1. Introduction
Despite the growing contribution of renewable energy sources, such as solar/wind energy or biomass, and respective technologies and devices for energy generation and storage, it is believed that for many decades, fossil resources (primarily oil and natural gas) will still form the basis of global energy consumption. An increase in the efficiency of using hydrocarbon raw materials while improving operational characteristics (an important factor for the final consumer) is associated with the use of fuel cells (FCs) and hydrogen storage technologies for the needs of distributed energy [
1,
2]. Therefore, fuel cells, which allow for converting the energy of chemical bonds into electrical energy with high efficiency even in compact and mobile systems, are one of the most dynamically developing energy sectors.
At the moment, the infrastructure for supplying hydrogen and logistics are not well developed to meet mass consumption needs: there is a lack of hydrogen gas stations, and long-term storage of large quantities of hydrogen remains problematic. In this regard, most of the world research centers involved in the development of fuel cell based power plants show interest in the use of widely available traditional logistic fuels (gasoline, propane-butane, kerosene and diesel fuel). Besides, in comparison with other hydrogen storage technologies available today, hydrocarbon fuels contain the largest amount of hydrogen per unit volume [
3]. Therefore, the use of liquid or easily liquefied hydrocarbon fuels with high energy density is relevant for fuel cells. However, such fuels cannot be directly oxidized in the anode space of the fuel cell, since they initiate the carbonization of the electrode and the failure of the fuel cell. In this case, the fuels are first converted to a hydrogen-containing gas and then oxidized in the fuel cell. The processes of steam conversion or steam reforming (SC), autothermal reforming (ATR) and partial oxidation (PO) are used to obtain synthesis gas from hydrocarbons. The most effective process is SC, but it requires high operating temperatures, consumes a significant amount of heat to evaporate water and preheats steam and needs a water purification system. Therefore, SC can be implemented only for stationary power plants based on high-power fuel cells. PO of liquid hydrocarbon fuels has not yet been implemented due to thermodynamic limitations, an undesirable side process of carbon formation and high operating temperatures.
In order to solve the problem of partial oxidation of C2–C10 hydrocarbons, reach high performance and minimize the amount of water to be vaporized, it is proposed to combine the pre-reforming of hydrocarbons and the partial oxidation of the resulting methane-containing mixture into synthesis gas. The first stage is carried out at low molar ratios of H2O/C = 0.7–1.5 and temperatures of 250–350 °C. At these conditions, the main reaction products are methane, hydrogen and carbon dioxide. The second stage (PO) provides a high performance for the overall process of synthesis gas production.
Although catalytic steam conversion of hydrocarbons has been studied quite well, in the vast majority of works, the process was studied at temperatures of 450–700 °C and a molar ratio of H
2O/C > 2 to obtain synthesis gas directly from hydrocarbons. SC of C
2+-hydrocarbons at H
2O/C < 1 and temperatures below 350 °C are poorly studied. However, there is a limited number of works [
4,
5,
6,
7] showing the feasibility of such an approach to obtain methane-hydrogen mixtures.
Nickel-based catalysts are usually used in hydrocarbons steam reforming because of their high activity and low cost compared to platinum metals. Currently, nickel catalysts for methanation of carbon oxides, the pre-reforming of C
2+-hydrocarbons and the steam conversion of natural gas to synthesis gas are available commercially. Natural gas steam reforming catalysts possess high mechanical strength, a Ni content of about 10–15 wt.% and can be used at temperatures of 700–900 °C. As a result, the catalysts for natural gas steam conversion have a low nickel surface area and low catalytic activity at low temperatures. Conventional pre-reforming catalysts contain 15–30 wt.% of Ni and can be used at temperatures of 450–600 °C. These catalysts were developed with particular attention to carbonization resistance [
8] rather than catalytic activity. CO/CO
2 methanation catalysts have a high nickel content and dispersion but lack thermal stability and are usually operated at temperatures of 300–320 °C. Since the pre-reforming of hydrocarbons at such low steam to carbon molar ratios is carried out at about 250–350 °C [
5], the use of methanation catalysts seems to be the optimal choice.
There are a lack of data on the reaction kinetics for pre-reforming at such low temperatures and the steam to carbon molar ratio. Ethane steam conversion was studied in methane excess at molar ratios of CH
4/C
2H
6 ≈ 20 and H
2O/C
C2+ = 0.6−6 over a Ni-Cr catalyst at 300–360 °C, 1 atm pressure [
9]. It was shown that ethane conversion rate could be described by a Langmuir–Hinshelwood type equation:
where k and A are constants dependable of temperature; P
C2H6, P
H2O and P
H2 are partial pressures of the components. The equation corresponds to a scheme with ethane adsorption being the rate-determining step.
A two-stage macrokinetic scheme was suggested for the low-temperature steam conversion of methane-propane mixtures over Ni-based catalysts [
10]. Conversion of the methane-pentane mixtures under 1 bar pressure was simulated using the scheme, which included irreversible reactions of the steam conversion of C
2+-hydrocarbons, Equation (2), with the formation of CO
2 and H
2, followed by reversible CO
2 methanation, Equation (3):
Kinetic parameters were determined: CO2 methanation was shown to occur in a quasi-equilibrium mode at T > 250 °C. The apparent reaction order with respect to C3H8 was close to one. The effect of the CH4, CO2, H2 and H2O concentrations on the C3H8 conversion was insignificant. It should be noted that the kinetics are valid for C2+-hydrocarbons partial pressure much lower that 1 bar. Thus, kinetics for a wider pressure range are required.
The kinetic study of heterogenous catalytic reactions is generally a challenging task as mechanisms are typically multistep and the majority of experimental data are obtained in a plug-flow reactor in which the reaction conditions and reaction rates are different at all points of the reactor. The kinetic parameters are determined by solving an inverse problem of chemical kinetics.
The pre-reforming of the propane reaction is a complex chemical reaction, in other words, a multi-stage one, and for each stage of the reaction, it is necessary to find the kinetic parameters (activation energies and preexponential factors). Moreover, as will be shown later, we will look for additional reaction parameters when solving the inverse problem. Thus, an ill-posed optimization problem with high dimensionality is formulated. For these reasons, an efficient multidimensional optimization technique such as genetic algorithms (GAs) are, therefore, necessary. Genetic algorithms were designed for the accelerated search of a global minimum in various optimization problems [
11,
12,
13,
14]. It was successfully applied for the kinetics elucidation of [
15,
16,
17,
18,
19].
In this work, the experiments on propane pre-reforming were carried out over an industrial nickel methanation catalyst NIAP-07-05, and a mathematical simulation was applied to describe the process. The effect of temperature and pressure was studied. The experiments data were fitted by the power law (PL) and Langmuir–Hinshelwood (LH) kinetics.
3. Results and Discussion
Propane pre-reforming experiments were carried out at 220–380 °C. The main reaction products were methane, hydrogen and carbon dioxide, as well as residual propane.
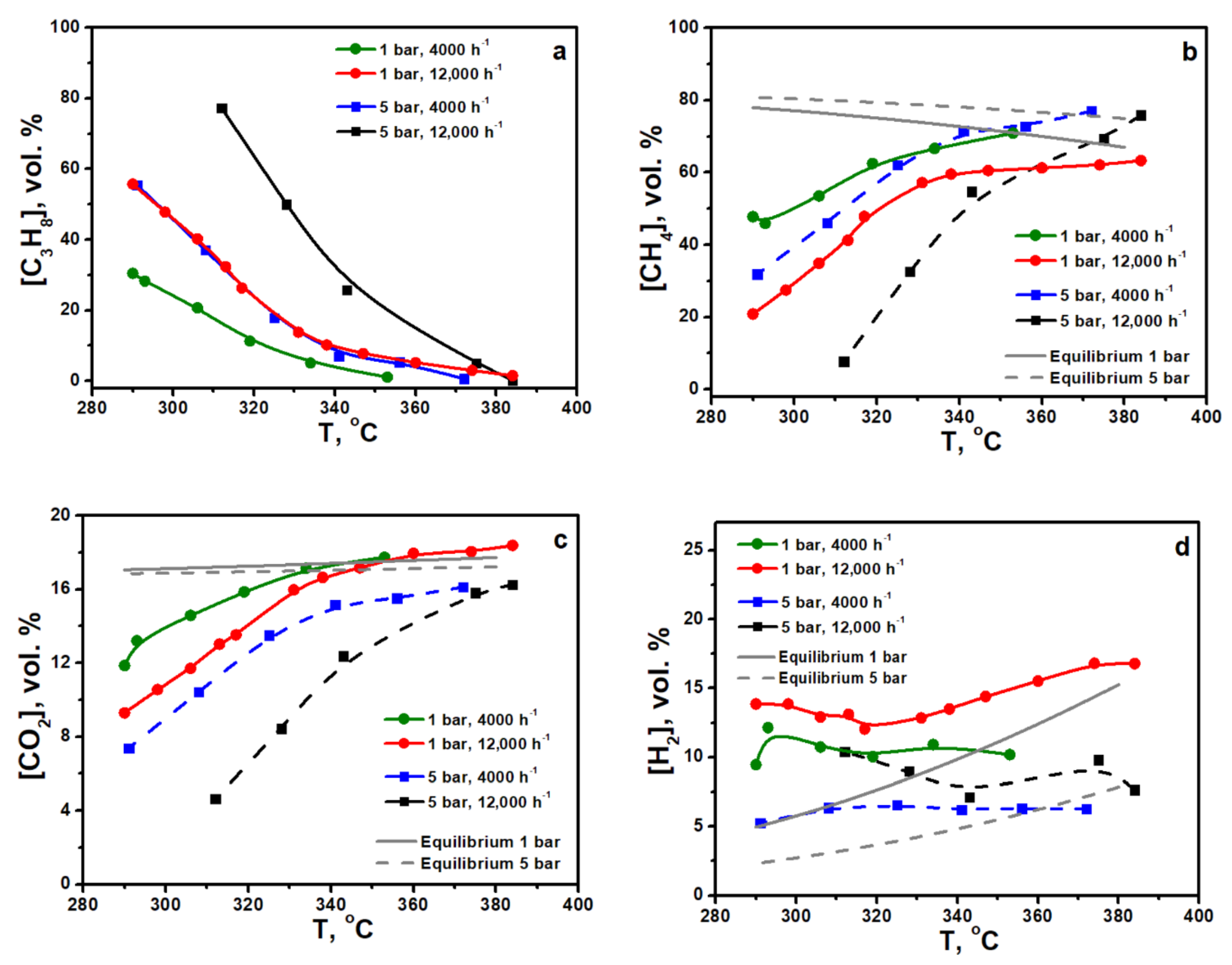
Figure 1a–d shows the temperature dependences of the outlet concentrations of propane, methane, carbon dioxide and hydrogen during propane pre-reforming under pressures 1 bar and 5 bar. The flow rates (GHSV) for both pressures were 4000 and 12,000 h
−1. It is seen that the conversion of propane proceeds more efficiently under 1 bar pressure. Complete propane conversion can be reached at temperatures above 350 °C.
It can be seen that at 1 bar pressure and a flow rate of 12,000 h−1, there is a maximum of hydrogen concentration at 290 °C, followed by a decrease to a minimum at 320 °C, and then a further increase. This can be explained that at low temperatures, hydrogen and CO2 were accumulating during the steam conversion reaction. With increasing temperature, hydrogen is consumed by the hydrogenolysis of propane. At high temperatures, the hydrogen concentration is determined by the equilibrium distribution of the products and increases according to the equilibrium of the CO2 methanation reaction.
The inverse problem was then solved for the experiments on propane pre-reforming. The reaction scheme consisted of two reactions: steam conversion of propane and methanation of CO
2.
Earlier, it was shown that this approach can be quite effective in the case of the low-temperature steam conversion of small concentrations of propane in methane excess [
10]. In this work, we use the following form of the kinetic equations:
where W
ref and W
met are the reaction rates (mol∙m
−3∙sec
−1); E
ref and E
met are the observed activation energies (J/mol); k
ref and k
met are pre-exponential factors ((mol∙m
3)
n−1∙sec
−1 and sec
−1). The indices ref and met refer to the pre-reforming and methanation reactions, respectively. C
C3H8 and C
H2 are concentrations of propane and hydrogen (mol/m
3); n is effective reaction order with respect to propane, which ranged from −2 to 2; K
eq is the equilibrium constant for CO
2 methanation reaction and P
CH4, P
H2O, P
CO2, P
H2 are partial pressures of the corresponding substances (bar).
As a result of solving the inverse task, the values of
,
,
,
and n were optimized. The obtained values are listed in
Table 1.
These parameters were substituted into Equations (7) and (8). Next, within the framework of the proposed reaction scheme and the model of a quasi-homogeneous isothermal plug flow reactor, the system of the material balance equations was solved to calculate the temperature dependences of the reaction product concentrations. The system of differential equations is written in the following form:
Here, G is the mass flow of the gas mixture (kg∙m−2∙sec−1); yi is the mass fraction of the i-th component, ; z is the coordinate along the catalyst bed (0 ≤ z ≤ 0.030 m); νi are stoichiometric coefficients for steam reforming reactions; Wref is the steam conversion rate of propane (mol∙m−3∙s−1) and ρi is the density of the i-th component (kg∙m−3). The boundary conditions were expressed as: yi = yi0 at z = 0.
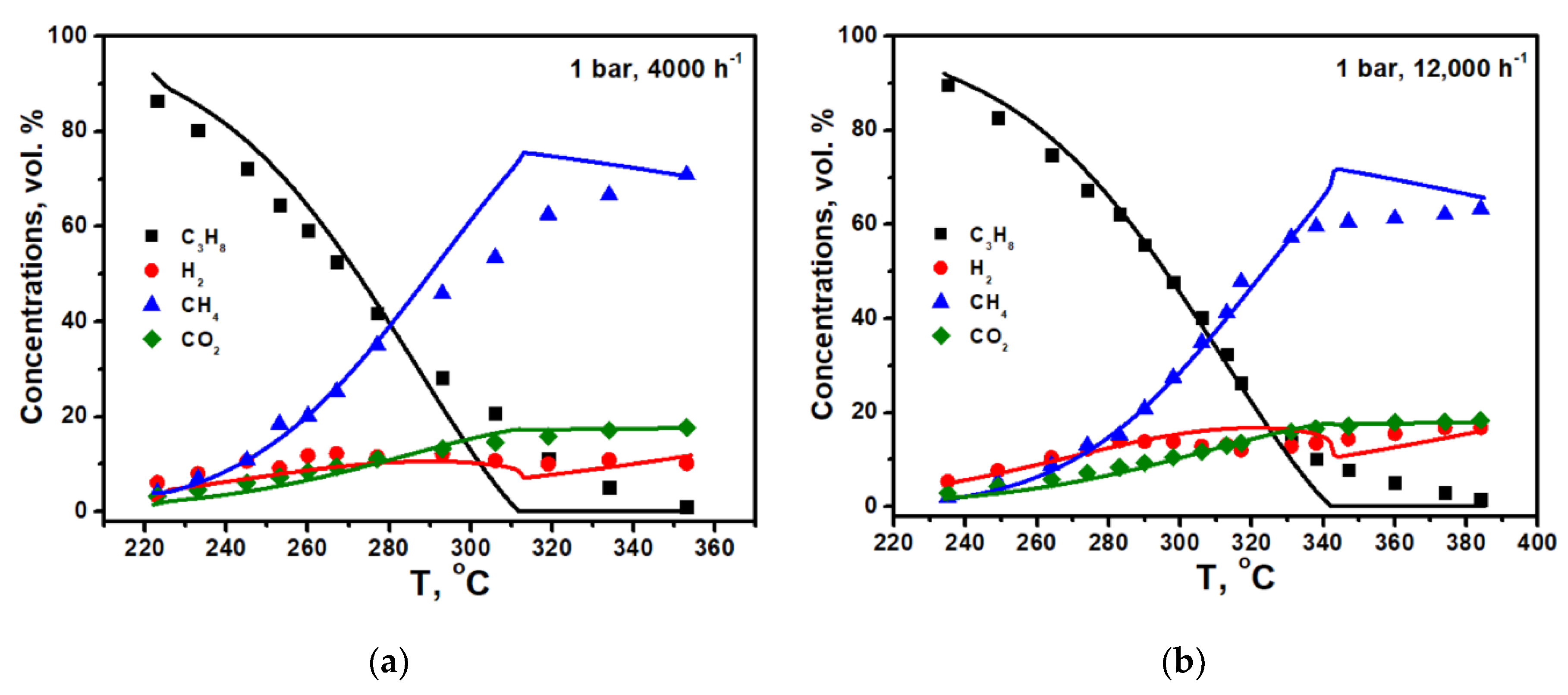
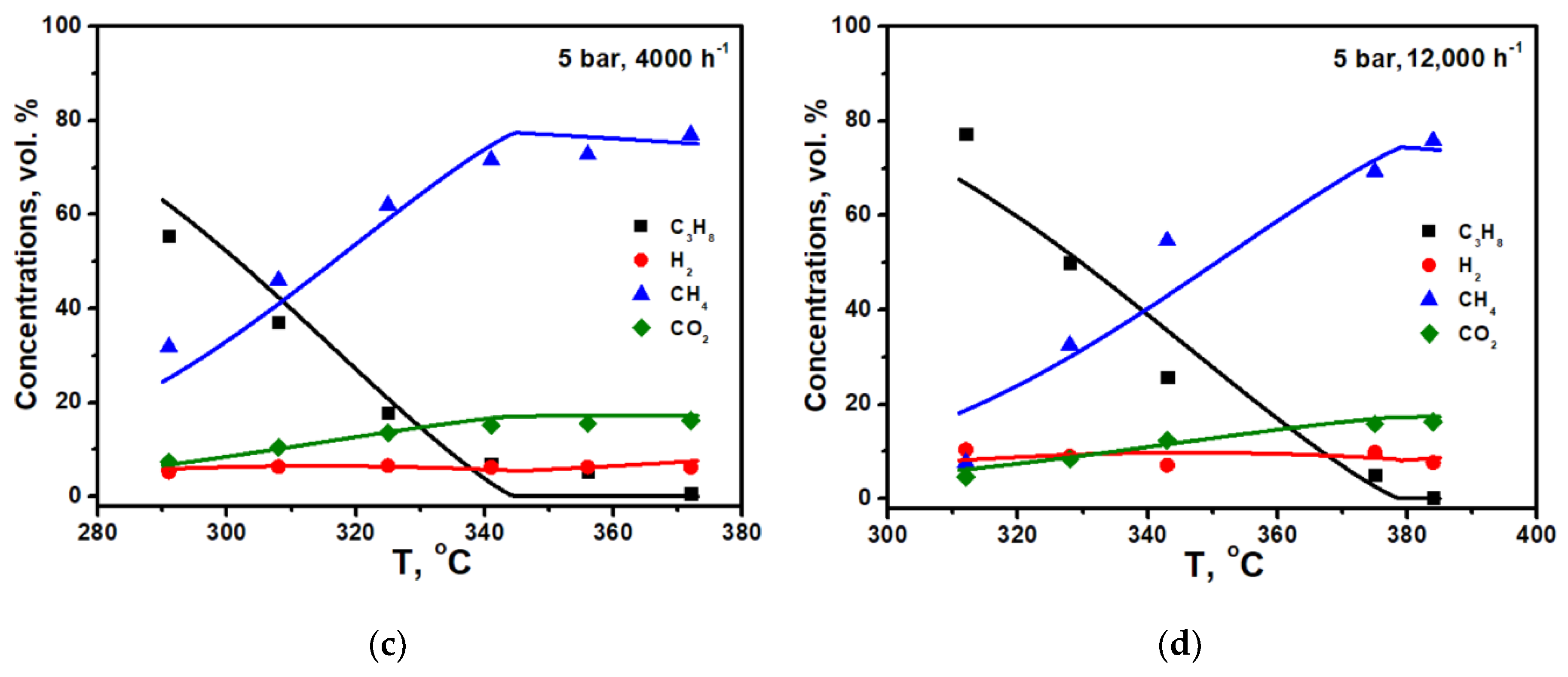
The calculation results for the experiments under 1 and 5 bar and flow rates of 4000 and 12,000 h
−1 are shown in
Figure 2a–d. It is seen that power low type kinetics correctly describes the experimental data at low temperatures for both pressures, while for high pressure, it predicts the reaching the equilibrium too fast. Note that the effective reaction order with respect to propane (n = 0.15) in the case of the pre-reforming of pure propane is less than or close to zero, in contrast to the conversion of diluted propane-methane mixtures [
10]. The reason for this is the high coverage of the catalyst surface with propane molecules (θ
C3H8), which, according to the Langmuir–Hinshelwood model, reduces the effective reaction order.
Aside from the power law dependence, attempts were made to find the solution of the inverse task in the form of the Langmuir–Hinshelwood (LH) model:
The value of B constant varied from 0 to 5, and the value of m varied from 0 to 2. When solving the inverse problem, the values of
,
,
and
also varied (the upper and lower restrictions were the same as for the power law model). The results are shown in
Table 2.
These values were used to solve the direct task.
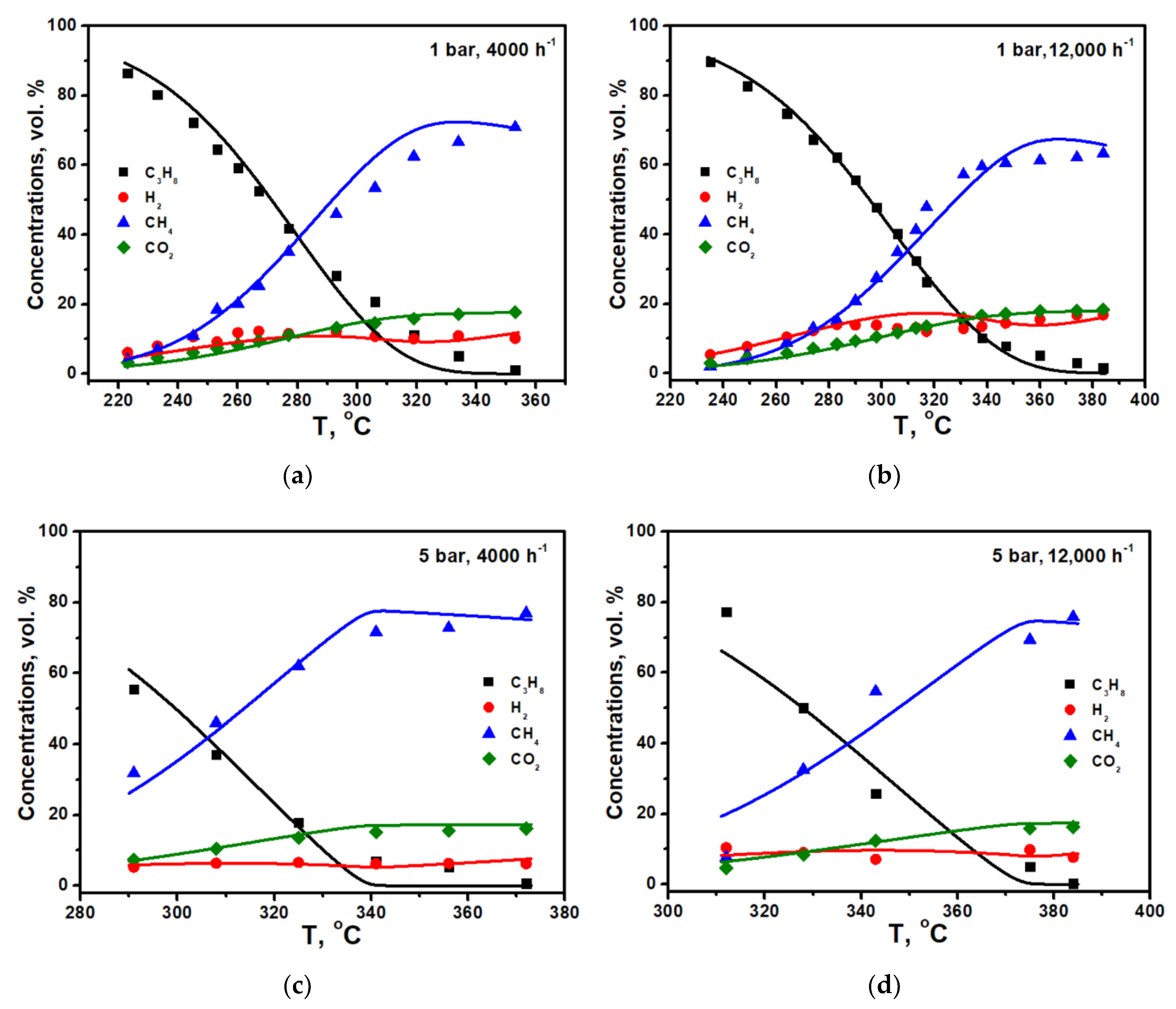
Figure 3a–d shows the results of the calculations for experiments on propane pre-reforming under 1 bar and 5 bar.
It is seen that the model based on the Langmuir–Hinshelwood mechanism correctly describes the experimental data. It is seen that apparent activation energies for both fits are quite close. The main difference is in the nature of the dependence of the rate of conversion of propane on its concentration.
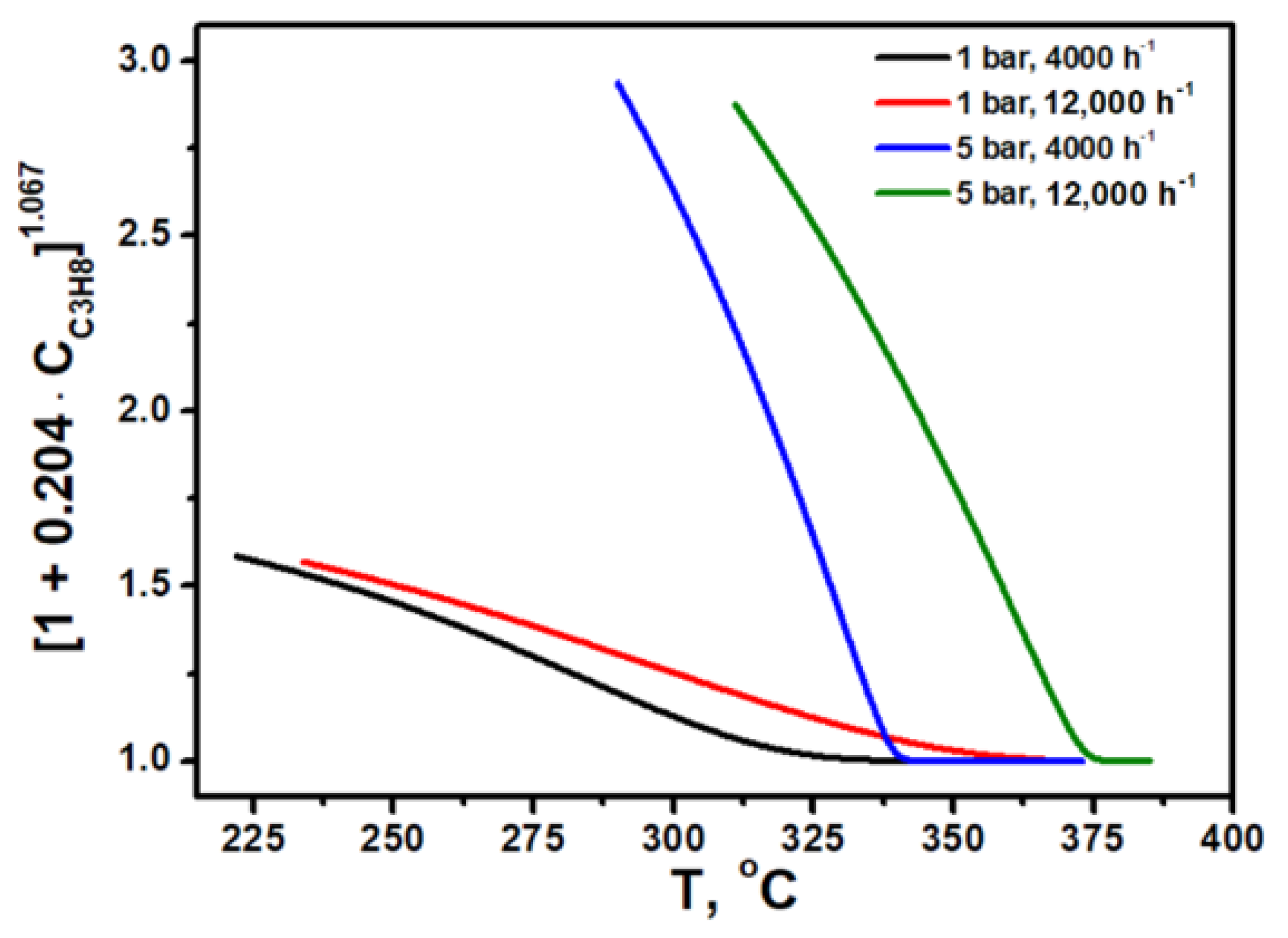
Figure 4 shows the temperature dependence of the denominator of Equation (10), which shows how the value of [1 + B∙C
C3H8]
m varies with temperature and pressure (with parameters B and m selected). It is seen that the denominator varies within the range of 1–3 under the conditions studied, and decreases with pressure decrease and temperature increase. The results obtained explain the inconsistency of the power law kinetics: at a low temperature, the apparent reaction order with respect to propane is close to zero, providing good agreement of experiment and fitting; however, with temperature increase, the apparent propane reaction order becomes closer to one, but power law kinetics cannot reflect this, therefore making it inappropriate.


 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}