
Enhanced Permeability and Retention Effect as a Ubiquitous and Epoch-Making Phenomenon for the Selective Drug Targeting of Solid Tumors


Abstract
:
1. Introduction
2. Discovery of the Concept of the EPR Effect
3. Criticisms and Misconceptions about the EPR Effect
4. The EPR Effect Is a Rational and Dynamic Phenomenon for Tumor-Selective Drug Delivery
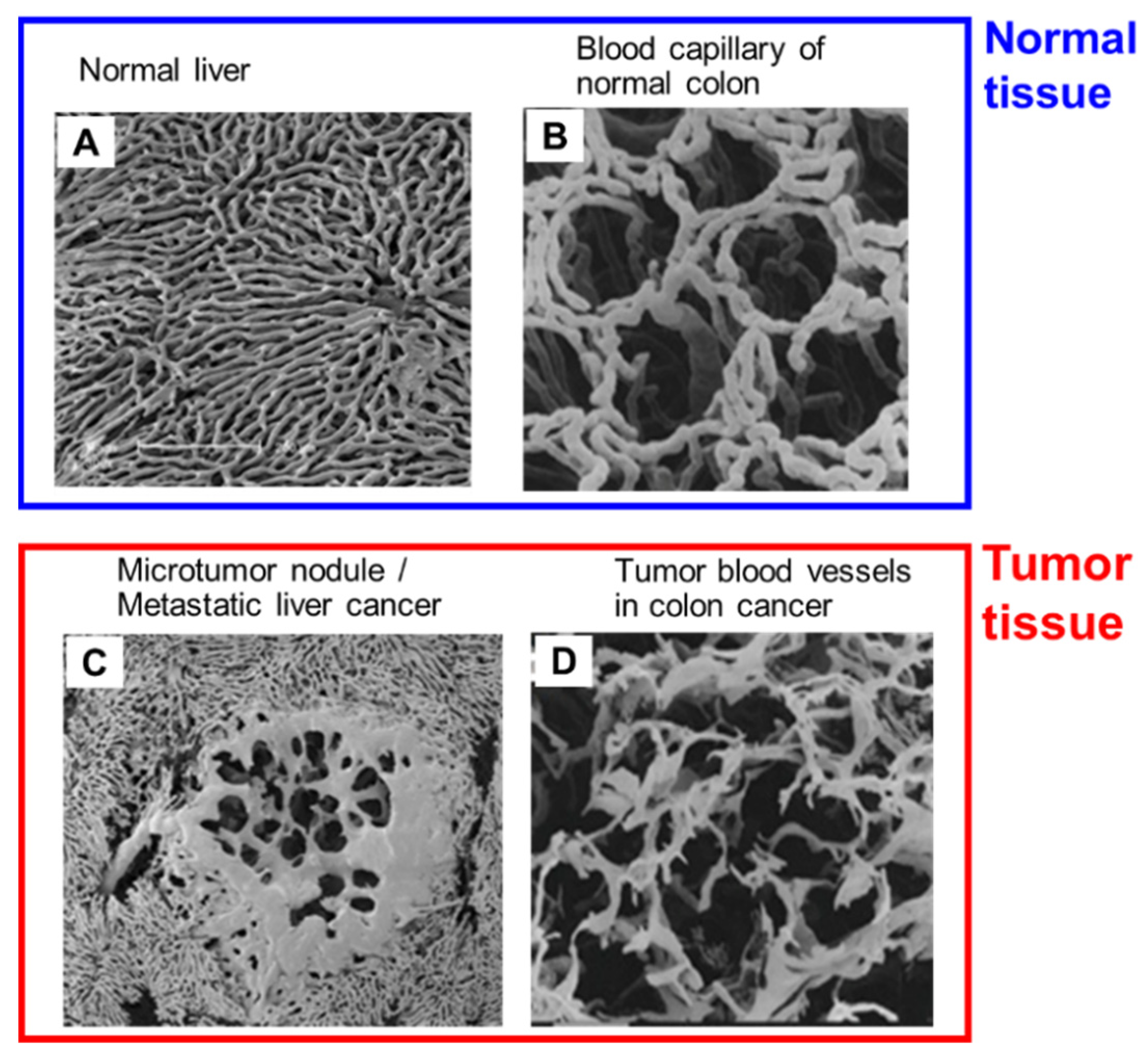
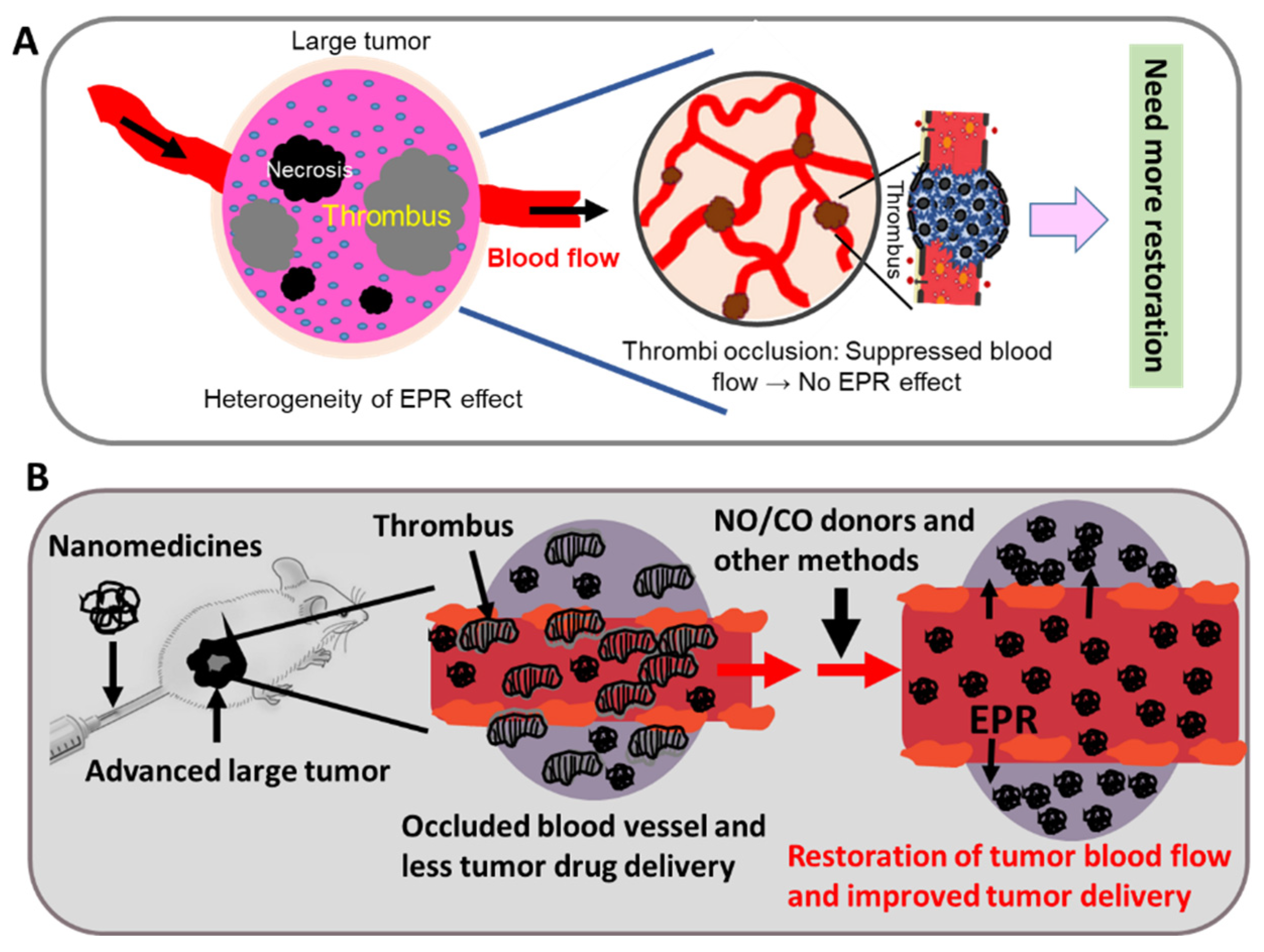
5. Heterogeneity of the EPR Effect: An Obstacle to Successful Nanomedicine Therapy in Clinical Settings
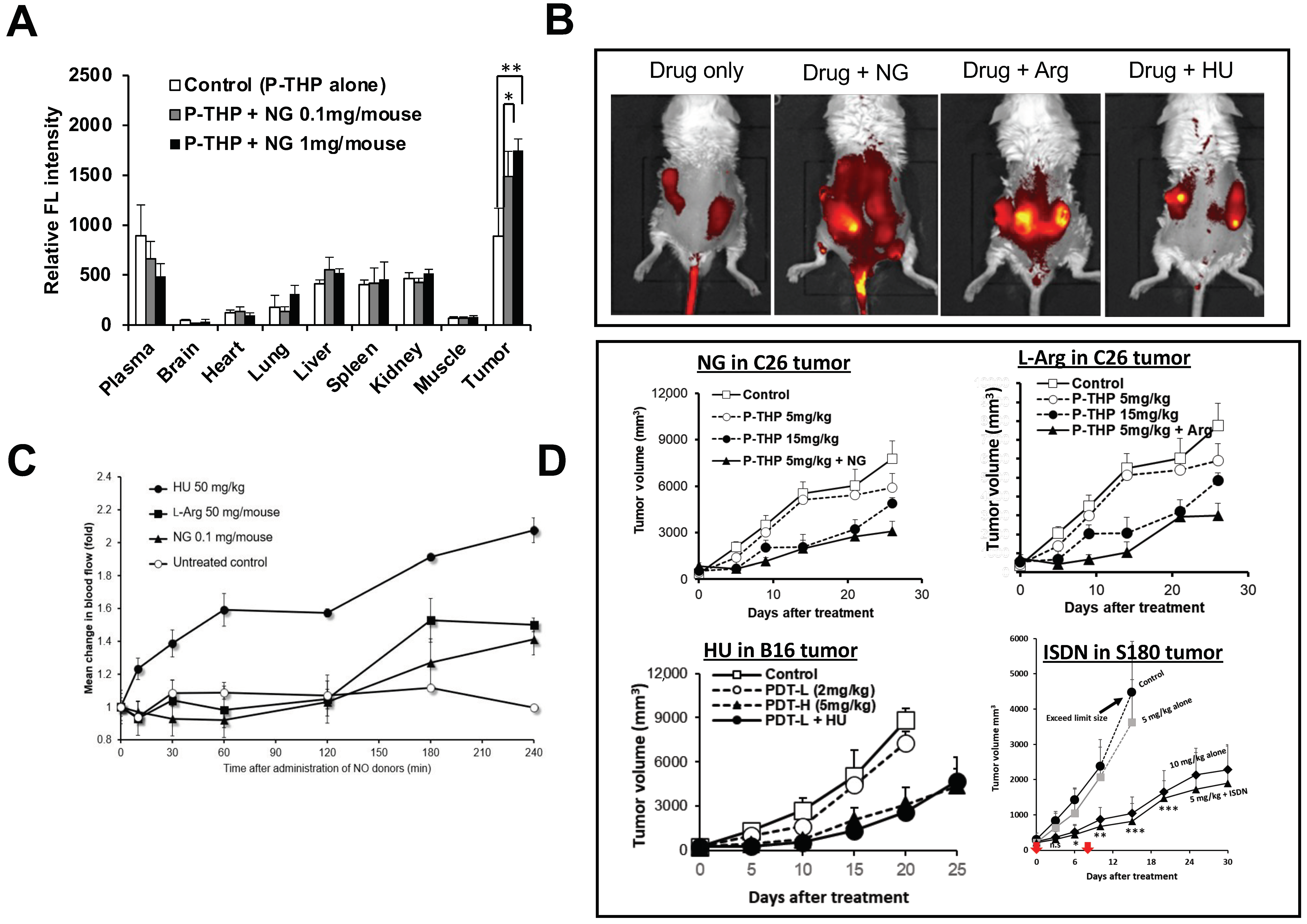
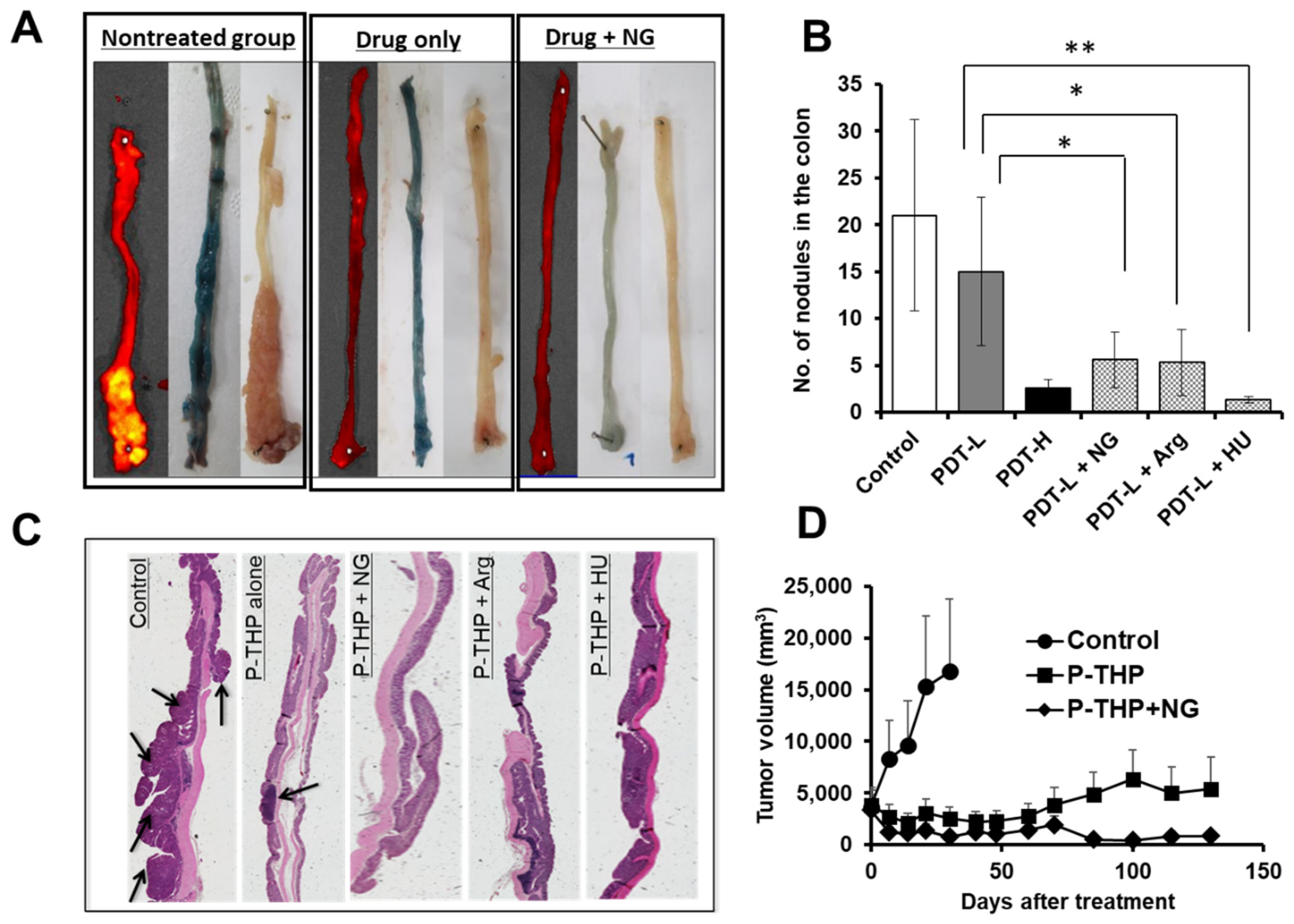
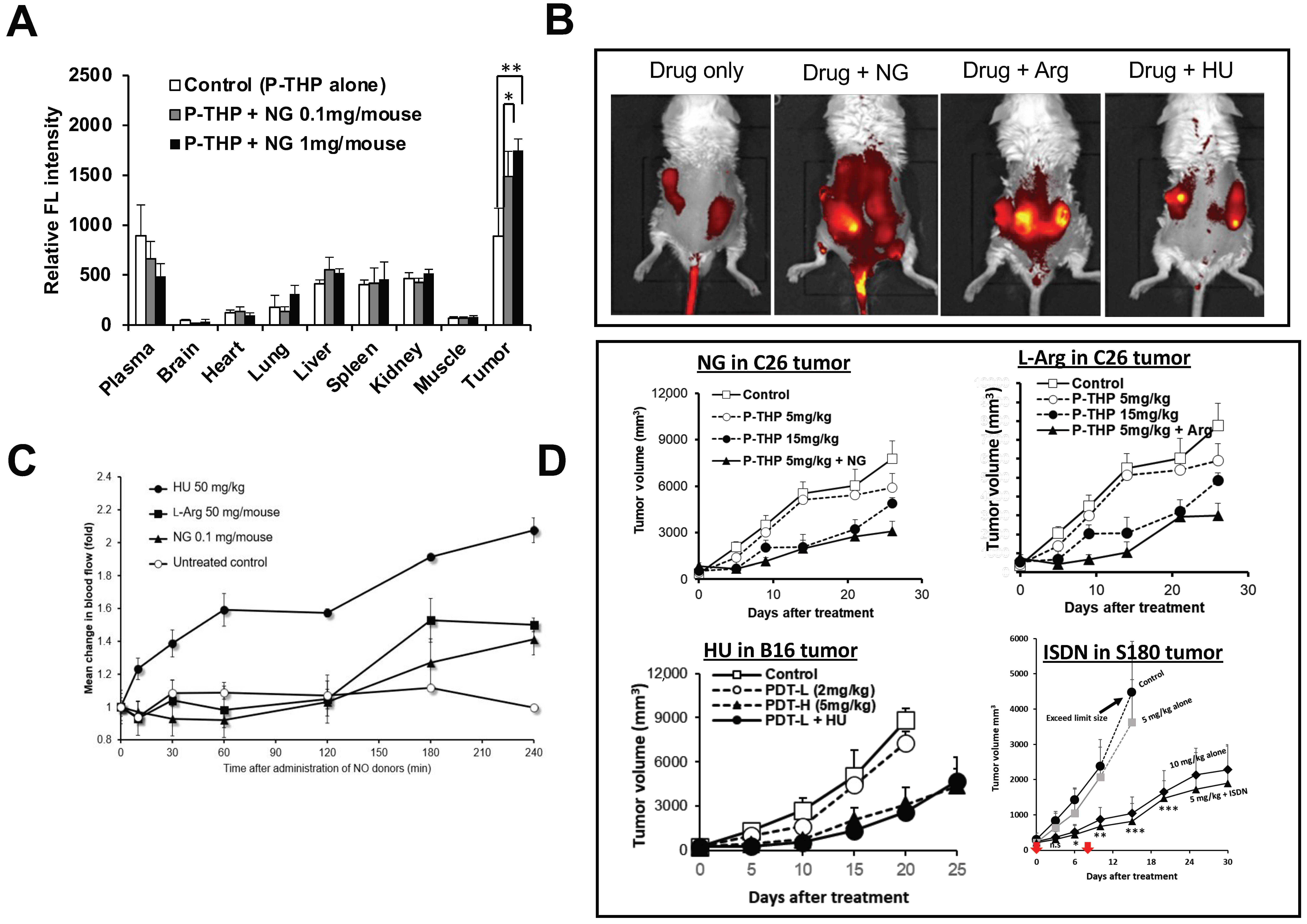
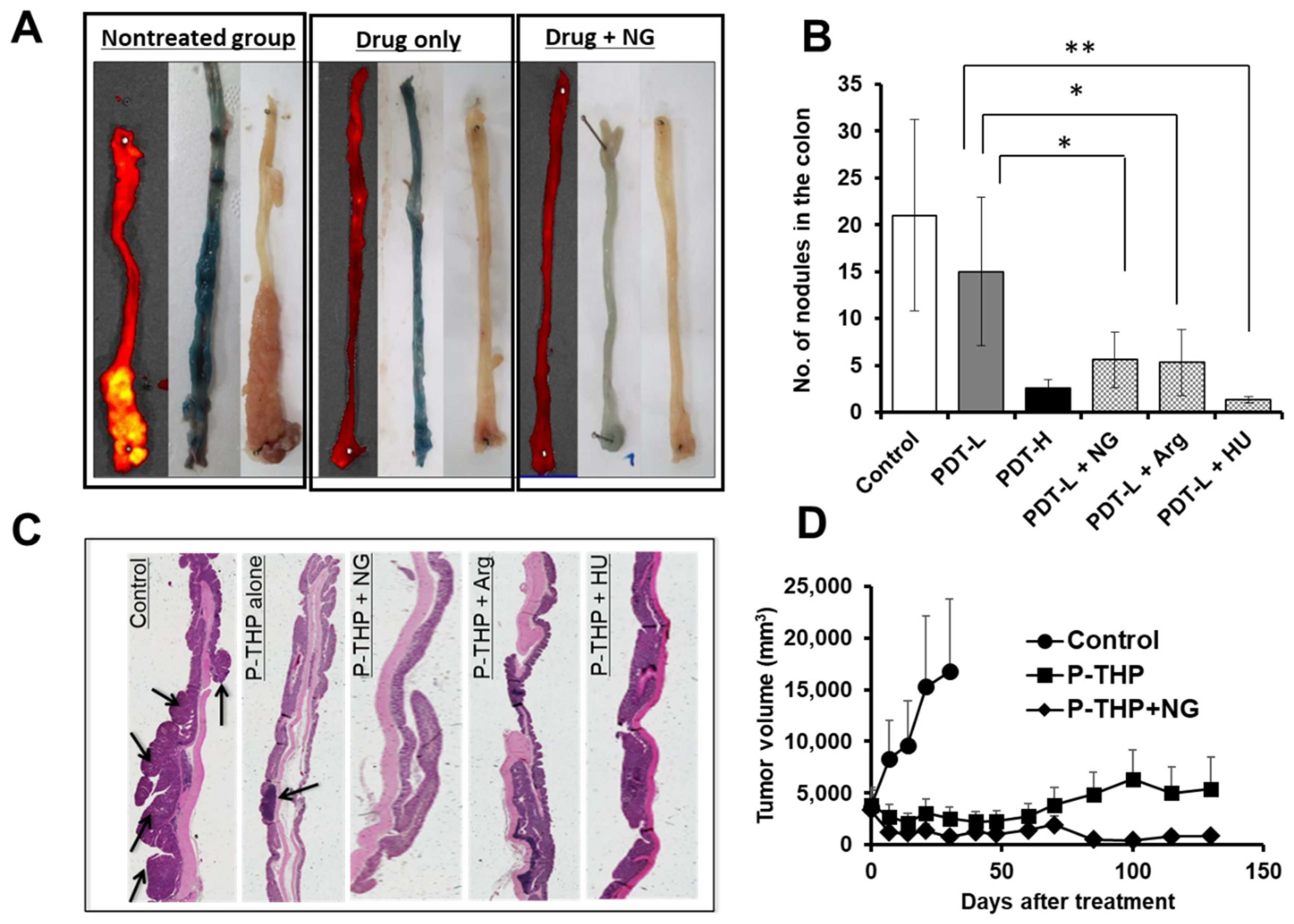
6. NO Donor-Induced Enhancement of Drug Delivery to Tumors as Well as of Therapeutic Effects
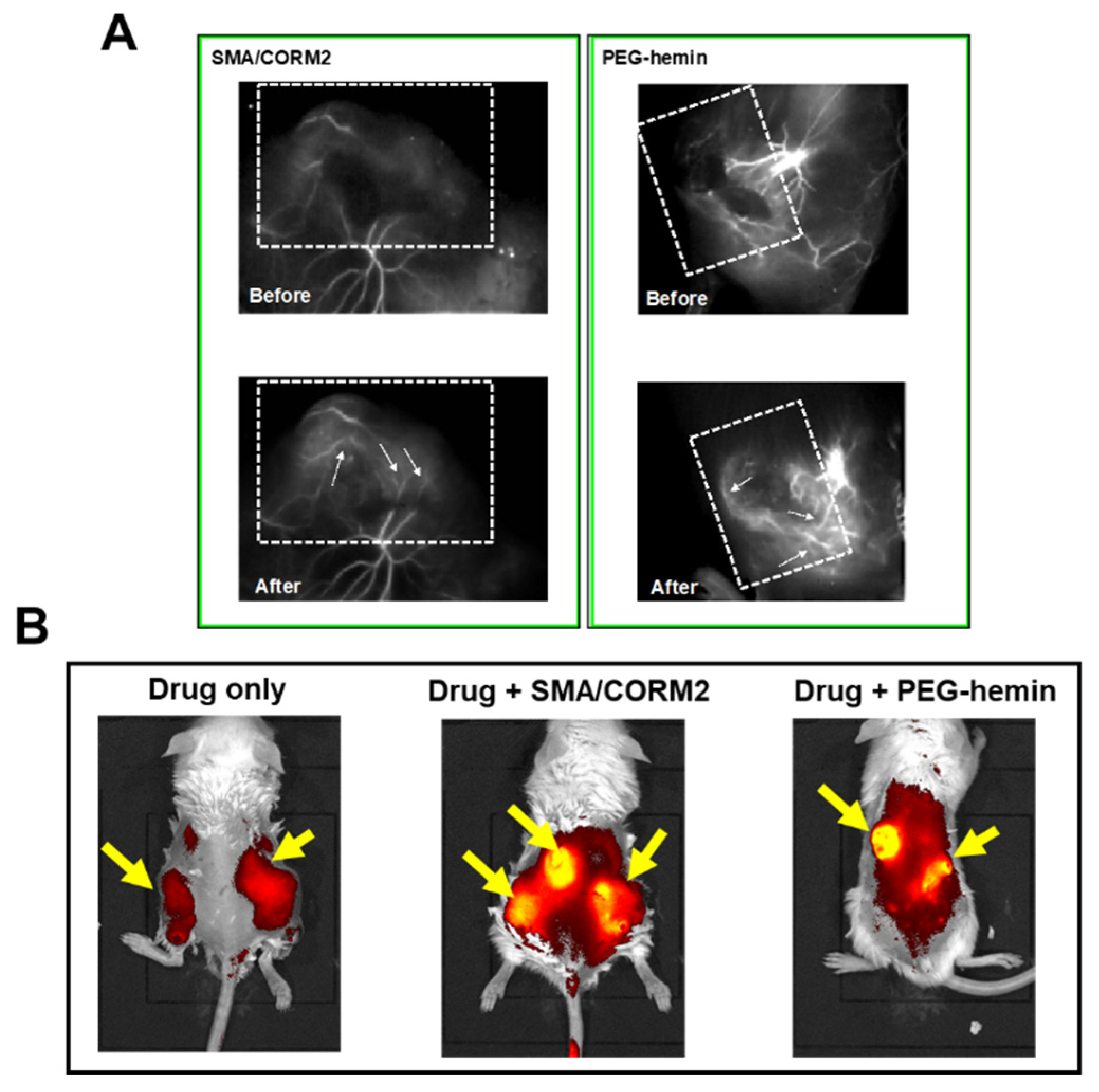
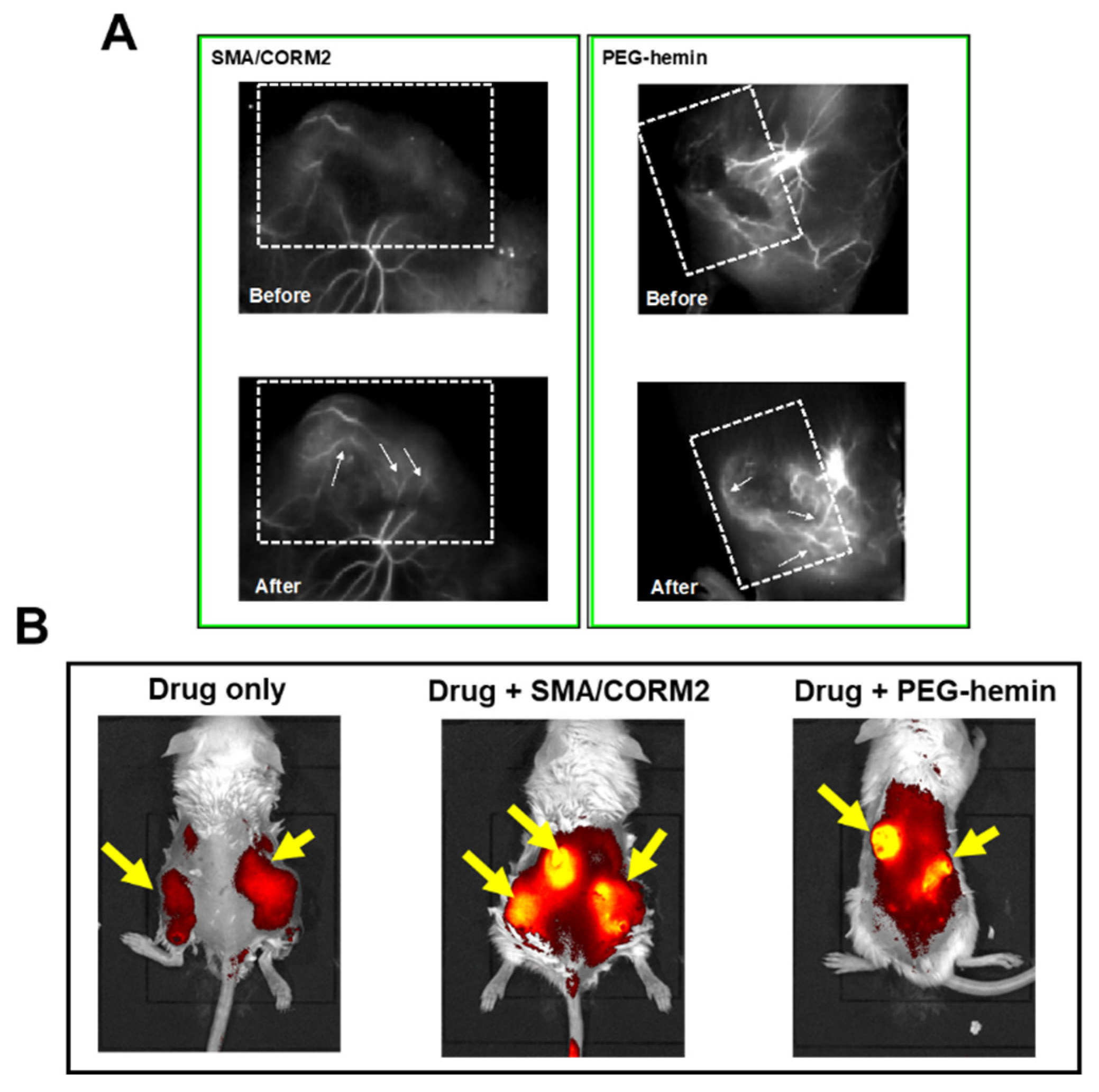
7. Enhancement of the Anticancer Effects of Drugs by Using CO Donors
8. Other EPR Effect Enhancers Used to Improve Drug Delivery to Tumors
9. Limitation of Using EPR Effect Enhancers
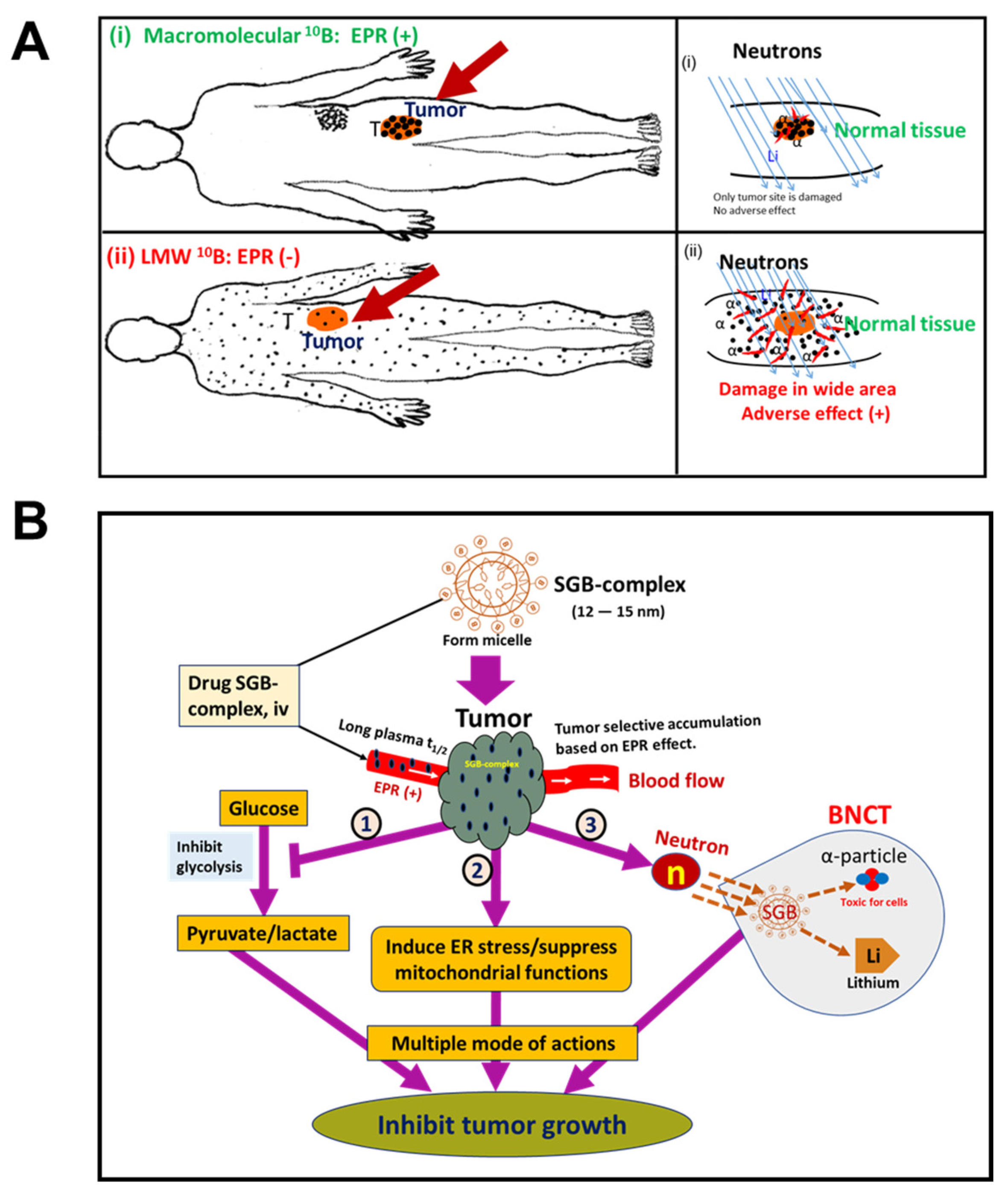
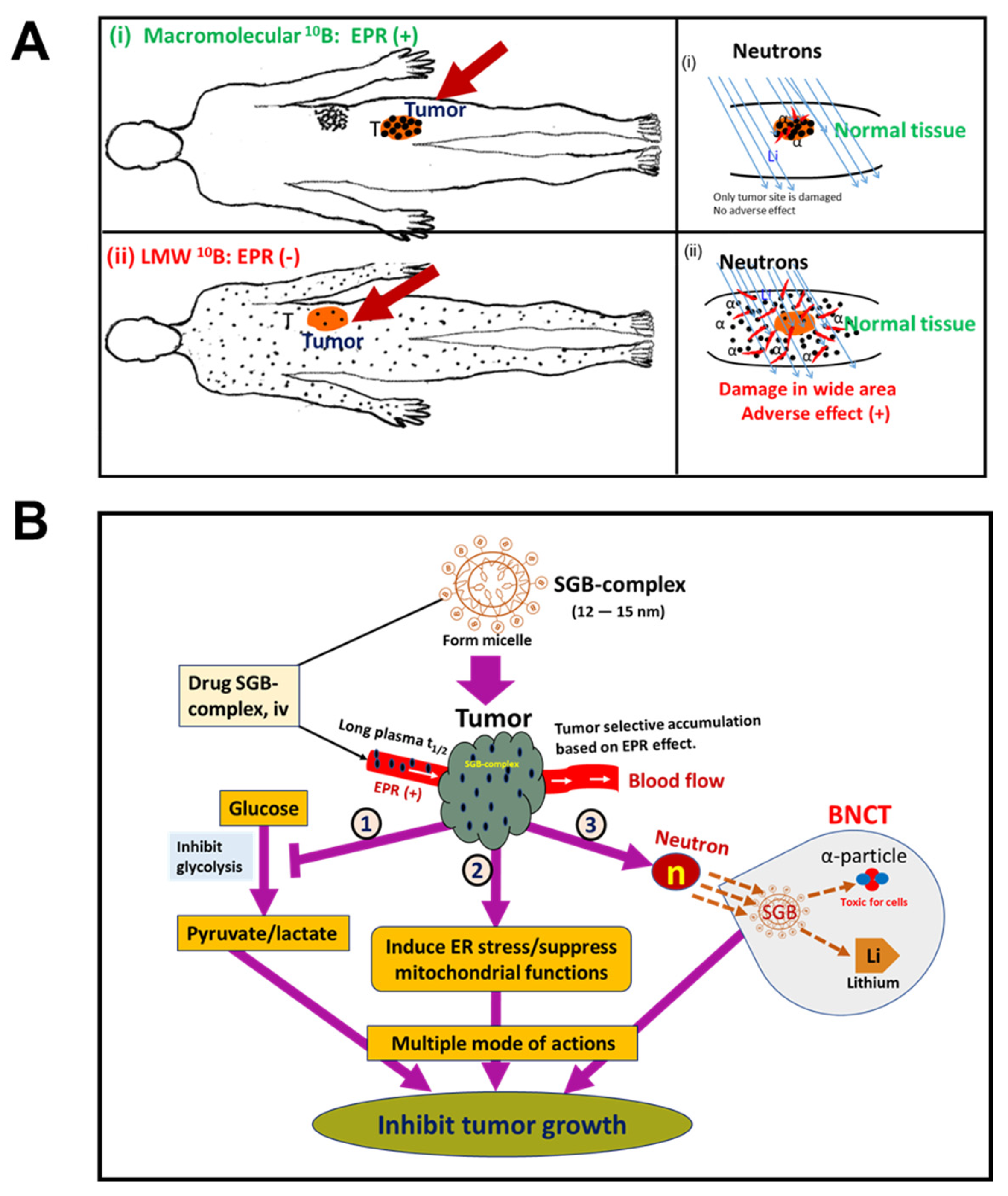
10. EPR-Based Nanomedicine Breakthrough in BNCT Used in Cancer Treatment
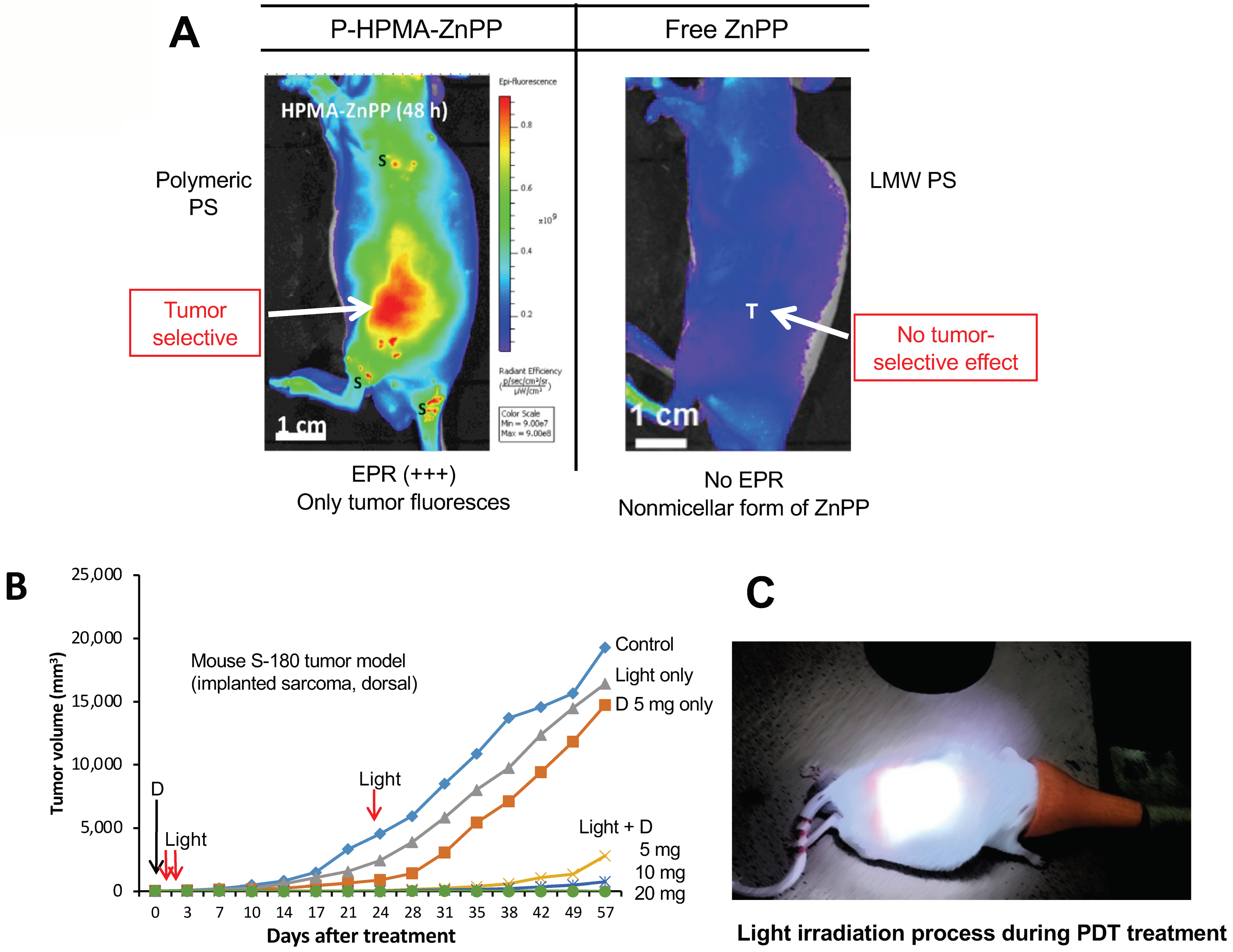
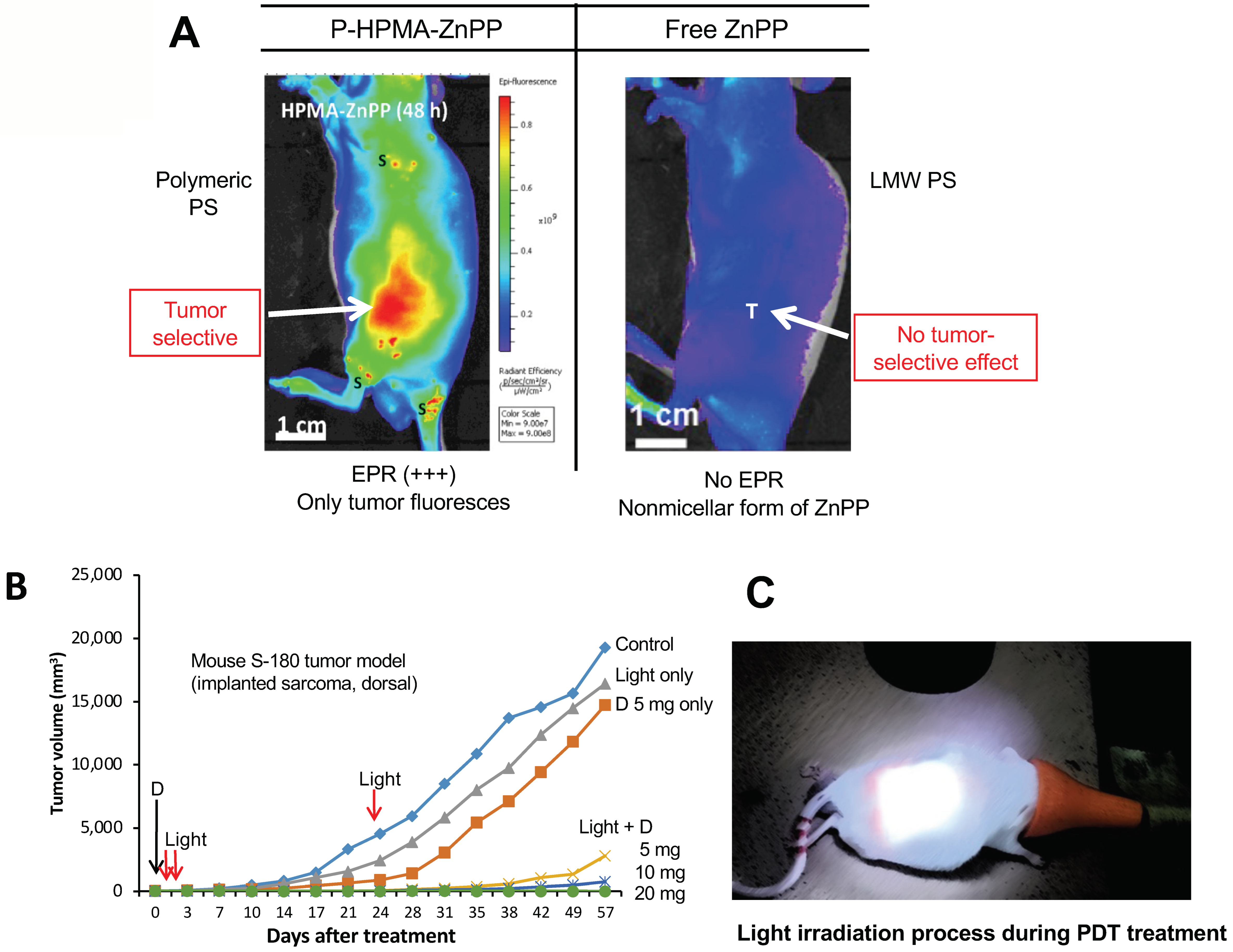
11. The Significant Role of EPR-Based Nanomedicine in PDT
12. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fang, J.; Nakamura, H.; Maeda, H. The EPR Effect: Unique Features of Tumor Blood Vessels for Drug Delivery, Factors Involved, and Limitations and Augmentation of the Effect. Adv. Drug Deliv. Rev. 2011, 63, 136–151. [Google Scholar] [CrossRef] [PubMed]
- Golombek, S.K.; May, J.-N.; Theek, B.; Appold, L.; Drude, N.; Kiessling, F.; Lammers, T. Tumor Targeting via EPR: Strategies to Enhance Patient Responses. Adv. Drug Deliv. Rev. 2018, 130, 17–38. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H. Polymer Therapeutics and the EPR Effect. J. Drug Target. 2017, 25, 781–785. [Google Scholar] [CrossRef]
- Maeda, H. Toward a Full Understanding of the EPR Effect in Primary and Metastatic Tumors as Well as Issues Related to Its Heterogeneity. Adv. Drug Deliv. Rev. 2015, 91, 3–6. [Google Scholar] [CrossRef]
- Maeda, H.; Tsukigawa, K.; Fang, J. A Retrospective 30 Years After Discovery of the Enhanced Permeability and Retention Effect of Solid Tumors: Next-Generation Chemotherapeutics and Photodynamic Therapy--Problems, Solutions, and Prospects. Microcirculation 2016, 23, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, Y.; Maeda, H. A New Concept for Macromolecular Therapeutics in Cancer Chemotherapy: Mechanism of Tumoritropic Accumulation of Proteins and the Antitumor Agent Smancs. Cancer Res. 1986, 46, 6387–6392. [Google Scholar]
- Fang, J.; Long, L.; Maeda, H. Enhancement of Tumor-Targeted Delivery of Bacteria with Nitroglycerin Involving Augmentation of the EPR Effect. Methods Mol. Biol. 2016, 1409, 9–23. [Google Scholar] [CrossRef]
- Fang, J.; Sawa, T.; Maeda, H. Factors and Mechanism of “EPR” Effect and the Enhanced Antitumor Effects of Macromolecular Drugs Including SMANCS. Adv. Exp. Med. Biol. 2003, 519, 29–49. [Google Scholar] [CrossRef]
- Folkman, J.; Klagsbrun, M. Angiogenic Factors. Science 1987, 235, 442–447. [Google Scholar] [CrossRef]
- Fang, J.; Islam, W.; Maeda, H. Exploiting the Dynamics of the EPR Effect and Strategies to Improve the Therapeutic Effects of Nanomedicines by Using EPR Effect Enhancers. Adv. Drug Deliv. Rev. 2020, 157, 142–160. [Google Scholar] [CrossRef]
- Maeda, H.; Islam, W. 3—Overcoming Barriers for Tumor-Targeted Drug Delivery: The Power of Macromolecular Anticancer Drugs with the EPR Effect and the Modulation of Vascular Physiology. In Polymer-Protein Conjugates; Pasut, G., Zalipsky, S., Eds.; Elsevier: Amsterdam, Netherlands, 2020; pp. 41–58. ISBN 978-0-444-64081-9. [Google Scholar]
- Daruwalla, J.; Greish, K.; Malcontenti-Wilson, C.; Muralidharan, V.; Iyer, A.; Maeda, H.; Christophi, C. Styrene Maleic Acid-Pirarubicin Disrupts Tumor Microcirculation and Enhances the Permeability of Colorectal Liver Metastases. J. Vasc. Res. 2009, 46, 218–228. [Google Scholar] [CrossRef]
- Lee, H.; Shields, A.F.; Siegel, B.A.; Miller, K.D.; Krop, I.; Ma, C.X.; LoRusso, P.M.; Munster, P.N.; Campbell, K.; Gaddy, D.F.; et al. 64Cu-MM-302 Positron Emission Tomography Quantifies Variability of Enhanced Permeability and Retention of Nanoparticles in Relation to Treatment Response in Patients with Metastatic Breast Cancer. Clin. Cancer Res. 2017, 23, 4190–4202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Y.; Xu, Y.; Yang, W.; Niu, P.; Li, X.; Chen, Y.; Li, Z.; Liu, Y.; An, Y.; Liu, Y.; et al. Investigating the EPR Effect of Nanomedicines in Human Renal Tumors via Ex Vivo Perfusion Strategy. Nano Today 2020, 35, 100970. [Google Scholar] [CrossRef]
- Duncan, R.; Sat-Klopsch, Y.-N.; Burger, A.M.; Bibby, M.C.; Fiebig, H.H.; Sausville, E.A. Validation of Tumour Models for Use in Anticancer Nanomedicine Evaluation: The EPR Effect and Cathepsin B-Mediated Drug Release Rate. Cancer Chemother. Pharmacol. 2013, 72, 417–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeda, H. The 35th Anniversary of the Discovery of EPR Effect: A New Wave of Nanomedicines for Tumor-Targeted Drug Delivery—Personal Remarks and Future Prospects. J. Pers. Med. 2021, 11, 229. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H. Macromolecular Therapeutics in Cancer Treatment: The EPR Effect and Beyond. J. Control. Release 2012, 164, 138–144. [Google Scholar] [CrossRef] [PubMed]
- MacConaill, L.E.; Campbell, C.D.; Kehoe, S.M.; Bass, A.J.; Hatton, C.; Niu, L.; Davis, M.; Yao, K.; Hanna, M.; Mondal, C.; et al. Profiling Critical Cancer Gene Mutations in Clinical Tumor Samples. PLoS ONE 2009, 4, e7887. [Google Scholar] [CrossRef]
- Jiao, X.; Wei, X.; Li, S.; Liu, C.; Chen, H.; Gong, J.; Li, J.; Zhang, X.; Wang, X.; Peng, Z.; et al. A Genomic Mutation Signature Predicts the Clinical Outcomes of Immunotherapy and Characterizes Immunophenotypes in Gastrointestinal Cancer. NPJ Precis. Oncol. 2021, 5, 36. [Google Scholar] [CrossRef]
- Fusco, M.J.; West, H.; Walko, C.M. Tumor Mutation Burden and Cancer Treatment. JAMA Oncol. 2021, 7, 316. [Google Scholar] [CrossRef]
- Navi, B.B.; Reiner, A.S.; Kamel, H.; Iadecola, C.; Okin, P.M.; Tagawa, S.T.; Panageas, K.S.; DeAngelis, L.M. Arterial Thromboembolic Events Preceding the Diagnosis of Cancer in Older Persons. Blood 2019, 133, 781–789. [Google Scholar] [CrossRef]
- Navi, B.B.; Reiner, A.S.; Kamel, H.; Iadecola, C.; Okin, P.M.; Elkind, M.S.V.; Panageas, K.S.; DeAngelis, L.M. Risk of Arterial Thromboembolism in Patients with Cancer. J. Am. Coll. Cardiol. 2017, 70, 926–938. [Google Scholar] [CrossRef]
- Young, A.; Chapman, O.; Connor, C.; Poole, C.; Rose, P.; Kakkar, A.K. Thrombosis and Cancer. Nat. Rev. Clin. Oncol. 2012, 9, 437–449. [Google Scholar] [CrossRef]
- Prabhakar, U.; Maeda, H.; Jain, R.K.; Sevick-Muraca, E.M.; Zamboni, W.; Farokhzad, O.C.; Barry, S.T.; Gabizon, A.; Grodzinski, P.; Blakey, D.C. Challenges and Key Considerations of the Enhanced Permeability and Retention (EPR) Effect for Nanomedicine Drug Delivery in Oncology. Cancer Res. 2013, 73, 2412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, H.Y.; Kano, M.R. Stromal Barriers to Nanomedicine Penetration in the Pancreatic Tumor Microenvironment. Cancer Sci. 2018, 109, 2085–2092. [Google Scholar] [CrossRef]
- Nel, A.; Ruoslahti, E.; Meng, H. New Insights into “Permeability” as in the Enhanced Permeability and Retention Effect of Cancer Nanotherapeutics. ACS Nano 2017, 11, 9567–9569. [Google Scholar] [CrossRef]
- Seki, T.; Fang, J.; Maeda, H. Enhanced Delivery of Macromolecular Antitumor Drugs to Tumors by Nitroglycerin Application. Cancer Sci. 2009, 100, 2426–2430. [Google Scholar] [CrossRef]
- Islam, W.; Fang, J.; Imamura, T.; Etrych, T.; Subr, V.; Ulbrich, K.; Maeda, H. Augmentation of the Enhanced Permeability and Retention Effect with Nitric Oxide-Generating Agents Improves the Therapeutic Effects of Nanomedicines. Mol. Cancer Ther. 2018, 17, 2643–2653. [Google Scholar] [CrossRef] [Green Version]
- Islam, W.; Kimura, S.; Islam, R.; Harada, A.; Ono, K.; Fang, J.; Niidome, T.; Sawa, T.; Maeda, H. EPR-Effect Enhancers Strongly Potentiate Tumor-Targeted Delivery of Nanomedicines to Advanced Cancers: Further Extension to Enhancement of the Therapeutic Effect. J. Pers. Med. 2021, 11, 487. [Google Scholar] [CrossRef]
- Fang, J.; Islam, R.; Islam, W.; Yin, H.; Subr, V.; Etrych, T.; Ulbrich, K.; Maeda, H. Augmentation of EPR Effect and Efficacy of Anticancer Nanomedicine by Carbon Monoxide Generating Agents. Pharmaceutics 2019, 11, 343. [Google Scholar] [CrossRef] [Green Version]
- Fang, J.; Qin, H.; Seki, T.; Nakamura, H.; Tsukigawa, K.; Shin, T.; Maeda, H. Therapeutic Potential of Pegylated Hemin for Reactive Oxygen Species-Related Diseases via Induction of Heme Oxygenase-1: Results from a Rat Hepatic Ischemia/Reperfusion Injury Model. J. Pharmacol. Exp. Ther. 2011, 339, 779–789. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Qin, H.; Nakamura, H.; Tsukigawa, K.; Shin, T.; Maeda, H. Carbon Monoxide, Generated by Heme Oxygenase-1, Mediates the Enhanced Permeability and Retention Effect in Solid Tumors. Cancer Sci. 2012, 103, 535–541. [Google Scholar] [CrossRef]
- Matsumoto, K.; Yamamoto, T.; Kamata, R.; Maeda, H. Pathogenesis of Serratial Infection: Activation of the Hageman Factor-Prekallikrein Cascade by Serratial Protease. J. Biochem. 1984, 96, 739–749. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H.; Yamamoto, T. Pathogenic Mechanisms Induced by Microbial Proteases in Microbial Infections. Biol. Chem. Hoppe-Seyler 1996, 377, 217–226. [Google Scholar] [CrossRef]
- Molla, A.; Matsumura, Y.; Yamamoto, T.; Okamura, R.; Maeda, H. Pathogenic Capacity of Proteases from Serratia Marcescens and Pseudomonas Aeruginosa and Their Suppression by Chicken Egg White Ovomacroglobulin. Infect. Immun. 1987, 55, 2509–2517. [Google Scholar] [CrossRef] [Green Version]
- Maeda, H.; Matsumura, Y.; Kato, H. Purification and Identification of [Hydroxyprolyl3]Bradykinin in Ascitic Fluid from a Patient with Gastric Cancer. J. Biol. Chem. 1988, 263, 16051–16054. [Google Scholar] [CrossRef]
- Matsumura, Y.; Maruo, K.; Kimura, M.; Yamamoto, T.; Konno, T.; Maeda, H. Kinin-Generating Cascade in Advanced Cancer Patients and in Vitro Study. Jpn. J. Cancer Res. 1991, 82, 732–741. [Google Scholar] [CrossRef]
- Maeda, H.; Noguchi, Y.; Sato, K.; Akaike, T. Enhanced Vascular Permeability in Solid Tumor Is Mediated by Nitric Oxide and Inhibited by Both New Nitric Oxide Scavenger and Nitric Oxide Synthase Inhibitor. Jpn. J. Cancer Res. 1994, 85, 331–334. [Google Scholar] [CrossRef]
- Maeda, H. SMANCS and Polymer-Conjugated Macromolecular Drugs: Advantages in Cancer Chemotherapy. Adv. Drug Deliv. Rev. 2001, 46, 169–185. [Google Scholar] [CrossRef]
- Seymour, L.W.; Olliff, S.P.; Poole, C.J.; De Takats, P.G.; Orme, R.; Ferry, D.R.; Maeda, H.; Konno, T.; Kerr, D.J. A Novel Dosage Approach for Evaluation of SMANCS [Poly-(Styrene-Co-Maleyl-Half-n-Butylate)—Neocarzinostatin] in the Treatment of Primary Hepatocellular Carcinoma. Int. J. Oncol. 1998, 12, 1217–1223. [Google Scholar] [CrossRef]
- Tsuchiya, K.; Uchida, T.; Kobayashi, M.; Maeda, H.; Konno, T.; Yamanaka, H. Tumor-Targeted Chemotherapy with SMANCS in Lipiodol for Renal Cell Carcinoma: Longer Survival with Larger Size Tumors. Urology 2000, 55, 495–500. [Google Scholar] [CrossRef]
- Maeda, H. Principle and therapeutic effect of lipophilic anticancer agent [SMANCS/lipiodol]: Selective targeting with oily contrast medium. Gan To Kagaku Ryoho 1989, 16, 3323–3331. [Google Scholar]
- Maeda, H. The Enhanced Permeability and Retention (EPR) Effect in Tumor Vasculature: The Key Role of Tumor-Selective Macromolecular Drug Targeting. Adv. Enzyme Regul. 2001, 41, 189–207. [Google Scholar] [CrossRef]
- Maeda, H. Vascular Permeability in Cancer and Infection as Related to Macromolecular Drug Delivery, with Emphasis on the EPR Effect for Tumor-Selective Drug Targeting. Proc. Jpn. Acad. Ser. B 2012, 88, 53–71. [Google Scholar] [CrossRef] [Green Version]
- Sindhwani, S.; Syed, A.M.; Ngai, J.; Kingston, B.R.; Maiorino, L.; Rothschild, J.; MacMillan, P.; Zhang, Y.; Rajesh, N.U.; Hoang, T.; et al. The Entry of Nanoparticles into Solid Tumours. Nat. Mater. 2020, 19, 566–575. [Google Scholar] [CrossRef]
- Park, K. Questions on the Role of the EPR Effect in Tumor Targeting. J. Control. Release 2013, 172, 391. [Google Scholar] [CrossRef]
- Nichols, J.W.; Bae, Y.H. EPR: Evidence and Fallacy. J. Control. Release 2014, 190, 451–464. [Google Scholar] [CrossRef] [PubMed]
- Danhier, F. To Exploit the Tumor Microenvironment: Since the EPR Effect Fails in the Clinic, What Is the Future of Nanomedicine? J. Control. Release 2016, 244, 108–121. [Google Scholar] [CrossRef] [PubMed]
- Kwon, I.K.; Lee, S.C.; Han, B.; Park, K. Analysis on the Current Status of Targeted Drug Delivery to Tumors. J. Control. Release 2012, 164, 108–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Islam, W.; Fang, J.; Etrych, T.; Chytil, P.; Ulbrich, K.; Sakoguchi, A.; Kusakabe, K.; Maeda, H. HPMA Copolymer Conjugate with Pirarubicin: In Vitro and Ex Vivo Stability and Drug Release Study. Int. J. Pharm. 2018, 536, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H.; Bharate, G.Y.; Daruwalla, J. Polymeric Drugs for Efficient Tumor-Targeted Drug Delivery Based on EPR-Effect. Eur. J. Pharm. Biopharm. 2009, 71, 409–419. [Google Scholar] [CrossRef]
- Chytil, P.; Koziolová, E.; Etrych, T.; Ulbrich, K. HPMA Copolymer-Drug Conjugates with Controlled Tumor-Specific Drug Release. Macromol. Biosci. 2018, 18, 1700209. [Google Scholar] [CrossRef] [PubMed]
- Peterson, C.M.; Shiah, J.-G.; Sun, Y.; Kopecková, P.; Minko, T.; Straight, R.C.; Kopecek, J. HPMA Copolymer Delivery of Chemotherapy and Photodynamic Therapy in Ovarian Cancer. Adv. Exp. Med. Biol. 2003, 519, 101–123. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, J.C.B.; Mochly-Rosen, D. Nitroglycerin Use in Myocardial Infarction Patients: Risks and Benefits. Circ. J. 2012, 76, 15–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, J.B.; Wink, D.A.; DeGraff, W.; Gamson, J.; Keefer, L.K.; Krishna, M.C. Hypoxic Mammalian Cell Radiosensitization by Nitric Oxide. Cancer Res. 1993, 53, 5845–5848. [Google Scholar] [PubMed]
- Chen, Z.; Stamler, J.S. Bioactivation of Nitroglycerin by the Mitochondrial Aldehyde Dehydrogenase. Trends Cardiovasc. Med. 2006, 16, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Divakaran, S.; Loscalzo, J. The Role of Nitroglycerin and Other Nitrogen Oxides in Cardiovascular Therapeutics. J. Am. Coll. Cardiol. 2017, 70, 2393–2410. [Google Scholar] [CrossRef] [PubMed]
- Pekarova, M.; Lojek, A.; Martiskova, H.; Vasicek, O.; Bino, L.; Klinke, A.; Lau, D.; Kuchta, R.; Kadlec, J.; Vrba, R.; et al. New Role for L-Arginine in Regulation of Inducible Nitric-Oxide-Synthase-Derived Superoxide Anion Production in Raw 264.7 Macrophages. Sci. World J. 2011, 11, 2443–2457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akaike, T.; Maeda, H. Nitric Oxide and Virus Infection. Immunology 2000, 101, 300–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gladwin, M.T.; Shelhamer, J.H.; Ognibene, F.P.; Pease-Fye, M.E.; Nichols, J.S.; Link, B.; Patel, D.B.; Jankowski, M.A.; Pannell, L.K.; Schechter, A.N.; et al. Nitric Oxide Donor Properties of Hydroxyurea in Patients with Sickle Cell Disease. Br. J. Haematol. 2002, 116, 436–444. [Google Scholar] [CrossRef]
- Sato, K.; Akaike, T.; Sawa, T.; Miyamoto, Y.; Suga, M.; Ando, M.; Maeda, H. Nitric Oxide Generation from Hydroxyurea via Copper-Catalyzed Peroxidation and Implications for Pharmacological Actions of Hydroxyurea. Jpn. J. Cancer Res. 1997, 88, 1199–1204. [Google Scholar] [CrossRef]
- Mantle, J.A.; Russell, R.O.; Moraski, R.E.; Rackley, C.E. Isosorbide Dinitrate for the Relief of Severe Heart Failure after Myocardial Infarction. Am. J. Cardiol. 1976, 37, 263–268. [Google Scholar] [CrossRef]
- Chavey, W.E.; Bleske, B.E.; Van Harrison, R.; Hogikyan, R.V.; Kesterson, S.K.; Nicklas, J.M. Pharmacologic Management of Heart Failure Caused by Systolic Dysfunction. Am. Fam. Physician 2008, 77, 957–964. [Google Scholar]
- Nakamura, H.; Etrych, T.; Chytil, P.; Ohkubo, M.; Fang, J.; Ulbrich, K.; Maeda, H. Two Step Mechanisms of Tumor Selective Delivery of N-(2-Hydroxypropyl)Methacrylamide Copolymer Conjugated with Pirarubicin via an Acid-Cleavable Linkage. J. Control. Release 2014, 174, 81–87. [Google Scholar] [CrossRef]
- Fang, J.; Šubr, V.; Islam, W.; Hackbarth, S.; Islam, R.; Etrych, T.; Ulbrich, K.; Maeda, H. N-(2-Hydroxypropyl)Methacrylamide Polymer Conjugated Pyropheophorbide-a, a Promising Tumor-Targeted Theranostic Probe for Photodynamic Therapy and Imaging. Eur. J. Pharm. Biopharm. 2018, 130, 165–176. [Google Scholar] [CrossRef]
- Nakamura, H.; Liao, L.; Hitaka, Y.; Tsukigawa, K.; Subr, V.; Fang, J.; Ulbrich, K.; Maeda, H. Micelles of Zinc Protoporphyrin Conjugated to N-(2-Hydroxypropyl)Methacrylamide (HPMA) Copolymer for Imaging and Light-Induced Antitumor Effects in Vivo. J. Control. Release 2013, 165, 191–198. [Google Scholar] [CrossRef]
- Saisyo, A.; Nakamura, H.; Fang, J.; Tsukigawa, K.; Greish, K.; Furukawa, H.; Maeda, H. PH-Sensitive Polymeric Cisplatin-Ion Complex with Styrene-Maleic Acid Copolymer Exhibits Tumor-Selective Drug Delivery and Antitumor Activity as a Result of the Enhanced Permeability and Retention Effect. Colloids Surf. B 2016, 138, 128–137. [Google Scholar] [CrossRef] [Green Version]
- Islam, W.; Matsumoto, Y.; Fang, J.; Harada, A.; Niidome, T.; Ono, K.; Tsutsuki, H.; Sawa, T.; Imamura, T.; Sakurai, K.; et al. Polymer-Conjugated Glucosamine Complexed with Boric Acid Shows Tumor-Selective Accumulation and Simultaneous Inhibition of Glycolysis. Biomaterials 2020, 269, 120631. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Akaike, T.; Maeda, H. Antiapoptotic Role of Heme Oxygenase (HO) and the Potential of HO as a Target in Anticancer Treatment. Apoptosis 2004, 9, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Fang, J.; Liao, L.; Nakamura, H.; Maeda, H. Styrene-Maleic Acid Copolymer-Encapsulated CORM2, a Water-Soluble Carbon Monoxide (CO) Donor with a Constant CO-Releasing Property, Exhibits Therapeutic Potential for Inflammatory Bowel Disease. J. Control. Release 2014, 187, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Seki, T.; Carroll, F.; Illingworth, S.; Green, N.; Cawood, R.; Bachtarzi, H.; Subr, V.; Fisher, K.D.; Seymour, L.W. Tumour Necrosis Factor-Alpha Increases Extravasation of Virus Particles into Tumour Tissue by Activating the Rho A/Rho Kinase Pathway. J. Control. Release 2011, 156, 381–389. [Google Scholar] [CrossRef]
- du Vernay, A.J.V.B.; De Faveri, R.; Nunes, R.; Steimbach, V.M.B.; Santin, J.R.; Quintão, N.L.M. The Role of Kinins in the Proliferation of Fibroblast Primed with TNF in Scratch Wound Assay: Kinins and Cell Proliferation. Int. Immunopharmacol. 2018, 65, 23–28. [Google Scholar] [CrossRef]
- Hisada, Y.; Ay, C.; Auriemma, A.C.; Cooley, B.C.; Mackman, N. Human Pancreatic Tumors Grown in Mice Release Tissue Factor-Positive Microvesicles That Increase Venous Clot Size. J. Thromb. Haemost. 2017, 15, 2208–2217. [Google Scholar] [CrossRef] [Green Version]
- Tsumura, R.; Manabe, S.; Takashima, H.; Koga, Y.; Yasunaga, M.; Matsumura, Y. Evaluation of the Antitumor Mechanism of Antibody-drug Conjugates against Tissue Factor in Stroma-rich Allograft Models. Cancer Sci. 2019, 110, 3296–3305. [Google Scholar] [CrossRef]
- Yasunaga, M.; Manabe, S.; Tsuji, A.; Furuta, M.; Ogata, K.; Koga, Y.; Saga, T.; Matsumura, Y. Development of Antibody-Drug Conjugates Using DDS and Molecular Imaging. Bioengineering 2017, 4, 78. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Jiang, T.; She, X.; Shen, S.; Wang, S.; Deng, J.; Shi, W.; Mei, H.; Hu, Y.; Pang, Z.; et al. Fibrin Degradation by RtPA Enhances the Delivery of Nanotherapeutics to A549 Tumors in Nude Mice. Biomaterials 2016, 96, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Mei, T.; Shashni, B.; Maeda, H.; Nagasaki, Y. Fibrinolytic Tissue Plasminogen Activator Installed Redox-Active Nanoparticles (t-PA@iRNP) for Cancer Therapy. Biomaterials 2020, 259, 120290. [Google Scholar] [CrossRef]
- Witte, L.; Hicklin, D.J.; Zhu, Z.; Pytowski, B.; Kotanides, H.; Rockwell, P.; Böhlen, P. Monoclonal Antibodies Targeting the VEGF Receptor-2 (Flk1/KDR) as an Anti-Angiogenic Therapeutic Strategy. Cancer Metastasis Rev. 1998, 17, 155–161. [Google Scholar] [CrossRef]
- Jain, R.K. Normalization of Tumor Vasculature: An Emerging Concept in Antiangiogenic Therapy. Science 2005, 307, 58–62. [Google Scholar] [CrossRef]
- Ikeda-Imafuku, M.; Wang, L.L.-W.; Rodrigues, D.; Shaha, S.; Zhao, Z.; Mitragotri, S. Strategies to Improve the EPR Effect: A Mechanistic Perspective and Clinical Translation. J. Control. Release 2022, 345, 512–536. [Google Scholar] [CrossRef]
- Mattheolabakis, G.; Mikelis, C.M. Nanoparticle Delivery and Tumor Vascular Normalization: The Chicken or The Egg? Front. Oncol. 2019, 9, 1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chauhan, V.P.; Stylianopoulos, T.; Martin, J.D.; Popović, Z.; Chen, O.; Kamoun, W.S.; Bawendi, M.G.; Fukumura, D.; Jain, R.K. Normalization of Tumour Blood Vessels Improves the Delivery of Nanomedicines in a Size-Dependent Manner. Nat. Nanotechnol. 2012, 7, 383–388. [Google Scholar] [CrossRef] [Green Version]
- Binnemars-Postma, K.A.; Ten Hoopen, H.W.; Storm, G.; Prakash, J. Differential Uptake of Nanoparticles by Human M1 and M2 Polarized Macrophages: Protein Corona as a Critical Determinant. Nanomedicine 2016, 11, 2889–2902. [Google Scholar] [CrossRef]
- Maeda, H.; Nakamura, H.; Fang, J. The EPR Effect for Macromolecular Drug Delivery to Solid Tumors: Improvement of Tumor Uptake, Lowering of Systemic Toxicity, and Distinct Tumor Imaging in Vivo. Adv. Drug Deliv. Rev. 2013, 65, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, V.P.; Martin, J.D.; Liu, H.; Lacorre, D.A.; Jain, S.R.; Kozin, S.V.; Stylianopoulos, T.; Mousa, A.S.; Han, X.; Adstamongkonkul, P.; et al. Angiotensin Inhibition Enhances Drug Delivery and Potentiates Chemotherapy by Decompressing Tumour Blood Vessels. Nat. Commun. 2013, 4, 2516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, P.-P.; Demircioglu, F.; Ghazaly, E.; Alrawashdeh, W.; Stratford, M.R.L.; Scudamore, C.L.; Cereser, B.; Crnogorac-Jurcevic, T.; McDonald, S.; Elia, G.; et al. Dual-Action Combination Therapy Enhances Angiogenesis While Reducing Tumor Growth and Spread. Cancer Cell 2015, 27, 123–137. [Google Scholar] [CrossRef] [Green Version]
- Mas-Moruno, C.; Rechenmacher, F.; Kessler, H. Cilengitide: The First Anti-Angiogenic Small Molecule Drug Candidate Design, Synthesis and Clinical Evaluation. Anticancer Agents Med. Chem. 2010, 10, 753–768. [Google Scholar] [CrossRef] [Green Version]
- Ojha, T.; Pathak, V.; Shi, Y.; Hennink, W.E.; Moonen, C.T.W.; Storm, G.; Kiessling, F.; Lammers, T. Pharmacological and Physical Vessel Modulation Strategies to Improve EPR-Mediated Drug Targeting to Tumors. Adv. Drug Deliv. Rev. 2017, 119, 44–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunjachan, S.; Kotb, S.; Pola, R.; Pechar, M.; Kumar, R.; Singh, B.; Gremse, F.; Taleeli, R.; Trichard, F.; Motto-Ros, V.; et al. Selective Priming of Tumor Blood Vessels by Radiation Therapy Enhances Nanodrug Delivery. Sci. Rep. 2019, 9, 15844. [Google Scholar] [CrossRef] [Green Version]
- Lubner, M.G.; Brace, C.L.; Hinshaw, J.L.; Lee, F.T. Microwave Tumor Ablation: Mechanism of Action, Clinical Results and Devices. J. Vas. Interv. Radiol. 2010, 21, S192–S203. [Google Scholar] [CrossRef] [Green Version]
- Kurokohchi, K.; Watanabe, S.; Masaki, T.; Hosomi, N.; Funaki, T.; Arima, K.; Yoshida, S.; Miyauchi, Y.; Kuriyama, S. Combined Use of Percutaneous Ethanol Injection and Radiofrequency Ablation for the Effective Treatment of Hepatocelluar Carcinoma. Int. J. Oncol. 2002, 21, 841–846. [Google Scholar] [CrossRef]
- Hijnen, N.M.; Heijman, E.; Köhler, M.O.; Ylihautala, M.; Ehnholm, G.J.; Simonetti, A.W.; Grüll, H. Tumour Hyperthermia and Ablation in Rats Using a Clinical MR-HIFU System Equipped with a Dedicated Small Animal Set-Up. Int. J. Hyperth. 2012, 28, 141–155. [Google Scholar] [CrossRef] [PubMed]
- Lentacker, I.; De Cock, I.; Deckers, R.; De Smedt, S.C.; Moonen, C.T.W. Understanding Ultrasound Induced Sonoporation: Definitions and Underlying Mechanisms. Adv. Drug Deliv. Rev. 2014, 72, 49–64. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Zhou, S.; Lv, N.; Zhen, Z.; Liu, T.; Gao, S.; Xie, J.; Ma, Q. Photosensitizer-Encapsulated Ferritins Mediate Photodynamic Therapy against Cancer-Associated Fibroblasts and Improve Tumor Accumulation of Nanoparticles. Mol. Pharm. 2018, 15, 3595–3599. [Google Scholar] [CrossRef] [PubMed]
- Zhen, Z.; Tang, W.; Chuang, Y.-J.; Todd, T.; Zhang, W.; Lin, X.; Niu, G.; Liu, G.; Wang, L.; Pan, Z.; et al. Tumor Vasculature Targeted Photodynamic Therapy for Enhanced Delivery of Nanoparticles. ACS Nano 2014, 8, 6004–6013. [Google Scholar] [CrossRef] [PubMed]
- Heber, E.M.; Hawthorne, M.F.; Kueffer, P.J.; Garabalino, M.A.; Thorp, S.I.; Pozzi, E.C.C.; Monti Hughes, A.; Maitz, C.A.; Jalisatgi, S.S.; Nigg, D.W.; et al. Therapeutic Efficacy of Boron Neutron Capture Therapy Mediated by Boron-Rich Liposomes for Oral Cancer in the Hamster Cheek Pouch Model. Proc. Natl. Acad. Sci. USA 2014, 111, 16077–16081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rees, D.D.; Palmer, R.M.; Moncada, S. Role of Endothelium-Derived Nitric Oxide in the Regulation of Blood Pressure. Proc. Natl. Acad. Sci. USA 1989, 86, 3375–3378. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Liu, L.Z.; Loizidou, M.; Ahmed, M.; Charles, I.G. The Role of Nitric Oxide in Cancer. Cell Res. 2002, 12, 311–320. [Google Scholar] [CrossRef] [Green Version]
- Fukumura, D.; Kashiwagi, S.; Jain, R.K. The Role of Nitric Oxide in Tumour Progression. Nat. Rev. Cancer 2006, 6, 521–534. [Google Scholar] [CrossRef] [PubMed]
- Kamba, T.; McDonald, D.M. Mechanisms of Adverse Effects of Anti-VEGF Therapy for Cancer. Br. J. Cancer 2007, 96, 1788–1795. [Google Scholar] [CrossRef]
- Bharadwaj, A.G.; Holloway, R.W.; Miller, V.A.; Waisman, D.M. Plasmin and Plasminogen System in the Tumor Microenvironment: Implications for Cancer Diagnosis, Prognosis, and Therapy. Cancers 2021, 13, 1838. [Google Scholar] [CrossRef]
- Baig, M.U.; Bodle, J. Thrombolytic Therapy. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Barth, R.F. Boron Neutron Capture Therapy at the Crossroads: Challenges and Opportunities. Appl. Radiat. Isot. 2009, 67, S3–S6. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhang, Z.; Miao, L.; Li, Y. Boron Neutron Capture Therapy: Current Status and Challenges. Front. Oncol. 2022, 12, 788770. [Google Scholar] [CrossRef] [PubMed]
- Koganei, H.; Ueno, M.; Tachikawa, S.; Tasaki, L.; Ban, H.S.; Suzuki, M.; Shiraishi, K.; Kawano, K.; Yokoyama, M.; Maitani, Y.; et al. Development of High Boron Content Liposomes and Their Promising Antitumor Effect for Neutron Capture Therapy of Cancers. Bioconjugate Chem. 2013, 24, 124–132. [Google Scholar] [CrossRef]
- Kawasaki, R.; Sasaki, Y.; Akiyoshi, K. Self-Assembled Nanogels of Carborane-Bearing Polysaccharides for Boron Neutron Capture Therapy. Chem. Lett. 2017, 46, 513–515. [Google Scholar] [CrossRef]
- Nedunchezhian, K.; Aswath, N.; Thiruppathy, M.; Thirugnanamurthy, S. Boron Neutron Capture Therapy—A Literature Review. J. Clin. Diagn Res. 2016, 10, ZE01–ZE04. [Google Scholar] [CrossRef]
- Matsumoto, Y.; Fukumitsu, N.; Ishikawa, H.; Nakai, K.; Sakurai, H. A Critical Review of Radiation Therapy: From Particle Beam Therapy (Proton, Carbon, and BNCT) to Beyond. J. Pers. Med. 2021, 11, 825. [Google Scholar] [CrossRef]
- Takano, S.; Islam, W.; Fujii, S.; Maeda, H.; Sakurai, K. Weak Interplay between Hydrophobic Part of Water-Soluble Polymers and Serum Protein. Chem. Lett. 2021, 50, 1392–1393. [Google Scholar] [CrossRef]
- Pavlides, S.; Whitaker-Menezes, D.; Castello-Cros, R.; Flomenberg, N.; Witkiewicz, A.K.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S.; et al. The Reverse Warburg Effect: Aerobic Glycolysis in Cancer Associated Fibroblasts and the Tumor Stroma. Cell Cycle 2009, 8, 3984–4001. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Dolmans, D.E.; Fukumura, D.; Jain, R.K. Photodynamic Therapy for Cancer. Nat. Rev. Cancer 2003, 3, 380–387. [Google Scholar] [CrossRef]
- Wilson, B.C. Photodynamic Therapy for Cancer: Principles. Can. J. Gastroenterol. 2002, 16, 393–396. [Google Scholar] [CrossRef] [PubMed]
- Baskaran, R.; Lee, J.; Yang, S.-G. Clinical Development of Photodynamic Agents and Therapeutic Applications. Biomater. Res. 2018, 22, 25. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Fang, J.; Gahininath, B.; Tsukigawa, K.; Maeda, H. Intracellular Uptake and Behavior of Two Types Zinc Protoporphyrin (ZnPP) Micelles, SMA-ZnPP and PEG-ZnPP as Anticancer Agents; Unique Intracellular Disintegration of SMA Micelles. J. Control. Release 2011, 155, 367–375. [Google Scholar] [CrossRef]
- Fang, J.; Tsukigawa, K.; Liao, L.; Yin, H.; Eguchi, K.; Maeda, H. Styrene-Maleic Acid-Copolymer Conjugated Zinc Protoporphyrin as a Candidate Drug for Tumor-Targeted Therapy and Imaging. J. Drug Target. 2016, 24, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Abe, S.; Otsuki, M. Styrene Maleic Acid Neocarzinostatin Treatment for Hepatocellular Carcinoma. Curr. Med. Chem. Anticancer Agents 2002, 2, 715–726. [Google Scholar] [CrossRef]
- Kubo, M.; Fuchigami, T.; Murata, S.; Konno, T.; Maeda, H. A case of massive hepatoma which responded to SMANCS/Lipiodol regimen with intra-arterial infusion. Gan To Kagaku Ryoho 1989, 16, 2953–2956. [Google Scholar]
- Sakaguchi, T.; Yoshimatsu, S.; Sagara, K.; Yamashita, Y.; Takahashi, M. Intra-arterial infusion of SMANCS for treatment of patients with hepatocellular carcinoma--adverse reactions and complications. Gan To Kagaku Ryoho 1998, 25 (Suppl. S1), 64–69. [Google Scholar]
- Dozono, H.; Yanazume, S.; Nakamura, H.; Etrych, T.; Chytil, P.; Ulbrich, K.; Fang, J.; Arimura, T.; Douchi, T.; Kobayashi, H.; et al. HPMA Copolymer-Conjugated Pirarubicin in Multimodal Treatment of a Patient with Stage IV Prostate Cancer and Extensive Lung and Bone Metastases. Target. Oncol. 2016, 11, 101–106. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Methods | Drugs/Agents | Tumor Model | Outcome (Augmentation) | Brief Mechanisms |
|---|---|---|---|---|
| Vascular mediators | NO generating (i) NG (ii) L-Arg (ii) HU (iv) ISDN (v) Sildenafil | Xenograft tumor S180, C26, B16, 4T1 Chemically induced AOM/DSS-induced colon tumor and DMBA-induced breast tumor | 2- to 5-fold | Open tumor blood vessels as a vasodilator and thus improve drug delivery to tumors [10,27,28,29,30,96] |
| CO generating (i) SMA/CORM2 (ii) PEG-hemin | S180, C26, B16 | 2- to 3-fold | Functions similar to those of NO donors [10,30] | |
| Others (i) Tumor necrosis factor-α (TNF-α) (ii) Anti-tissue factor-antibody drug conjugate (anti-TF-ADC) (iii) Tissue plasminogen activator (tPA) (iv) anti-VEGF receptor 2 (v) Angiotensin II receptor blockers | (i) EL4 (ii) Pancreatic cancer (iii) A549 (iv) Breast tumor (v) 4T1, AK4.4, E0771, Pan-02 | (i) 2- to 3-fold (ii) Significantly (iii) 2- to 3-fold (iv) 3-fold (v) Significantly | (i) Increase endothelial cell permeability [71] (ii) Enhance penetration capacity [74] (iii) Restore blood flow via fibrinolysis [76] (iv) Normalized disorganized tumor vessels by pruning immature vessels [78,79,80,81,82] (v) promote vessel permeability and dilation through the loosening of the fasciae adherents [2,79,80,81,82,83,84,85] | |
| Physical methods | (i) Radiation therapy (ii) Hyperthermia (iii) Ultrasound (US) with microbubbles (MBs) (iv) PDT | (i) h-PDAC, R3230 (ii) SK-VO-3, DU145 (iii) A431, BxPC-3 (iv) 4T1, U87MG, MDA-MB-435S, and PC-3 | (i) 2-fold (ii) 2-fold (iii) Significantly (iv) 18- to 20-fold | (i) Induce physical vascular damage related to photoelectric interaction [80,88] (ii) Improve perfusion, vasodilation, and vascular permeability [80,88] (iii) Disrupt endothelium [80,88] (iv) Damage tumor associated fibroblasts [80,88] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Islam, W.; Niidome, T.; Sawa, T. Enhanced Permeability and Retention Effect as a Ubiquitous and Epoch-Making Phenomenon for the Selective Drug Targeting of Solid Tumors. J. Pers. Med. 2022, 12, 1964. https://doi.org/10.3390/jpm12121964
Islam W, Niidome T, Sawa T. Enhanced Permeability and Retention Effect as a Ubiquitous and Epoch-Making Phenomenon for the Selective Drug Targeting of Solid Tumors. Journal of Personalized Medicine. 2022; 12(12):1964. https://doi.org/10.3390/jpm12121964
Chicago/Turabian StyleIslam, Waliul, Takuro Niidome, and Tomohiro Sawa. 2022. "Enhanced Permeability and Retention Effect as a Ubiquitous and Epoch-Making Phenomenon for the Selective Drug Targeting of Solid Tumors" Journal of Personalized Medicine 12, no. 12: 1964. https://doi.org/10.3390/jpm12121964
APA StyleIslam, W., Niidome, T., & Sawa, T. (2022). Enhanced Permeability and Retention Effect as a Ubiquitous and Epoch-Making Phenomenon for the Selective Drug Targeting of Solid Tumors. Journal of Personalized Medicine, 12(12), 1964. https://doi.org/10.3390/jpm12121964