
Impaired Peripheral Vascular Function Following Ischemic Stroke in Mice: Potential Insights into Blood Pressure Variations in the Post-Stroke Patient



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Ischemic Stroke Model
2.3. Wire Myography
2.4. Statistical Analysis
3. Results
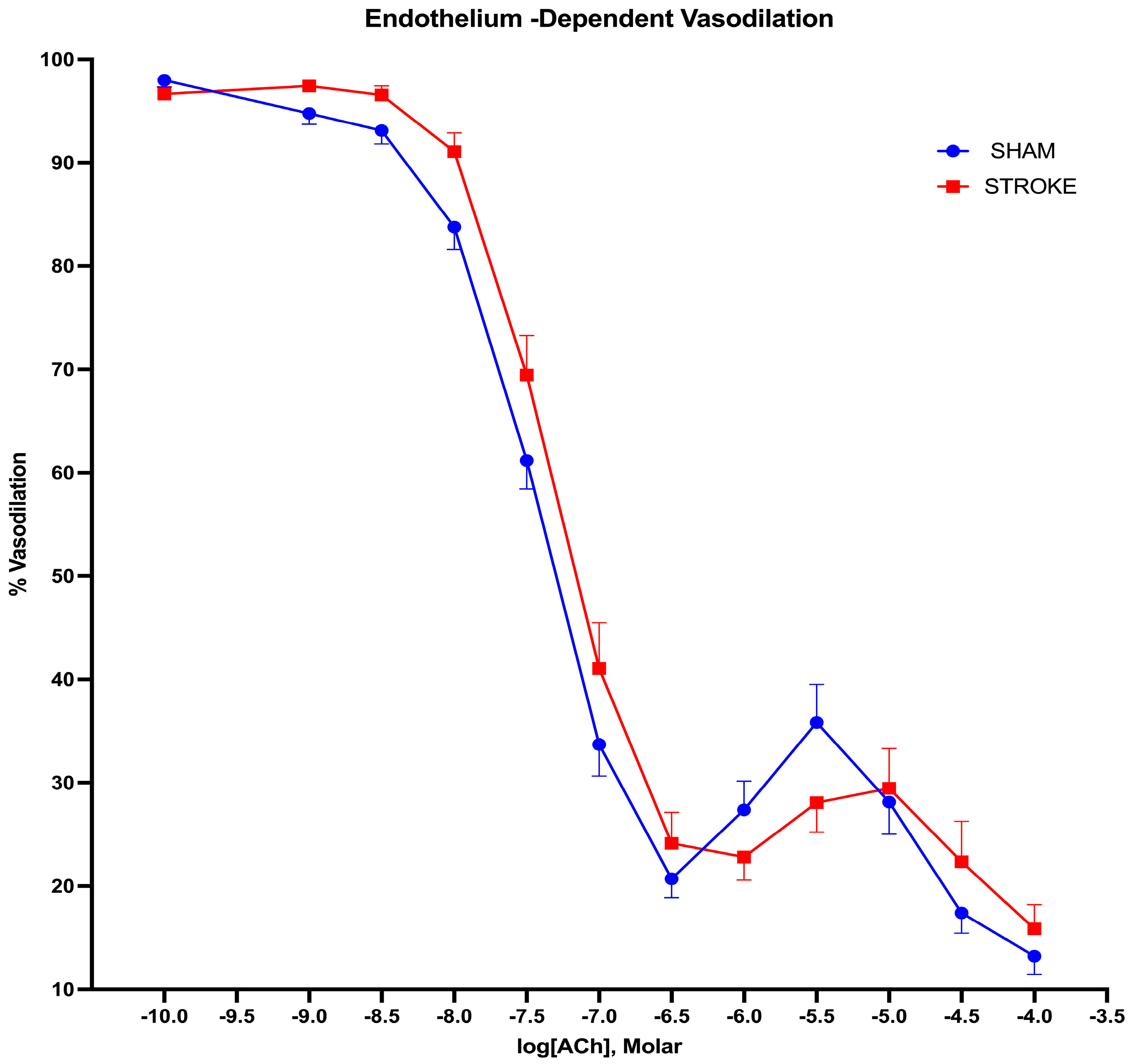
3.1. Endothelium-Dependent Vasodilation Is Impaired Following Ischemic Stroke
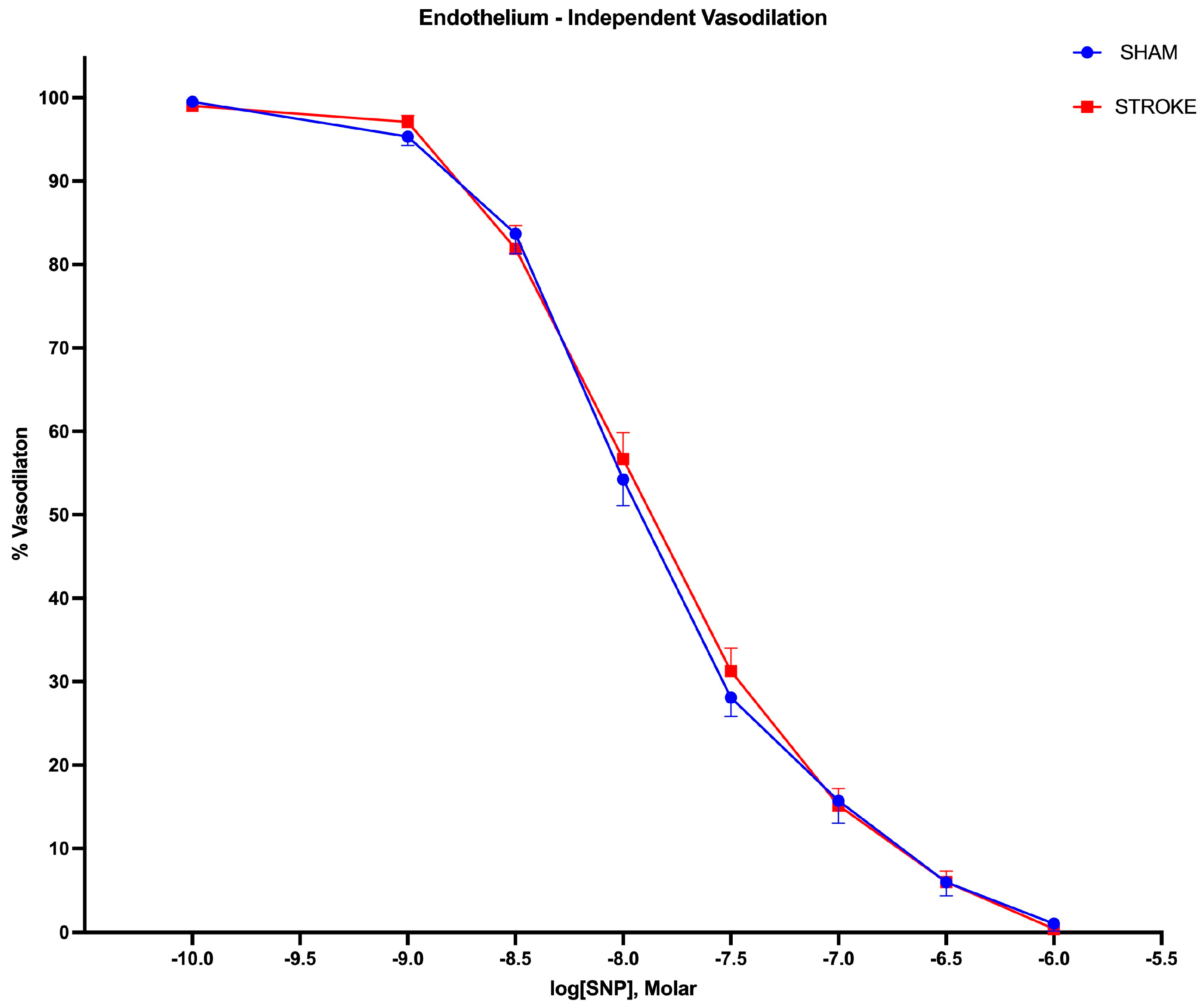
3.2. Endothelium-Independent Vasodilation Is Preserved Following Ischemic Stroke
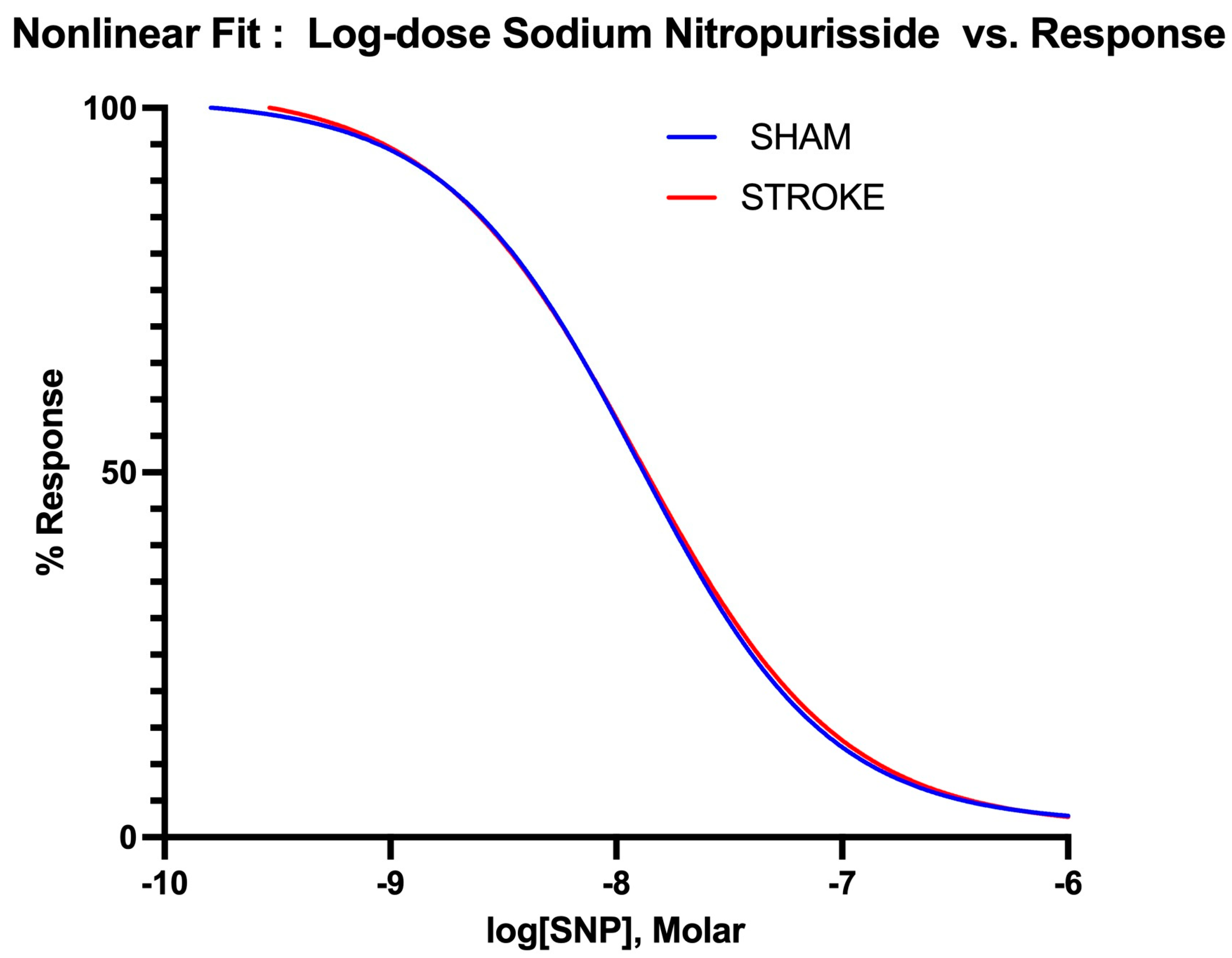
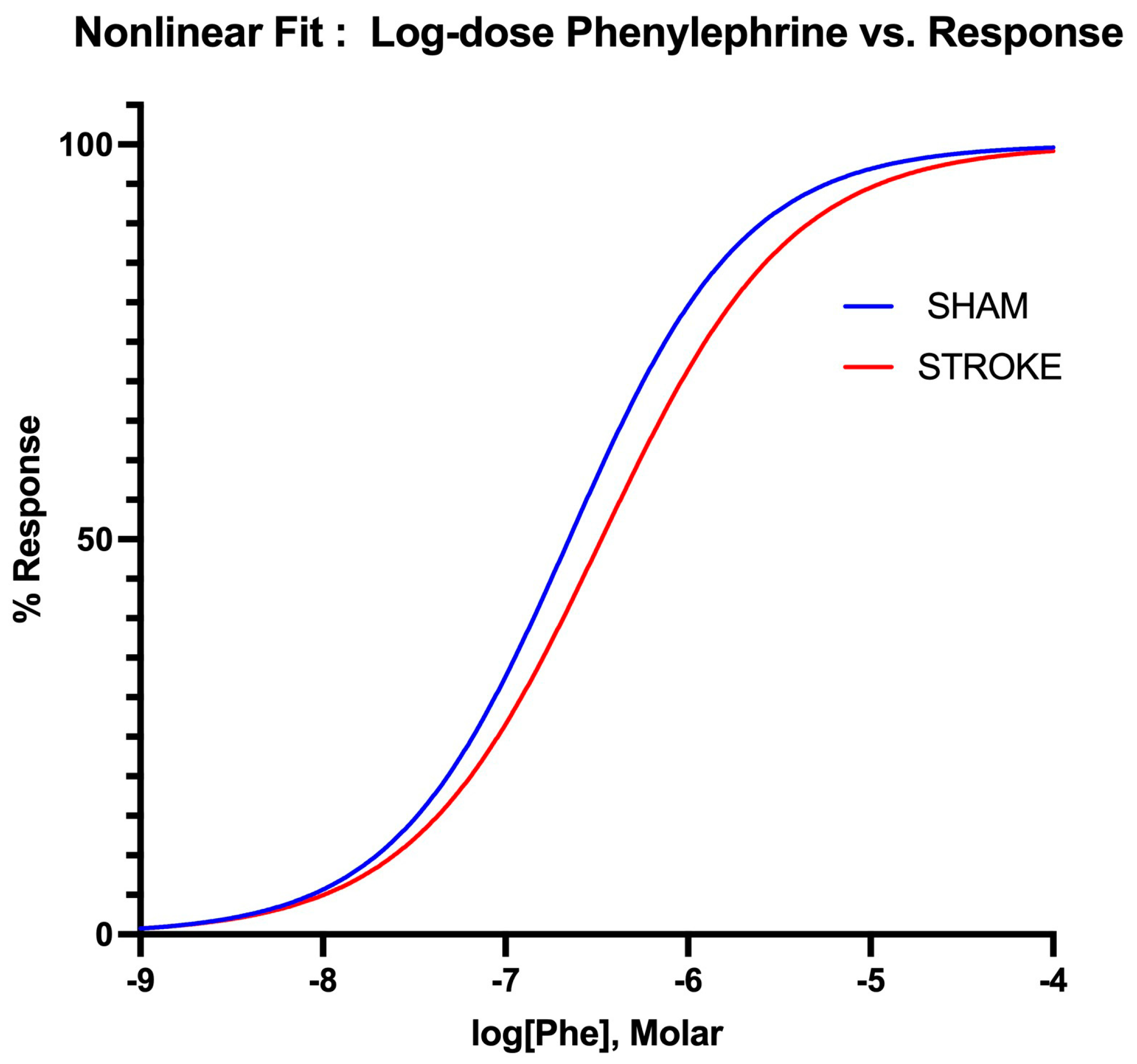
3.3. Vascular Smooth Muscle Contraction Is Impaired Following Ischemic Stroke
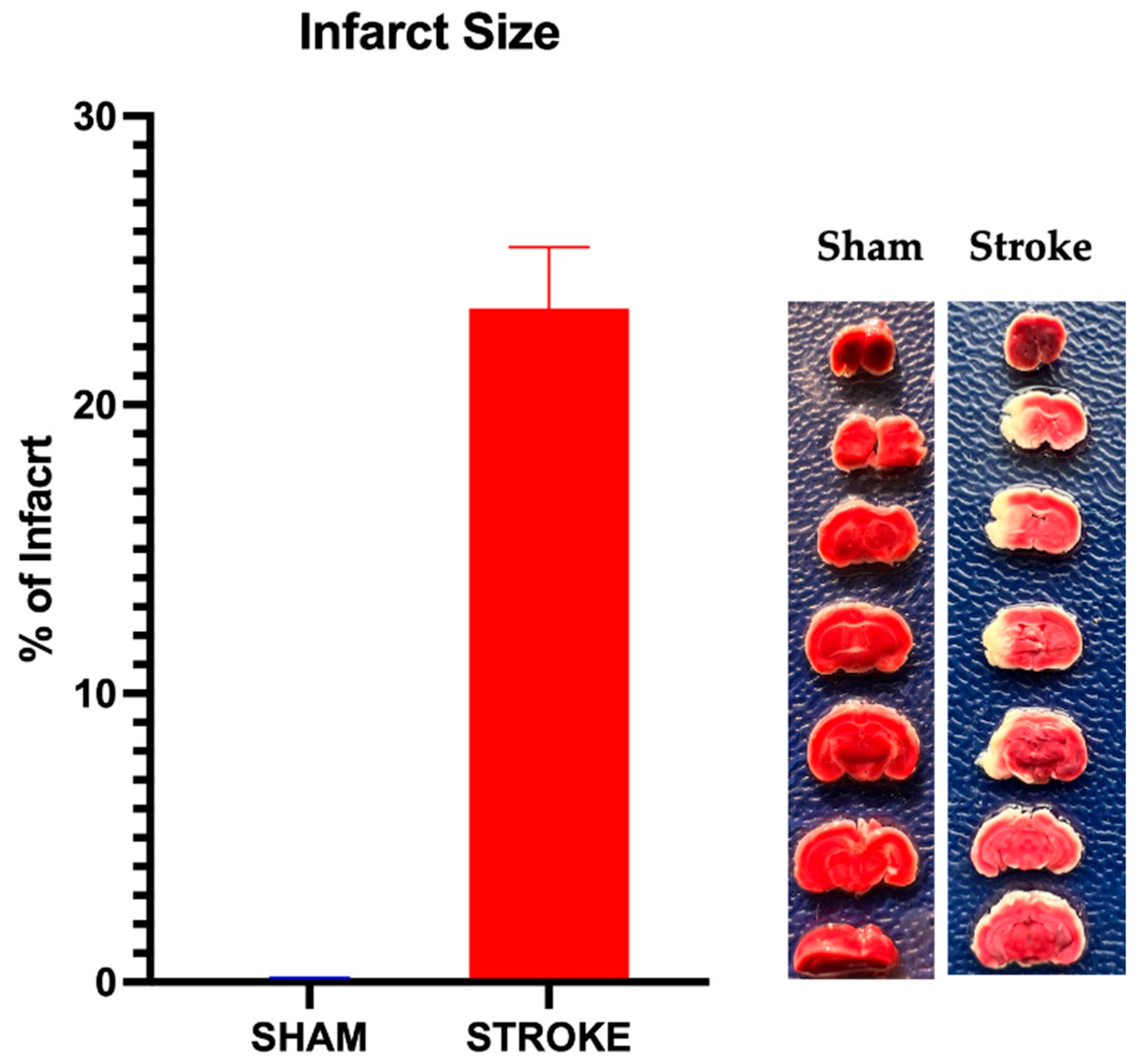
3.4. Infarct Size
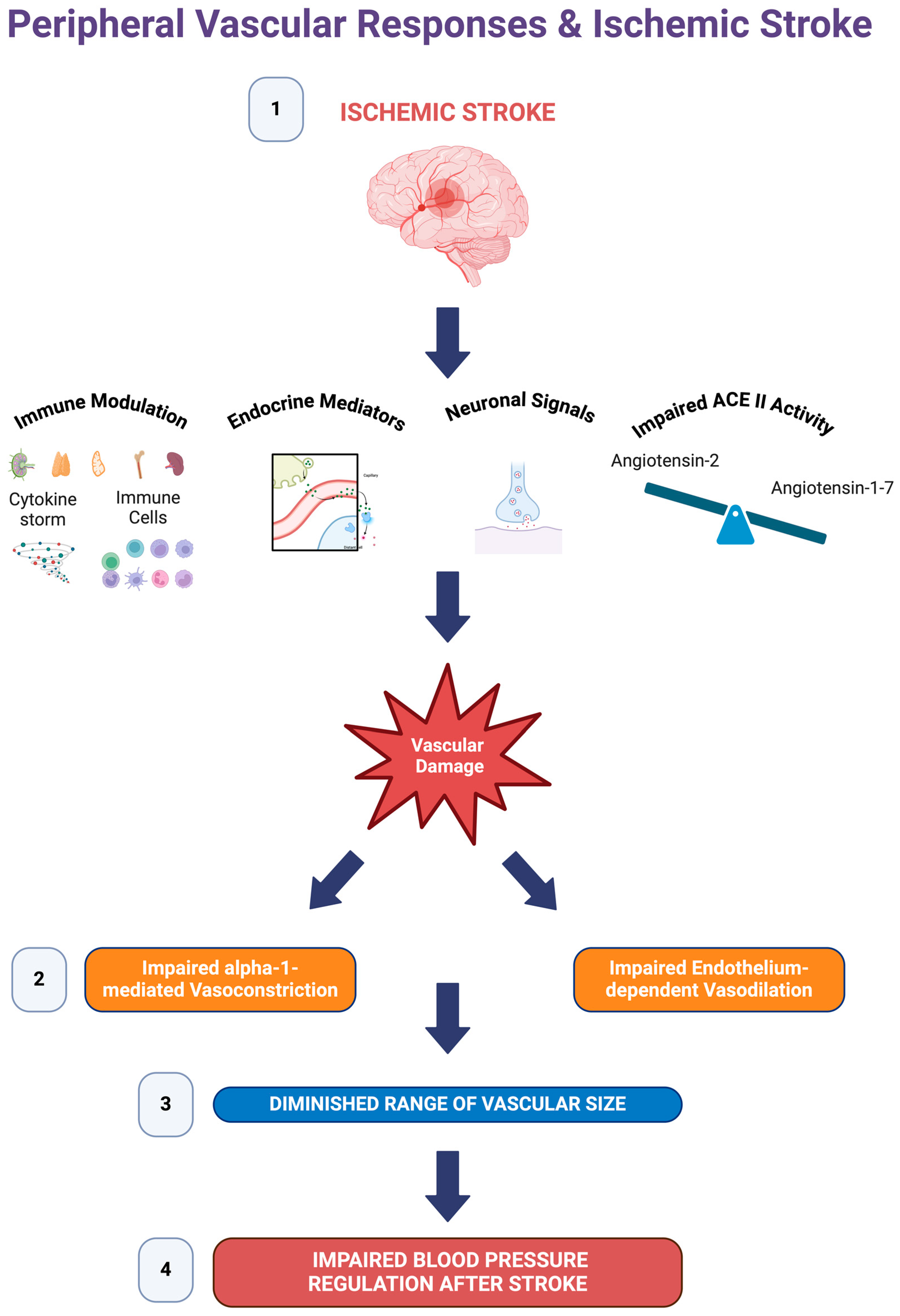
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Virani, S.S.; Alonso, A.; Aparicio, H.J.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics-2021 Update: A Report from the American Heart Association. Circulation 2021, 143, e254–e743. [Google Scholar] [CrossRef] [PubMed]
- Tsao, C.W.; Aday, A.W.; Almarzooq, Z.I.; Alonso, A.; Beaton, A.Z.; Bittencourt, M.S.; Boehme, A.K.; Buxton, A.E.; Carson, A.P.; Commodore-Mensah, Y.; et al. Heart Disease and Stroke Statistics-2022 Update: A Report from the American Heart Association. Circulation 2022, 145, e153–e639. [Google Scholar] [CrossRef]
- Campbell, B.C.; Mitchell, P.J.; Kleinig, T.J.; Dewey, H.M.; Churilov, L.; Yassi, N.; Yan, B.; Dowling, R.J.; Parsons, M.W.; Oxley, T.J.; et al. Endovascular therapy for ischemic stroke with perfusion-imaging selection. N. Engl. J. Med. 2015, 372, 1009–1018. [Google Scholar] [CrossRef]
- O’Collins, V.E.; Macleod, M.R.; Donnan, G.A.; Horky, L.L.; van der Worp, B.H.; Howells, D.W. 1,026 experimental treatments in acute stroke. Ann. Neurol. 2006, 59, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Cipolla, M.J.; Liebeskind, D.S.; Chan, S.L. The importance of comorbidities in ischemic stroke: Impact of hypertension on the cerebral circulation. J. Cereb. Blood Flow Metab. 2018, 38, 2129–2149. [Google Scholar] [CrossRef] [PubMed]
- Gallacher, K.I.; Jani, B.D.; Hanlon, P.; Nicholl, B.I.; Mair, F.S. Multimorbidity in Stroke. Stroke 2019, 50, 1919–1926. [Google Scholar] [CrossRef]
- Manning, L.S.; Rothwell, P.M.; Potter, J.F.; Robinson, T.G. Prognostic Significance of Short-Term Blood Pressure Variability in Acute Stroke: Systematic Review. Stroke 2015, 46, 2482–2490. [Google Scholar] [CrossRef]
- Hawkes, M.A.; Anderson, C.S.; Rabinstein, A.A. Blood Pressure Variability After Cerebrovascular Events: A Possible New Therapeutic Target: A Narrative Review. Neurology 2022, 99, 150–160. [Google Scholar] [CrossRef]
- Lattanzi, S.; Silvestrini, M.; Provinciali, L. Elevated blood pressure in the acute phase of stroke and the role of Angiotensin receptor blockers. Int. J. Hypertens. 2013, 2013, 941783. [Google Scholar] [CrossRef]
- de Havenon, A.; Bennett, A.; Stoddard, G.J.; Smith, G.; Chung, L.; O’Donnell, S.; McNally, J.S.; Tirschwell, D.; Majersik, J.J. Determinants of the impact of blood pressure variability on neurological outcome after acute ischaemic stroke. Stroke Vasc. Neurol. 2017, 2, 1–6. [Google Scholar] [CrossRef]
- Chung, J.-W.; Kim, N.; Kang, J.; Park, S.H.; Kim, W.-J.; Ko, Y.; Park, J.H.; Lee, J.S.; Lee, J.; Yang, M.H.; et al. Blood pressure variability and the development of early neurological deterioration following acute ischemic stroke. J. Hypertens. 2015, 33, 2099–2106. [Google Scholar] [CrossRef] [PubMed]
- de Havenon, A.; Stoddard, G.; Saini, M.; Wong, K.H.; Tirschwell, D.; Bath, P.; Collaborators, V.I.-A. Increased blood pressure variability after acute ischemic stroke increases the risk of death: A secondary analysis of the Virtual International Stroke Trial Archive. JRSM Cardiovasc. Dis. 2019, 8, 2048004019856496. [Google Scholar] [CrossRef]
- Xiong, L.; Tian, G.; Leung, H.; Soo, Y.O.Y.; Chen, X.; Ip, V.H.L.; Mok, V.C.T.; Chu, W.C.W.; Wong, K.S.; Leung, T.W.H. Autonomic Dysfunction Predicts Clinical Outcomes After Acute Ischemic Stroke. Stroke 2018, 49, 215–218. [Google Scholar] [CrossRef]
- Iadecola, C.; Buckwalter, M.S.; Anrather, J. Immune responses to stroke: Mechanisms, modulation, and therapeutic potential. J. Clin. Investig. 2020, 130, 2777–2788. [Google Scholar] [CrossRef]
- Boysen, G.; Christensen, H. Stroke severity determines body temperature in acute stroke. Stroke 2001, 32, 413–417. [Google Scholar] [CrossRef]
- Stenborg, A.; Terent, A.; Lind, L. Endothelium-dependent vasodilatation in forearm is impaired in stroke patients. J. Intern. Med. 2006, 259, 569–575. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.L.; Wang, P.Y.; Sheu, W.H.; Chen, Y.T.; Ho, Y.P.; Hu, H.H.; Hsu, H.Y. Changes of brachial flow-mediated vasodilation in different ischemic stroke subtypes. Neurology 2006, 67, 1056–1058. [Google Scholar] [CrossRef]
- Billinger, S.A.; Sisante, J.V.; Mattlage, A.E.; Alqahtani, A.S.; Abraham, M.G.; Rymer, M.M.; Camarata, P.J. The relationship of pro-inflammatory markers to vascular endothelial function after acute stroke. Int. J. Neurosci. 2017, 127, 486–492. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, G.; Alexander, J.S.; Erkuran Yilmaz, C.; Granger, D.N. Induction of neuro-protective/regenerative genes in stem cells infiltrating post-ischemic brain tissue. Exp. Transl. Stroke Med. 2010, 2, 11. [Google Scholar] [CrossRef]
- del Campo, L.; Ferrer, M. Wire Myography to Study Vascular Tone and Vascular Structure of Isolated Mouse Arteries. Methods Mol. Biol. 2015, 1339, 255–276. [Google Scholar] [CrossRef]
- Wenceslau, C.F.; McCarthy, C.G.; Earley, S.; England, S.K.; Filosa, J.A.; Goulopoulou, S.; Gutterman, D.D.; Isakson, B.E.; Kanagy, N.L.; Martinez-Lemus, L.A.; et al. Guidelines for the measurement of vascular function and structure in isolated arteries and veins. Am. J. Physiol. Heart Circ. Physiol. 2021, 321, H77–H111. [Google Scholar] [CrossRef] [PubMed]
- Monica, F.Z.; Bian, K.; Murad, F. The Endothelium-Dependent Nitric Oxide-cGMP Pathway. Adv. Pharmacol. 2016, 77, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.B.; Marshall, I. Characterization of α1-adrenoceptor subtypes mediating contractions to phenylephrine in rat thoracic aorta, mesenteric artery and pulmonary artery. Br. J. Pharmacol. 1997, 122, 849–858. [Google Scholar] [CrossRef] [PubMed]
- Vanhoutte, P.M.; Zhao, Y.; Xu, A.; Leung, S.W. Thirty Years of Saying NO: Sources, Fate, Actions, and Misfortunes of the Endothelium-Derived Vasodilator Mediator. Circ. Res. 2016, 119, 375–396. [Google Scholar] [CrossRef]
- Panza, J.A.; Quyyumi, A.A.; Brush, J.E., Jr.; Epstein, S.E. Abnormal endothelium-dependent vascular relaxation in patients with essential hypertension. N. Engl. J. Med. 1990, 323, 22–27. [Google Scholar] [CrossRef]
- Panza, J.A.; Garcia, C.E.; Kilcoyne, C.M.; Quyyumi, A.A.; Cannon, R.O., 3rd. Impaired endothelium-dependent vasodilation in patients with essential hypertension. Evidence that nitric oxide abnormality is not localized to a single signal transduction pathway. Circulation 1995, 91, 1732–1738. [Google Scholar] [CrossRef]
- Bath, P.; Chalmers, J.; Powers, W.; Beilin, L.; Davis, S.; Lenfant, C.; Mancia, G.; Neal, B.; Whitworth, J.; Zanchetti, A.; et al. International Society of Hypertension (ISH): Statement on the management of blood pressure in acute stroke. J. Hypertens. 2003, 21, 665–672. [Google Scholar] [CrossRef]
- Pretnar-Oblak, J.; Sabovic, M.; Pogacnik, T.; Sebestjen, M.; Zaletel, M. Flow-mediated dilatation and intima-media thickness in patients with lacunar infarctions. Acta Neurol. Scand 2006, 113, 273–277. [Google Scholar] [CrossRef]
- Leonardi-Bee, J.; Bath, P.M.; Phillips, S.J.; Sandercock, P.A.; Group, I.S.T.C. Blood pressure and clinical outcomes in the International Stroke Trial. Stroke 2002, 33, 1315–1320. [Google Scholar] [CrossRef]
- Jillella, D.V.; Calder, C.S.; Uchino, K.; Qeadan, F.; Ikram, A.; Casul, Y.R.; Tran, H.Q. Blood Pressure and Hospital Discharge Outcomes in Acute Ischemic Stroke Patients Undergoing Reperfusion Therapy. J. Stroke Cerebrovasc. Dis. 2020, 29, 105211. [Google Scholar] [CrossRef]
- Liesz, A.; Hagmann, S.; Zschoche, C.; Adamek, J.; Zhou, W.; Sun, L.; Hug, A.; Zorn, M.; Dalpke, A.; Nawroth, P.; et al. The spectrum of systemic immune alterations after murine focal ischemia: Immunodepression versus immunomodulation. Stroke 2009, 40, 2849–2858. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, G.; Arumugam, T.V.; Stokes, K.Y.; Granger, D.N. Role of T lymphocytes and interferon-gamma in ischemic stroke. Circulation 2006, 113, 2105–2112. [Google Scholar] [CrossRef]
- Lucaciu, A.; Kuhn, H.; Trautmann, S.; Ferreiros, N.; Steinmetz, H.; Pfeilschifter, J.; Brunkhorst, R.; Pfeilschifter, W.; Subburayalu, J.; Vutukuri, R. A Sphingosine 1-Phosphate Gradient Is Linked to the Cerebral Recruitment of T Helper and Regulatory T Helper Cells during Acute Ischemic Stroke. Int. J. Mol. Sci. 2020, 21, 6242. [Google Scholar] [CrossRef]
- Terao, S.; Yilmaz, G.; Stokes, K.Y.; Russell, J.; Ishikawa, M.; Kawase, T.; Granger, D.N. Blood cell-derived RANTES mediates cerebral microvascular dysfunction, inflammation, and tissue injury after focal ischemia-reperfusion. Stroke 2008, 39, 2560–2570. [Google Scholar] [CrossRef] [PubMed]
- Canavero, I.; Sherburne, H.A.; Tremble, S.M.; Clark, W.M.; Cipolla, M.J. Effects of Acute Stroke Serum on Non-Ischemic Cerebral and Mesenteric Vascular Function. Transl. Stroke Res. 2016, 7, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Puertas-Umbert, L.; Puig, N.; Camacho, M.; Dantas, A.P.; Marín, R.; Martí-Fàbregas, J.; Jiménez-Xarrié, E.; Benitez, S.; Camps-Renom, P.; Jiménez-Altayó, F. Serum from Stroke Patients with High-Grade Carotid Stenosis Promotes Cyclooxygenase-Dependent Endothelial Dysfunction in Non-ischemic Mice Carotid Arteries. Transl. Stroke Res. 2024, 15, 140–152. [Google Scholar] [CrossRef]
- Asano, S.; O’Connell, G.C.; Lemaster, K.C.; DeVallance, E.R.; Branyan, K.W.; Simpkins, J.W.; Frisbee, J.C.; Barr, T.L.; Chantler, P.D. Circulating leucocytes perpetuate stroke-induced aortic dysfunction. Exp. Physiol. 2017, 102, 1321–1331. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Q.; Wu, H.; Du, H.; Liu, L.; Shi, H.; Wang, C.; Xia, Y.; Guo, X.; Li, C.; et al. Blood Neutrophil to Lymphocyte Ratio as a Predictor of Hypertension. Am. J. Hypertens. 2015, 28, 1339–1346. [Google Scholar] [CrossRef]
- Yilmaz, G.; Granger, D.N. Cell adhesion molecules and ischemic stroke. Neurol. Res. 2008, 30, 783–793. [Google Scholar] [CrossRef]
- van Langen, J.T.; Van Hove, C.E.; Schrijvers, D.M.; Martinet, W.; De Meyer, G.R.; Fransen, P.; Bult, H. Contribution of alpha-adrenoceptor stimulation by phenylephrine to basal nitric oxide production in the isolated mouse aorta. J. Cardiovasc. Pharmacol. 2013, 61, 318–323. [Google Scholar] [CrossRef]
- Zhao, Y.; Vanhoutte, P.M.; Leung, S.W. alpha1 -Adrenoceptor activation of PKC-epsilon causes heterologous desensitization of thromboxane receptors in the aorta of spontaneously hypertensive rats. Br. J. Pharmacol. 2015, 172, 3687–3701. [Google Scholar] [CrossRef] [PubMed]
- Ohkawa, F.; Ikeda, U.; Kanbe, T.; Kawasaki, K.; Shimada, K. Effects of inflammatory cytokines on vascular tone. Cardiovasc. Res. 1995, 30, 711–715. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Wu, M.; Wang, H.; Zheng, Y.; Zhang, S.; Wang, X.; Wang, S.; Fang, Z. Daily blood pressure variability in relation to neurological functional outcomes after acute ischemic stroke. Front. Neurol. 2022, 13, 958166. [Google Scholar] [CrossRef]
- Stromsnes, T.A.; Kaugerud Hagen, T.J.; Ouyang, M.; Wang, X.; Chen, C.; Rygg, S.E.; Hewson, D.; Lenthall, R.; McConachie, N.; Izzath, W.; et al. Pressor therapy in acute ischaemic stroke: An updated systematic review. Eur. Stroke J. 2022, 7, 99–116. [Google Scholar] [CrossRef]
- Bang, O.Y.; Chung, J.W.; Kim, S.K.; Kim, S.J.; Lee, M.J.; Hwang, J.; Seo, W.K.; Ha, Y.S.; Sung, S.M.; Kim, E.G.; et al. Therapeutic-induced hypertension in patients with noncardioembolic acute stroke. Neurology 2019, 93, e1955–e1963. [Google Scholar] [CrossRef]
- Kim, H.J.; Kang, D.W. Induced hypertensive therapy in an acute ischemic stroke patient with early neurological deterioration. J. Clin. Neurol. 2007, 3, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Bates, J.N.; Baker, M.T.; Guerra, R., Jr.; Harrison, D.G. Nitric oxide generation from nitroprusside by vascular tissue. Evidence that reduction of the nitroprusside anion and cyanide loss are required. Biochem. Pharmacol. 1991, 42 (Suppl. S1), S157–S165. [Google Scholar] [CrossRef] [PubMed]
- Rudner, X.L.; Berkowitz, D.E.; Booth, J.V.; Funk, B.L.; Cozart, K.L.; D’Amico, E.B.; El-Moalem, H.; Page, S.O.; Richardson, C.D.; Winters, B.; et al. Subtype specific regulation of human vascular alpha(1)-adrenergic receptors by vessel bed and age. Circulation 1999, 100, 2336–2343. [Google Scholar] [CrossRef]
- Chen, J.; Cui, C.; Yang, X.; Xu, J.; Venkat, P.; Zacharek, A.; Yu, P.; Chopp, M. MiR-126 Affects Brain-Heart Interaction after Cerebral Ischemic Stroke. Transl. Stroke Res. 2017, 8, 374–385. [Google Scholar] [CrossRef]
- Rasmussen, M.; Schonenberger, S.; Henden, P.L.; Valentin, J.B.; Espelund, U.S.; Sorensen, L.H.; Juul, N.; Uhlmann, L.; Johnsen, S.P.; Rentzos, A.; et al. Blood Pressure Thresholds and Neurologic Outcomes after Endovascular Therapy for Acute Ischemic Stroke: An Analysis of Individual Patient Data from 3 Randomized Clinical Trials. JAMA Neurol. 2020, 77, 622–631. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yilmaz, G.; Alexander, J.S. Impaired Peripheral Vascular Function Following Ischemic Stroke in Mice: Potential Insights into Blood Pressure Variations in the Post-Stroke Patient. Pathophysiology 2024, 31, 488-501. https://doi.org/10.3390/pathophysiology31030036
Yilmaz G, Alexander JS. Impaired Peripheral Vascular Function Following Ischemic Stroke in Mice: Potential Insights into Blood Pressure Variations in the Post-Stroke Patient. Pathophysiology. 2024; 31(3):488-501. https://doi.org/10.3390/pathophysiology31030036
Chicago/Turabian StyleYilmaz, Gokhan, and Jonathan Steven Alexander. 2024. "Impaired Peripheral Vascular Function Following Ischemic Stroke in Mice: Potential Insights into Blood Pressure Variations in the Post-Stroke Patient" Pathophysiology 31, no. 3: 488-501. https://doi.org/10.3390/pathophysiology31030036