
Efficient Selection of Enhancers and Promoters from MIA PaCa-2 Pancreatic Cancer Cells by ChIP-lentiMPRA

 and
and
Abstract
:1. Introduction
2. Results
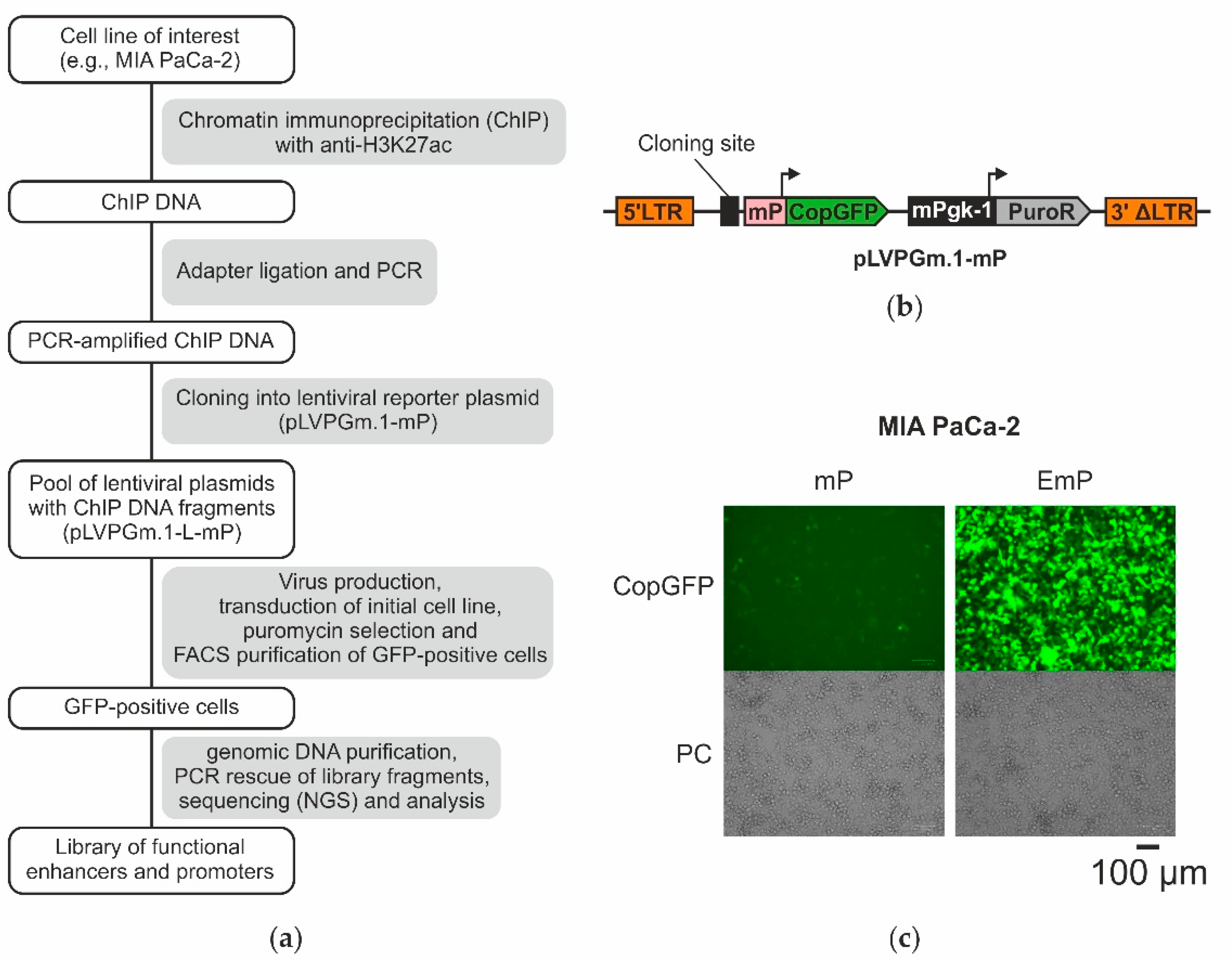
2.1. The Design of the Lentiviral System for Selection of Active Enhancers and Promoters
2.2. Bidirectional Transcription as a Probable Cause of Reduced Representation of Functional Elements in Lentiviral Libraries
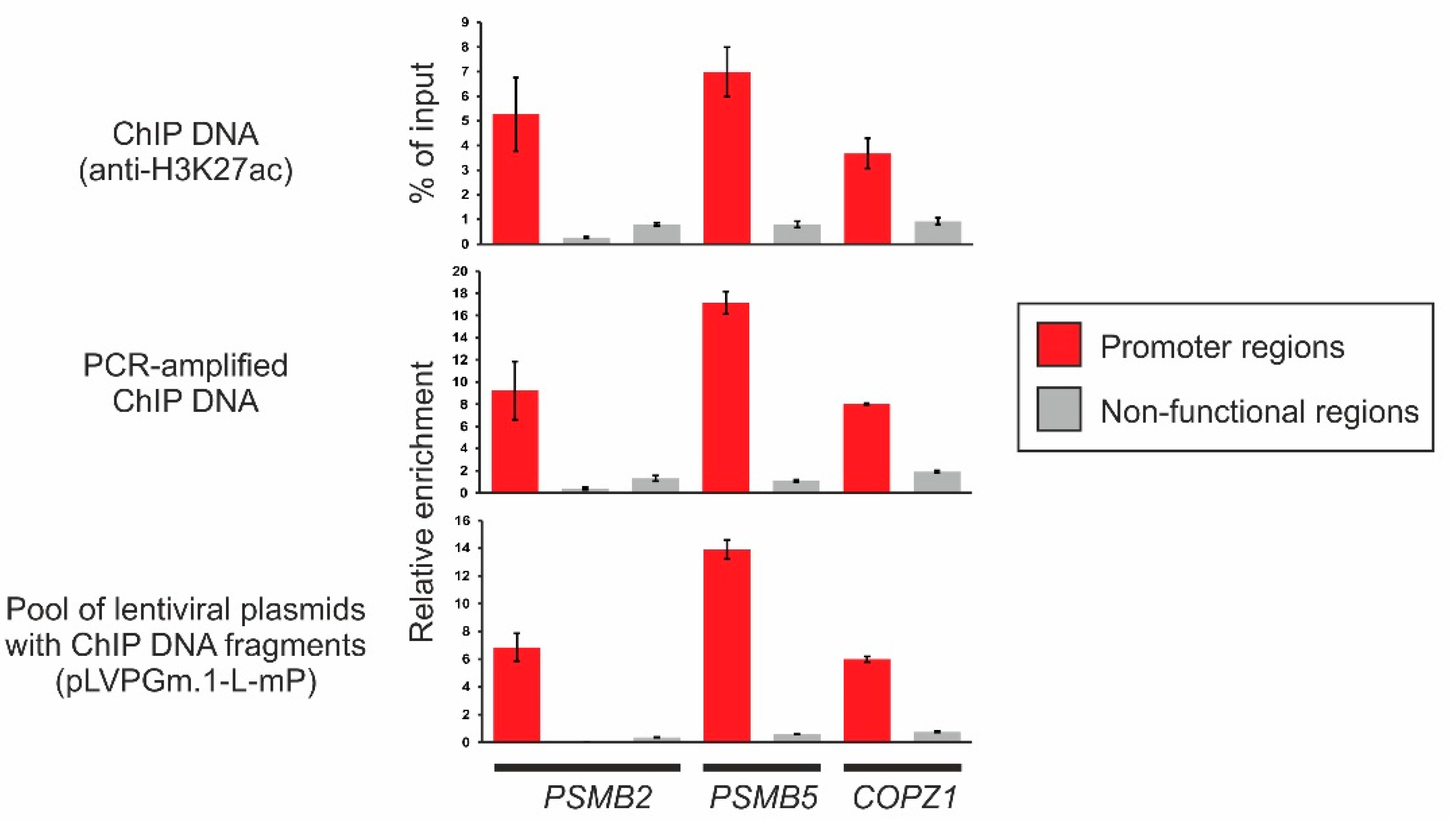
2.3. Obtaining a Primary Library Using ChIP and Cloning It into the Lentiviral Plasmid pLVPGm.1-L-mP
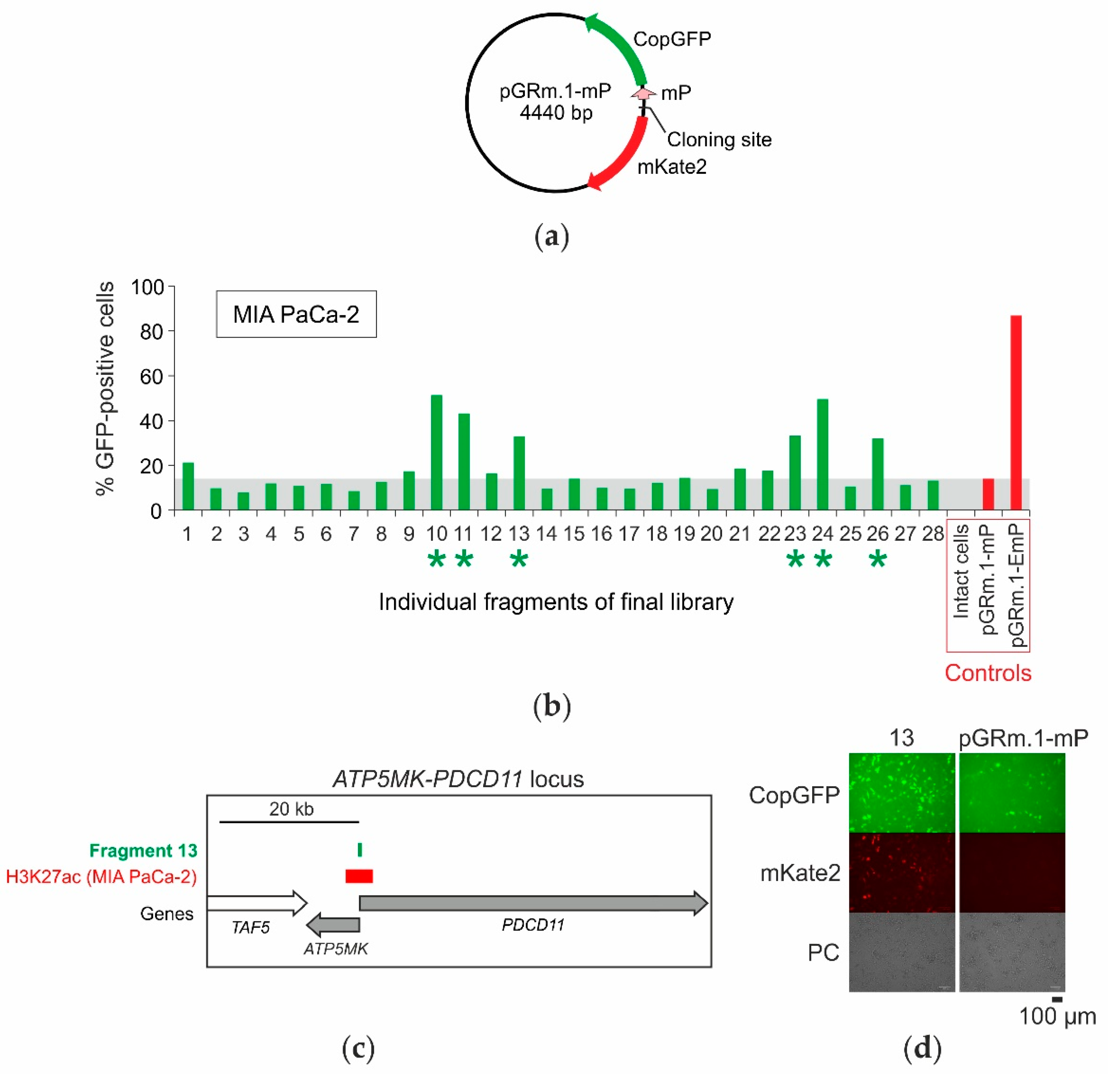
2.4. Selection of Enhancers and Promoters Active in MIA PaCa-2 Cells
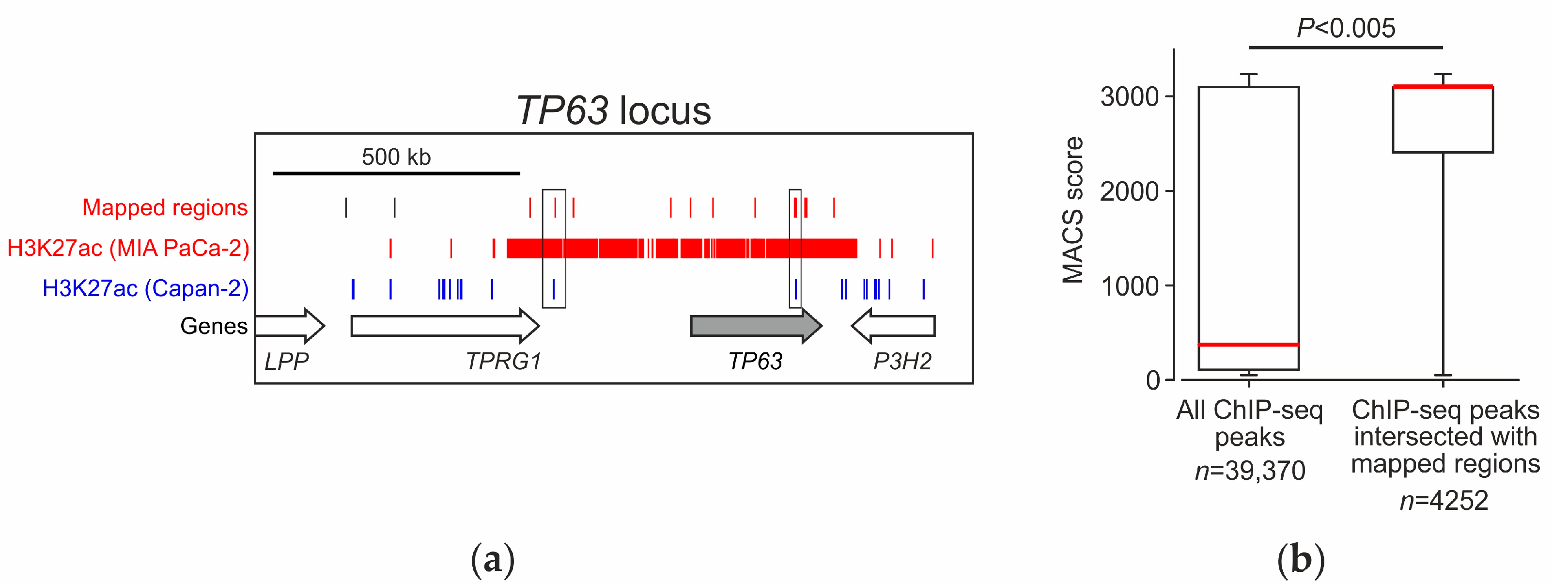
2.5. Analysis of the Distribution of Identified Fragments in the Genome
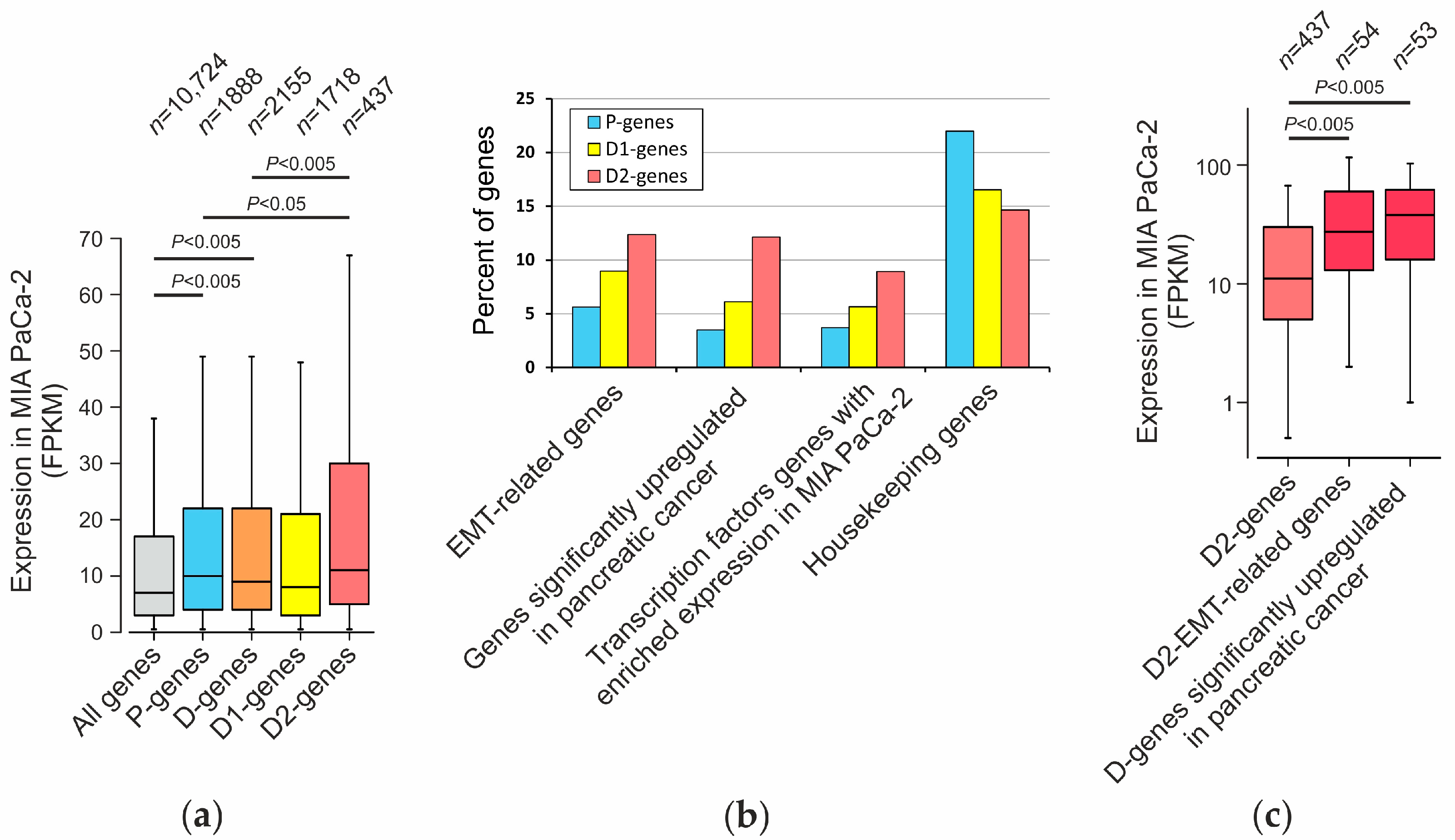
2.6. Analysis of Genes Located near Mapped Regions
3. Discussion
4. Materials and Methods
4.1. ChIP DNA Preparation and Cloning
4.2. Lentivirus Production
4.3. FACS-Based Titration of Control Lentiviral Vector pLVPGm.1-EmP
4.4. Adapter Ligation and PCR
4.5. ChIP DNA Cloning
4.6. Massive Functional Selection of Enhancers and Promoters, Active in MIA PaCa-2 Cells
4.6.1. Transduction of MIA PaCa-2 Cells
4.6.2. PCR Amplification of Inserts and Functional Assay
4.6.3. Libraries Preparation and Next-Generation Sequencing
4.6.4. Paired-End Reads Mapping
4.7. Bioinformatic Data Analysis
4.8. Gene Ontology Annotation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Ibragimov, A.N.; Bylino, O.V.; Shidlovskii, Y.V. Molecular Basis of the Function of Transcriptional Enhancers. Cells 2020, 9, 1620. [Google Scholar] [CrossRef] [PubMed]
- Haberle, V.; Stark, A. Eukaryotic core promoters and the functional basis of transcription initiation. Nat. Rev. Mol. Cell Biol. 2018, 19, 621–637. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.K.; Shiekhattar, R. Architectural and Functional Commonalities between Enhancers and Promoters. Cell 2015, 162, 948–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersson, R.; Sandelin, A. Determinants of enhancer and promoter activities of regulatory elements. Nat. Rev. Genet. 2020, 21, 71–87. [Google Scholar] [CrossRef]
- Dao, L.T.M.; Spicuglia, S. Transcriptional regulation by promoters with enhancer function. Transcription 2018, 9, 307–314. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Sharp, P.A. Divergent transcription: A driving force for new gene origination? Cell 2013, 155, 990–996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carelli, F.N.; Liechti, A.; Halbert, J.; Warnefors, M.; Kaessmann, H. Repurposing of promoters and enhancers during mammalian evolution. Nat. Commun. 2018, 9, 4066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersson, R.; Gebhard, C.; Miguel-Escalada, I.; Hoof, I.; Bornholdt, J.; Boyd, M.; Chen, Y.; Zhao, X.; Schmidl, C.; Suzuki, T.; et al. An atlas of active enhancers across human cell types and tissues. Nature 2014, 507, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Notani, D.; Rosenfeld, M.G. Enhancers as non-coding RNA transcription units: Recent insights and future perspectives. Nat. Rev. Genet. 2016, 17, 207–223. [Google Scholar] [CrossRef]
- Sartorelli, V.; Lauberth, S.M. Enhancer RNAs are an important regulatory layer of the epigenome. Nat. Struct. Mol. Biol. 2020, 27, 521–528. [Google Scholar] [CrossRef]
- Didych, D.A.; Shamsutdinov, M.F.; Smirnov, N.A.; Akopov, S.B.; Monastyrskaya, G.S.; Uspenskaya, N.Y.; Nikolaev, L.G.; Sverdlov, E.D. Human PSENEN and U2AF1L4 genes are concertedly regulated by a genuine bidirectional promoter. Gene 2013, 515, 34–41. [Google Scholar] [CrossRef]
- Kashkin, K.N.; Sverdlov, E.D. Properties, functions, and therapeutic prospects of enhancer RNAs. Russ. J. Bioorg. Chem. 2016, 42, 473–478. [Google Scholar] [CrossRef]
- Kowalczyk, M.S.; Hughes, J.R.; Garrick, D.; Lynch, M.D.; Sharpe, J.A.; Sloane-Stanley, J.A.; McGowan, S.J.; De Gobbi, M.; Hosseini, M.; Vernimmen, D.; et al. Intragenic enhancers act as alternative promoters. Mol. Cell 2012, 45, 447–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skvortsova, Y.V.; Kondratieva, S.A.; Zinovyeva, M.V.; Nikolaev, L.G.; Azhikina, T.L.; Gainetdinov, I.V. Intragenic Locus in Human PIWIL2 Gene Shares Promoter and Enhancer Functions. PLoS ONE 2016, 11, e0156454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.; Ruan, X.; Auerbach, R.K.; Sandhu, K.S.; Zheng, M.; Wang, P.; Poh, H.M.; Goh, Y.; Lim, J.; Zhang, J.; et al. Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation. Cell 2012, 148, 84–98. [Google Scholar] [CrossRef] [Green Version]
- Engreitz, J.M.; Haines, J.E.; Perez, E.M.; Munson, G.; Chen, J.; Kane, M.; McDonel, P.E.; Guttman, M.; Lander, E.S. Local regulation of gene expression by lncRNA promoters, transcription and splicing. Nature 2016, 539, 452–455. [Google Scholar] [CrossRef] [Green Version]
- Paralkar, V.R.; Taborda, C.C.; Huang, P.; Yao, Y.; Kossenkov, A.V.; Prasad, R.; Luan, J.; Davies, J.O.; Hughes, J.R.; Hardison, R.C.; et al. Unlinking an lncRNA from Its Associated cis Element. Mol. Cell 2016, 62, 104–110. [Google Scholar] [CrossRef] [Green Version]
- Roadmap Epigenomics Consortium; Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J.; et al. Integrative analysis of 111 reference human epigenomes. Nature 2015, 518, 317–330. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Sun, L.; Chen, Z.; Whitaker, J.W.; Wang, T.; Wang, W. Predicting enhancer transcription and activity from chromatin modifications. Nucleic Acids Res. 2013, 41, 10032–10043. [Google Scholar] [CrossRef]
- Stueve, T.R.; Marconett, C.N.; Zhou, B.; Borok, Z.; Laird-Offringa, I.A. The importance of detailed epigenomic profiling of different cell types within organs. Epigenomics 2016, 8, 817–829. [Google Scholar] [CrossRef]
- Pradeepa, M.M. Causal role of histone acetylations in enhancer function. Transcription 2017, 8, 40–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagy, G.; Daniel, B.; Jonas, D.; Nagy, L.; Barta, E. A novel method to predict regulatory regions based on histone mark landscapes in macrophages. Immunobiology 2013, 218, 1416–1427. [Google Scholar] [CrossRef] [PubMed]
- Calo, E.; Wysocka, J. Modification of enhancer chromatin: What, how, and why? Mol. Cell 2013, 49, 825–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, C.M.; Henikoff, S. Histone variants: Dynamic punctuation in transcription. Genes Dev. 2014, 28, 672–682. [Google Scholar] [CrossRef] [Green Version]
- Inoue, F.; Ahituv, N. Decoding enhancers using massively parallel reporter assays. Genomics 2015, 106, 159–164. [Google Scholar] [CrossRef]
- Dailey, L. High throughput technologies for the functional discovery of mammalian enhancers: New approaches for understanding transcriptional regulatory network dynamics. Genomics 2015, 106, 151–158. [Google Scholar] [CrossRef]
- Trauernicht, M.; Martinez-Ara, M.; van Steensel, B. Deciphering Gene Regulation Using Massively Parallel Reporter Assays. Trends Biochem. Sci. 2020, 45, 90–91. [Google Scholar] [CrossRef]
- Santiago-Algarra, D.; Dao, L.T.M.; Pradel, L.; Espana, A.; Spicuglia, S. Recent advances in high-throughput approaches to dissect enhancer function. F1000Research 2017, 6, 939. [Google Scholar] [CrossRef] [Green Version]
- Inoue, F.; Kreimer, A.; Ashuach, T.; Ahituv, N.; Yosef, N. Identification and Massively Parallel Characterization of Regulatory Elements Driving Neural Induction. Cell Stem Cell 2019, 25, 713–727.e710. [Google Scholar] [CrossRef]
- Chernov, I.; Stukacheva, E.; Akopov, S.; Didych, D.; Nikolaev, L.; Sverdlov, E. A new technique for selective identification and mapping of enhancers within long genomic sequences. BioTechniques 2008, 44, 775–784. [Google Scholar] [CrossRef]
- Murtha, M.; Tokcaer-Keskin, Z.; Tang, Z.; Strino, F.; Chen, X.; Wang, Y.; Xi, X.; Basilico, C.; Brown, S.; Bonneau, R.; et al. FIREWACh: High-throughput functional detection of transcriptional regulatory modules in mammalian cells. Nat. Chem. Biol. 2014, 11, 559–565. [Google Scholar] [CrossRef] [Green Version]
- Inoue, F.; Kircher, M.; Martin, B.; Cooper, G.M.; Witten, D.M.; McManus, M.T.; Ahituv, N.; Shendure, J. A systematic comparison reveals substantial differences in chromosomal versus episomal encoding of enhancer activity. Genome Res. 2017, 27, 38–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, M.G.; Inoue, F.; Martin, B.; Schubach, M.; Agarwal, V.; Whalen, S.; Feng, S.; Zhao, J.; Ashuach, T.; Ziffra, R.; et al. lentiMPRA and MPRAflow for high-throughput functional characterization of gene regulatory elements. Nat. Protoc. 2020, 15, 2387–2412. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, C.S.; Ganem, D. A virus-encoded inhibitor that blocks RNA interference in mammalian cells. J. Virol. 2005, 79, 7371–7379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlabach, M.R.; Hu, J.K.; Li, M.; Elledge, S.J. Synthetic design of strong promoters. Proc. Natl. Acad. Sci. USA 2010, 107, 2538–2543. [Google Scholar] [CrossRef] [Green Version]
- Maetzig, T.; Galla, M.; Brugman, M.H.; Loew, R.; Baum, C.; Schambach, A. Mechanisms controlling titer and expression of bidirectional lentiviral and gammaretroviral vectors. Gene Ther. 2010, 17, 400–411. [Google Scholar] [CrossRef]
- Consortium, E.P.; Moore, J.E.; Purcaro, M.J.; Pratt, H.E.; Epstein, C.B.; Shoresh, N.; Adrian, J.; Kawli, T.; Davis, C.A.; Dobin, A.; et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 2020, 583, 699–710. [Google Scholar] [CrossRef]
- Diaferia, G.R.; Balestrieri, C.; Prosperini, E.; Nicoli, P.; Spaggiari, P.; Zerbi, A.; Natoli, G. Dissection of transcriptional and cis-regulatory control of differentiation in human pancreatic cancer. EMBO J. 2016, 35, 595–617. [Google Scholar] [CrossRef] [Green Version]
- Rosenbloom, K.R.; Armstrong, J.; Barber, G.P.; Casper, J.; Clawson, H.; Diekhans, M.; Dreszer, T.R.; Fujita, P.A.; Guruvadoo, L.; Haeussler, M.; et al. The UCSC Genome Browser database: 2015 update. Nucleic Acids Res. 2015, 43, D670–D681. [Google Scholar] [CrossRef] [Green Version]
- The FANTOM Consortium and the RIKEN PMI and CLST (DGT); Forrest, A.R.; Kawaji, H.; Rehli, M.; Baillie, J.K.; de Hoon, M.J.; Haberle, V.; Lassmann, T.; Kulakovskiy, I.V.; Lizio, M.; et al. A promoter-level mammalian expression atlas. Nature 2014, 507, 462–470. [Google Scholar] [CrossRef]
- Whyte, W.A.; Orlando, D.A.; Hnisz, D.; Abraham, B.J.; Lin, C.Y.; Kagey, M.H.; Rahl, P.B.; Lee, T.I.; Young, R.A. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 2013, 153, 307–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hnisz, D.; Abraham, B.J.; Lee, T.I.; Lau, A.; Saint-Andre, V.; Sigova, A.A.; Hoke, H.A.; Young, R.A. Super-enhancers in the control of cell identity and disease. Cell 2013, 155, 934–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLean, C.Y.; Bristor, D.; Hiller, M.; Clarke, S.L.; Schaar, B.T.; Lowe, C.B.; Wenger, A.M.; Bejerano, G. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 2010, 28, 495–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petryszak, R.; Keays, M.; Tang, Y.A.; Fonseca, N.A.; Barrera, E.; Burdett, T.; Fullgrabe, A.; Fuentes, A.M.; Jupp, S.; Koskinen, S.; et al. Expression Atlas update--an integrated database of gene and protein expression in humans, animals and plants. Nucleic Acids Res. 2016, 44, D746–D752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goonesekere, N.C.; Wang, X.; Ludwig, L.; Guda, C. A meta analysis of pancreatic microarray datasets yields new targets as cancer genes and biomarkers. PLoS ONE 2014, 9, e93046. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Kong, L.; Liu, Y.; Qu, H. dbEMT: An epithelial-mesenchymal transition associated gene resource. Sci. Rep. 2015, 5, 11459. [Google Scholar] [CrossRef] [Green Version]
- Abugessaisa, I.; Shimoji, H.; Sahin, S.; Kondo, A.; Harshbarger, J.; Lizio, M.; Hayashizaki, Y.; Carninci, P.; The FANTOM consortium; Forrest, A.; et al. FANTOM5 transcriptome catalog of cellular states based on Semantic MediaWiki. Database 2016, 2016, 1–10. [Google Scholar] [CrossRef]
- Chang, C.W.; Cheng, W.C.; Chen, C.R.; Shu, W.Y.; Tsai, M.L.; Huang, C.L.; Hsu, I.C. Identification of human housekeeping genes and tissue-selective genes by microarray meta-analysis. PLoS ONE 2011, 6, e22859. [Google Scholar] [CrossRef] [Green Version]
- Zabidi, M.A.; Arnold, C.D.; Schernhuber, K.; Pagani, M.; Rath, M.; Frank, O.; Stark, A. Enhancer-core-promoter specificity separates developmental and housekeeping gene regulation. Nature 2015, 518, 556–559. [Google Scholar] [CrossRef]
- Ferrer, C.C.; Berthenet, K.; Ichim, G. Apoptosis–Fueling the oncogenic fire. FEBS J. 2021, 288, 4445–4463. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Wyckoff, J.; Condeelis, J. Cell migration in tumors. Curr. Opin. Cell Biol. 2005, 17, 559–564. [Google Scholar] [CrossRef]
- Esteban-Villarrubia, J.; Soto-Castillo, J.J.; Pozas, J.; San Roman-Gil, M.; Orejana-Martin, I.; Torres-Jimenez, J.; Carrato, A.; Alonso-Gordoa, T.; Molina-Cerrillo, J. Tyrosine Kinase Receptors in Oncology. Int. J. Mol. Sci. 2020, 21, 8529. [Google Scholar] [CrossRef] [PubMed]
- Kontomanolis, E.N.; Koutras, A.; Syllaios, A.; Schizas, D.; Mastoraki, A.; Garmpis, N.; Diakosavvas, M.; Angelou, K.; Tsatsaris, G.; Pagkalos, A.; et al. Role of Oncogenes and Tumor-suppressor Genes in Carcinogenesis: A Review. Anticancer Res. 2020, 40, 6009–6015. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Grant, T.J.; Hua, K.; Singh, A. Molecular Pathogenesis of Pancreatic Cancer. Prog. Mol. Biol. Transl. Sci. 2016, 144, 241–275. [Google Scholar] [CrossRef] [PubMed]
- Reyes, C.M.; Dogruoz, A.; Istvanffy, R.; Friess, H.; Ceyhan, G.O.; Demir, I.E. Molecular Profiling in Pancreatic Cancer: Current Role and Its Impact on Primary Surgery. Visc. Med. 2022, 38, 37–41. [Google Scholar] [CrossRef]
- Collisson, E.A.; Bailey, P.; Chang, D.K.; Biankin, A.V. Molecular subtypes of pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 207–220. [Google Scholar] [CrossRef]
- Mizrahi, J.D.; Surana, R.; Valle, J.W.; Shroff, R.T. Pancreatic cancer. Lancet 2020, 395, 2008–2020. [Google Scholar] [CrossRef]
- Kang, Y.; Kim, Y.W.; Kang, J.; Kim, A. Histone H3K4me1 and H3K27ac play roles in nucleosome eviction and eRNA transcription, respectively, at enhancers. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2021, 35, e21781. [Google Scholar] [CrossRef]
- Khambata-Ford, S.; Liu, Y.; Gleason, C.; Dickson, M.; Altman, R.B.; Batzoglou, S.; Myers, R.M. Identification of promoter regions in the human genome by using a retroviral plasmid library-based functional reporter gene assay. Genome Res. 2003, 13, 1765–1774. [Google Scholar] [CrossRef]
- Dickel, D.E.; Zhu, Y.; Nord, A.S.; Wylie, J.N.; Akiyama, J.A.; Afzal, V.; Plajzer-Frick, I.; Kirkpatrick, A.; Gottgens, B.; Bruneau, B.G.; et al. Function-based identification of mammalian enhancers using site-specific integration. Nat. Methods 2014, 11, 566–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kashkin, K.N.; Chernov, I.P.; Stukacheva, E.A.; Kopantzev, E.P.; Monastyrskaya, G.S.; Uspenskaya, N.Y.; Sverdlov, E.D. Cancer specificity of promoters of the genes involved in cell proliferation control. Acta Nat. 2013, 5, 79–83. [Google Scholar] [CrossRef]
- Herz, H.M. Enhancer deregulation in cancer and other diseases. BioEssays News Rev. Mol. Cell. Dev. Biol. 2016, 38, 1003–1015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dietlein, F.; Wang, A.B.; Fagre, C.; Tang, A.; Besselink, N.J.M.; Cuppen, E.; Li, C.; Sunyaev, S.R.; Neal, J.T.; Van Allen, E.M. Genome-wide analysis of somatic noncoding mutation patterns in cancer. Science 2022, 376, eabg5601. [Google Scholar] [CrossRef]
- Zhang, T.; Zhang, Z.; Dong, Q.; Xiong, J.; Zhu, B. Histone H3K27 acetylation is dispensable for enhancer activity in mouse embryonic stem cells. Genome Biol. 2020, 21, 45. [Google Scholar] [CrossRef] [Green Version]
- Naldini, L.; Blomer, U.; Gage, F.H.; Trono, D.; Verma, I.M. Efficient transfer, integration, and sustained long-term expression of the transgene in adult rat brains injected with a lentiviral vector. Proc. Natl. Acad. Sci. USA 1996, 93, 11382–11388. [Google Scholar] [CrossRef] [Green Version]
- Zufferey, R.; Nagy, D.; Mandel, R.J.; Naldini, L.; Trono, D. Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nat. Biotechnol. 1997, 15, 871–875. [Google Scholar] [CrossRef]
- Blankenberg, D.; Gordon, A.; Von Kuster, G.; Coraor, N.; Taylor, J.; Nekrutenko, A.; Galaxy, T. Manipulation of FASTQ data with Galaxy. Bioinformatics 2010, 26, 1783–1785. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Barnett, D.W.; Garrison, E.K.; Quinlan, A.R.; Stromberg, M.P.; Marth, G.T. BamTools: A C++ API and toolkit for analyzing and managing BAM files. Bioinformatics 2011, 27, 1691–1692. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Y.; Zhang, S.; Shang, S.; Zhang, B.; Li, S.; Wang, X.; Wang, F.; Su, J.; Wu, Q.; Liu, H.; et al. SEA: A super-enhancer archive. Nucleic Acids Res. 2016, 44, D172–D179. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Qian, F.; Bai, X.; Liu, Y.; Wang, Q.; Ai, B.; Han, X.; Shi, S.; Zhang, J.; Li, X.; et al. SEdb: A comprehensive human super-enhancer database. Nucleic Acids Res. 2019, 47, D235–D243. [Google Scholar] [CrossRef]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets--update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [Green Version]
- Afgan, E.; Baker, D.; van den Beek, M.; Blankenberg, D.; Bouvier, D.; Cech, M.; Chilton, J.; Clements, D.; Coraor, N.; Eberhard, C.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res. 2016, 44, W3–W10. [Google Scholar] [CrossRef] [Green Version]
- Supek, F.; Bosnjak, M.; Skunca, N.; Smuc, T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | Genome Position (hg38) | Length, bp | Position Relative to Genes | DNase I HS 1 | TFBS 2 | SEA SE 3 | H3K27ac 4 | CAGE 5 | Downstream Gene 6 | GeneHancer |
|---|---|---|---|---|---|---|---|---|---|---|
| 10 | chr13:99,498,575- 99,499,229 | 655 | LINC01232 and TM9SF2 promoter | yes | yes | - | yes | - | LINC01232 | GH04J041933 |
| 11 | chr4:41,934,622- 41,935,299 | 678 | ENSG00000249771 and TMEM33 promoter | yes | yes | - | yes | 4 | TMEM33 | GH04J041933 |
| 13 | chr10:103,396,330- 103,396,860 | 531 | ATP5MK and PDCD11 promoter | yes | yes | - | yes | 3 | PDCD11 | GH10J103394 |
| 23 | chr6:26,171,938- 26,172,533 | 596 | ENSG00000283064 and H2BC6 1st exons | yes | yes | yes | yes | 2 | ENSG00000283064 | GH06J026170 |
| 24 | chr2:20,822,774- 20,823,142 | 369 | LDAH promoter | yes | yes | - | yes | 2 | LDAH | GH02J020821 |
| 26 | chr3:9,791,789- 9,792,567 | 779 | TADA3 and ARPC4/ARPC4-TTLL3 promoter | yes | yes | - | yes | 4 | ARPC4/ARPC4-TTLL | GH03J009791 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kashkin, K.N.; Kotova, E.S.; Alekseenko, I.V.; Bulanenkova, S.S.; Akopov, S.B.; Kopantzev, E.P.; Nikolaev, L.G.; Chernov, I.P.; Didych, D.A. Efficient Selection of Enhancers and Promoters from MIA PaCa-2 Pancreatic Cancer Cells by ChIP-lentiMPRA. Int. J. Mol. Sci. 2022, 23, 15011. https://doi.org/10.3390/ijms232315011
Kashkin KN, Kotova ES, Alekseenko IV, Bulanenkova SS, Akopov SB, Kopantzev EP, Nikolaev LG, Chernov IP, Didych DA. Efficient Selection of Enhancers and Promoters from MIA PaCa-2 Pancreatic Cancer Cells by ChIP-lentiMPRA. International Journal of Molecular Sciences. 2022; 23(23):15011. https://doi.org/10.3390/ijms232315011
Chicago/Turabian StyleKashkin, Kirill Nikitich, Elena Sergeevna Kotova, Irina Vasilievna Alekseenko, Svetlana Sergeevna Bulanenkova, Sergey Borisovich Akopov, Eugene Pavlovich Kopantzev, Lev Grigorievich Nikolaev, Igor Pavlovich Chernov, and Dmitry Alexandrovich Didych. 2022. "Efficient Selection of Enhancers and Promoters from MIA PaCa-2 Pancreatic Cancer Cells by ChIP-lentiMPRA" International Journal of Molecular Sciences 23, no. 23: 15011. https://doi.org/10.3390/ijms232315011
APA StyleKashkin, K. N., Kotova, E. S., Alekseenko, I. V., Bulanenkova, S. S., Akopov, S. B., Kopantzev, E. P., Nikolaev, L. G., Chernov, I. P., & Didych, D. A. (2022). Efficient Selection of Enhancers and Promoters from MIA PaCa-2 Pancreatic Cancer Cells by ChIP-lentiMPRA. International Journal of Molecular Sciences, 23(23), 15011. https://doi.org/10.3390/ijms232315011