
Impact of Copper Loading on NH3-Selective Catalytic Reduction, Oxidation Reactions and N2O Formation over Cu/SAPO-34

 ,
,
Abstract
:
1. Introduction
2. Results
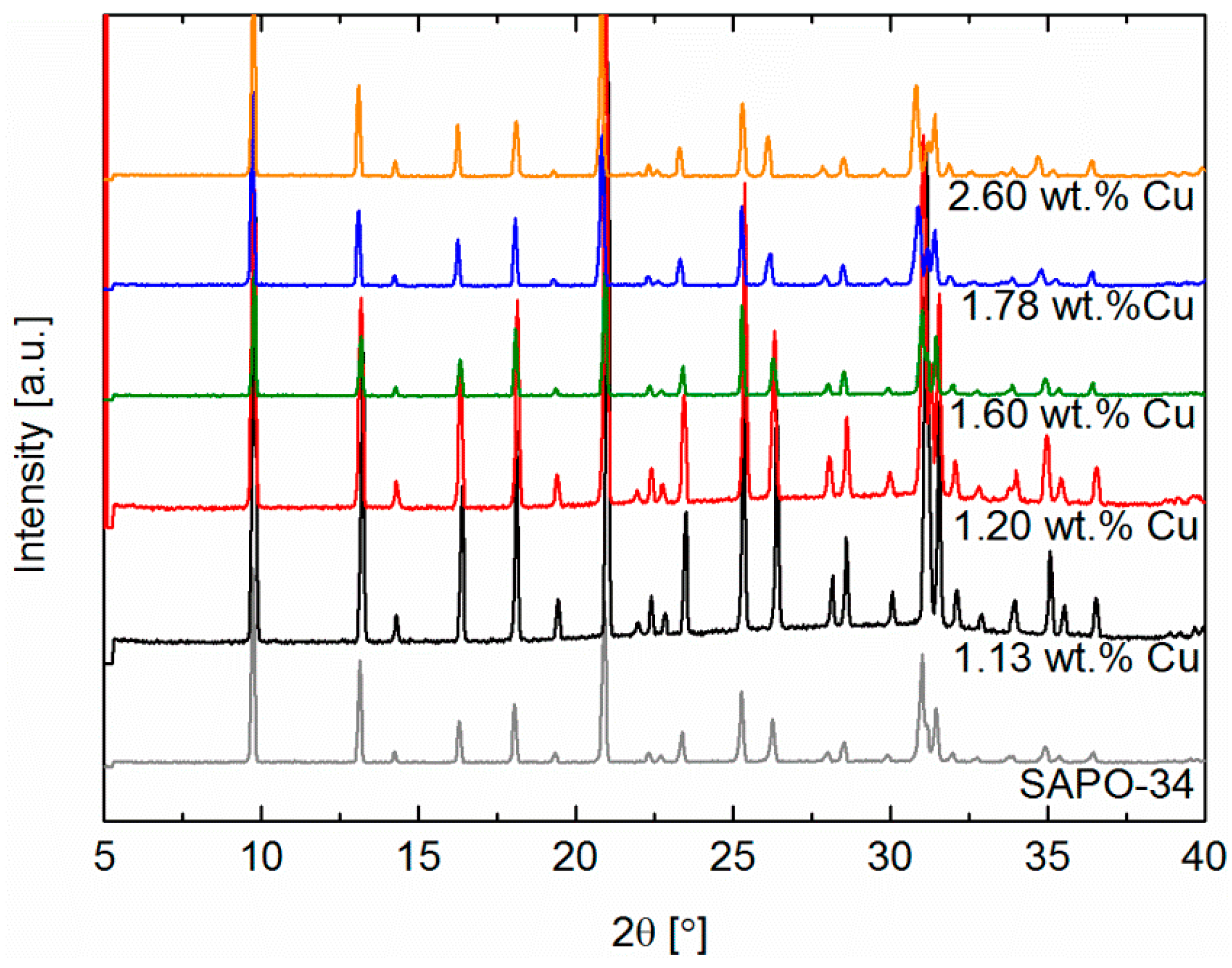
2.1. Characterisation by X-ray Diffraction (XRD)
2.2. Characterisation by N2 Adsorption
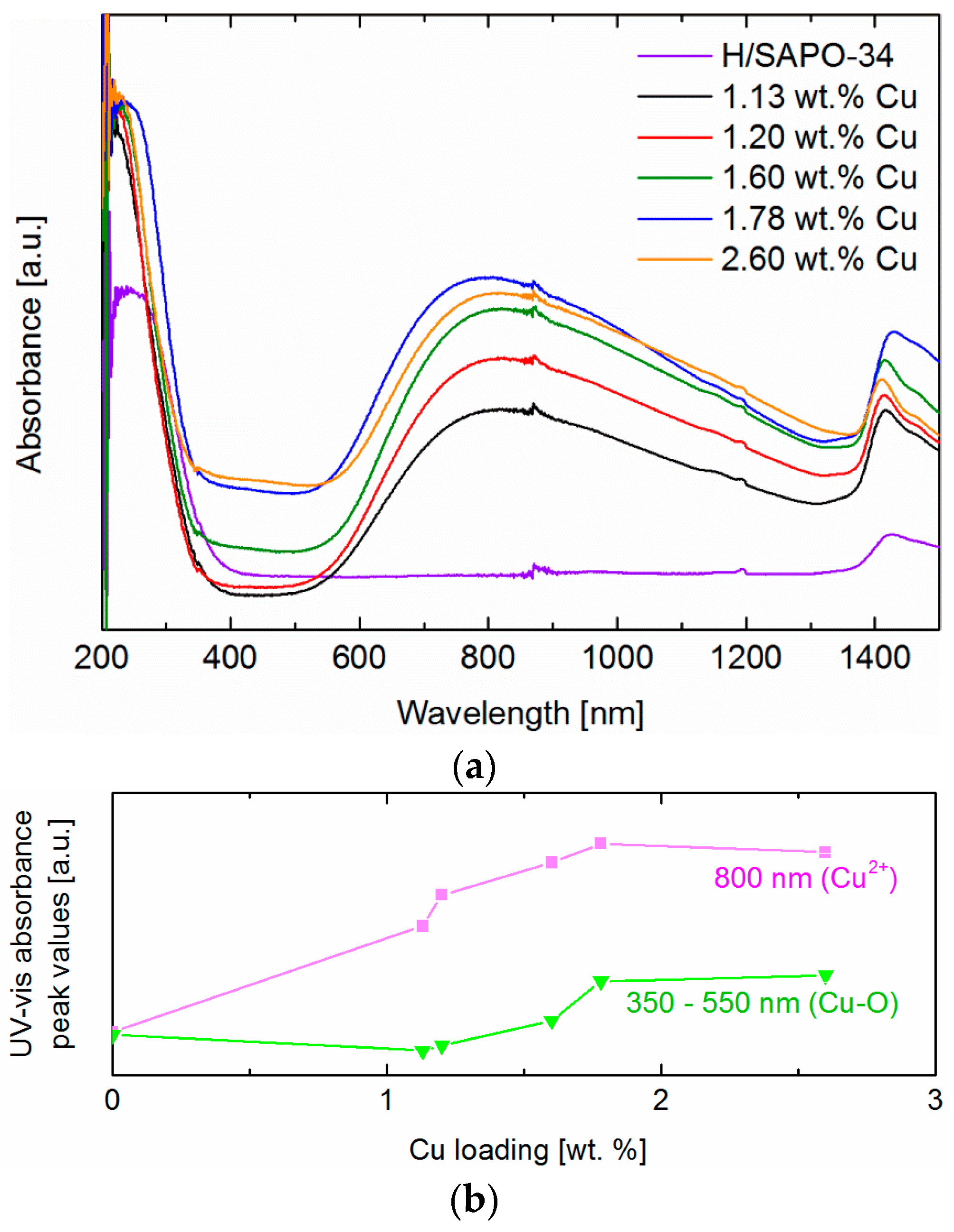
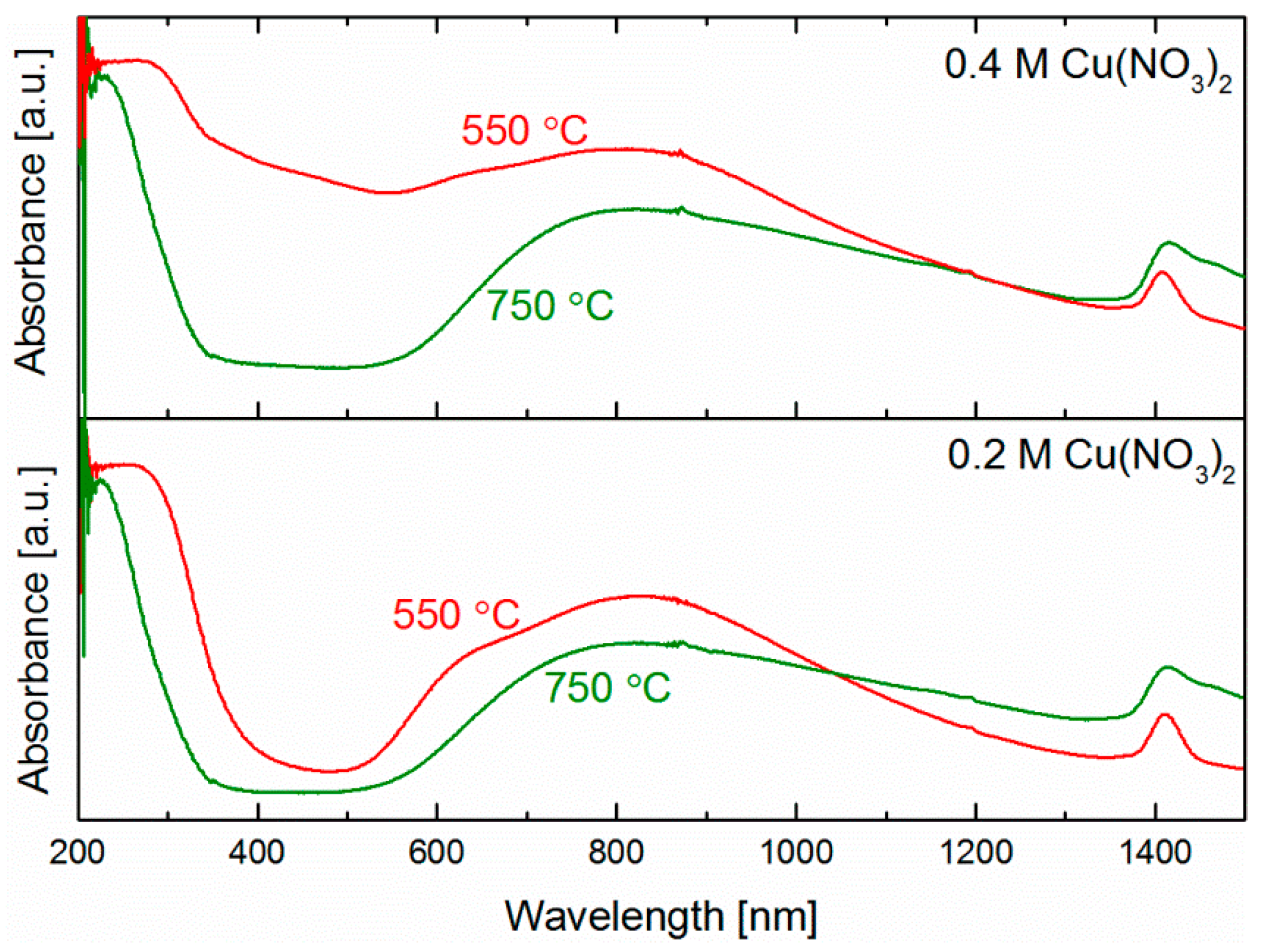
2.3. Catalyst Composition and Copper Species
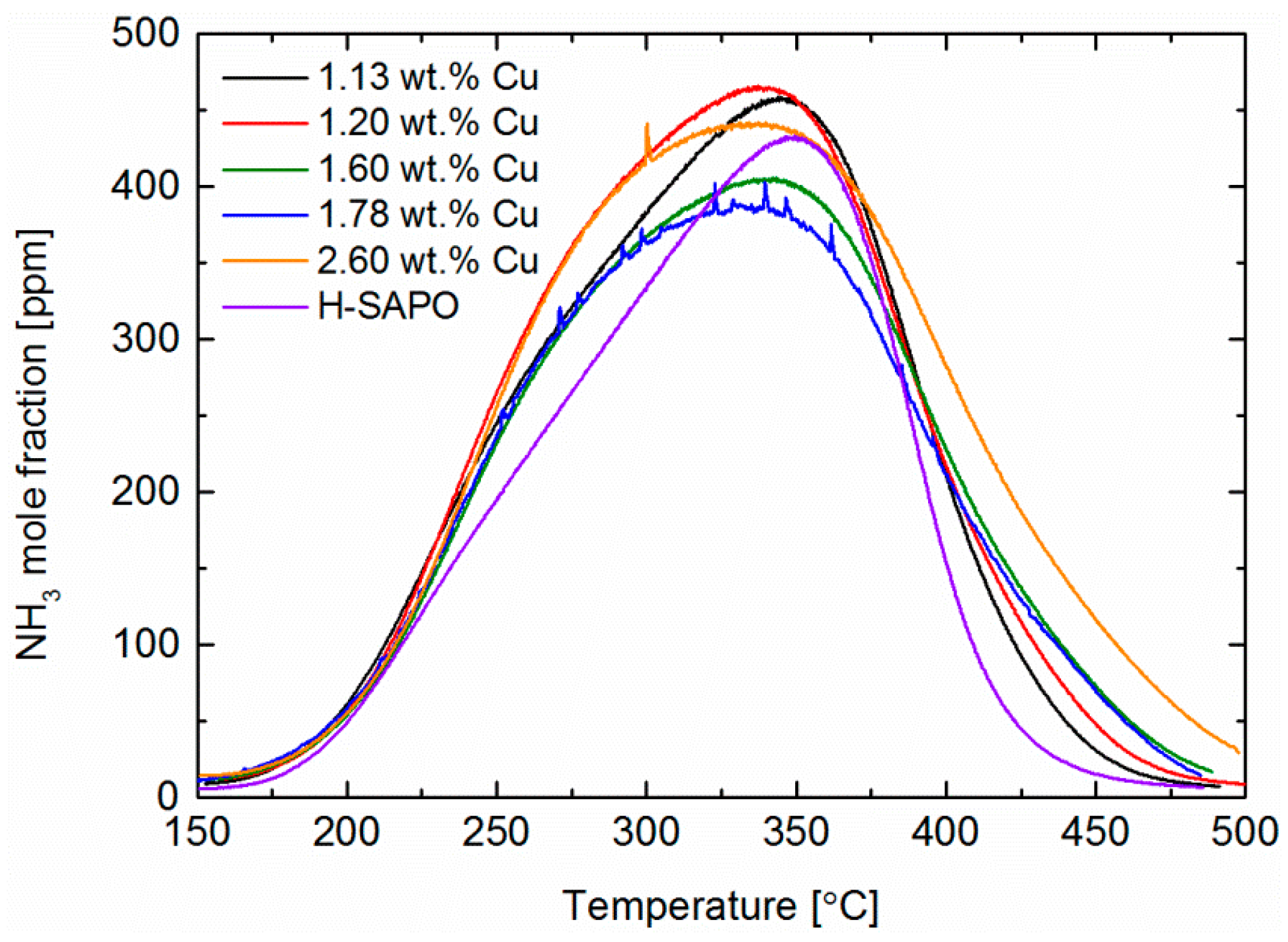
2.4. Characterisation by NH3 Adsorption
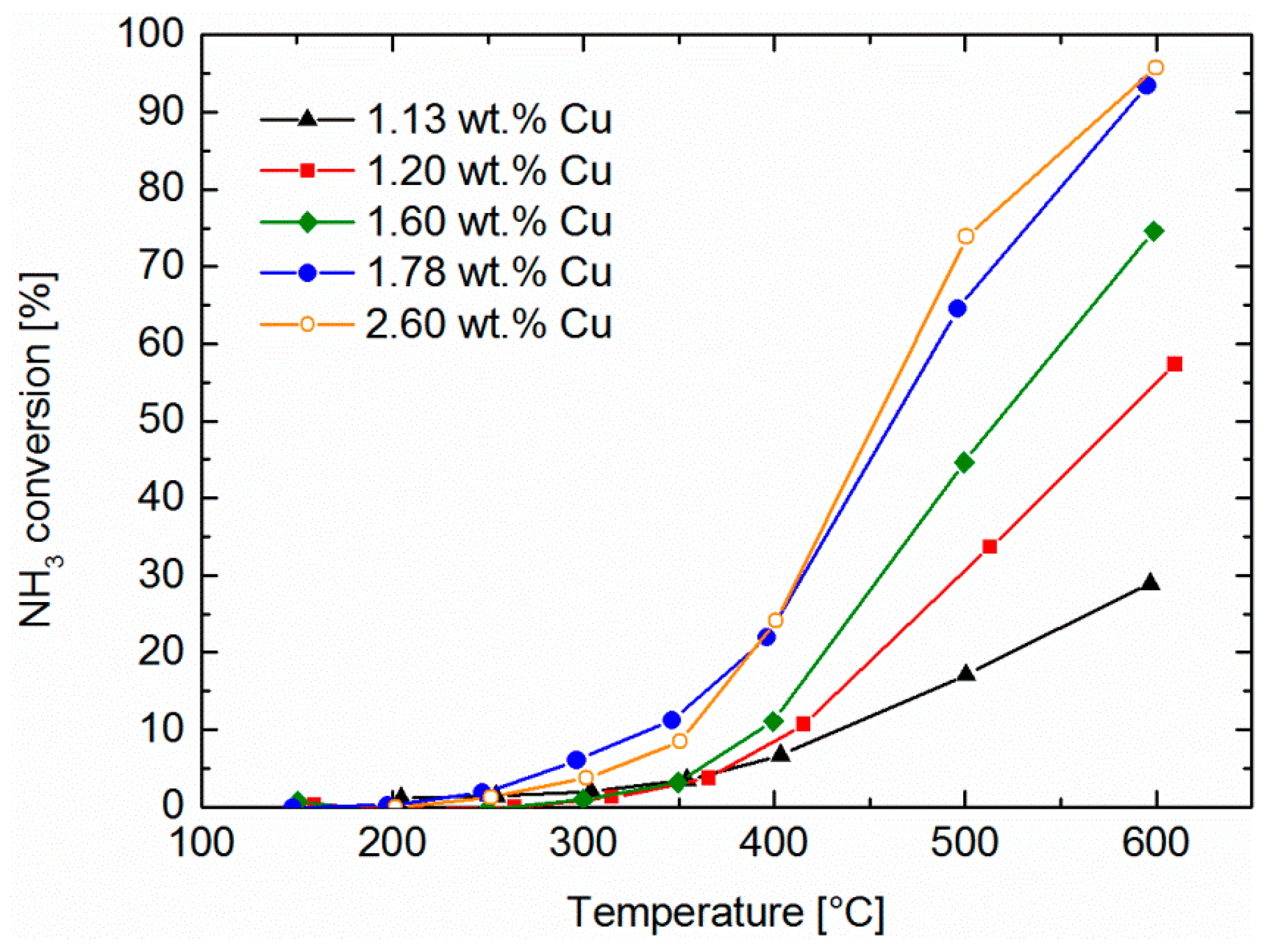
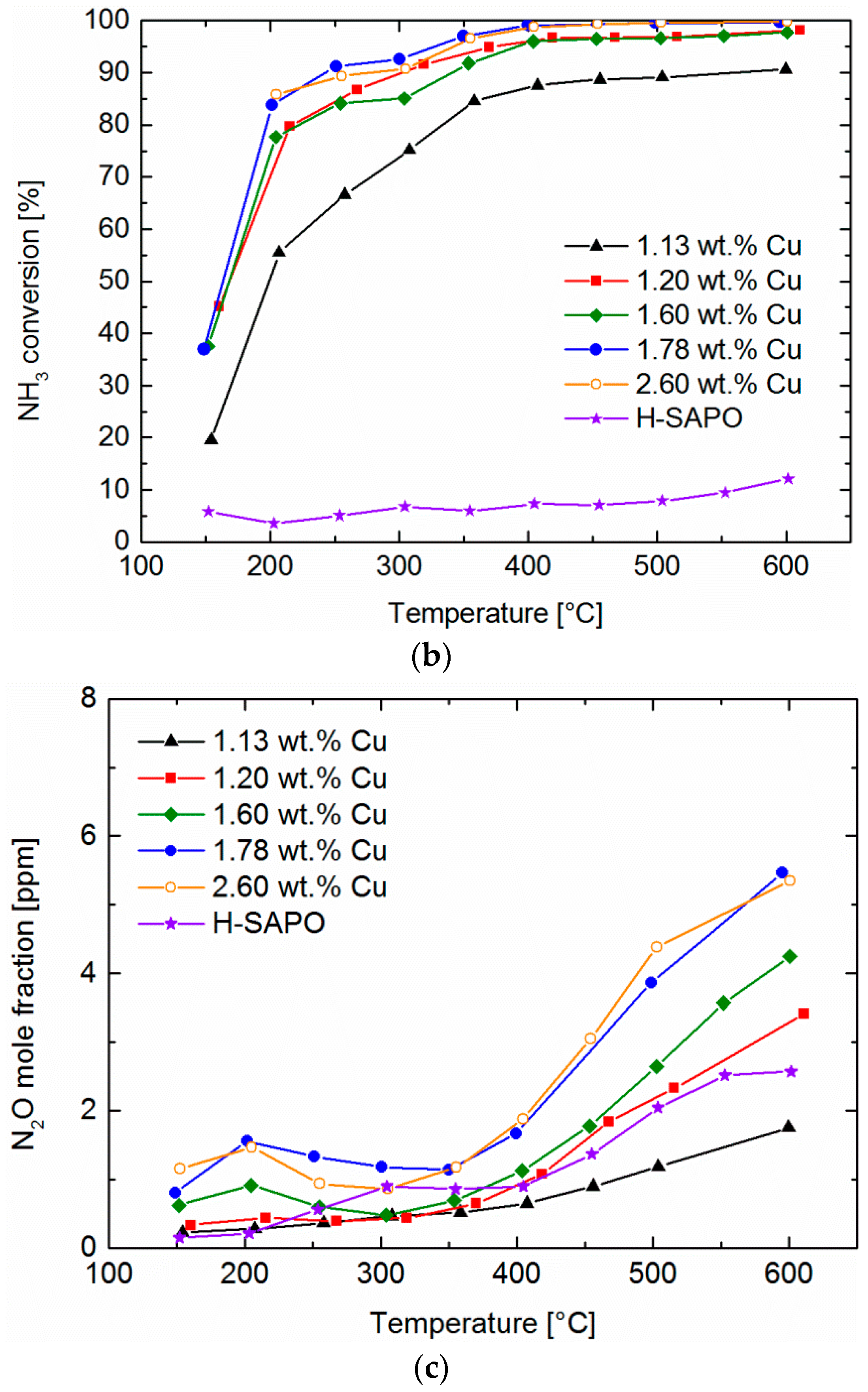
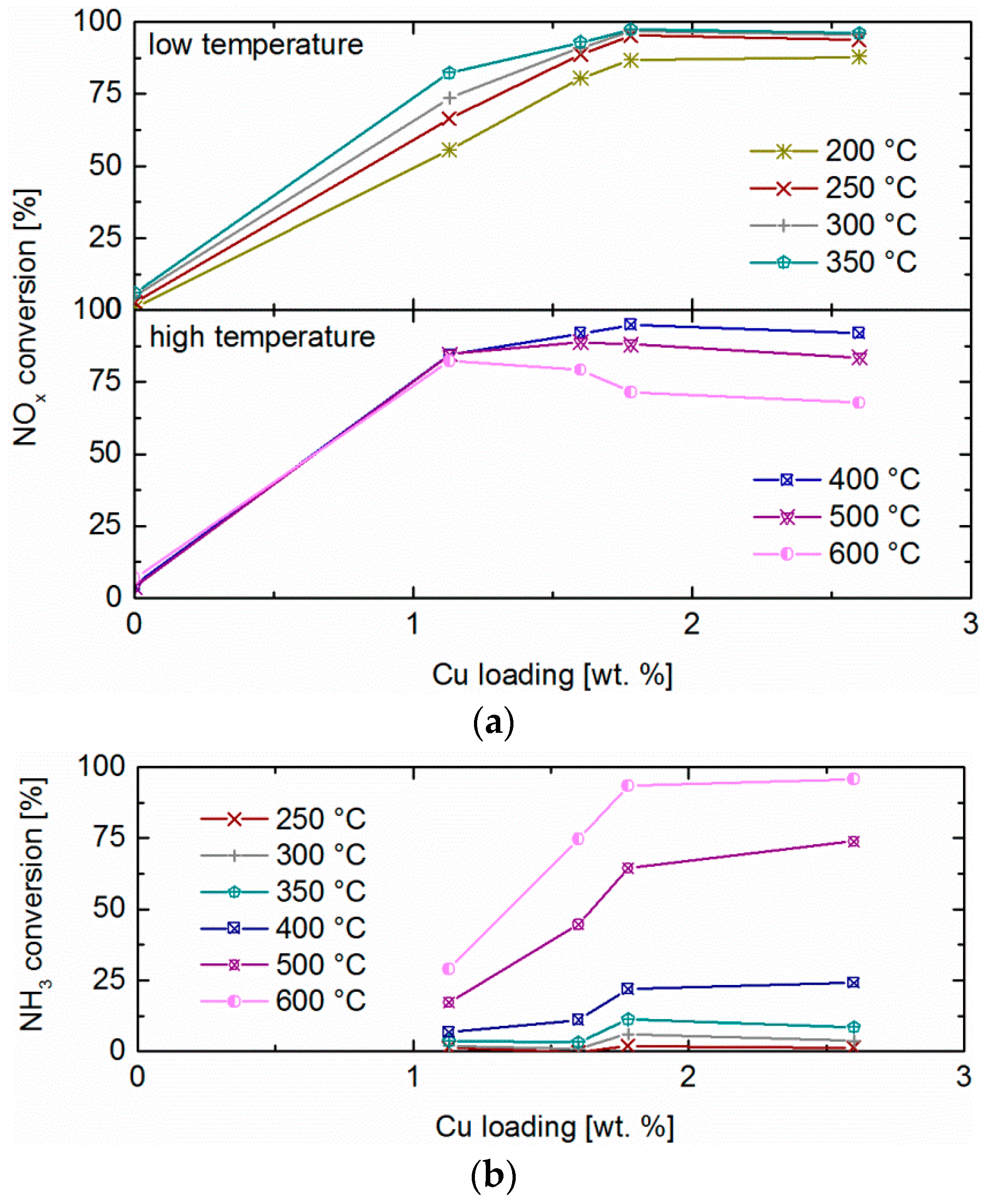
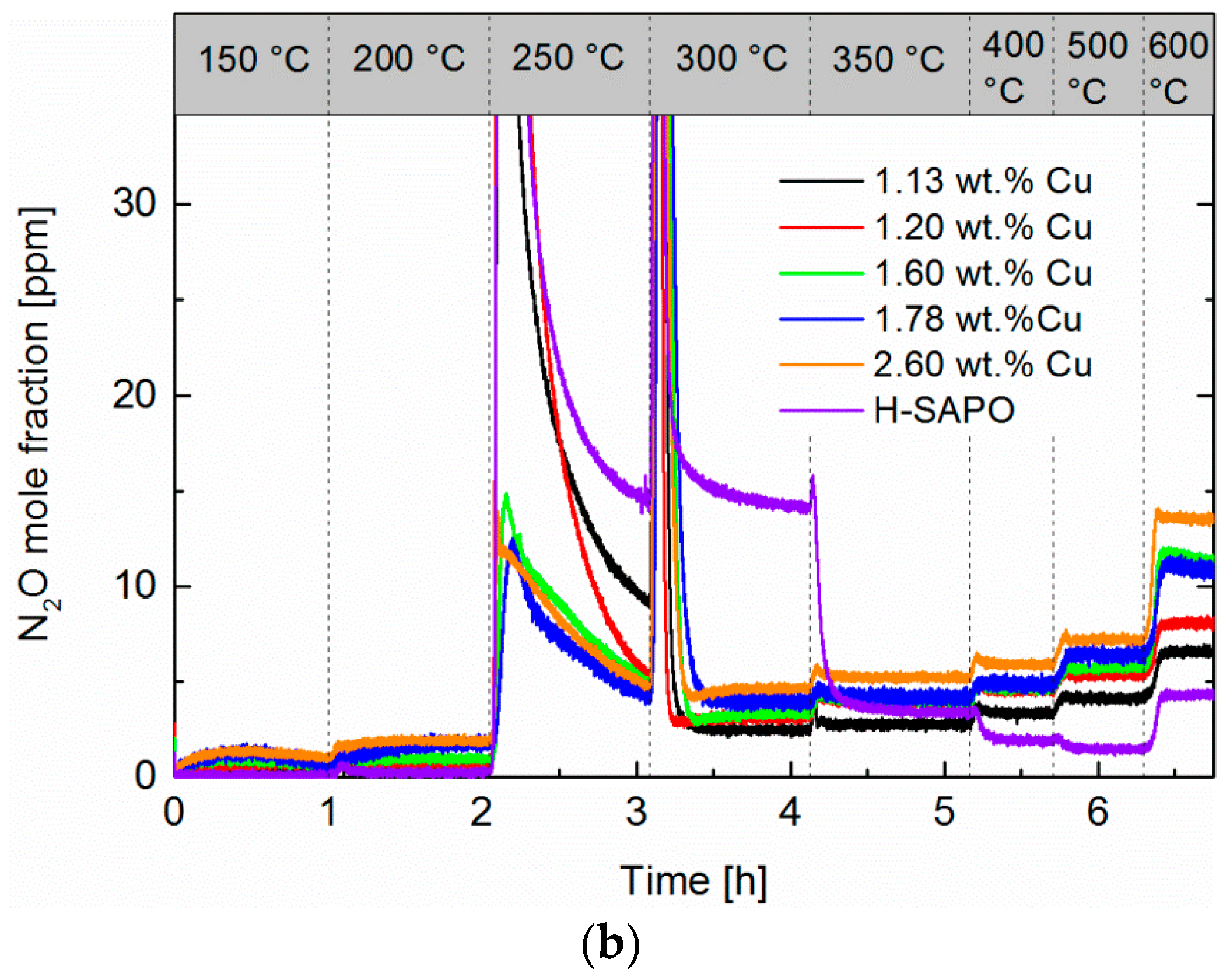
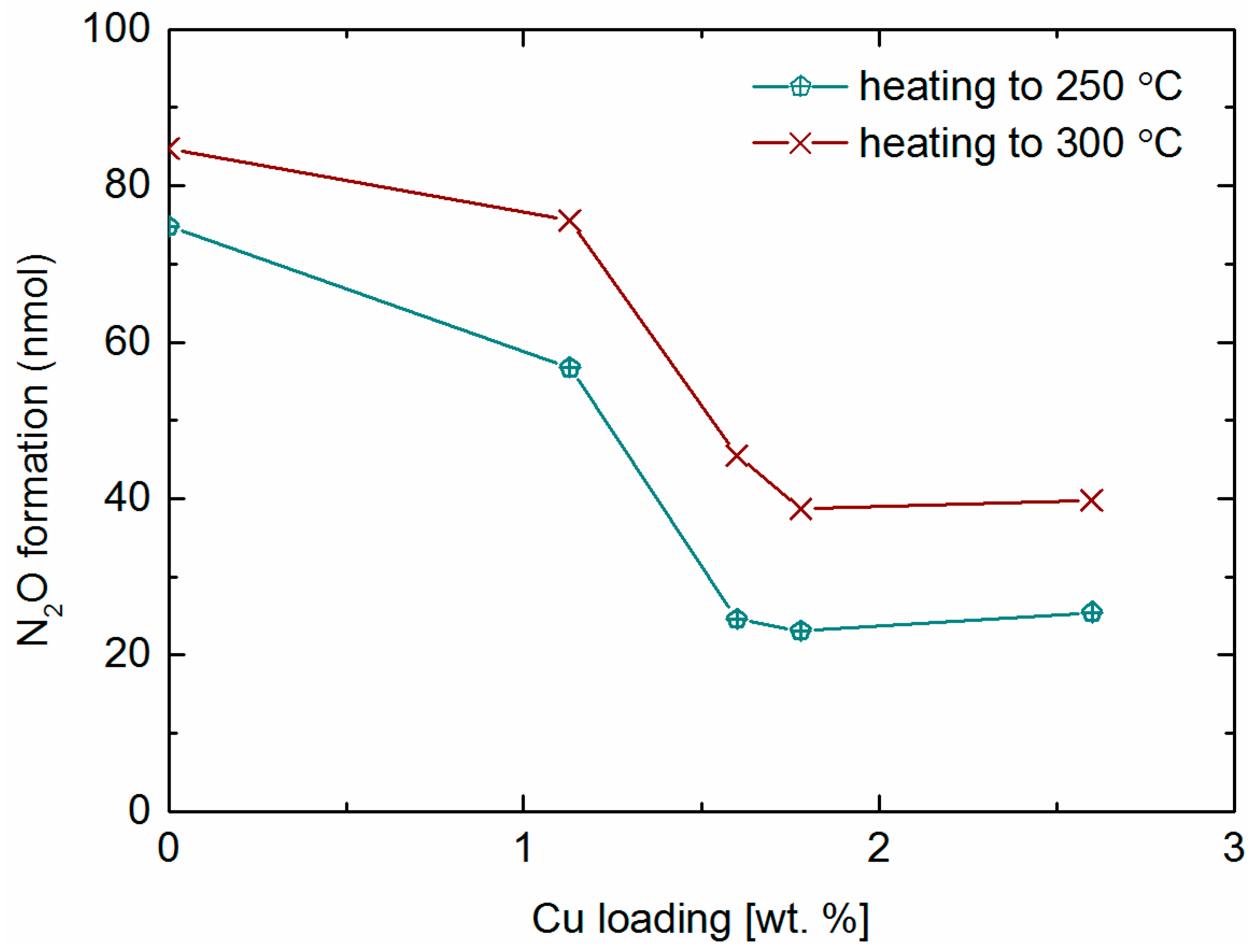
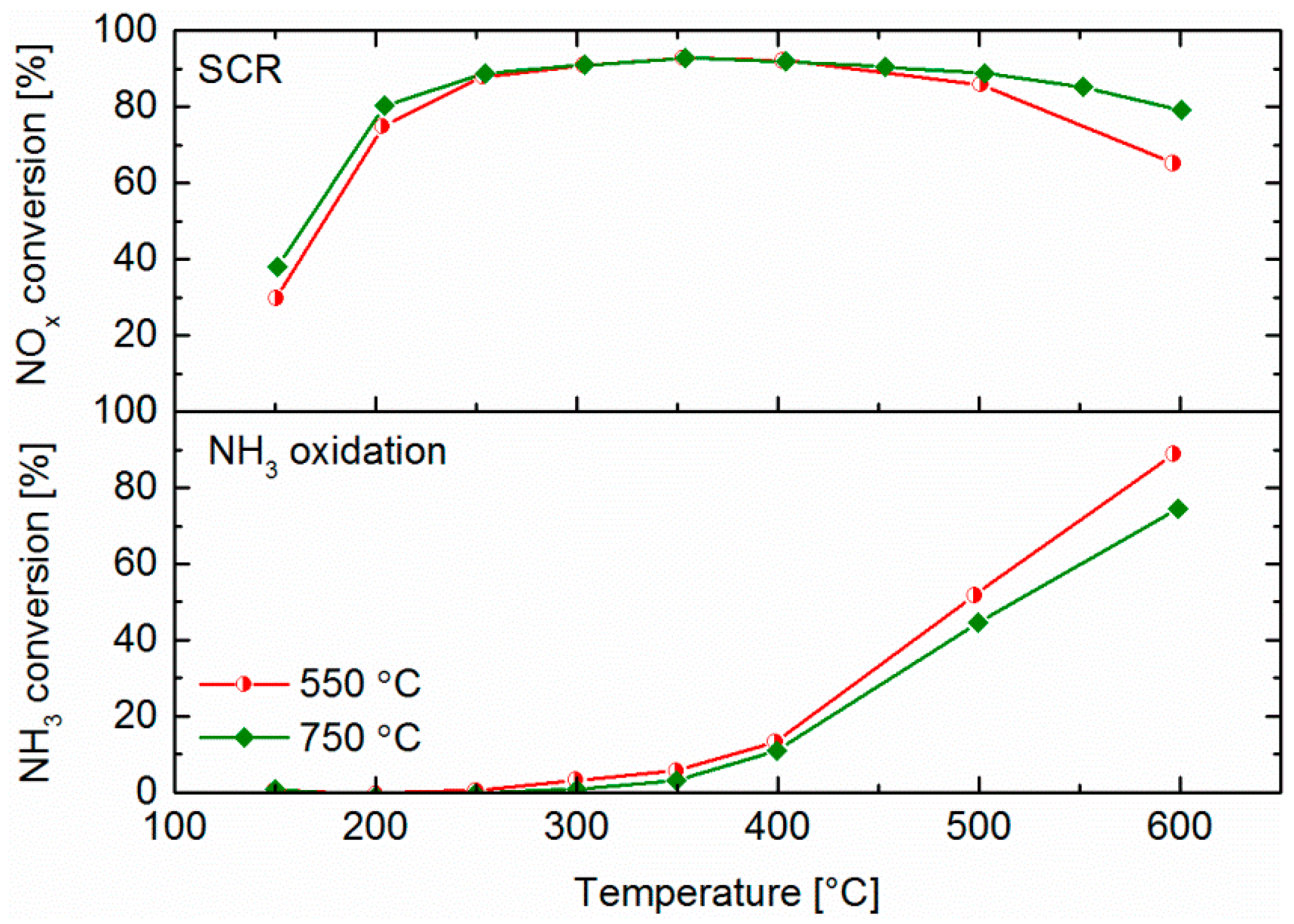
2.5. NH3 and NO Oxidation
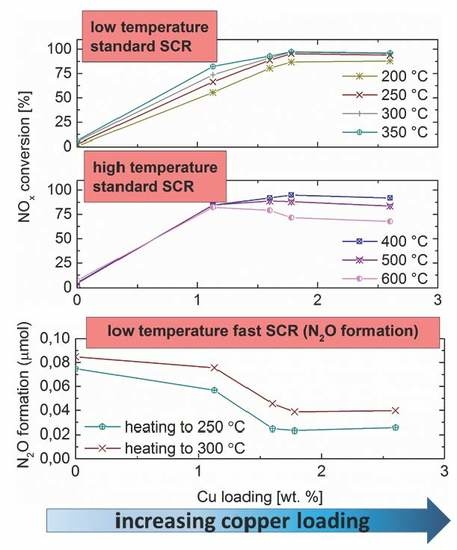
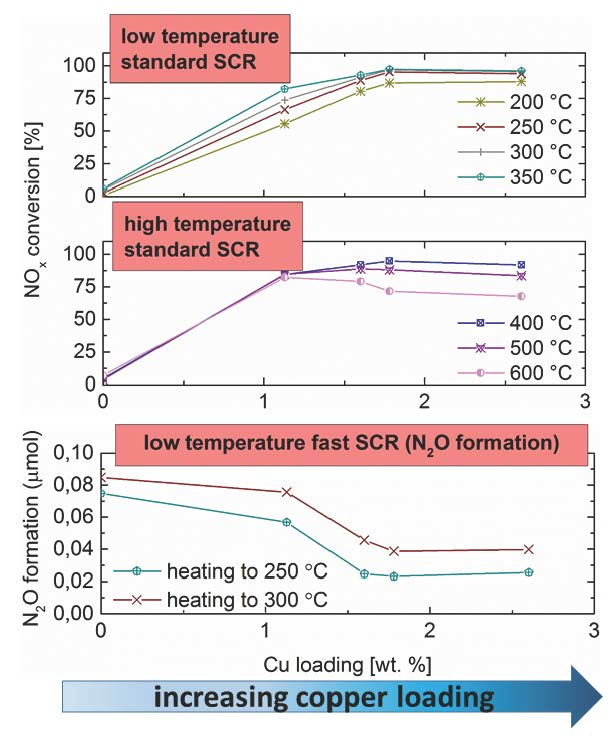
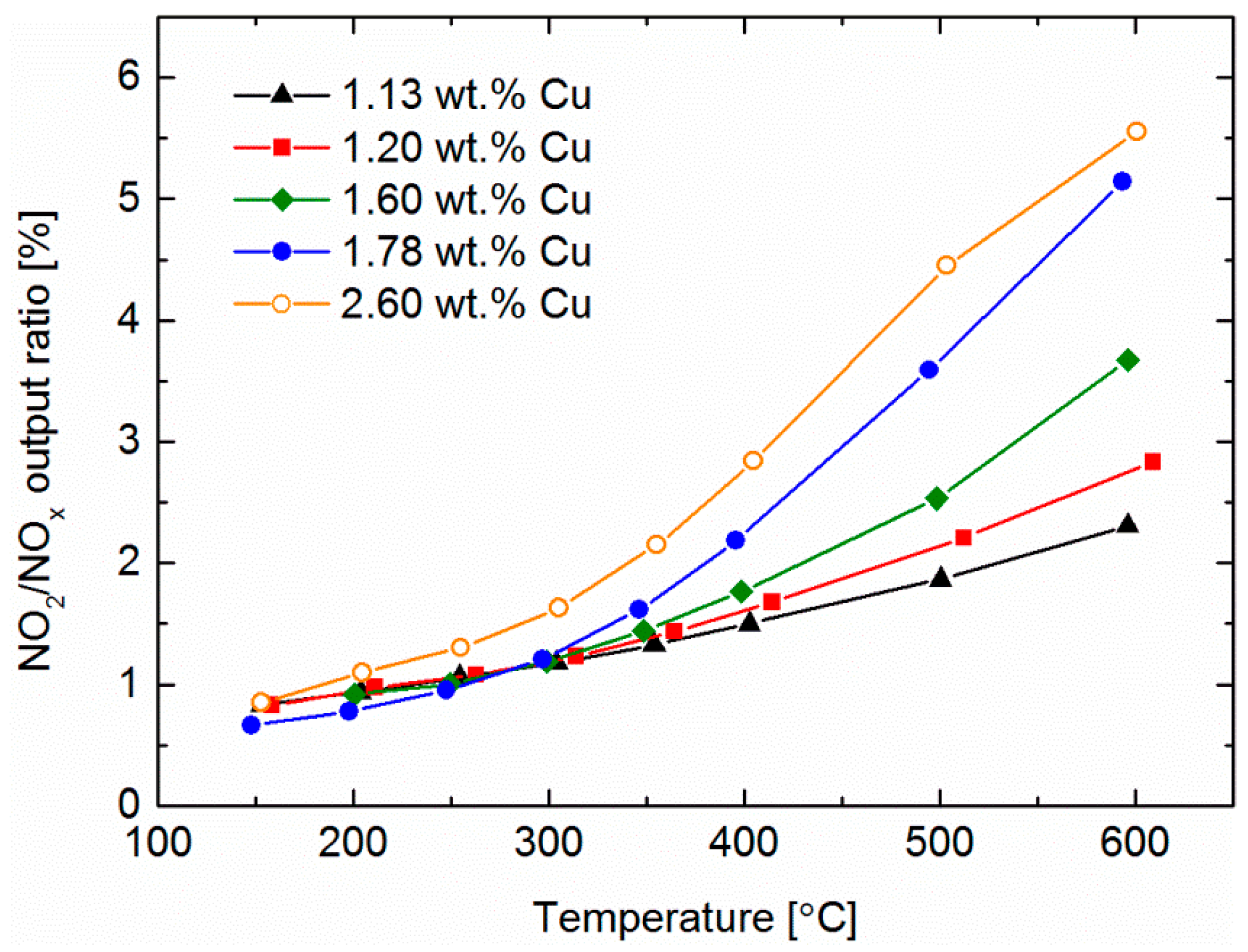
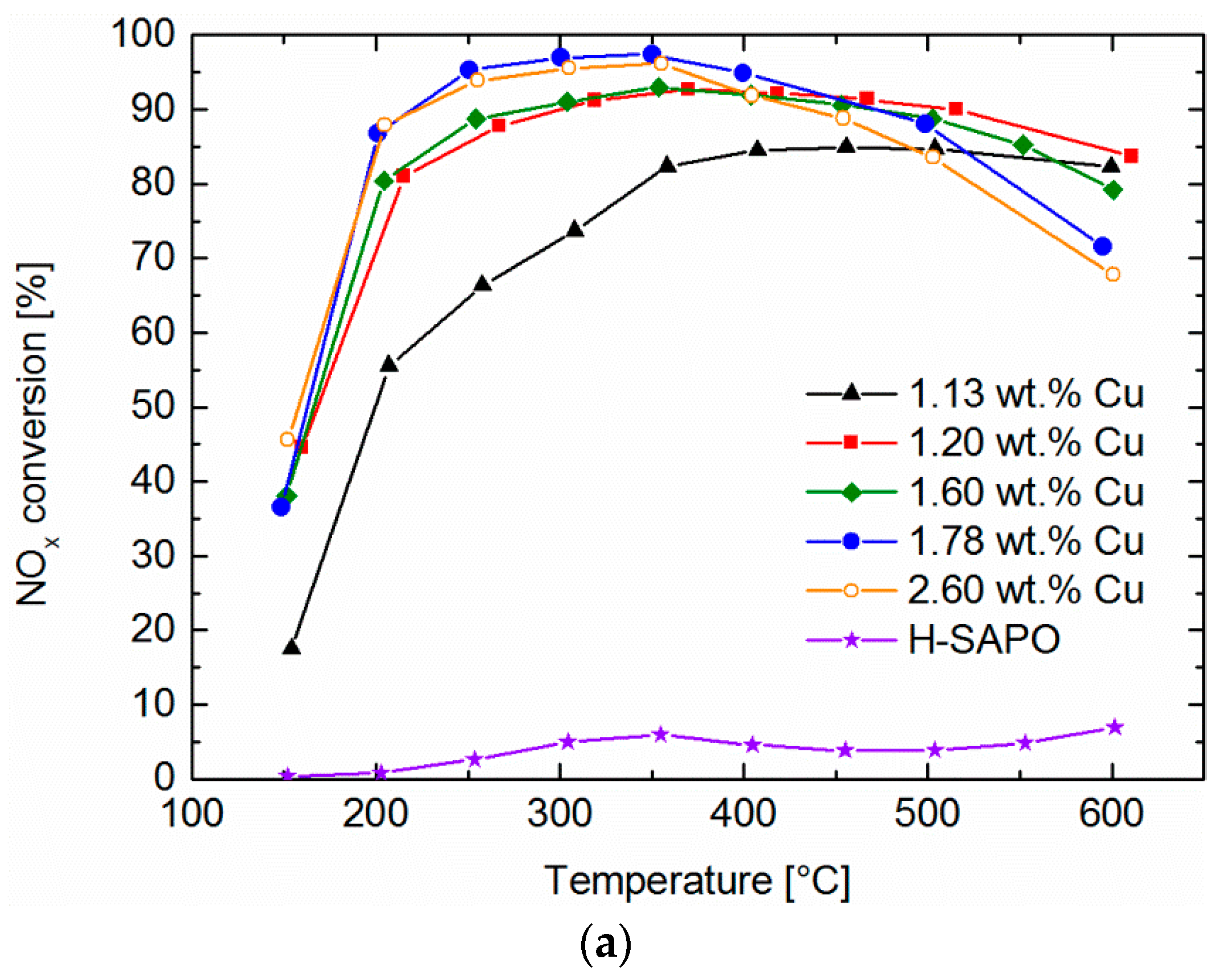
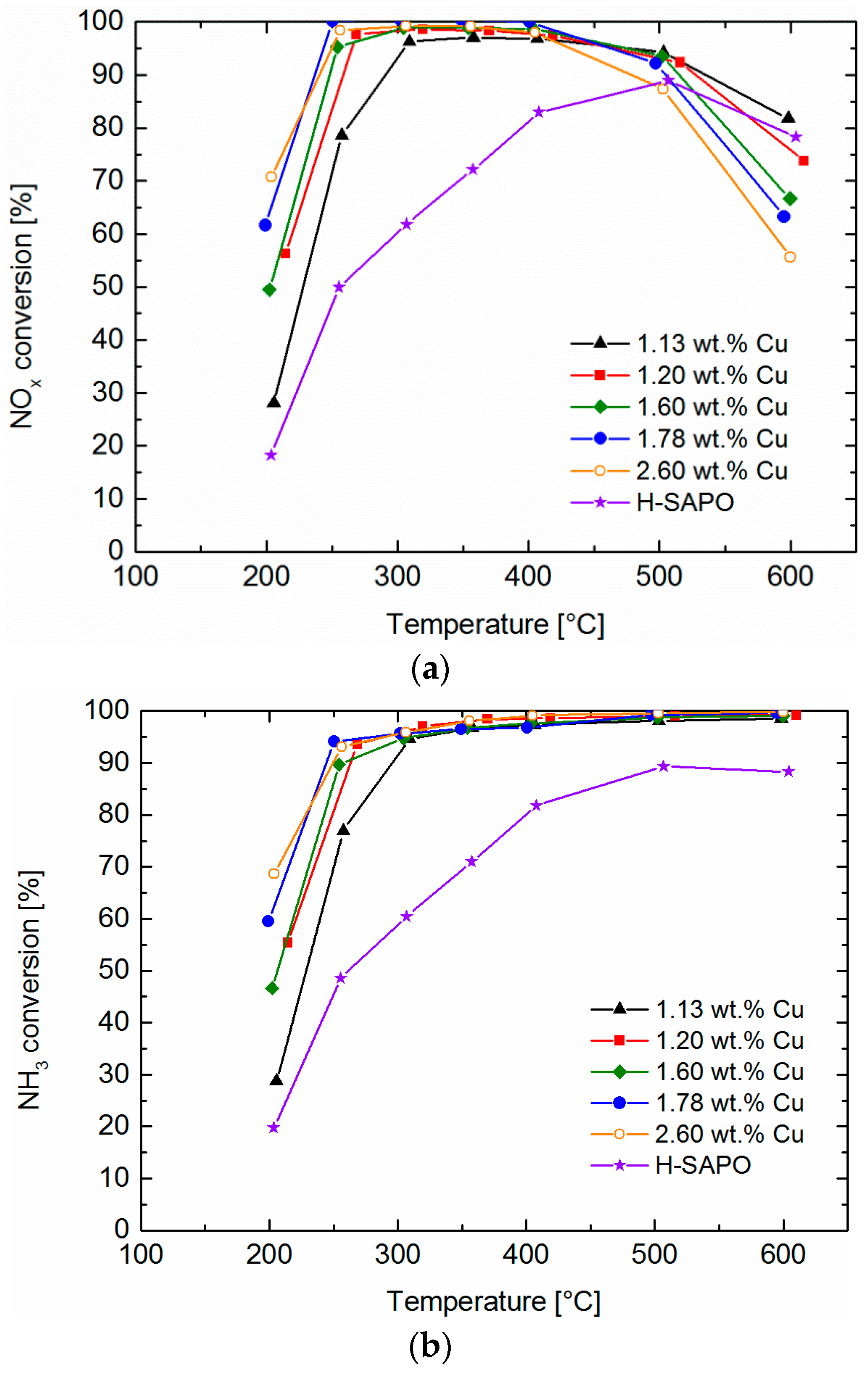
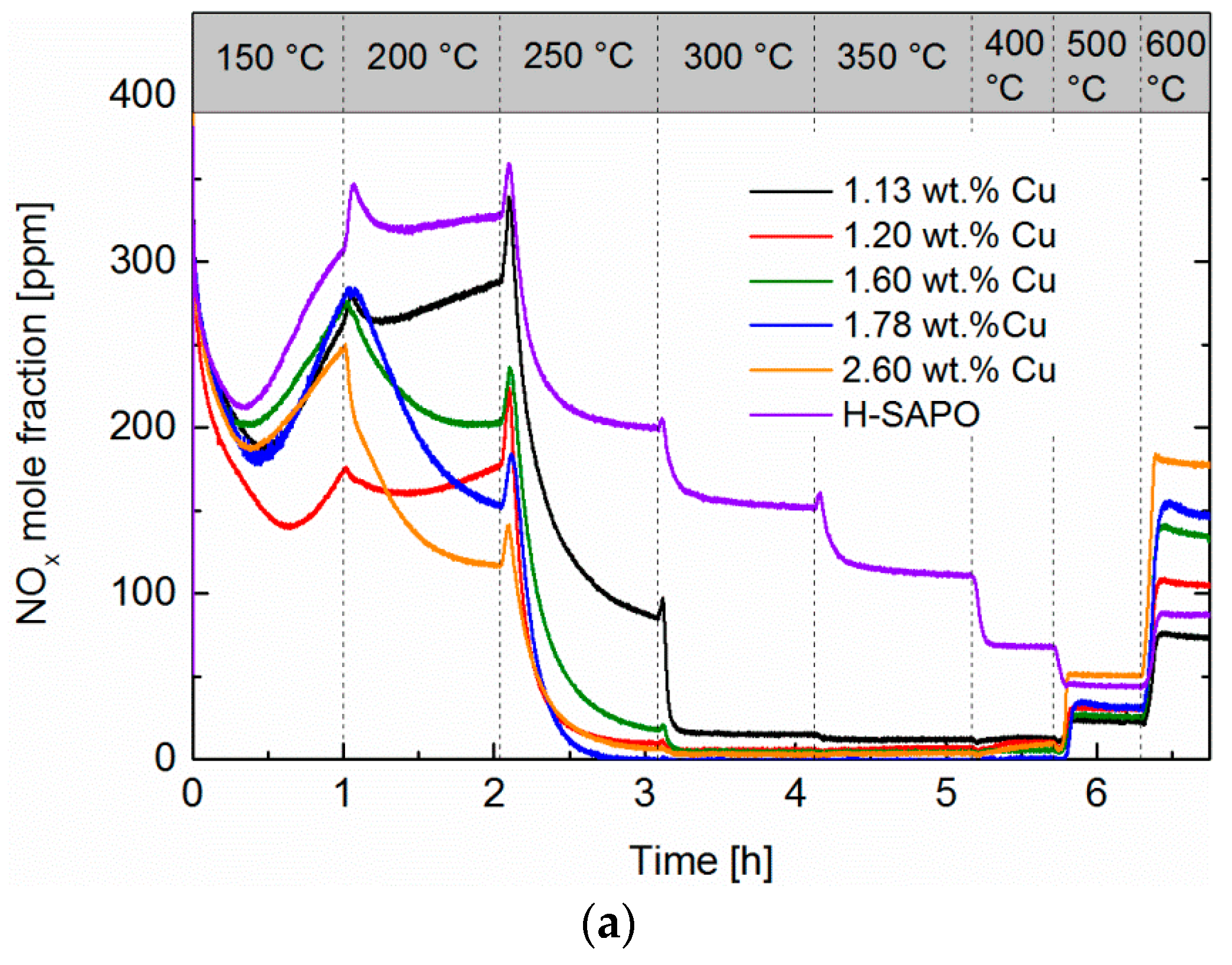
2.6. Standard SCR
2.7. Fast SCR
2.8. Impact of Calcination Temperature
3. Discussion
4. Materials and Methods
4.1. Catalyst Preparation
4.2. Activity and Selectivity Measurements
4.3. Catalyst Characterisation
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Forzatti, P. Present status and perspectives in de-NOx SCR catalysis. Appl. Catal. A Gen. 2001, 222, 221–236. [Google Scholar] [CrossRef]
- Deutschmann, O.; Knözinger, H.; Kochloefl, K.; Turek, T. Heterogeneous Catalysis and Solid Catalysts, 1. Fundamentals. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2000. [Google Scholar]
- Wang, J.; Zhao, H.; Haller, G.; Li, Y. Recent advances in the selective catalytic reduction of NOx with NH3 on Cu-Chabazite catalysts. Appl. Catal. B Environ. 2017, 202, 346–354. [Google Scholar] [CrossRef]
- Dosda, S.; Berthout, D.; Mauviot, G.; Nogre, A. Modeling of a DOC SCR-F SCR Exhaust Line for Design Optimization Taking into Account Performance Degradation due to Hydrothermal Aging. SAE Int. J. Fuels Lubr. 2016, 9, 621–632. [Google Scholar] [CrossRef]
- Pereira, M.V.L.; Nicolle, A.; Berthout, D. Hydrothermal aging effects on Cu-zeolite NH 3-SCR catalyst. Catal. Today 2015, 258, 424–431. [Google Scholar] [CrossRef]
- Fu, M.; Li, C.; Lu, P.; Qu, L.; Zhang, M.; Zhou, Y.; Yu, M.; Fang, Y. A review on selective catalytic reduction of NOx by supported catalysts at 100–300 C—Catalysts, mechanism, kinetics. Catal. Sci. Technol. 2014, 4, 14–25. [Google Scholar] [CrossRef]
- Toops, T.J.; Pihl, J.A.; Partridge, W.P. Fe-Zeolite Functionality, Durability, and Deactivation Mechanisms in the Selective Catalytic Reduction (SCR) of NOx with Ammonia. In Urea-SCR Technology for deNOx after Treatment of Diesel Exhausts; Nova, I., Tronconi, E., Eds.; Springer: New York, NY, USA, 2014; pp. 97–121. [Google Scholar]
- Niu, C.; Shi, X.; Liu, F.; Liu, K.; Xie, L.; You, Y.; He, H. High hydrothermal stability of Cu-SAPO-34 catalysts for the NH 3-SCR of NOx. Chem. Eng. J. 2016, 294, 254–263. [Google Scholar] [CrossRef]
- Liu, F.; Xie, L.; Shi, X.; He, H. Emerging Applications of Environmentally Friendly Zeolites in the Selective Catalytic Reduction of Nitrogen Oxides. In Zeolites in Sustainable Chemistry; Springer: Berlin/Heidelberg, Germany, 2016; pp. 393–434. [Google Scholar]
- Gao, F.; Walter, E.D.; Washton, N.M.; Szanyi, J.; Peden, C.H.F. Synthesis and Evaluation of Cu-SAPO-34 Catalysts for Ammonia Selective Catalytic Reduction. 1. Aqueous Solution Ion Exchange. ACS Catal. 2013, 3, 2083–2093. [Google Scholar] [CrossRef]
- Gao, F.; Walter, E.D.; Washton, N.M.; Szanyi, J.; Peden, C.H.F. Synthesis and evaluation of Cu/SAPO-34 catalysts for NH3-SCR 2: Solid-state ion exchange and one-pot synthesis. Appl. Catal. B Environ. 2015, 162, 501–514. [Google Scholar] [CrossRef]
- Ma, L.; Cheng, Y.; Cavataio, G.; McCabe, R.W.; Fu, L.; Li, J. In situ DRIFTS and temperature-programmed technology study on NH3-SCR of NOx over Cu-SSZ-13 and Cu-SAPO-34 catalysts. Appl. Catal. B Environ. 2014, 156–157, 428–437. [Google Scholar] [CrossRef]
- Kwak, J.H.; Tran, D.; Szanyi, J.; Peden, C.H.F.; Lee, J.H. The Effect of Copper Loading on the Selective Catalytic Reduction of Nitric Oxide by Ammonia Over Cu-SSZ-13. Catal. Lett. 2012, 142, 295–301. [Google Scholar] [CrossRef]
- Dědeček, J.; Sobalik, Z.; Tvaruazkova, Z.; Kaucky, D.; Wichterlová, B. Coordination of Cu Ions in High-Silica Zeolite Matrixes. Cu+ Photoluminescence, IR of NO Adsorbed on Cu2+, and Cu2+ ESR Study. J. Phys. Chem. 1995, 99, 16327–16337. [Google Scholar] [CrossRef]
- Gao, F.; Walter, E.D.; Karp, E.M.; Luo, J.; Tonkyn, R.G.; Kwak, J.H.; Szanyi, J.; Peden, C.H.F. Structure–activity relationships in NH3-SCR over Cu-SSZ-13 as probed by reaction kinetics and EPR studies. J. Catal. 2013, 300, 20–29. [Google Scholar] [CrossRef]
- Mihai, O.; Widyastuti, C.R.; Andonova, S.; Kamasamudram, K.; Li, J.; Joshi, S.Y.; Currier, N.W.; Yezerets, A.; Olsson, L. The effect of Cu-loading on different reactions involved in NH3-SCR over Cu-BEA catalysts. J. Catal. 2014, 311, 170–181. [Google Scholar] [CrossRef]
- Moden, B.; Donohue, J.M.; Cormier, W.E.; Li, H.X. Effect of Cu-loading and structure on the activity of Cu-exchanged zeolites for NH3-SCR. In Studies in Surface Science and Catalysis; Antoine Gédéon, P.M., Florence, B., Eds.; Elsevier: Amsterdam, The Netherlands, 2008; Volume 174, Part B; pp. 1219–1222. [Google Scholar]
- Shelef, M. Selective Catalytic Reduction of NOx with N-Free Reductants. Chem. Rev. 1995, 95, 209–225. [Google Scholar] [CrossRef]
- Kwak, J.H.; Zhu, H.; Lee, J.H.; Peden, C.H.F.; Szanyi, J. Two different cationic positions in Cu-SSZ-13? Chem. Commun. 2012, 48, 4758–4760. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zhang, L.; Li, J.; Kamasamudram, K.; Epling, W.S. NH3-SCR over Cu/SAPO-34—Zeolite acidity and Cu structure changes as a function of Cu loading. Catal. Today 2014, 231, 64–74. [Google Scholar] [CrossRef]
- Xue, J.; Wang, X.; Qi, G.; Wang, J.; Shen, M.; Li, W. Characterization of copper species over Cu/SAPO-34 in selective catalytic reduction of NOx with ammonia: Relationships between active Cu sites and de-NOx performance at low temperature. J. Catal. 2013, 297, 56–64. [Google Scholar] [CrossRef]
- Fan, S.; Xue, J.; Yu, T.; Fan, D.; Hao, T.; Shen, M.; Li, W. The effect of synthesis methods on Cu species and active sites over Cu/SAPO-34 for NH3-SCR reaction. Catal. Sci. Technol. 2013, 3, 2357–2364. [Google Scholar] [CrossRef]
- Wang, L.; Li, W.; Qi, G.; Weng, D. Location and nature of Cu species in Cu/SAPO-34 for selective catalytic reduction of NO with NH3. J. Catal. 2012, 289, 21–29. [Google Scholar] [CrossRef]
- Thommes, M. Textural Characterization of Zeolites and Ordered Mesoporous Materials. In Introduction to Zeolite Molecular Sieves, 3rd ed.; Čejka, J., van Bekkum, H., Corma, A., Schüth, F., Eds.; Elsevier: Amsterdam, The Netherlands, 2007; Volume 168. [Google Scholar]
- Fickel, D.W.; D’Addio, E.; Lauterbach, J.A.; Lobo, R.F. The ammonia selective catalytic reduction activity of copper-exchanged small-pore zeolites. Appl. Catal. B Environ. 2011, 102, 441–448. [Google Scholar] [CrossRef]
- Kwak, J.H.; Tonkyn, R.G.; Kim, D.H.; Szanyi, J.; Peden, C.H.F. Excellent activity and selectivity of Cu-SSZ-13 in the selective catalytic reduction of NOx with NH3. J. Catal. 2010, 275, 187–190. [Google Scholar] [CrossRef]
- Park, J.-H.; Park, H.J.; Baik, J.H.; Nam, I.-S.; Shin, C.-H.; Lee, J.-H.; Cho, B.K.; Oh, S.H. Hydrothermal stability of CuZSM5 catalyst in reducing NO by NH3 for the urea selective catalytic reduction process. J. Catal. 2006, 240, 47–57. [Google Scholar] [CrossRef]
- Ma, L.; Cheng, Y.; Cavataio, G.; McCabe, R.W.; Fu, L.; Li, J. Characterization of commercial Cu-SSZ-13 and Cu-SAPO-34 catalysts with hydrothermal treatment for NH3-SCR of NOx in diesel exhaust. Chem. Eng. J. 2013, 225, 323–330. [Google Scholar] [CrossRef]
- Bates, S.A.; Verma, A.A.; Paolucci, C.; Parekh, A.A.; Anggara, T.; Yezerets, A.; Schneider, W.F.; Miller, J.T.; Delgass, W.N.; Ribeiro, F.H. Identification of the active Cu site in standard selective catalytic reduction with ammonia on Cu-SSZ-13. J. Catal. 2014, 312, 87–97. [Google Scholar] [CrossRef]
- Dědeček, J.; Wichterlová, B. Role of Hydrated Cu Ion Complexes and Aluminum Distribution in the Framework on the Cu Ion Siting in ZSM-5. J. Phys. Chem. B 1997, 101, 10233–10240. [Google Scholar] [CrossRef]
- Wilken, N.; Nedyalkova, R.; Kamasamudram, K.; Li, J.; Currier, N.; Vedaiyan, R.; Yezerets, A.; Olsson, L. Investigation of the Effect of Accelerated Hydrothermal Aging on the Cu Sites in a Cu-BEA Catalyst for NH3-SCR Applications. Top. Catal. 2013, 56, 317–322. [Google Scholar] [CrossRef]
- Kortüm, G. Reflectance Spectroscopy: Principles, Methods, Applications; Springer: New York, NY, USA, 1969. [Google Scholar]
- Wang, J.; Yu, T.; Wang, X.; Qi, G.; Xue, J.; Shen, M.; Li, W. The influence of silicon on the catalytic properties of Cu/SAPO-34 for NOx reduction by ammonia-SCR. Appl. Catal. B Environ. 2012, 127, 137–147. [Google Scholar] [CrossRef]
- Yu, T.; Fan, D.; Hao, T.; Wang, J.; Shen, M.; Li, W. The effect of various templates on the NH3-SCR activities over Cu/SAPO-34 catalysts. Chem. Eng. J. 2014, 243, 159–168. [Google Scholar] [CrossRef]
- Briend, M.; Vomscheid, R.; Peltre, M.J.; Man, P.P.; Barthomeuf, D. Influence of the Choice of the Template on the Short- and Long-Term Stability of SAPO-34 Zeolite. J. Phys. Chem. 1995, 99, 8270–8276. [Google Scholar] [CrossRef]
- Prakash, A.; Unnikrishnan, S. Synthesis of SAPO-34: High silicon incorporation in the presence of morpholine as template. J. Chem. Soc. Faraday Trans. 1994, 90, 2291–2296. [Google Scholar] [CrossRef]
- Weitkamp, J. Zeolites and catalysis. Solid State Ion. 2000, 131, 175–188. [Google Scholar] [CrossRef]
- Leistner, K.; Mihai, O.; Wijayanti, K.; Kumar, A.; Kamasamudram, K.; Currier, N.W.; Yezerets, A.; Olsson, L. Comparison of Cu/BEA, Cu/SSZ-13 and Cu/SAPO-34 for ammonia-SCR reactions. Catal. Today 2015, 258, 49–55. [Google Scholar] [CrossRef]
- Yu, T.; Wang, J.; Shen, M.; Li, W. NH3-SCR over Cu/SAPO-34 catalysts with various acid contents and low Cu loading. Catal. Sci. Technol. 2013, 3, 3234–3241. [Google Scholar] [CrossRef]
- Devadas, M.; Kröcher, O.; Elsener, M.; Wokaun, A.; Söger, N.; Pfeifer, M.; Demel, Y.; Mussmann, L. Influence of NO2 on the selective catalytic reduction of NO with ammonia over Fe-ZSM5. Appl. Catal. B Environ. 2006, 67, 187–196. [Google Scholar] [CrossRef]
- Wilken, N.; Wijayanti, K.; Kamasamudram, K.; Currier, N.W.; Vedaiyan, R.; Yezerets, A.; Olsson, L. Mechanistic investigation of hydrothermal aging of Cu-Beta for ammonia SCR. Appl. Catal. B Environ. 2012, 111–112, 58–66. [Google Scholar] [CrossRef]
- Olsson, L.; Wijayanti, K.; Leistner, K.; Kumar, A.; Joshi, S.; Kamasamudram, K.; Currier, N.W.; Yezerets, A. A multi-site kinetic model for NH3-SCR over Cu/SSZ-13. Appl. Catal. B Environ. 2015, 174–175, 212. [Google Scholar] [CrossRef]
- Grossale, A.; Nova, I.; Tronconi, E.; Chatterjee, D.; Weibel, M. The chemistry of the NO/NO2–NH3 “fast” SCR reaction over Fe-ZSM5 investigated by transient reaction analysis. J. Catal. 2008, 256, 312–322. [Google Scholar] [CrossRef]
- Forzatti, P.; Lietti, L.; Nova, I.; Tronconi, E. Diesel NOx aftertreatment catalytic technologies: Analogies in LNT and SCR catalytic chemistry. Catal. Today 2010, 151, 202–211. [Google Scholar] [CrossRef]
- Gao, F.; Wang, Y.; Kollár, M.; Washton, N.M.; Szanyi, J.; Peden, C.H. A comparative kinetics study between Cu/SSZ-13 and Fe/SSZ-13 SCR catalysts. Catal. Today 2015, 258, 347–358. [Google Scholar] [CrossRef]
- Mihai, O.; Widyastuti, C.; Kumar, A.; Li, J.; Joshi, S.; Kamasamudram, K.; Currier, N.; Yezerets, A.; Olsson, L. The Effect of NO2/NOx Feed Ratio on the NH3-SCR System Over Cu–Zeolites with Varying Copper Loading. Catal. Lett. 2014, 144, 70–80. [Google Scholar] [CrossRef]
- Eng, J.; Bartholomew, C.H. Kinetic and Mechanistic Study of NOx Reduction by NH3 over H-Form Zeolites. J. Catal. 1997, 171, 27–44. [Google Scholar] [CrossRef]
- Schwidder, M.; Heikens, S.; De Toni, A.; Geisler, S.; Berndt, M.; Brückner, A.; Grünert, W. The role of NO2 in the selective catalytic reduction of nitrogen oxides over Fe-ZSM-5 catalysts: Active sites for the conversion of NO and of NO/NO2 mixtures. J. Catal. 2008, 259, 96–103. [Google Scholar] [CrossRef]
- Wang, J.; Huang, Y.; Yu, T.; Zhu, S.; Shen, M.; Li, W.; Wang, J. The migration of Cu species over Cu-SAPO-34 and its effect on NH3 oxidation at high temperature. Catal. Sci. Technol. 2014, 4, 3004–3012. [Google Scholar] [CrossRef]
- Wang, L.; Gaudet, J.R.; Li, W.; Weng, D. Migration of Cu species in Cu/SAPO-34 during hydrothermal aging. J. Catal. 2013, 306, 68–77. [Google Scholar] [CrossRef]
- Vennestrøm, P.N.R.; Katerinopoulou, A.; Tiruvalam, R.R.; Kustov, A.; Moses, P.G.; Concepcion, P.; Corma, A. Migration of Cu Ions in SAPO-34 and Its Impact on Selective Catalytic Reduction of NOx with NH3. ACS Catal. 2013, 3, 2158–2161. [Google Scholar] [CrossRef]
- Leistner, K.; Olsson, L. Deactivation of Cu/SAPO-34 during low-temperature NH3-SCR. Appl. Catal. B Environ. 2015, 165, 192–199. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Batch | Type | Molarity Cu(NO3)2 Solution (M) | Cu wt % | Cu/Si (mol/mol) | Calcination Temperature (°C) | BET Surface Area (m2/g) | Micropore Volume (cm3/g) |
|---|---|---|---|---|---|---|---|
| 1 | SAPO-34 | 0 | 530.0 | 0.243 | |||
| Cu/SAPO-34 | 0.6 | 1.78 | 0.15 | 750 | 551.1 | 0.257 | |
| Cu/SAPO-34 | 0.4 | 1.49 | 0.12 | 550 | 622.3 | 0.290 | |
| 2 | SAPO-34 | 0 | 570.6 | 0.265 | |||
| H/SAPO-34 | 0 | 750 | 513.9 | 0.237 | |||
| Cu/SAPO-34 | 0.4 | 1.60 | 0.15 | 750 | 588.2 | 0.277 | |
| 3 | SAPO-34 | 0 | 587.8 | 0.294 | |||
| Cu/SAPO-34 | 0.8 | 2.60 | 0.20 | 750 | 544.2 | 0.255 | |
| 4 | SAPO-34 | 0 | 574.5 | 0.284 | |||
| Cu/SAPO-34 | 0.05 | 1.13 | 0.11 | 750 | 568.9 | 0.282 | |
| Cu/SAPO-34 | 0.2 | 1.20 | 0.11 | 750 | 536.6 | 0.253 | |
| 5 | SAPO-34 | 0 | 584.2 | 0.289 | |||
| Cu/SAPO-34 | 0.2 | 1.27 | 0.12 | 550 | 581.9 | 0.274 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leistner, K.; Brüsewitz, F.; Wijayanti, K.; Kumar, A.; Kamasamudram, K.; Olsson, L. Impact of Copper Loading on NH3-Selective Catalytic Reduction, Oxidation Reactions and N2O Formation over Cu/SAPO-34. Energies 2017, 10, 489. https://doi.org/10.3390/en10040489
Leistner K, Brüsewitz F, Wijayanti K, Kumar A, Kamasamudram K, Olsson L. Impact of Copper Loading on NH3-Selective Catalytic Reduction, Oxidation Reactions and N2O Formation over Cu/SAPO-34. Energies. 2017; 10(4):489. https://doi.org/10.3390/en10040489
Chicago/Turabian StyleLeistner, Kirsten, Florian Brüsewitz, Kurnia Wijayanti, Ashok Kumar, Krishna Kamasamudram, and Louise Olsson. 2017. "Impact of Copper Loading on NH3-Selective Catalytic Reduction, Oxidation Reactions and N2O Formation over Cu/SAPO-34" Energies 10, no. 4: 489. https://doi.org/10.3390/en10040489
APA StyleLeistner, K., Brüsewitz, F., Wijayanti, K., Kumar, A., Kamasamudram, K., & Olsson, L. (2017). Impact of Copper Loading on NH3-Selective Catalytic Reduction, Oxidation Reactions and N2O Formation over Cu/SAPO-34. Energies, 10(4), 489. https://doi.org/10.3390/en10040489