
SnCl4 Promoted Efficient Cleavage of Acetal/Ketal Groups with the Assistance of Water in CH2Cl2

Abstract
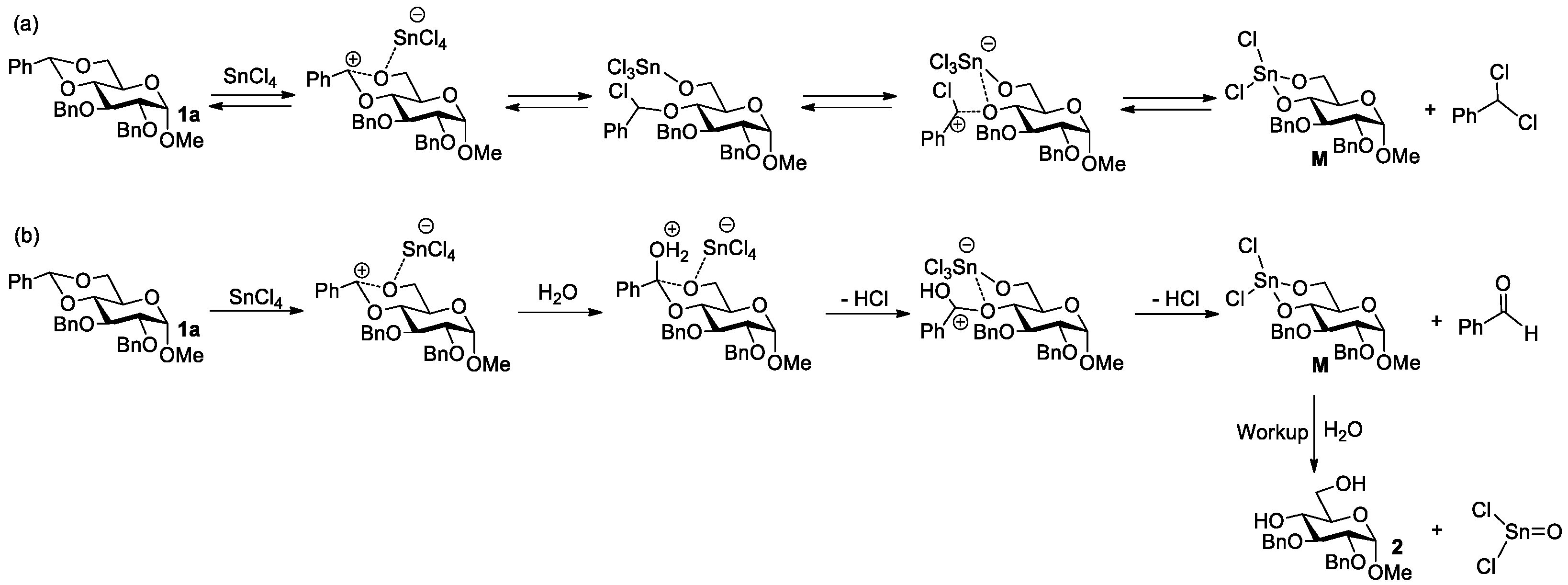
:1. Introduction
2. Results
3. Conclusions
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, H.-Y.; Blaszczyk, S.A.; Xiao, G.-Z.; Tang, W.-P. Chiral reagents in glycosylation and modification of carbohydrates. Chem. Soc. Rev. 2018, 47, 681–701. [Google Scholar] [CrossRef] [PubMed]
- Dimakos, V.; Taylor, M.S. Site-Selective Functionalization of Hydroxyl Groups in Carbohydrate Derivatives. Chem. Rev. 2018, 118, 11457–11517. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.; Liu, C.-Y.; Guo, Y.-F.; Feng, G.-J.; Dong, H. SnCl2-Catalyzed Acetalation/Selective Benzoylation Sequence for the Synthesis of Orthogonally Protected Glycosyl Acceptors. Eur. J. Org. Chem. 2022, 2022, e202101565. [Google Scholar] [CrossRef]
- Guo, Y.-F.; Luo, T.; Feng, G.-J.; Liu, C.-Y.; Dong, H. Efficient Synthesis of 2-OH Thioglycosides from Glycals Based on the Reduction of Aryl Disulfides by NaBH4. Molecules 2022, 27, 5980. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.; Yu, J.-C.; Feng, G.-J.; Luo, T.; Dong, H. Stannous chloride as a low toxicity and extremely cheap catalyst for regio-/site-selective acylation with unusually broad substrate scope. Green Chem. 2020, 22, 6936–6942. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, F.-L.; Luo, T.; Pei, Z.; Dong, H. Regio/Stereoselective Glycosylation of Diol and Polyol Acceptors in Efficient Synthesis of Neu5Ac-α-2,3-LacNPhth Trisaccharide. Chem. Asian J. 2019, 14, 223–234. [Google Scholar] [CrossRef]
- Ramadan, S.; Yang, W.-Z.; Zhang, Z.-R.; Huang, X.-F. Synthesis of Chondroitin Sulfate A Bearing Syndecan 1 Glycopeptide. Org. Lett. 2017, 19, 4838–4841. [Google Scholar] [CrossRef]
- Lv, J.; Zhu, J.-J.; Liu, Y.; Dong, H. Regioselective Sulfonylation/Acylation of Carbohydrates Catalyzed by FeCl3 Combined with Benzoyltrifluoroacetone and Its Mechanism Study. J. Org. Chem. 2020, 85, 3307–3319. [Google Scholar] [CrossRef]
- Lv, J.; Liu, Y.; Zhu, J.-J.; Zou, D.; Dong, H. Regio/site-selective alkylation of substrates containing a cis-, 1,2- or 1,3-diol with ferric chloride and dipivaloylmethane as the catalytic system. Green Chem. 2020, 22, 1139–1144. [Google Scholar] [CrossRef]
- Lv, J.; Ge, J.-T.; Luo, T.; Dong, H. An inexpensive catalyst, Fe(acac)3, for regio/siteselective acylation of diols and carbohydrates containing a 1,2-cis-diol. Green Chem. 2018, 20, 1987–1991. [Google Scholar] [CrossRef]
- Xu, H.-F.; Ren, B.; Zhao, W.; Xin, X.-T.; Lu, Y.-C.; Pei, Y.-X.; Dong, H.; Pei, Z.-C. Regioselective mono and multiple alkylation of diols and polyols catalyzed by organotin and its applications on the synthesis of value-added carbohydrate intermediates. Tetrahedron 2016, 72, 3490–3499. [Google Scholar] [CrossRef]
- Traboni, S.; Bedini, E.; Giordano, M.; Iadonisi, A. Three Solvent-Free Catalytic Approaches to the Acetal Functionalization of Carbohydrates and Their Applicability to One-Pot Generation of Orthogonally Protected Building Blocks. Adv. Synth. Catal. 2015, 357, 3562–3572. [Google Scholar] [CrossRef]
- Tran, A.T.; Jones, R.A.; Pastor, J.; Boisson, J.; Smith, N.; Galan, M.C. Copper(II) Triflate: A Versatile Catalyst for the One-Pot Preparation of Orthogonally Protected Glycosides. Adv. Synth. Catal. 2011, 353, 2593–2598. [Google Scholar] [CrossRef]
- Jones, R.A.; Davidson, R.; Tran, A.T.; Smith, N.; Galan, M.C. Iodine-catalyzed one-pot acetalation-esterification reaction for the preparation of orthogonally protected glycosides. Carbohydr. Res. 2010, 345, 1842–1845. [Google Scholar] [CrossRef]
- Vohra, Y.; Vasan, M.; Venot, A.; Boons, G.J. One-Pot Synthesis of Oligosaccharides by Combining Reductive Openings of Benzylidene Acetals and Glycosylations. Org. Lett. 2008, 10, 3247–3250. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.-C.; Lee, J.-C.; Luo, S.-Y.; Kulkarni, S.S.; Huang, Y.-W.; Lee, C.-C.; Chang, K.-L.; Hung, S.-C. Regioselective one-pot protection of carbohydrates. Nature 2007, 446, 896–899. [Google Scholar] [CrossRef]
- Shie, C.-R.; Tzeng, Z.-H.; Kulkarni, S.S.; Uang, B.-J.; Hsu, C.-Y.; Hung, S.-C. Cu(OTf)2 as an Efficient and Dual-Purpose Catalyst in the Regioselective Reductive Ring Opening of Benzylidene Acetals. Angew. Chem. Int. Ed. 2005, 44, 1665–1668. [Google Scholar] [CrossRef]
- Garegg, P.J.; Kvarnstrom, I.; Niklasson, A.; Niklasson, G.; Svensson, S.C.T. Partial Substitution of Thioglycosides by Phase Transfer Catalyzed Benzoylation and Benzylation. J. Carbohydr. Chem. 1993, 12, 933–953. [Google Scholar] [CrossRef]
- Abronina, P.I.; Malysheva, N.N.; Litvinenko, V.V.; Zinin, A.I.; Kolotyrkina, N.G.; Kononov, L.O. A Ring Contraction of 2,3-Di-O-Silylated Thiopyranosides To Give Thiofuranosides under Mildly Acidic Conditions. Org. Lett. 2018, 20, 6051–6054. [Google Scholar] [CrossRef]
- Yan, M.-C.; Chen, Y.-N.; Wu, H.-T.; Lin, C.-C.; Chen, C.-T.; Lin, C.-C. Removal of Acid-Labile Protecting Groups on Carbohydrates Using Water-Tolerant and Recoverable Vanadyl Triflate Catalyst. J. Org. Chem. 2007, 72, 299–302. [Google Scholar] [CrossRef]
- Procopio, A.; Dalpozzo, R.; Nino, A.D.; Maiuolo, L.; Nardi, M.; Romeo, G. Mild and efficient method for the cleavage of benzylidene acetals by using erbium (III) triflate. Org. Biomol. Chem. 2005, 3, 4129–4133. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Hui, Y.-Z. A Convenient Method for Highly Selective Deprotection of Benzylidene Acetals from Sugars. Synth. Commun. 1996, 26, 881–886. [Google Scholar] [CrossRef]
- Michigami, K.; Terauchi, M.; Hayashi, M. Cleavage of 4,6-O-Benzylidene Acetal Using Sodium Hydrogen Sulfate Monohydrate. Synthesis 2013, 45, 1519–1523. [Google Scholar]
- Szarek, W.A.; Zamojski, A.; Tiwari, K.N.; Ison, E.R. A new, facile method for cleavage of acetals and dithioacetals in carbohydrate derivatives. Tetrahedron Lett. 1986, 27, 3827–3830. [Google Scholar] [CrossRef]
- Chen, C.-T.; Lin, Y.-D.; Liu, C.-Y. Catalytic carbon–sulfur bond formation by amphoteric vanadyl triflate: Exploring with thia-Michael addition, thioacetalization, and transthioacetalization reactions. Tetrahedron 2009, 65, 10470–10476. [Google Scholar] [CrossRef]
- Liu, Y.; Zeng, J.; Sun, J.-C.; Cai, L.; Zhao, Y.-Q.; Fang, J.; Hu, B.; Shu, P.-H.; Meng, L.-K.; Wan, Q. 1,4-Dithiothreitol mediated cleavage of the acetal and ketal type of diol protecting groups. Org. Chem. Front. 2018, 5, 2427–2431. [Google Scholar] [CrossRef]
- Kim, K.-S.; Song, Y.-H.; Lee, B.-H.; Hahn, C.-S. Efficient and Selective Cleavage of Acetals and Ketals Using Ferric Chloride Adsorbed on Silica Gel. J. Org. Chem. 1986, 51, 404–407. [Google Scholar] [CrossRef]
- Niu, Y.-H.; Wang, N.; Cao, X.-P.; Ye, X.-S. Efficient Formation and Cleavage of Benzylidene Acetals by Sodium Hydrogen Sulfate Supported on Silica Gel. Synlett 2007, 13, 2116–2120. [Google Scholar] [CrossRef]
- Agnihotri, G.; Misra, A.K. Mild and efficient method for the cleavage of benzylidene acetals using HClO4-SiO2 and direct conversion of acetals to acetates. Tetrahedron Lett. 2006, 47, 3653–3658. [Google Scholar] [CrossRef]
- Roy, B.; Verma, P.; Mukhopadhyay, B. H2SO4-silica-promoted ‘on-column’ removal of benzylidene, isopropylidene, trityl and tert-butyldimethylsilyl groups. Carbohydr. Res. 2009, 344, 145–148. [Google Scholar] [CrossRef]
- Kumar, P.S.; Kumar, G.D.K.; Baskaran, S. Truly Catalytic and Chemoselective Cleavage of Benzylidene Acetal with Phosphomolybdic Acid Supported on Silica Gel. Eur. J. Org. Chem. 2008, 2008, 6063–6067. [Google Scholar] [CrossRef]
- Couri, M.R.C.; Evangelista, E.A.; Alves, R.B.; Prado, M.A.F.; Gil, R.P.F.; De Almeida, M.V.; Raslan, D.S. Microwave-Assisted Rapid Deacetalation of Carbohydrates. Synth. Commun. 2005, 35, 2025–2031. [Google Scholar] [CrossRef]
- Santra, A.; Ghosh, T.; Misra, A.K. Removal of benzylidene acetal and benzyl ether in carbohydrate derivatives using triethylsilane and Pd/C. Beilstein J. Org. Chem. 2013, 9, 74–78. [Google Scholar] [CrossRef] [Green Version]
- Hori, H.; Nishida, Y.; Ohrui, H.; Meguro, H. Regioselective De-O-benzylation with Lewis Acids. J. Org. Chem. 1989, 54, 1346–1353. [Google Scholar] [CrossRef]
- Doyle, L.M.; O’Sullivan, S.; Di Salvo, C.; McKinney, M.; McArdle, P.; Murphy, P.V. Stereoselective Epimerizations of Glycosyl Thiols. Org. Lett. 2017, 19, 5802–5805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kartha, K.P.R.; Kiso, M.; Hasegawa, A.; Jennings, H.J. Novel Selectivity in Carbohydrate Reactions III. Selective Deprotection of p-Methoxybenzyl (PMBn) Ethers of Carbohydrates by Tin(IV) Chloride. J. Carbohydr. Chem. 1998, 17, 811–817. [Google Scholar] [CrossRef]
- Sawama, Y.; Masuda, M.; Asai, S.; Goto, R.; Nagata, S.; Nishimura, S.; Monguchi, Y.; Sajiki, H. FeCl3-Catalyzed Self-Cleaving Deprotection of Methoxyphenylmethyl-Protected Alcohols. Org. Lett. 2015, 17, 434–437. [Google Scholar] [CrossRef] [PubMed]
- Kern, N.; Dombray, T.; Blanc, A.; Weibel, J.M.; Pale, P. Silver(I)-Catalyzed Deprotection of p-Methoxybenzyl Ethers: A Mild and Chemoselective Method. J. Org. Chem. 2012, 77, 9227–9235. [Google Scholar] [CrossRef]
- Ren, B.; Gan, L.; Zhang, L.; Yan, N.-N.; Dong, H. Diisopropylethylamine-triggered, highly efficient, self-catalyzed regioselective acylation of carbohydrates and diols. Org. Biomol. Chem. 2018, 16, 5591–5597. [Google Scholar] [CrossRef]
- Matwiejuk, M.; Thiem, J. Defining oxyanion reactivities in base-promoted glycosylations. Chem. Commun. 2011, 47, 8379–8381. [Google Scholar] [CrossRef]
- Rocheleau, S.; Pottel, J.; Huskić, I.; Moitessier, N. Highly Regioselective Monoacylation of Unprotected Glucopyranoside Using Transient Directing-Protecting Groups. Eur. J. Org. Chem. 2017, 2017, 646–656. [Google Scholar] [CrossRef]
- Medgyes, G.; Jerkovich, G.; Kuszmann, J.; Fügedi, P. Synthesis of sorbistin analogues. Carbohydr. Res. 1989, 186, 225–239. [Google Scholar] [CrossRef] [PubMed]
- Moen, A.R.; Anthonsen, T. Screening of the regioselectivity of acetyl xylan esterase from Bacillus pumilus as a catalyst for the deacetylation of glycoside acetates. Biocatal. Biotransformation 2009, 27, 226–236. [Google Scholar] [CrossRef]
- Attouche, A.; Urban, D.; Beau, J.M. A Tin-Free Regioselective Radical De-O-benzylation by an Intramolecular Hydrogen Atom Transfer on Carbohydrate Templates. Angew. Chem. Int. Ed. 2013, 52, 9572–9575. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.-Z.; Wang, K.-X.; Ma, K.-R.; Zhao, W.; Zhang, G.-Q. Preparation of rare L-idose derivatives from D-glucofuranose via neighboring acyl group assistance. Tetrahedron Lett. 2021, 73, 153135–153139. [Google Scholar] [CrossRef]
- Kajihara, Y.; Kodam, H.; Endo, T.; Hashimoto, H. Novel features of acceptor recognition by β-(1→4)-galactosyltransferase. Carbohydr. Res. 1998, 306, 361–378. [Google Scholar] [CrossRef]
- Fujiki, K.; Tanaka, K. Exploration of the Fluoride Reactivity of Aryltrifluoroborate on Selective Cleavage of Diphenylmethylsilyl Groups. Eur. J. Org. Chem. 2020, 29, 4616–4620. [Google Scholar] [CrossRef]
- Cavedon, C.; Sletten, E.T.; Madani, A.; Niemeyer, O.; Seeberger, P.H.; Pieber, B. Visible-Light-Mediated Oxidative Debenzylation Enables the Use of Benzyl Ethers as Temporary Protecting Groups. Org. Lett. 2021, 23, 514–518. [Google Scholar] [CrossRef]
- Johnsson, R.; Ohlin, M.; Ellervik, U. Reductive Openings of Benzylidene Acetals Revisited: A Mechanistic Scheme for Regio- and Stereoselectivity. J. Org. Chem. 2010, 75, 8003–8011. [Google Scholar] [CrossRef]
- Borén, H.B.; Garegg, P.J.; Pilotti, Å.; Swahn, C.-G. NMR Spectra of Some Glycoside Acetates in the Presence of Tris(dipivaloylmethanato)europium. Acta Chem. Scand. 1972, 26, 3261–3268. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Ramstrom, O.; Dong, H. Organosilicon-mediated regioselective acetylation of carbohydrates. Chem. Commun. 2012, 48, 5370–5372. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Senthilkumar, S.; Baskaran, S. Benzylidene acetal protecting group as a carboxylic acid surrogate: Synthesis of functionalized uronic acids and sugar amino acids. Chem. Eur. J. 2016, 22, 902–906. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-J.; Dayoub, W.; Chen, G.-R.; Lemaire, M. TMDS as a Dual-Purpose Reductant in the Regioselective Ring Cleavage of Hexopyranosyl Acetals to Ethers. Eur. J. Org. Chem. 2012, 2012, 1960–1966. [Google Scholar] [CrossRef]
- Seitz, A.; Wende, R.C.; Roesner, E.; Niedek, D.; Topp, C.; Colgan, A.C. McGarrigle, E.M.; Schreiner, P.R. Site-Selective Acylation of Pyranosides with Oligopeptide Catalysts. J. Org. Chem. 2021, 86, 3907–3922. [Google Scholar] [CrossRef] [PubMed]
- van der Vorm, S.; Hansen, T.; Overkleeft, H.S.; van der Marel, G.A.; Codée, J.D.C. The Influence of Acceptor Nucleophilicity on the Glycosylation Reaction Mechanism. Chem. Sci. 2017, 8, 1867–1875. [Google Scholar] [CrossRef] [Green Version]
- Maki, Y.; Nomura, K.; Okamoto, R.; Izumi, M.; Mizutani, Y.; Kajihara, Y. Acceleration and Deceleration Factors on the Hydrolysis Reaction of 4,6-O-Benzylidene Acetal Group. J. Org. Chem. 2020, 85, 15849–15856. [Google Scholar] [CrossRef]
- Emmadi, M.; Kulkarni, S.S. Synthesis of Rare Deoxy Amino Sugar Building Blocks Enabled the Total Synthesis of a Polysaccharide Repeating Unit Analogue from the LPS of Psychrobacter cryohalolentis K5T. J. Org. Chem. 2018, 83, 14323–14337. [Google Scholar] [CrossRef]
- Ye, D.-F.; Liu, Z.-Y.; Chen, H.; Sessler, J.L.; Lei, C.-H. Cesium Carbonate Catalyzed Esterification of N-Benzyl-N-Bocamides under Ambient Conditions. Org. Lett. 2019, 21, 6888–6892. [Google Scholar] [CrossRef]
- Bauder, C. A Convenient synthesis of orthogonally protected 2-deoxystreptamine (2-DOS) as an aminocyclitol scaffold for the development of novel aminoglycoside antibiotic derivatives against bacterial resistance. Org. Biomol. Chem. 2008, 6, 2952–2960. [Google Scholar] [CrossRef]
- Babu, R.B.R.; Sørensen, M.D. Parmar, V.S.; Harrit, N.H.; Wengel, J. Oligodeoxynucleotides containing α-L-ribo confifigured LNA-type C-aryl nucleotides. Org. Biomol. Chem. 2004, 2, 80–89. [Google Scholar]
- Viuffa, A.H.; Heuckendorfa, M.; Jensen, H.H. p-Chlorobenzyl Ether: A p-Methoxybenzyl Ether in Disguise. Org. Lett. 2016, 18, 5773–5775. [Google Scholar] [CrossRef] [PubMed]
- Crich, D.; Banerjee, A.; Yao, Q.-J. Direct Chemical Synthesis of the β-d-Mannans: The β-(1→2) and β-(1→4) Series. J. Am. Chem. Soc. 2004, 126, 14930–14934. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, Y.; Kawada, T.; Rosenau, T.; Kosma, P. Synthesis of methyl 4′-O-methyl-13C12-β-D-cellobioside from 13C6-D-glucose. Part 1: Reaction optimization and synthesis. Carbohydr. Res. 2005, 340, 2428–2435. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Reaction Conditions | Lit. |
|---|---|---|
| 1 | AcOH/H2O, 100 °C, 0.5–1 h | [18,19] |
| 2 | TFA/H2O, CH2Cl2, rt, 0.5–1 h | [18,19] |
| 3 | VO(OTf)2, MeOH/CH2Cl2, 55 °C, 16–18 h | [20] |
| 4 | Er(OTf)3, CH3CN, rt, 2–24 h | [21] |
| 5 | SnCl2, CH2Cl2, rt, 12 h | [22] |
| 6 | NaHSO4, MeOH/CH2Cl2, rt, 1–24 h | [23] |
| 7 | I2, CH3OH, rt/reflux, 24/2.5 h | [24] |
| 8 | VO(OTf)2/HS(CH2)3SH, CH3CN/CH2Cl2, rt, 1 h | [25] |
| 9 | CSA/DTT, CH2Cl2, rt, 2 h | [26] |
| 10 | FeCl3-SiO2, CHCl3, rt, 10 h | [27] |
| 11 | NaHSO4-SiO2, MeOH/CH2Cl2, rt, 10–20 h | [28] |
| 12 | HClO4-SiO2, CH3CN, rt, 0.5 h | [29] |
| 13 | H2SO4-SiO2, CH2Cl2, rt, 0.5 h | [30] |
| 14 | PMA-SiO2, CH2Cl2, 0.5–5 h | [31] |
| 15 | AcOH/H2O-SiO2, microwave, 10 min | [32] |
| 16 | Et3SiH, Pd/C, CH3OH, rt, 0.5–2 h | [33] |

| Entry | Additives (equiv) | Reaction Conditions | Yield of 2 (%) |
|---|---|---|---|
| 1 | SnCl4 (0.2\0.5) | DCM, rt. 0.5 h | 19\47 |
| 2 | SnCl4 (1.0\1.5) | DCM, rt. 0.5 h | 69\79 |
| 3 | SnCl4 (2.0\2.5) | DCM, rt. 0.5 h | 93\95 |
| 4 | SnCl4 (2.5) | MeOH\MeCN, rt. 0.5 h | 20\40 |
| 5 b | SnCl4 (1.2), H2O (0.5\1.0) | DCM, rt. 0.5 h | 85\92 |
| 6 | SnCl4 (1.2), H2O (0.5\1.0) | DCM, rt. 0.5 h | 80\91 |
| 7 | SnCl4 (0.5\1.0), H2O (1.0) | DCM, rt. 0.5 h | 47\75 |
| 8 | SnCl4 (1.2\1.5), H2O (1.5\1.0) | DCM, rt. 10 min | 93\95 |
| 9 | SnCl4 (1.5), MeOH (2.0\3.0) | DCM, rt. 0.5 h | 37\41 |
| 10 | HCl (36%, 0.3) | DCM, rt. 0.5 h | 32 |
| 11 | SnCl2\FeCl3 (1.2), H2O (1.0) | DCM, rt. 0.5 h | 63\71 |
| 12 | CuCl2\Cu(OTf)2 (1.2), H2O (1.0) | DCM, rt. 0.5 h | 0\10 |
| 13 | SnCl4 (1.2), AcCl (1.2\2.2) | DCM, rt. 0.5 h | - |
| 14 | SnCl4 (1.2), Ac2O (1.2\2.2) | DCM, rt. 0.5 h | 2a: 30\78 |
| Entry | Substrate | Product | Yield |
|---|---|---|---|
| 1 |  |  | 92% from 1b 90% from 1c |
| 2 |  | 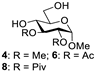 | 4: 91% 6: 98% 8: 97% |
| 3 |  | 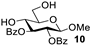 | 95% |
| 4 |  | 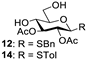 | 12: 90% 14: 94% |
| 5 |  | 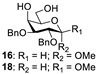 | 16: 89% 18: 90% |
| 6 |  |  | 93% |
| 7 |  |  | 97% |
| 8 | 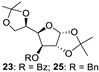 | 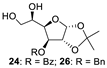 | 25: 94% 27: 95% |
| 9 b |  |  | 92% |
| 10 b |  |  | 98% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, T.; Xu, T.-T.; Guo, Y.-F.; Dong, H. SnCl4 Promoted Efficient Cleavage of Acetal/Ketal Groups with the Assistance of Water in CH2Cl2. Molecules 2022, 27, 8258. https://doi.org/10.3390/molecules27238258
Luo T, Xu T-T, Guo Y-F, Dong H. SnCl4 Promoted Efficient Cleavage of Acetal/Ketal Groups with the Assistance of Water in CH2Cl2. Molecules. 2022; 27(23):8258. https://doi.org/10.3390/molecules27238258
Chicago/Turabian StyleLuo, Tao, Tian-Tian Xu, Yang-Fan Guo, and Hai Dong. 2022. "SnCl4 Promoted Efficient Cleavage of Acetal/Ketal Groups with the Assistance of Water in CH2Cl2" Molecules 27, no. 23: 8258. https://doi.org/10.3390/molecules27238258
APA StyleLuo, T., Xu, T.-T., Guo, Y.-F., & Dong, H. (2022). SnCl4 Promoted Efficient Cleavage of Acetal/Ketal Groups with the Assistance of Water in CH2Cl2. Molecules, 27(23), 8258. https://doi.org/10.3390/molecules27238258