

Strategizing Drug Therapies in Pulmonary Hypertension for Improved Outcomes


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Physiology
Classification
3. Group 1 PH: Pulmonary Arterial Hypertension
3.1. Genetics
3.2. Pathogenesis
3.2.1. Endothelin
3.2.2. Nitric Oxide
3.2.3. Prostacyclin
3.3. Diagnosing Pulmonary Arterial Hypertension
3.3.1. Clinical Assessment
3.3.2. Staging PAH
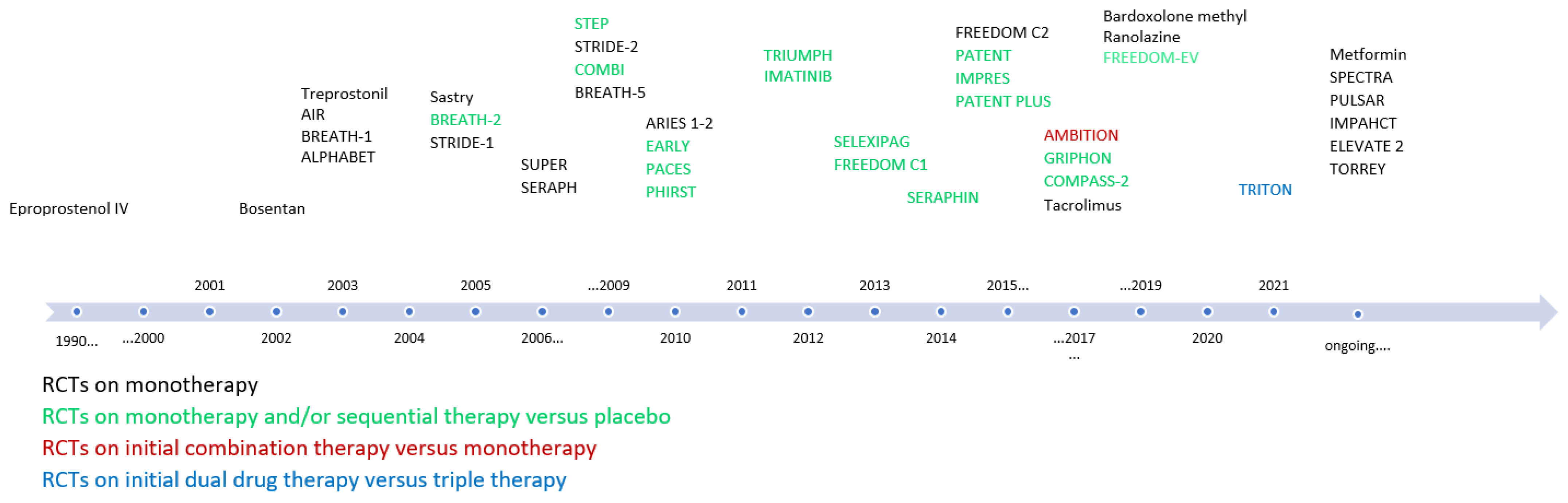
3.4. Current Medical Therapies for PAH
3.4.1. Vasoreactivity Testing and Calcium Channel Blockers
3.4.2. Prostacyclin Pathway
3.4.3. Evidence for Prostacyclins
3.4.4. Endothelin Receptor Antagonists
3.4.5. Evidence for the ERAs
3.4.6. The Nitric Oxide pathway
3.4.7. Evidence PDE5i and sGC simulators
3.4.8. Monotherapy vs. Combination Therapy
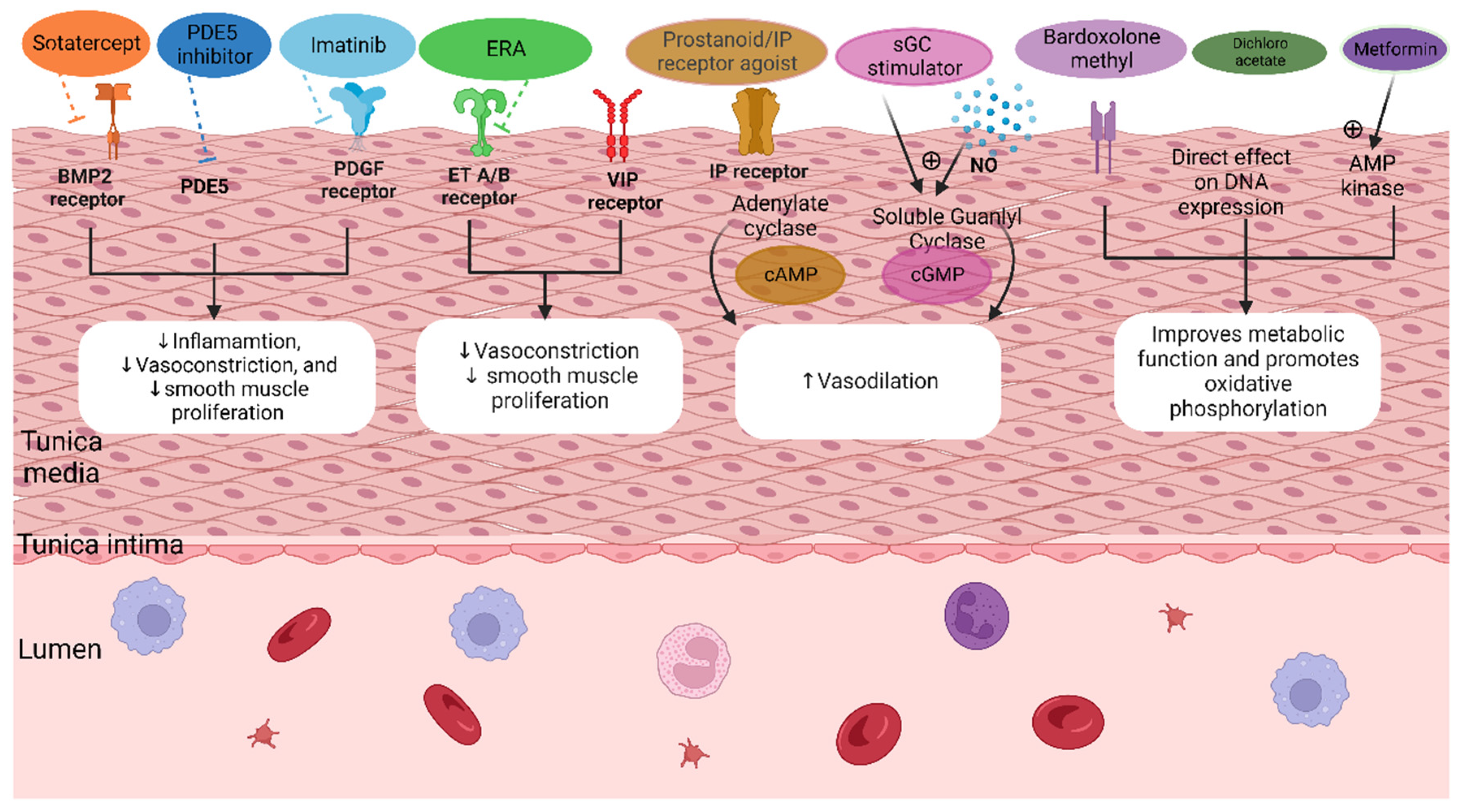
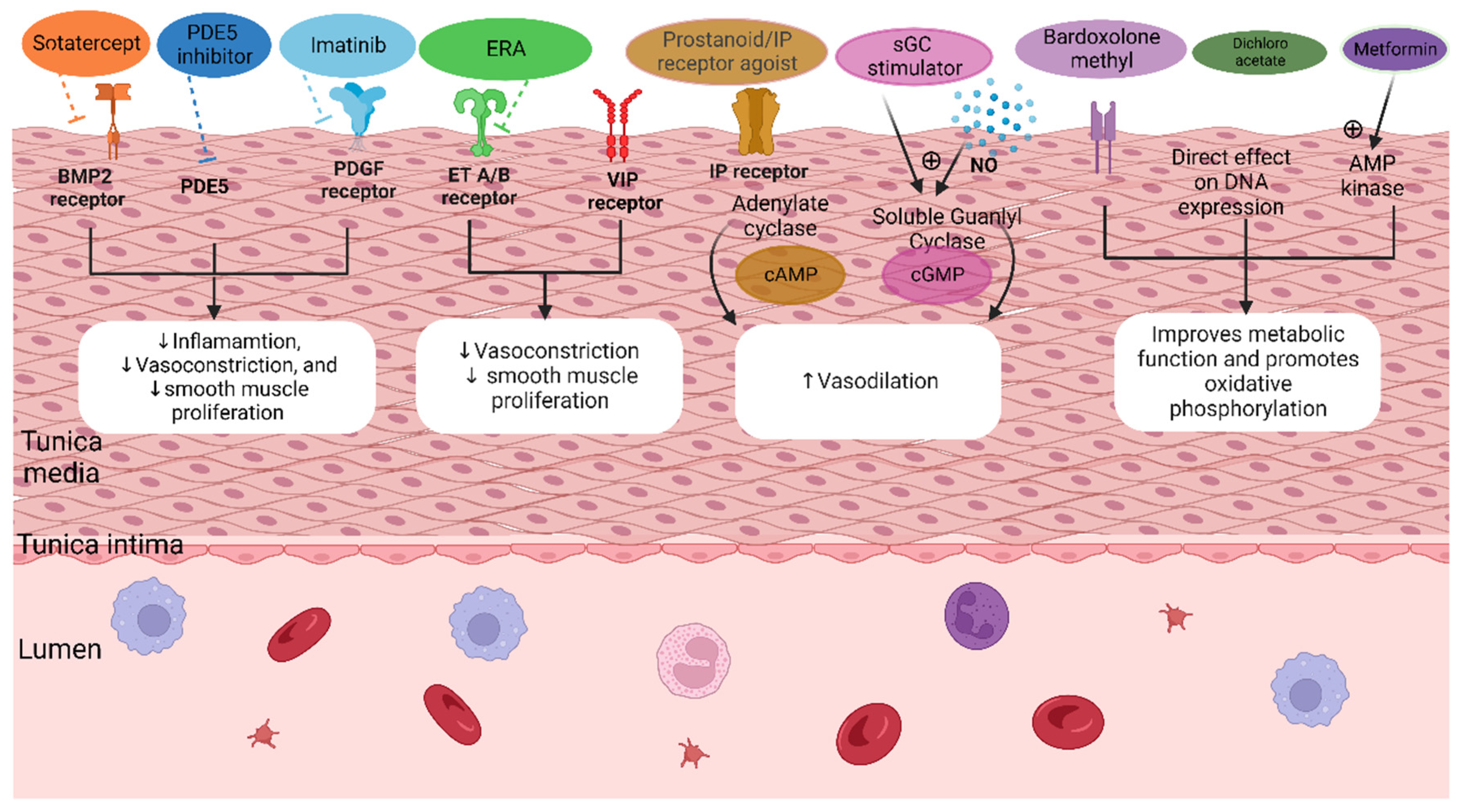
3.5. Novel Treatment Therapies
3.5.1. BMPR2 Modulators
3.5.2. Platelet-Derived Growth Factor (TK Inhibitor Imatinib)
3.5.3. Mitochondrial Dysfunction and Oxidative Stress
3.5.4. Renin–Angiotensin–Aldosterone System
3.5.5. Other Novel Pharmacological Targets
4. Group 2 PH: Pulmonary Hypertension Due to Left-Heart Disease (PH-LHD)
4.1. Definition
4.2. Epidemiology
4.3. Pathophysiology
4.4. Treatment
4.4.1. Epoprostenol
4.4.2. Endothelin Receptor Antagonists (ERA)
4.4.3. Phosphodiesterase 5 Inhibition
4.4.4. Soluble Guanylate Cyclase Stimulators
4.4.5. Metformin
5. Group 3 PH: Respiratory Diseases and Treatments
5.1. Pathophysiology and Etiology
5.2. Classifications
5.3. Diagnosis
5.4. Treatment
6. Group 4 PH: CTEPH
6.1. Pathogenesis and Etiology
6.2. Making the Diagnosis
6.3. Surgical Options and Indications
6.4. Medical Management and Current Therapies in Nonsurgical Candidates
7. Group 5 PH: Unclear or Multifactorial Causes
7.1. Hematologic Disorders
7.2. Systemic and Metabolic Disorders
7.3. Metabolic Disorders
7.4. Other Disorders
7.5. Current Therapies
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 6MWD | 6 min walk distance |
| AV | Arterio-venous |
| BMI | Body Mass Index |
| BMPR | Bone morphogenic protein receptor |
| cAMP | Cyclic adenosine monophosphate |
| CCB | Calcium channel blockers |
| cGMP | Cyclic guanosine monophosphate |
| CI | Cardiac index |
| CO | Cardiac output |
| COPD | Chronic obstructive pulmonary disease |
| COX | Cyclo-oxygenase |
| Cpc | Combined post and precapillary |
| CTD | Connective tissue diseases |
| CTEPH | Chronic thromboembolic pulmonary hypertension |
| DCA | Dichloroacetate |
| DOACS | Direct oral anticoagulants |
| ERA | Endothelin receptor antagonist |
| ET | Endothelin |
| FC | Functional class |
| FDA | Food and Drug Association |
| GD | Gaucher disease |
| HFpEF | Heart failure with preserved ejection fracture |
| HFrEF | Heart failure with reduced ejection fracture |
| ILD | Interstitial lung disease |
| IPAH | Idiopathic pulmonary arterial hypertension |
| Ipc | Isolated postcapillary |
| JVD | Jugular venous distension |
| Kv | Potassium gated ion channel |
| LAP | Left-atrial pressure |
| LHC | Left-heart catheterization |
| LHD | Left-heart disease |
| LV | Left ventricle |
| LVEDP | Left-ventricular end-diastolic pressure |
| mPAP | Mean pulmonary artery pressure |
| NO | Nitric oxide |
| NT-pro BNP | N-terminal pro-B-type natriuretic peptide |
| OSA | Obstructive sleep apnea |
| PA | Pulmonary artery |
| PCA | Prostacyclin analog/agonist |
| PCWP | Pulmonary capillary wedge pressure |
| PDE-5 | Phosphodiesterase 5 |
| PDGF | Platelet-derived growth factor |
| PGI2 | Prostaglandin I2 |
| PLCH | Pulmonary Langerhans cell histiocytosis |
| PTE | Pulmonary thromboendoarterectomy |
| PVR | Pulmonary vascular resistance |
| RCT | Randomized control trial |
| RV | Right ventricle |
| SAPH | Sarcoidosis-associated PH |
| SCD | Scleroderma |
| SQ | Subcutaneous |
| TGF-B | Transforming growth factor-beta |
| TTCW | Time to clinical worsening |
| TTE | Transthoracic echocardiography |
| V/Q | Ventilation and perfusion |
| WHO | World Health Organization |
References
- Ruopp, N.F.; Farber, H.W. The New World Symposium on Pulmonary Hypertension Guidelines: Should Twenty-One Be the New Twenty-Five? Circulation 2019, 140, 1134–1136. [Google Scholar] [CrossRef] [PubMed]
- Naeije, R.; Richter, M.J.; Rubin, L.J. The Physiological Basis of Pulmonary Arterial Hypertension. Eur. Respir. J. 2022, 59, 2102334. [Google Scholar] [CrossRef]
- Hsu, S. Coupling Right Ventricular-Pulmonary Arterial Research to the Pulmonary Hypertension Patient Bedside. Circ. Heart Fail. 2019, 12, e005715. [Google Scholar] [CrossRef] [PubMed]
- Brown, L.M.; Chen, H.; Halpern, S.; Taichman, D.; McGoon, M.D.; Farber, H.W.; Frost, A.E.; Liou, T.G.; Turner, M.; Feldkircher, K.; et al. Delay in Recognition of Pulmonary Arterial Hypertension: Factors Identified from the REVEAL Registry. Chest 2011, 140, 19–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humbert, M.; Kovacs, G.; Hoeper, M.M.; Badagliacca, R.; Berger, R.M.F.; Brida, M.; Carlsen, J.; Coats, A.J.S.; Escribano-Subias, P.; Ferrari, P.; et al. 2022 ESC/ERS Guidelines for the Diagnosis and Treatment of Pulmonary Hypertension: Developed by the Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS). Endorsed by the International Society for Heart and Lung Transplantation (ISHLT) and the European Reference Network on Rare Respiratory Diseases (ERN-LUNG). Eur. Heart J. 2022, ehac237. [Google Scholar] [CrossRef]
- Rosenkranz, S.; Howard, L.S.; Gomberg-Maitland, M.; Hoeper, M.M. Systemic Consequences of Pulmonary Hypertension and Right-Sided Heart Failure. Circulation 2020, 141, 678–693. [Google Scholar] [CrossRef] [PubMed]
- Tuder, R.M. Pulmonary Vascular Remodeling in Pulmonary Hypertension. Cell Tissue Res. 2017, 367, 643–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemnes, A.R.; Humbert, M. Pathobiology of Pulmonary Arterial Hypertension: Understanding the Roads Less Travelled. Eur. Respir. Rev. 2017, 26, 170093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dieffenbach, P.B.; Maracle, M.; Tschumperlin, D.J.; Fredenburgh, L.E. Mechanobiological Feedback in Pulmonary Vascular Disease. Front. Physiol. 2018, 9, 951. [Google Scholar] [CrossRef] [PubMed]
- Sanz, J.; Kariisa, M.; Dellegrottaglie, S.; Prat-González, S.; Garcia, M.J.; Fuster, V.; Rajagopalan, S. Evaluation of Pulmonary Artery Stiffness in Pulmonary Hypertension with Cardiac Magnetic Resonance. JACC Cardiovasc. Imaging 2009, 2, 286–295. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E.; Grignola, J.C.; Aguilar, R.; Messeguer, M.L.; Roman, A. Pulmonary Arterial Wall Disease in COPD and Interstitial Lung Diseases Candidates for Lung Transplantation. Respir. Res. 2017, 18, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertero, T.; Cottrill, K.A.; Lu, Y.; Haeger, C.M.; Dieffenbach, P.; Annis, S.; Hale, A.; Bhat, B.; Kaimal, V.; Zhang, Y.-Y.; et al. Matrix Remodeling Promotes Pulmonary Hypertension through Feedback Mechanoactivation of the YAP/TAZ-MiR-130/301 Circuit. Cell Rep. 2015, 13, 1016–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galiè, N.; Humbert, M.; Vachiery, J.-L.; Gibbs, S.; Lang, I.; Torbicki, A.; Simonneau, G.; Peacock, A.; Vonk Noordegraaf, A.; Beghetti, M.; et al. 2015 ESC/ERS Guidelines for the Diagnosis and Treatment of Pulmonary Hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS)Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur. Respir. J. 2015, 46, 903–975. [Google Scholar] [CrossRef]
- McLaughlin, V.V.; Shah, S.J.; Souza, R.; Humbert, M. Management of Pulmonary Arterial Hypertension. J. Am. Coll. Cardiol. 2015, 65, 1976–1997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic Definitions and Updated Clinical Classification of Pulmonary Hypertension. Eur. Respir. J. 2019, 53, 1801913. [Google Scholar] [CrossRef] [PubMed]
- Douschan, P.; Kovacs, G.; Avian, A.; Foris, V.; Gruber, F.; Olschewski, A.; Olschewski, H. Mild Elevation of Pulmonary Arterial Pressure as a Predictor of Mortality. Am. J. Respir. Crit. Care Med. 2018, 197, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Zolty, R. Pulmonary Arterial Hypertension Specific Therapy: The Old and the New. Pharmacol. Ther. 2020, 214, 107576. [Google Scholar] [CrossRef] [PubMed]
- Thenappan, T.; Ormiston, M.L.; Ryan, J.J.; Archer, S.L. Pulmonary Arterial Hypertension: Pathogenesis and Clinical Management. BMJ 2018, 360, j5492. [Google Scholar] [CrossRef]
- Qaiser, K.N.; Tonelli, A.R. Novel Treatment Pathways in Pulmonary Arterial Hypertension. Methodist Debakey Cardiovasc. J. 2021, 17, 106–114. [Google Scholar] [CrossRef]
- Correale, M.; Ferraretti, A.; Monaco, I.; Grazioli, D.; Di Biase, M.; Brunetti, N.D. Endothelin-Receptor Antagonists in the Management of Pulmonary Arterial Hypertension: Where Do We Stand? Vasc. Health Risk Manag. 2018, 14, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Sommer, N.; Ghofrani, H.A.; Pak, O.; Bonnet, S.; Provencher, S.; Sitbon, O.; Rosenkranz, S.; Hoeper, M.M.; Kiely, D.G. Current and Future Treatments of Pulmonary Arterial Hypertension. Br. J. Pharmacol. 2021, 178, 6–30. [Google Scholar] [CrossRef] [Green Version]
- Galiè, N.; Manes, A.; Branzi, A. Prostanoids for Pulmonary Arterial Hypertension. Am. J. Respir. Med. 2003, 2, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.A.; Ahmetaj-Shala, B.; Kirkby, N.S.; Wright, W.R.; Mackenzie, L.S.; Reed, D.M.; Mohamed, N. Role of Prostacyclin in Pulmonary Hypertension. Glob. Cardiol. Sci. Pract. 2014, 2014, 382–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaine, S.; Sitbon, O.; Channick, R.N.; Chin, K.M.; Sauter, R.; Galiè, N.; Hoeper, M.M.; McLaughlin, V.V.; Preiss, R.; Rubin, L.J.; et al. Relationship Between Time from Diagnosis and Morbidity/Mortality in Pulmonary Arterial Hypertension: Results from the Phase III GRIPHON Study. Chest 2021, 160, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Highland, K.B.; Crawford, R.; Classi, P.; Morrison, R.; Doward, L.; Nelsen, A.C.; Castillo, H.; Mathai, S.C.; DuBrock, H.M. Development of the Pulmonary Hypertension Functional Classification Self-Report: A Patient Version Adapted from the World Health Organization Functional Classification Measure. Health Qual. Life Outcomes 2021, 19, 202. [Google Scholar] [CrossRef] [PubMed]
- Benza, R.L.; Miller, D.P.; Barst, R.J.; Badesch, D.B.; Frost, A.E.; McGoon, M.D. An Evaluation of Long-Term Survival from Time of Diagnosis in Pulmonary Arterial Hypertension from the REVEAL Registry. Chest 2012, 142, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Klings, E.S.; Machado, R.F.; Barst, R.J.; Morris, C.R.; Mubarak, K.K.; Gordeuk, V.R.; Kato, G.J.; Ataga, K.I.; Gibbs, J.S.; Castro, O.; et al. An Official American Thoracic Society Clinical Practice Guideline: Diagnosis, Risk Stratification, and Management of Pulmonary Hypertension of Sickle Cell Disease. Am. J. Respir. Crit. Care Med. 2014, 189, 727–740. [Google Scholar] [CrossRef] [Green Version]
- Sitbon, O.; Humbert, M.; Jaïs, X.; Ioos, V.; Hamid, A.M.; Provencher, S.; Garcia, G.; Parent, F.; Hervé, P.; Simonneau, G. Long-Term Response to Calcium Channel Blockers in Idiopathic Pulmonary Arterial Hypertension. Circulation 2005, 111, 3105–3111. [Google Scholar] [CrossRef] [Green Version]
- Rich, S.; Kaufmann, E.; Levy, P.S. The Effect of High Doses of Calcium-Channel Blockers on Survival in Primary Pulmonary Hypertension. N. Engl. J. Med. 1992, 327, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Sadushi-Koliçi, R.; Skoro-Sajer, N.; Zimmer, D.; Bonderman, D.; Schemper, M.; Klepetko, W.; Glatz, J.; Jakowitsch, J.; Lang, I.M. Long-Term Treatment, Tolerability, and Survival with Sub-Cutaneous Treprostinil for Severe Pulmonary Hypertension. J. Heart Lung Transplant. 2012, 31, 735–743. [Google Scholar] [CrossRef]
- Jing, Z.-C.; Parikh, K.; Pulido, T.; Jerjes-Sanchez, C.; White, R.J.; Allen, R.; Torbicki, A.; Xu, K.-F.; Yehle, D.; Laliberte, K.; et al. Efficacy and Safety of Oral Treprostinil Monotherapy for the Treatment of Pulmonary Arterial Hypertension: A Randomized, Controlled Trial. Circulation 2013, 127, 624–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, R.J.; Jerjes-Sanchez, C.; Bohns Meyer, G.M.; Pulido, T.; Sepulveda, P.; Wang, K.Y.; Grünig, E.; Hiremath, S.; Yu, Z.; Gangcheng, Z.; et al. Combination Therapy with Oral Treprostinil for Pulmonary Arterial Hypertension. A Double-Blind Placebo-Controlled Clinical Trial. Am. J. Respir. Crit. Care Med. 2020, 201, 707–717. [Google Scholar] [CrossRef]
- Spikes, L.A.; Bajwa, A.A.; Burger, C.D.; Desai, S.V.; Eggert, M.S.; El-Kersh, K.A.; Fisher, M.R.; Johri, S.; Joly, J.M.; Mehta, J.; et al. BREEZE: Open-label Clinical Study to Evaluate the Safety and Tolerability of Treprostinil Inhalation Powder as Tyvaso DPITM in Patients with Pulmonary Arterial Hypertension. Pulm. Circ. 2022, 12, e12063. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Chen, X.; Li, J.; Su, S.; Ding, T.; Shi, C.; Jiang, Y.; Zhu, Z. Efficacy and Safety of Pulmonary Arterial Hypertension-Specific Therapy in Pulmonary Arterial Hypertension: A Meta-Analysis of Randomized Controlled Trials. Chest 2016, 150, 353–366. [Google Scholar] [CrossRef] [PubMed]
- Torres, F.; Farber, H.; Ristic, A.; McLaughlin, V.; Adams, J.; Zhang, J.; Klassen, P.; Shanahan, W.; Grundy, J.; Hoffmann, I.; et al. Efficacy and Safety of Ralinepag, a Novel Oral IP Agonist, in PAH Patients on Mono or Dual Background Therapy: Results from a Phase 2 Randomised, Parallel Group, Placebo-Controlled Trial. Eur. Respir. J. 2019, 54, 1901030. [Google Scholar] [CrossRef] [PubMed]
- Galiè, N.; Olschewski, H.; Oudiz, R.J.; Torres, F.; Frost, A.; Ghofrani, H.A.; Badesch, D.B.; McGoon, M.D.; McLaughlin, V.V.; Roecker, E.B.; et al. Ambrisentan for the Treatment of Pulmonary Arterial Hypertension: Results of the Ambrisentan in Pulmonary Arterial Hypertension, Randomized, Double-Blind, Placebo-Controlled, Multicenter, Efficacy (ARIES) Study 1 and 2. Circulation 2008, 117, 3010–3019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pulido, T.; Adzerikho, I.; Channick, R.N.; Delcroix, M.; Galiè, N.; Ghofrani, H.-A.; Jansa, P.; Jing, Z.-C.; Le Brun, F.-O.; Mehta, S.; et al. Macitentan and Morbidity and Mortality in Pulmonary Arterial Hypertension. N. Engl. J. Med. 2013, 369, 809–818. [Google Scholar] [CrossRef] [Green Version]
- Humbert, M. Combination of Bosentan with Epoprostenol in Pulmonary Arterial Hypertension: BREATHE-2. Eur. Respir. J. 2004, 24, 353–359. [Google Scholar] [CrossRef] [Green Version]
- Rivera-Lebron, B.N.; Risbano, M.G. Ambrisentan: A Review of Its Use in Pulmonary Arterial Hypertension. Ther. Adv. Respir. Dis. 2017, 11, 233–244. [Google Scholar] [CrossRef]
- Galiè, N.; Barberà, J.A.; Frost, A.E.; Ghofrani, H.-A.; Hoeper, M.M.; McLaughlin, V.V.; Peacock, A.J.; Simonneau, G.; Vachiery, J.-L.; Grünig, E.; et al. Initial Use of Ambrisentan plus Tadalafil in Pulmonary Arterial Hypertension. N. Engl. J. Med. 2015, 373, 834–844. [Google Scholar] [CrossRef]
- Ghofrani, H.-A.; D’Armini, A.M.; Grimminger, F.; Hoeper, M.M.; Jansa, P.; Kim, N.H.; Mayer, E.; Simonneau, G.; Wilkins, M.R.; Fritsch, A.; et al. Riociguat for the Treatment of Chronic Thromboembolic Pulmonary Hypertension. N. Engl. J. Med. 2013, 369, 319–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubin, L.J.; Badesch, D.B.; Fleming, T.R.; Galiè, N.; Simonneau, G.; Ghofrani, H.A.; Oakes, M.; Layton, G.; Serdarevic-Pehar, M.; McLaughlin, V.V.; et al. Long-Term Treatment with Sildenafil Citrate in Pulmonary Arterial Hypertension. Chest 2011, 140, 1274–1283. [Google Scholar] [CrossRef] [PubMed]
- Macchia, A.; Marchioli, R.; Marfisi, R.; Scarano, M.; Levantesi, G.; Tavazzi, L.; Tognoni, G. A Meta-Analysis of Trials of Pulmonary Hypertension: A Clinical Condition Looking for Drugs and Research Methodology. Am. Heart J. 2007, 153, 1037–1047. [Google Scholar] [CrossRef] [PubMed]
- Hoeper, M.M.; Al-Hiti, H.; Benza, R.L.; Chang, S.-A.; Corris, P.A.; Gibbs, J.S.R.; Grünig, E.; Jansa, P.; Klinger, J.R.; Langleben, D.; et al. Switching to Riociguat versus Maintenance Therapy with Phosphodiesterase-5 Inhibitors in Patients with Pulmonary Arterial Hypertension (REPLACE): A Multicentre, Open-Label, Randomised Controlled Trial. Lancet Respir. Med. 2021, 9, 573–584. [Google Scholar] [CrossRef]
- McLaughlin, V.; Channick, R.N.; Ghofrani, H.-A.; Lemarié, J.-C.; Naeije, R.; Packer, M.; Souza, R.; Tapson, V.F.; Tolson, J.; Al Hiti, H.; et al. Bosentan Added to Sildenafil Therapy in Patients with Pulmonary Arterial Hypertension. Eur. Respir. J. 2015, 46, 405–413. [Google Scholar] [CrossRef] [Green Version]
- Chin, K.M.; Sitbon, O.; Doelberg, M.; Feldman, J.; Gibbs, J.S.R.; Grünig, E.; Hoeper, M.M.; Martin, N.; Mathai, S.C.; McLaughlin, V.V.; et al. Three- Versus Two-Drug Therapy for Patients with Newly Diagnosed Pulmonary Arterial Hypertension. J. Am. Coll. Cardiol. 2021, 78, 1393–1403. [Google Scholar] [CrossRef]
- Badesch, D.; Gibbs, S.; Gomberg-Maitland, M.; Humbert, M.; Mclaughlin, V.; Preston, I.; Souza, R.; Waxman, A.; De Oliveira Pena, J.; Barnes, J.; et al. PULSAR: A Phase 2, Randomized, Double-Blind, Placebo-Controlled Study to Assess the Efficacy and Safety of Sotatercept (ACE-011) when Added to Standard of Care (SOC) for Treatment of Pulmonary Arterial Hypertension (PAH). Eur. Respir. J. 2019, 54, PA4750. [Google Scholar] [CrossRef]
- Humbert, M.; McLaughlin, V.; Gibbs, J.S.R.; Gomberg-Maitland, M.; Hoeper, M.M.; Preston, I.R.; Souza, R.; Waxman, A.; Escribano Subias, P.; Feldman, J.; et al. Sotatercept for the Treatment of Pulmonary Arterial Hypertension. N. Engl. J. Med. 2021, 384, 1204–1215. [Google Scholar] [CrossRef]
- Spiekerkoetter, E.; Sung, Y.K.; Sudheendra, D.; Bill, M.; Aldred, M.A.; van de Veerdonk, M.C.; Vonk Noordegraaf, A.; Long-Boyle, J.; Dash, R.; Yang, P.C.; et al. Low-Dose FK506 (Tacrolimus) in End-Stage Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2015, 192, 254–257. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Cui, X.; Qian, Z.; Li, Y.; Kang, K.; Qu, J.; Li, L.; Gou, D. Multi-Omics Analysis Reveals Regulators of the Response to PDGF-BB Treatment in Pulmonary Artery Smooth Muscle Cells. BMC Genom. 2016, 17, 781. [Google Scholar] [CrossRef]
- Hoeper, M.M.; Barst, R.J.; Bourge, R.C.; Feldman, J.; Frost, A.E.; Galié, N.; Gómez-Sánchez, M.A.; Grimminger, F.; Grünig, E.; Hassoun, P.M.; et al. Imatinib Mesylate as Add-on Therapy for Pulmonary Arterial Hypertension: Results of the Randomized IMPRES Study. Circulation 2013, 127, 1128–1138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frantz, R.P.; Benza, R.L.; Channick, R.N.; Chin, K.; Howard, L.S.; McLaughlin, V.V.; Sitbon, O.; Zamanian, R.T.; Hemnes, A.R.; Cravets, M.; et al. TORREY, a Phase 2 Study to Evaluate the Efficacy and Safety of Inhaled Seralutinib for the Treatment of Pulmonary Arterial Hypertension. Pulm. Circ. 2021, 11, 20458940211057071. [Google Scholar] [CrossRef] [PubMed]
- Frantz, R.P.; Highland, K.B.; McConnell, J.W.; Burger, C.D.; Roscigno, R.F.; Cravets, M.; McCaffrey, R.; Zisman, L.S.; Howard, L. A Phase 1b, Multi-Center, Randomized, Placebo-Controlled Trial of Inhaled Seralutinib in Subjects with WHO Group 1 Pulmonary Arterial Hypertension. In TP82. TP082 Let It Be—Clinical Advances in Pulmonary Vascular Disease: Pah and beyond, May; American Thoracic Society: New York, NY, USA, 2021; p. A3602. [Google Scholar]
- Archer, S.L. Acquired Mitochondrial Abnormalities, Including Epigenetic Inhibition of Superoxide Dismutase 2, in Pulmonary Hypertension and Cancer: Therapeutic Implications. In Hypoxia: Translation in Progress; Roach, R.C., Hackett, P.H., Wagner, P.D., Eds.; Springer: Boston, MA, USA, 2016; pp. 29–53. ISBN 978-1-4899-7678-9. [Google Scholar]
- Dean, A.; Nilsen, M.; Loughlin, L.; Salt, I.P.; MacLean, M.R. Metformin Reverses Development of Pulmonary Hypertension via Aromatase Inhibition. Hypertension 2016, 68, 446–454. [Google Scholar] [CrossRef] [Green Version]
- Safdar, Z.; Cho, E. Effect of Spironolactone Use in Pulmonary Arterial Hypertension—Analysis from Pivotal Trial Databases. Pulm. Circ. 2021, 11, 20458940211045616. [Google Scholar] [CrossRef] [PubMed]
- Lazarus, H.; Denning, J.; Kamau-Kelley, W.; Wring, S.; Palacios, M.; Humbert, M. ELEVATE 2: A Multicenter Study of Rodatristat Ethyl in Patients with WHO Group 1 Pulmonary Arterial Hypertension (PAH). Eur. Respir. J. 2021, 58, PA1919. [Google Scholar] [CrossRef]
- Lazarus, H.M.; Denning, J.; Wring, S.; Palacios, M.; Hoffman, S.; Crizer, K.; Kamau-Kelley, W.; Symonds, W.; Feldman, J. A Trial Design to Maximize Knowledge of the Effects of Rodatristat Ethyl in the Treatment of Pulmonary Arterial Hypertension (ELEVATE 2). Pulm. Circ. 2022, 12, e12088. [Google Scholar] [CrossRef]
- Al-Omary, M.S.; Sugito, S.; Boyle, A.J.; Sverdlov, A.L.; Collins, N.J. Pulmonary Hypertension Due to Left Heart Disease: Diagnosis, Pathophysiology, and Therapy. Hypertension 2020, 75, 1397–1408. [Google Scholar] [CrossRef]
- Fang, J.C.; DeMarco, T.; Givertz, M.M.; Borlaug, B.A.; Lewis, G.D.; Rame, J.E.; Gomberg-Maitland, M.; Murali, S.; Frantz, R.P.; McGlothlin, D.; et al. World Health Organization Pulmonary Hypertension Group 2: Pulmonary Hypertension due to Left Heart Disease in the Adult—A Summary Statement from the Pulmonary Hypertension Council of the International Society for Heart and Lung Transplantation. J. Heart Lung Transplant. 2012, 31, 913–933. [Google Scholar] [CrossRef]
- Tedford, R.J.; Hassoun, P.M.; Mathai, S.C.; Girgis, R.E.; Russell, S.D.; Thiemann, D.R.; Cingolani, O.H.; Mudd, J.O.; Borlaug, B.A.; Redfield, M.M.; et al. Pulmonary Capillary Wedge Pressure Augments Right Ventricular Pulsatile Loading. Circulation 2012, 125, 289–297. [Google Scholar] [CrossRef] [Green Version]
- Melenovsky, V.; Andersen, M.J.; Andress, K.; Reddy, Y.N.; Borlaug, B.A. Lung Congestion in Chronic Heart Failure: Haemodynamic, Clinical, and Prognostic Implications: Congestion and Pulmonary Haemodynamics in HF. Eur. J. Heart Fail. 2015, 17, 1161–1171. [Google Scholar] [CrossRef]
- Yancy, C.W.; Jessup, M.; Bozkurt, B.; Butler, J.; Casey, D.E.; Drazner, M.H.; Fonarow, G.C.; Geraci, S.A.; Horwich, T.; Januzzi, J.L.; et al. 2013 ACCF/AHA Guideline for the Management of Heart Failure: A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 2013, 128, e147–e239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Califf, R.M.; Adams, K.F.; McKenna, W.J.; Gheorghiade, M.; Uretsky, B.F.; McNulty, S.E.; Darius, H.; Schulman, K.; Zannad, F.; Handberg-Thurmond, E.; et al. A Randomized Controlled Trial of Epoprostenol Therapy for Severe Congestive Heart Failure: The Flolan International Randomized Survival Trial (FIRST). Am. Heart J. 1997, 134, 44–54. [Google Scholar] [CrossRef]
- Anand, I.; McMurray, J.; Cohn, J.N.; Konstam, M.A.; Notter, T.; Quitzau, K.; Ruschitzka, F.; Lüscher, T.F. Long-Term Effects of Darusentan on Left-Ventricular Remodelling and Clinical Outcomes in the Endothelin A Receptor Antagonist Trial in Heart Failure (EARTH): Randomised, Double-Blind, Placebo-Controlled Trial. Lancet 2004, 364, 347–354. [Google Scholar] [CrossRef]
- Vachiéry, J.-L.; Delcroix, M.; Al-Hiti, H.; Efficace, M.; Hutyra, M.; Lack, G.; Papadakis, K.; Rubin, L.J. Macitentan in Pulmonary Hypertension Due to Left Ventricular Dysfunction. Eur. Respir. J. 2018, 51, 1701886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kramer, T.; Dumitrescu, D.; Gerhardt, F.; Orlova, K.; ten Freyhaus, H.; Hellmich, M.; Baldus, S.; Rosenkranz, S. Therapeutic Potential of Phosphodiesterase Type 5 Inhibitors in Heart Failure with Preserved Ejection Fraction and Combined Post- and Pre-Capillary Pulmonary Hypertension. Int. J. Cardiol. 2019, 283, 152–158. [Google Scholar] [CrossRef]
- Ghofrani, H.A.; Grimminger, F. Soluble Guanylate Cyclase Stimulation: An Emerging Option in Pulmonary Hypertension Therapy. Eur. Respir. Rev. 2009, 18, 35–41. [Google Scholar] [CrossRef]
- Bonderman, D.; Ghio, S.; Felix, S.B.; Ghofrani, H.-A.; Michelakis, E.; Mitrovic, V.; Oudiz, R.J.; Boateng, F.; Scalise, A.-V.; Roessig, L.; et al. Riociguat for Patients with Pulmonary Hypertension Caused by Systolic Left Ventricular Dysfunction: A Phase IIb Double-Blind, Randomized, Placebo-Controlled, Dose-Ranging Hemodynamic Study. Circulation 2013, 128, 502–511. [Google Scholar] [CrossRef] [Green Version]
- Gheorghiade, M.; Greene, S.J.; Butler, J.; Filippatos, G.; Lam, C.S.P.; Maggioni, A.P.; Ponikowski, P.; Shah, S.J.; Solomon, S.D.; Kraigher-Krainer, E.; et al. Effect of Vericiguat, a Soluble Guanylate Cyclase Stimulator, on Natriuretic Peptide Levels in Patients with Worsening Chronic Heart Failure and Reduced Ejection Fraction: The Socrates-Reduced Randomized Trial. JAMA 2015, 314, 2251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brittain, E.L.; Niswender, K.; Agrawal, V.; Chen, X.; Fan, R.; Pugh, M.E.; Rice, T.W.; Robbins, I.M.; Song, H.; Thompson, C.; et al. Mechanistic Phase II Clinical Trial of Metformin in Pulmonary Arterial Hypertension. JAHA 2020, 9, e018349. [Google Scholar] [CrossRef] [PubMed]
- McGettrick, M.; Peacock, A. Group 3 Pulmonary Hypertension: Challenges and Opportunities. Glob. Cardiol. Sci. Pract. 2020, 2020, e202006. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Dorfmüller, P.; Shlobin, O.A.; Ventetuolo, C.E. Group 3 Pulmonary Hypertension: From Bench to Bedside. Circ. Res. 2022, 130, 1404–1422. [Google Scholar] [CrossRef]
- Chaouat, A.; Naeije, R.; Weitzenblum, E. Pulmonary Hypertension in COPD. Eur. Respir. J. 2008, 32, 1371–1385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minai, O.A.; Ricaurte, B.; Kaw, R.; Hammel, J.; Mansour, M.; McCarthy, K.; Golish, J.A.; Stoller, J.K. Frequency and Impact of Pulmonary Hypertension in Patients with Obstructive Sleep Apnea Syndrome. Am. J. Cardiol. 2009, 104, 1300–1306. [Google Scholar] [CrossRef]
- King, C.S.; Shlobin, O.A. The Trouble with Group 3 Pulmonary Hypertension in Interstitial Lung Disease: Dilemmas in Diagnosis and the Conundrum of Treatment. Chest 2020, 158, 1651–1664. [Google Scholar] [CrossRef]
- King, T.E.; Brown, K.K.; Raghu, G.; du Bois, R.M.; Lynch, D.A.; Martinez, F.; Valeyre, D.; Leconte, I.; Morganti, A.; Roux, S.; et al. BUILD-3: A Randomized, Controlled Trial of Bosentan in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2011, 184, 92–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, T.E.; Behr, J.; Brown, K.K.; du Bois, R.M.; Lancaster, L.; de Andrade, J.A.; Stähler, G.; Leconte, I.; Roux, S.; Raghu, G. BUILD-1: A Randomized Placebo-Controlled Trial of Bosentan in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2008, 177, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Corte, T.J.; Keir, G.J.; Dimopoulos, K.; Howard, L.; Corris, P.A.; Parfitt, L.; Foley, C.; Yanez-Lopez, M.; Babalis, D.; Marino, P.; et al. Bosentan in Pulmonary Hypertension Associated with Fibrotic Idiopathic Interstitial Pneumonia. Am. J. Respir. Crit. Care Med. 2014, 190, 208–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raghu, G.; Million-Rousseau, R.; Morganti, A.; Perchenet, L.; Behr, J.; MUSIC Study Group. Macitentan for the Treatment of Idiopathic Pulmonary Fibrosis: The Randomised Controlled MUSIC Trial. Eur. Respir. J. 2013, 42, 1622–1632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raghu, G.; Behr, J.; Brown, K.K.; Egan, J.J.; Kawut, S.M.; Flaherty, K.R.; Martinez, F.J.; Nathan, S.D.; Wells, A.U.; Collard, H.R.; et al. Treatment of Idiopathic Pulmonary Fibrosis with Ambrisentan: A Parallel, Randomized Trial. Ann. Intern. Med. 2013, 158, 641–649. [Google Scholar] [CrossRef]
- Zisman, D.A.; Schwarz, M.; Anstrom, K.J. A Controlled Trial of Sildenafil in Advanced Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2010, 363, 620–628. [Google Scholar] [CrossRef] [PubMed]
- Nathan, S.D.; Behr, J.; Collard, H.R.; Cottin, V.; Hoeper, M.M.; Martinez, F.J.; Corte, T.J.; Keogh, A.M.; Leuchte, H.; Mogulkoc, N.; et al. Riociguat for Idiopathic Interstitial Pneumonia-Associated Pulmonary Hypertension (RISE-IIP): A Randomised, Placebo-Controlled Phase 2b Study. Lancet Respir. Med. 2019, 7, 780–790. [Google Scholar] [CrossRef]
- Waxman, A.; Restrepo-Jaramillo, R.; Thenappan, T.; Ravichandran, A.; Engel, P.; Bajwa, A.; Allen, R.; Feldman, J.; Argula, R.; Smith, P.; et al. Inhaled Treprostinil in Pulmonary Hypertension Due to Interstitial Lung Disease. N. Engl. J. Med. 2021, 384, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.; Jiang, X.; Jing, Z.-C. How Should a Physician Approach the Pharmacological Management of Chronic Thromboembolic Pulmonary Hypertension? Expert Opin. Pharmacother. 2021, 22, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.H.; Delcroix, M.; Jais, X.; Madani, M.M.; Matsubara, H.; Mayer, E.; Ogo, T.; Tapson, V.F.; Ghofrani, H.-A.; Jenkins, D.P. Chronic Thromboembolic Pulmonary Hypertension. Eur. Respir. J. 2019, 53, 1801915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simonneau, G.; Torbicki, A.; Dorfmüller, P.; Kim, N. The Pathophysiology of Chronic Thromboembolic Pulmonary Hypertension. Eur. Respir. Rev. 2017, 26, 160112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaïs, X.; D’Armini, A.M.; Jansa, P.; Torbicki, A.; Delcroix, M.; Ghofrani, H.A.; Hoeper, M.M.; Lang, I.M.; Mayer, E.; Pepke-Zaba, J.; et al. Bosentan for Treatment of Inoperable Chronic Thromboembolic Pulmonary Hypertension: BENEFiT (Bosentan Effects in INopErable Forms of ChronIc Thromboembolic Pulmonary Hypertension), a Randomized, Placebo-Controlled Trial. J. Am. Coll. Cardiol. 2008, 52, 2127–2134. [Google Scholar] [CrossRef] [PubMed]
- Escribano-Subias, P.; Bendjenana, H.; Curtis, P.S.; Lang, I.; Noordegraaf, A.V. Ambrisentan for Treatment of Inoperable Chronic Thromboembolic Pulmonary Hypertension (CTEPH). Pulm. Circ. 2019, 9, 2045894019846433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghofrani, H.-A.; Simonneau, G.; D’Armini, A.M.; Fedullo, P.; Howard, L.S.; Jaïs, X.; Jenkins, D.P.; Jing, Z.-C.; Madani, M.M.; Martin, N.; et al. Macitentan for the Treatment of Inoperable Chronic Thromboembolic Pulmonary Hypertension (MERIT-1): Results from the Multicentre, Phase 2, Randomised, Double-Blind, Placebo-Controlled Study. Lancet Respir. Med. 2017, 5, 785–794. [Google Scholar] [CrossRef]
- Sadushi-Kolici, R.; Jansa, P.; Kopec, G.; Torbicki, A.; Skoro-Sajer, N.; Campean, I.-A.; Halank, M.; Simkova, I.; Karlocai, K.; Steringer-Mascherbauer, R.; et al. Subcutaneous Treprostinil for the Treatment of Severe Non-Operable Chronic Thromboembolic Pulmonary Hypertension (CTREPH): A Double-Blind, Phase 3, Randomised Controlled Trial. Lancet Respir. Med. 2019, 7, 239–248. [Google Scholar] [CrossRef]
- Anthi, A.; Tsangaris, I.; Hamodraka, E.S.; Lekakis, J.; Armaganidis, A.; Orfanos, S.E. Treatment with Bosentan in a Patient with Thalassemia Intermedia and Pulmonary Arterial Hypertension. Blood 2012, 120, 1531–1532. [Google Scholar] [CrossRef] [PubMed]
- Morris, C.R.; Kim, H.-Y.; Wood, J.; Porter, J.B.; Klings, E.S.; Trachtenberg, F.L.; Sweeters, N.; Olivieri, N.F.; Kwiatkowski, J.L.; Virzi, L.; et al. Sildenafil Therapy in Thalassemia Patients with Doppler-Defined Risk of Pulmonary Hypertension. Haematologica 2013, 98, 1359–1367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalantari, S.; Gomberg-Maitland, M. Group 5 Pulmonary Hypertension: The Orphan’s Orphan Disease. Cardiol. Clin. 2016, 34, 443–449. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beckmann, T.; Shelley, P.; Patel, D.; Vorla, M.; Kalra, D.K. Strategizing Drug Therapies in Pulmonary Hypertension for Improved Outcomes. Pharmaceuticals 2022, 15, 1242. https://doi.org/10.3390/ph15101242
Beckmann T, Shelley P, Patel D, Vorla M, Kalra DK. Strategizing Drug Therapies in Pulmonary Hypertension for Improved Outcomes. Pharmaceuticals. 2022; 15(10):1242. https://doi.org/10.3390/ph15101242
Chicago/Turabian StyleBeckmann, Taylor, Patrisha Shelley, Darshan Patel, Mounica Vorla, and Dinesh K. Kalra. 2022. "Strategizing Drug Therapies in Pulmonary Hypertension for Improved Outcomes" Pharmaceuticals 15, no. 10: 1242. https://doi.org/10.3390/ph15101242
APA StyleBeckmann, T., Shelley, P., Patel, D., Vorla, M., & Kalra, D. K. (2022). Strategizing Drug Therapies in Pulmonary Hypertension for Improved Outcomes. Pharmaceuticals, 15(10), 1242. https://doi.org/10.3390/ph15101242