. Nature 2022, 612, 148–155. [Google Scholar] [CrossRef]
- Kalkat, M.; Resetca, D.; Lourenco, C.; Chan, P.K.; Wei, Y.; Shiah, Y.J.; Vitkin, N.; Tong, Y.; Sunnerhagen, M.; Done, S.J.; et al. MYC Protein Interactome Profiling Reveals Functionally Distinct Regions that Cooperate to Drive Tumorigenesis. Mol. Cell 2018, 72, 836–848.e837. [Google Scholar] [CrossRef]
- Su, Y.; Pelz, C.; Huang, T.; Torkenczy, K.; Wang, X.; Cherry, A.; Daniel, C.J.; Liang, J.; Nan, X.; Dai, M.S.; et al. Post-translational modification localizes MYC to the nuclear pore basket to regulate a subset of target genes involved in cellular responses to environmental signals. Genes Dev. 2018, 32, 1398–1419. [Google Scholar] [CrossRef] [PubMed]
- Krenning, L.; van den Berg, J.; Medema, R.H. Life or Death after a Break: What Determines the Choice? Mol. Cell 2019, 76, 346–358. [Google Scholar] [CrossRef] [PubMed]
- Eischen, C.M.; Weber, J.D.; Roussel, M.F.; Sherr, C.J.; Cleveland, J.L. Disruption of the ARF-Mdm2-p53 tumor suppressor pathway in Myc-induced lymphomagenesis. Genes Dev. 1999, 13, 2658–2669. [Google Scholar] [CrossRef] [PubMed]
- Gravina, G.L.; Festuccia, C.; Popov, V.M.; Di Rocco, A.; Colapietro, A.; Sanità, P.; Monache, S.D.; Musio, D.; De Felice, F.; Di Cesare, E.; et al. c-Myc Sustains Transformed Phenotype and Promotes Radioresistance of Embryonal Rhabdomyosarcoma Cell Lines. Radiat. Res. 2016, 185, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Pyndiah, S.; Tanida, S.; Ahmed, K.M.; Cassimere, E.K.; Choe, C.; Sakamuro, D. c-MYC suppresses BIN1 to release poly (ADP-ribose) polymerase 1: A mechanism by which cancer cells acquire cisplatin resistance. Sci. Signal. 2011, 4, ra19. [Google Scholar] [CrossRef]
- Walker, T.; White, J.; Esdale, W.; Burton, M.; DeCruz, E. Tumour cells surviving in vivo cisplatin chemotherapy display elevated c-myc expression. Br. J. Cancer 1996, 73, 610–614. [Google Scholar] [CrossRef]
- Leonetti, C.; Biroccio, A.; Benassi, B.; Stringaro, A.; Stoppacciaro, A.; Semple, S.C.; Zupi, G. Encapsulation of c-myc antisense oligodeoxynucleotides in lipid particles improves antitumoral efficacy in vivo in a human melanoma line. Cancer Gene Ther. 2001, 8, 459–468. [Google Scholar] [CrossRef]
- Luoto, K.R.; Meng, A.X.; Wasylishen, A.R.; Zhao, H.; Coackley, C.L.; Penn, L.Z.; Bristow, R.G. Tumor cell kill by c-MYC depletion: Role of MYC-regulated genes that control DNA double-strand break repair. Cancer Res. 2010, 70, 8748–8759. [Google Scholar] [CrossRef]
- Cui, F.; Fan, R.; Chen, Q.; He, Y.; Song, M.; Shang, Z.; Zhang, S.; Zhu, W.; Cao, J.; Guan, H. The involvement of c-Myc in the DNA double-strand break repair via regulating radiation-induced phosphorylation of ATM and DNA-PKcs activity. Mol. Cell. Biochem. 2015, 406, 43–51. [Google Scholar] [CrossRef]
- Sodir, N.M.; Pellegrinet, L.; Kortlever, R.M.; Campos, T.; Kwon, Y.-W.; Kim, S.; Garcia, D.; Perfetto, A.; Anastasiou, P.; Swigart, L.B.; et al. Reversible Myc hypomorphism identifies a key Myc-dependency in early cancer evolution. Nat. Commun. 2022, 13, 6782. [Google Scholar] [CrossRef]
- Zheng, P.; Li, W. Crosstalk Between Mesenchymal Stromal Cells and Tumor-Associated Macrophages in Gastric Cancer. Front. Oncol. 2020, 10, 571516. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Weng, Z.; Zhou, X.; Xu, Z.; Cao, B.; Wang, B.; Li, J. Mesenchymal stromal cells promote the drug resistance of gastrointestinal stromal tumors by activating the PI3K-AKT pathway via TGF-β2. J. Transl. Med. 2023, 21, 219. [Google Scholar] [CrossRef] [PubMed]
- Meškytė, E.M.; Keskas, S.; Ciribilli, Y. MYC as a Multifaceted Regulator of Tumor Microenvironment Leading to Metastasis. Int. J. Mol. Sci. 2020, 21, 7710. [Google Scholar] [CrossRef]
- Sodir, N.M.; Kortlever, R.M.; Barthet, V.J.A.; Campos, T.; Pellegrinet, L.; Kupczak, S.; Anastasiou, P.; Swigart, L.B.; Soucek, L.; Arends, M.J.; et al. MYC Instructs and Maintains Pancreatic Adenocarcinoma Phenotype. Cancer Discov. 2020, 10, 588–607. [Google Scholar] [CrossRef] [PubMed]
- Sodir, N.M.; Swigart, L.B.; Karnezis, A.N.; Hanahan, D.; Evan, G.I.; Soucek, L. Endogenous Myc maintains the tumor microenvironment. Genes Dev. 2011, 25, 907–916. [Google Scholar] [CrossRef] [PubMed]
- Barrett, R.L.; Puré, E. Cancer-associated fibroblasts and their influence on tumor immunity and immunotherapy. Elife 2020, 9, e57243. [Google Scholar] [CrossRef]
- Beatty, G.L.; Li, Y.; Long, K.B. Cancer immunotherapy: Activating innate and adaptive immunity through CD40 agonists. Expert. Rev. Anticancer. Ther. 2017, 17, 175–186. [Google Scholar] [CrossRef]
- Pello, O.M.; De Pizzol, M.; Mirolo, M.; Soucek, L.; Zammataro, L.; Amabile, A.; Doni, A.; Nebuloni, M.; Swigart, L.B.; Evan, G.I. Role of c-MYC in alternative activation of human macrophages and tumor-associated macrophage biology. Blood J. Am. Soc. Hematol. 2012, 119, 411–421. [Google Scholar] [CrossRef]
- Yan, W.; Wu, X.; Zhou, W.; Fong, M.Y.; Cao, M.; Liu, J.; Liu, X.; Chen, C.-H.; Fadare, O.; Pizzo, D.P. Cancer-cell-secreted exosomal miR-105 promotes tumour growth through the MYC-dependent metabolic reprogramming of stromal cells. Nat. Cell Biol. 2018, 20, 597–609. [Google Scholar] [CrossRef]
- Soucek, L.; Lawlor, E.R.; Soto, D.; Shchors, K.; Swigart, L.B.; Evan, G.I. Mast cells are required for angiogenesis and macroscopic expansion of Myc-induced pancreatic islet tumors. Nat. Med. 2007, 13, 1211–1218. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Oon, C.; Kothari, A.; Horton, W.; Link, J.; Sears, R.C.; Sherman, M.H. Acidic fibroblast growth factor underlies microenvironmental regulation of MYC in pancreatic cancer. J. Exp. Med. 2020, 217, e20191805. [Google Scholar] [CrossRef] [PubMed]
- Shchors, K.; Shchors, E.; Rostker, F.; Lawlor, E.R.; Brown-Swigart, L.; Evan, G.I. The Myc-dependent angiogenic switch in tumors is mediated by interleukin 1β. Genes Dev. 2006, 20, 2527–2538. [Google Scholar] [CrossRef] [PubMed]
- Riabov, V.; Gudima, A.; Wang, N.; Mickley, A.; Orekhov, A.; Kzhyshkowska, J. Role of tumor associated macrophages in tumor angiogenesis and lymphangiogenesis. Front. Physiol. 2014, 5, 75. [Google Scholar] [CrossRef] [PubMed]
- Mundim, F.G.L.; Pasini, F.S.; Brentani, M.M.; Soares, F.A.; Nonogaki, S.; Waitzberg, A.F.L. MYC is expressed in the stromal and epithelial cells of primary breast carcinoma and paired nodal metastases. Mol. Clin. Oncol. 2015, 3, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Mezquita, P.; Parghi, S.S.; Brandvold, K.A.; Ruddell, A. Myc regulates VEGF production in B cells by stimulating initiation of VEGF mRNA translation. Oncogene 2005, 24, 889–901. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef]
- Baudino, T.A.; McKay, C.; Pendeville-Samain, H.; Nilsson, J.A.; Maclean, K.H.; White, E.L.; Davis, A.C.; Ihle, J.N.; Cleveland, J.L. c-Myc is essential for vasculogenesis and angiogenesis during development and tumor progression. Genes Dev. 2002, 16, 2530–2543. [Google Scholar] [CrossRef]
- Dang, C.V.; Le, A.; Gao, P. MYC-Induced Cancer Cell Energy Metabolism and Therapeutic Opportunities Targeting MYC-Induced Cancer Cell Energy. Clin. Cancer Res. 2009, 15, 6479–6483. [Google Scholar] [CrossRef]
- Pavlides, S.; Whitaker-Menezes, D.; Castello-Cros, R.; Flomenberg, N.; Witkiewicz, A.K.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S. The reverse Warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009, 8, 3984–4001. [Google Scholar] [CrossRef]
- Casacuberta-Serra, S.; Soucek, L. Myc and Ras, the Bonnie and Clyde of immune evasion. Transl. Cancer Res. 2018, 7, S457. [Google Scholar] [CrossRef]
- Wang, R.; Dillon, C.P.; Shi, L.Z.; Milasta, S.; Carter, R.; Finkelstein, D.; McCormick, L.L.; Fitzgerald, P.; Chi, H.; Munger, J. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 2011, 35, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Maddipati, R.; Norgard, R.J.; Baslan, T.; Rathi, K.S.; Zhang, A.; Saeid, A.; Higashihara, T.; Wu, F.; Kumar, A.; Annamalai, V.; et al. MYC Levels Regulate Metastatic Heterogeneity in Pancreatic Adenocarcinoma. Cancer Discov. 2022, 12, 542–561. [Google Scholar] [CrossRef]
- Liston, A.; Gray, D.H. Homeostatic control of regulatory T cell diversity. Nat. Rev. Immunol. 2014, 14, 154–165. [Google Scholar] [CrossRef] [PubMed]
- Kurniawan, H.; Soriano-Baguet, L.; Brenner, D. Regulatory T cell metabolism at the intersection between autoimmune diseases and cancer. Eur. J. Immunol. 2020, 50, 1626–1642. [Google Scholar] [CrossRef] [PubMed]
- Michalek, R.D.; Gerriets, V.A.; Jacobs, S.R.; Macintyre, A.N.; MacIver, N.J.; Mason, E.F.; Sullivan, S.A.; Nichols, A.G.; Rathmell, J.C. Cutting edge: Distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J. Immunol. 2011, 186, 3299–3303. [Google Scholar] [CrossRef]
- Li, J.; Dong, T.; Wu, Z.; Zhu, D.; Gu, H. The effects of MYC on tumor immunity and immunotherapy. Cell Death Discov. 2023, 9, 103. [Google Scholar] [CrossRef]
- Sarkar, T.; Dhar, S.; Sa, G. Tumor-infiltrating T-regulatory cells adapt to altered metabolism to promote tumor-immune escape. Curr. Res. Immunol. 2021, 2, 132–141. [Google Scholar] [CrossRef]
- Trikha, P.; Carson, W.E., 3rd. Signaling pathways involved in MDSC regulation. Biochim. Biophys. Acta 2014, 1846, 55–65. [Google Scholar]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef]
- Talmadge, J.E.; Donkor, M.; Scholar, E. Inflammatory cell infiltration of tumors: Jekyll or Hyde. Cancer Metastasis Rev. 2007, 26, 373–400. [Google Scholar] [CrossRef]
- Pello, O.M. Macrophages and c-Myc cross paths. Oncoimmunology 2016, 5, e1151991. [Google Scholar] [CrossRef] [PubMed]
- Jablonski, K.A.; Amici, S.A.; Webb, L.M.; Ruiz-Rosado, J.d.D.; Popovich, P.G.; Partida-Sanchez, S.; Guerau-de-Arellano, M. Novel markers to delineate murine M1 and M2 macrophages. PLoS ONE 2015, 10, e0145342. [Google Scholar] [CrossRef] [PubMed]
- Hadjidaniel, M.D.; Muthugounder, S.; Hung, L.T.; Sheard, M.A.; Shirinbak, S.; Chan, R.Y.; Nakata, R.; Borriello, L.; Malvar, J.; Kennedy, R.J. Tumor-associated macrophages promote neuroblastoma via STAT3 phosphorylation and up-regulation of c-MYC. Oncotarget 2017, 8, 91516. [Google Scholar] [CrossRef] [PubMed]
- Layer, J.P.; Kronmüller, M.T.; Quast, T.; van den Boorn-Konijnenberg, D.; Effern, M.; Hinze, D.; Althoff, K.; Schramm, A.; Westermann, F.; Peifer, M.; et al. Amplification of N-Myc is associated with a T-cell-poor microenvironment in metastatic neuroblastoma restraining interferon pathway activity and chemokine expression. Oncoimmunology 2017, 6, e1320626. [Google Scholar] [CrossRef]
- Liang, M.Q.; Yu, F.Q.; Chen, C. C-Myc regulates PD-L1 expression in esophageal squamous cell carcinoma. Am. J. Transl. Res. 2020, 12, 379–388. [Google Scholar]
- Zou, W.; Wolchok, J.D.; Chen, L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci. Transl. Med. 2016, 8, 328rv4. [Google Scholar] [CrossRef]
- Aguadé-Gorgorió, G.; Solé, R. Genetic instability as a driver for immune surveillance. J. ImmunoTherapy Cancer 2019, 7, 345. [Google Scholar] [CrossRef]
- Yang, C.; Liu, Y.; Hu, Y.; Fang, L.; Huang, Z.; Cui, H.; Xie, J.; Hong, Y.; Chen, W.; Xiao, N.; et al. Myc inhibition tips the immune balance to promote antitumor immunity. Cell Mol. Immunol. 2022, 19, 1030–1041. [Google Scholar] [CrossRef]
- Casey, S.C.; Li, Y.; Felsher, D.W. An essential role for the immune system in the mechanism of tumor regression following targeted oncogene inactivation. Immunol. Res. 2014, 58, 282–291. [Google Scholar] [CrossRef]
- Jiang, K.; Zhang, Q.; Fan, Y.; Li, J.; Zhang, J.; Wang, W.; Fan, J.; Guo, Y.; Liu, S.; Hao, D.; et al. MYC inhibition reprograms tumor immune microenvironment by recruiting T lymphocytes and activating the CD40/CD40L system in osteosarcoma. Cell Death Discov. 2022, 8, 117. [Google Scholar] [CrossRef]
- Topper, M.J.; Vaz, M.; Chiappinelli, K.B.; Shields, C.E.D.; Niknafs, N.; Yen, R.-W.C.; Wenzel, A.; Hicks, J.; Ballew, M.; Stone, M. Epigenetic therapy ties MYC depletion to reversing immune evasion and treating lung cancer. Cell 2017, 171, 1284–1300.e1221. [Google Scholar] [CrossRef] [PubMed]
- Felsher, D.W.; Bishop, J.M. Reversible tumorigenesis by MYC in hematopoietic lineages. Mol. cell 1999, 4, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Restifo, N.P. Can antitumor immunity help to explain “oncogene addiction”? Cancer Cell 2010, 18, 403–405. [Google Scholar] [CrossRef][Green Version]
- Li, X.; Tang, L.; Chen, Q.; Cheng, X.; Liu, Y.; Wang, C.; Zhu, C.; Xu, K.; Gao, F.; Huang, J.; et al. Inhibition of MYC suppresses programmed cell death ligand-1 expression and enhances immunotherapy in triple-negative breast cancer. Chin. Med. J. 2022, 135, 2436–2445. [Google Scholar] [CrossRef] [PubMed]
- Whitfield, J.R.; Beaulieu, M.E.; Soucek, L. Strategies to Inhibit Myc and Their Clinical Applicability. Front. Cell Dev. Biol. 2017, 5, 10. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Liu, H.; Qing, G. Targeting oncogenic Myc as a strategy for cancer treatment. Signal Transduct. Target. Ther. 2018, 3, 5. [Google Scholar] [CrossRef]
- Ahmadi, S.E.; Rahimi, S.; Zarandi, B.; Chegeni, R.; Safa, M. MYC: A multipurpose oncogene with prognostic and therapeutic implications in blood malignancies. J. Hematol. Oncol. 2021, 14, 121. [Google Scholar] [CrossRef]
- Whitfield, J.R.; Soucek, L. The long journey to bring a Myc inhibitor to the clinic. J. Cell Biol. 2021, 220, e202103090. [Google Scholar] [CrossRef]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.E.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 2011, 146, 904–917. [Google Scholar] [CrossRef]
- Puissant, A.; Frumm, S.M.; Alexe, G.; Bassil, C.F.; Qi, J.; Chanthery, Y.H.; Nekritz, E.A.; Zeid, R.; Gustafson, W.C.; Greninger, P.; et al. Targeting MYCN in neuroblastoma by BET bromodomain inhibition. Cancer Discov. 2013, 3, 308–323. [Google Scholar] [CrossRef]
- Garcia-Cuellar, M.P.; Füller, E.; Mäthner, E.; Breitinger, C.; Hetzner, K.; Zeitlmann, L.; Borkhardt, A.; Slany, R.K. Efficacy of cyclin-dependent-kinase 9 inhibitors in a murine model of mixed-lineage leukemia. Leukemia 2014, 28, 1427–1435. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Hurley, L.H. Structure of the biologically relevant G-quadruplex in the c-MYC promoter. Nucleosides Nucleotides Nucleic Acids 2006, 25, 951–968. [Google Scholar] [CrossRef] [PubMed]
- Brooks, T.A.; Kendrick, S.; Hurley, L. Making sense of G-quadruplex and i-motif functions in oncogene promoters. FEBS J. 2010, 277, 3459–3469. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.V.; Danford, F.L.; Gokhale, V.; Hurley, L.H.; Brooks, T.A. Demonstration that drug-targeted down-regulation of MYC in non-Hodgkins lymphoma is directly mediated through the promoter G-quadruplex. J. Biol. Chem. 2011, 286, 41018–41027. [Google Scholar] [CrossRef]
- Allen-Petersen, B.L.; Risom, T.; Feng, Z.; Wang, Z.; Jenny, Z.P.; Thoma, M.C.; Pelz, K.R.; Morton, J.P.; Sansom, O.J.; Lopez, C.D.; et al. Activation of PP2A and Inhibition of mTOR Synergistically Reduce MYC Signaling and Decrease Tumor Growth in Pancreatic Ductal Adenocarcinoma. Cancer Res. 2019, 79, 209–219. [Google Scholar] [CrossRef]
- Bjornsti, M.A.; Houghton, P.J. The TOR pathway: A target for cancer therapy. Nat. Rev. Cancer 2004, 4, 335–348. [Google Scholar] [CrossRef]
- Ogami, K.; Hosoda, N.; Funakoshi, Y.; Hoshino, S. Antiproliferative protein Tob directly regulates c-myc proto-oncogene expression through cytoplasmic polyadenylation element-binding protein CPEB. Oncogene 2014, 33, 55–64. [Google Scholar] [CrossRef]
- Fernández-Miranda, G.; Méndez, R. The CPEB-family of proteins, translational control in senescence and cancer. Ageing Res. Rev. 2012, 11, 460–472. [Google Scholar] [CrossRef]
- Yada, M.; Hatakeyama, S.; Kamura, T.; Nishiyama, M.; Tsunematsu, R.; Imaki, H.; Ishida, N.; Okumura, F.; Nakayama, K.; Nakayama, K.I. Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7. EMBO J. 2004, 23, 2116–2125. [Google Scholar] [CrossRef]
- Welcker, M.; Orian, A.; Jin, J.; Grim, J.E.; Harper, J.W.; Eisenman, R.N.; Clurman, B.E. The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proc. Natl. Acad. Sci. USA 2004, 101, 9085–9090. [Google Scholar] [CrossRef]
- Popov, N.; Wanzel, M.; Madiredjo, M.; Zhang, D.; Beijersbergen, R.; Bernards, R.; Moll, R.; Elledge, S.J.; Eilers, M. The ubiquitin-specific protease USP28 is required for MYC stability. Nat. Cell Biol. 2007, 9, 765–774. [Google Scholar] [CrossRef]
- Sun, X.X.; He, X.; Yin, L.; Komada, M.; Sears, R.C.; Dai, M.S. The nucleolar ubiquitin-specific protease USP36 deubiquitinates and stabilizes c-Myc. Proc. Natl. Acad. Sci. USA 2015, 112, 3734–3739. [Google Scholar] [CrossRef]
- Brockmann, M.; Poon, E.; Berry, T.; Carstensen, A.; Deubzer, H.E.; Rycak, L.; Jamin, Y.; Thway, K.; Robinson, S.P.; Roels, F.; et al. Small molecule inhibitors of aurora-a induce proteasomal degradation of N-myc in childhood neuroblastoma. Cancer Cell 2013, 24, 75–89. [Google Scholar] [CrossRef] [PubMed]
- Xiao, D.; Yue, M.; Su, H.; Ren, P.; Jiang, J.; Li, F.; Hu, Y.; Du, H.; Liu, H.; Qing, G. Polo-like Kinase-1 Regulates Myc Stabilization and Activates a Feedforward Circuit Promoting Tumor Cell Survival. Mol. Cell 2016, 64, 493–506. [Google Scholar] [CrossRef] [PubMed]
- Perrotti, D.; Neviani, P. Protein phosphatase 2A: A target for anticancer therapy. Lancet Oncol. 2013, 14, e229–e238. [Google Scholar] [CrossRef]
- Shah, V.M.; English, I.A.; Sears, R.C. Select Stabilization of a Tumor-Suppressive PP2A Heterotrimer. Trends Pharmacol. Sci. 2020, 41, 595–597. [Google Scholar] [CrossRef] [PubMed]
- Arnold, H.K.; Sears, R.C. A tumor suppressor role for PP2A-B56alpha through negative regulation of c-Myc and other key oncoproteins. Cancer Metastasis Rev. 2008, 27, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Sangodkar, J.; Perl, A.; Tohme, R.; Kiselar, J.; Kastrinsky, D.B.; Zaware, N.; Izadmehr, S.; Mazhar, S.; Wiredja, D.D.; O’Connor, C.M.; et al. Activation of tumor suppressor protein PP2A inhibits KRAS-driven tumor growth. J. Clin. Investig. 2017, 127, 2081–2090. [Google Scholar] [CrossRef] [PubMed]
- Leonard, D.; Huang, W.; Izadmehr, S.; O’Connor, C.M.; Wiredja, D.D.; Wang, Z.; Zaware, N.; Chen, Y.; Schlatzer, D.M.; Kiselar, J. Selective PP2A enhancement through biased heterotrimer stabilization. Cell 2020, 181, 688–701.e616. [Google Scholar] [CrossRef]
- Adhikary, S.; Eilers, M. Transcriptional regulation and transformation by Myc proteins. Nat. Rev. Mol. Cell Biol. 2005, 6, 635–645. [Google Scholar] [CrossRef]
- Wang, H.; Hammoudeh, D.I.; Follis, A.V.; Reese, B.E.; Lazo, J.S.; Metallo, S.J.; Prochownik, E.V. Improved low molecular weight Myc-Max inhibitors. Mol. Cancer Ther. 2007, 6, 2399–2408. [Google Scholar] [CrossRef] [PubMed]
- Berg, T.; Cohen, S.B.; Desharnais, J.; Sonderegger, C.; Maslyar, D.J.; Goldberg, J.; Boger, D.L.; Vogt, P.K. Small-molecule antagonists of Myc/Max dimerization inhibit Myc-induced transformation of chicken embryo fibroblasts. Proc. Natl. Acad. Sci. USA 2002, 99, 3830–3835. [Google Scholar] [CrossRef] [PubMed]
- Soucek, L.; Helmer-Citterich, M.; Sacco, A.; Jucker, R.; Cesareni, G.; Nasi, S. Design and properties of a Myc derivative that efficiently homodimerizes. Oncogene 1998, 17, 2463–2472. [Google Scholar] [CrossRef] [PubMed]
- Dockery, L.; Gunderson, C.; Moore, K. Rucaparib: The past, present, and future of a newly approved PARP inhibitor for ovarian cancer. OncoTargets Ther. 2017, 10, 3029. [Google Scholar] [CrossRef]
- Lok, B.H.; Gardner, E.E.; Schneeberger, V.E.; Ni, A.; Desmeules, P.; Rekhtman, N.; De Stanchina, E.; Teicher, B.A.; Riaz, N.; Powell, S.N. PARP inhibitor activity correlates with SLFN11 expression and demonstrates synergy with temozolomide in small cell lung cancer. Clin. Cancer Res. 2017, 23, 523–535. [Google Scholar] [CrossRef]
- Min, A.; Im, S.-A. PARP inhibitors as therapeutics: Beyond modulation of PARylation. Cancers 2020, 12, 394. [Google Scholar] [CrossRef]
- Illuzzi, G.; O’Connor, M.J.; Leo, E. A novel assay for PARP-DNA trapping provides insights into the mechanism of action (MoA) of clinical PARP inhibitors (PARPi). Cancer Res. 2019, 79, 2077. [Google Scholar] [CrossRef]
- Murai, J.; Huang, S.-y.N.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef]
- Murai, J.; Huang, S.-Y.N.; Renaud, A.; Zhang, Y.; Ji, J.; Takeda, S.; Morris, J.; Teicher, B.; Doroshow, J.H.; Pommier, Y. Stereospecific PARP trapping by BMN 673 and comparison with olaparib and rucaparib. Mol. Cancer Ther. 2014, 13, 433–443. [Google Scholar] [CrossRef]
- Rose, M.; Burgess, J.T.; O’Byrne, K.; Richard, D.J.; Bolderson, E. PARP Inhibitors: Clinical Relevance, Mechanisms of Action and Tumor Resistance. Front. Cell Dev. Biol. 2020, 8, 564601. [Google Scholar] [CrossRef]
- Pishvaian, M.J.; Biankin, A.V.; Bailey, P.; Chang, D.K.; Laheru, D.; Wolfgang, C.L.; Brody, J.R. BRCA2 secondary mutation-mediated resistance to platinum and PARP inhibitor-based therapy in pancreatic cancer. Br. J. Cancer 2017, 116, 1021–1026. [Google Scholar] [CrossRef] [PubMed]
- Chand, S.; O’Hayer, K.; Blanco, F.F.; Winter, J.M.; Brody, J.R. The landscape of pancreatic cancer therapeutic resistance mechanisms. Int. J. Biol. Sci. 2016, 12, 273. [Google Scholar] [CrossRef] [PubMed]
- Shroff, R.T.; Hendifar, A.; McWilliams, R.R.; Geva, R.; Epelbaum, R.; Rolfe, L.; Goble, S.; Lin, K.K.; Biankin, A.V.; Giordano, H.; et al. Rucaparib Monotherapy in Patients With Pancreatic Cancer and a Known Deleterious BRCA Mutation. JCO Precis. Oncol. 2018, 2, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Wiegmans, A.P.; Al-Ejeh, F.; Chee, N.; Yap, P.-Y.; Gorski, J.J.; Da Silva, L.; Bolderson, E.; Chenevix-Trench, G.; Anderson, R.; Simpson, P.T. Rad51 supports triple negative breast cancer metastasis. Oncotarget 2014, 5, 3261. [Google Scholar] [CrossRef] [PubMed]
- Carey, J.P.W.; Karakas, C.; Bui, T.; Chen, X.; Vijayaraghavan, S.; Zhao, Y.; Wang, J.; Mikule, K.; Litton, J.K.; Hunt, K.K.; et al. Synthetic Lethality of PARP Inhibitors in Combination with MYC Blockade Is Independent of BRCA Status in Triple-Negative Breast Cancer. Cancer Res. 2018, 78, 742–757. [Google Scholar] [CrossRef]
- Barchiesi, G.; Roberto, M.; Verrico, M.; Vici, P.; Tomao, S.; Tomao, F. Emerging Role of PARP Inhibitors in Metastatic Triple Negative Breast Cancer. Current Scenario and Future Perspectives. Front. Oncol. 2021, 11, 769280. [Google Scholar] [CrossRef]
- Stover, E.H.; Konstantinopoulos, P.A.; Matulonis, U.A.; Swisher, E.M. Biomarkers of Response and Resistance to DNA Repair Targeted Therapies. Clin. Cancer Res. 2016, 22, 5651–5660. [Google Scholar] [CrossRef]
- Rosen, M.N.; Goodwin, R.A.; Vickers, M.M. BRCA mutated pancreatic cancer: A change is coming. World J. Gastroenterol. 2021, 27, 1943–1958. [Google Scholar] [CrossRef]
- Sun, C.; Fang, Y.; Yin, J.; Chen, J.; Ju, Z.; Zhang, D.; Chen, X.; Vellano, C.P.; Jeong, K.J.; Ng, P.K.; et al. Rational combination therapy with PARP and MEK inhibitors capitalizes on therapeutic liabilities in RAS mutant cancers. Sci. Transl. Med. 2017, 9, eaal5148. [Google Scholar] [CrossRef]
- Chiang, Y.C.; Teng, S.C.; Su, Y.N.; Hsieh, F.J.; Wu, K.J. c-Myc directly regulates the transcription of the NBS1 gene involved in DNA double-strand break repair. J. Biol. Chem. 2003, 278, 19286–19291. [Google Scholar] [CrossRef]
- Leonetti, C.; Biroccio, A.; Candiloro, A.; Citro, G.; Fornari, C.; Mottolese, M.; Bufalo, D.D.; Zupi, G. Increase of cisplatin sensitivity by c-myc antisense oligodeoxynucleotides in a human metastatic melanoma inherently resistant to cisplatin. Clin. Cancer Res. 1999, 5, 2588–2595. [Google Scholar] [PubMed]
- Bouvard, C.; Lim, S.M.; Ludka, J.; Yazdani, N.; Woods, A.K.; Chatterjee, A.K.; Schultz, P.G.; Zhu, S. Small molecule selectively suppresses MYC transcription in cancer cells. Proc. Natl. Acad. Sci. USA 2017, 114, 3497–3502. [Google Scholar] [CrossRef] [PubMed]
- Stellas, D.; Szabolcs, M.; Koul, S.; Li, Z.; Polyzos, A.; Anagnostopoulos, C.; Cournia, Z.; Tamvakopoulos, C.; Klinakis, A.; Efstratiadis, A. Therapeutic effects of an anti-Myc drug on mouse pancreatic cancer. J. Natl. Cancer Inst. 2014, 106, dju320. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).