Experimental and Computational Evaluation of Heavy Metal Cation Adsorption for Molecular Design of Hydrothermal Char

 and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Hydrochar Synthesis
2.3. Hydrochar Characterization
2.4. Hydrochar Adsorption Tests
2.5. Computational Modeling
3. Results and Discussion
3.1. Glucose Hydrochar Sorption Capacity and Characterization
3.2. DFT Simulations of the Metal-Carboxylate Interactions
3.3. Custom-Synthesis of Hydrochar for Heavy Metal Adsorption
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Christian-Smith, J.; Gleick, P.H.; Cooley, H. Twenty-first century US water policy. In 21st Century US Water Policy; Oxford University Press: Oxford, UK, 2012. [Google Scholar]
- Bellinger, D.C. Lead contamination in Flint—An abject failure to protect public health. N. Engl. J. Med. 2016, 374, 1101–1103. [Google Scholar] [CrossRef]
- Retief, F.P.; Cilliers, L. Lead poisoning in ancient Rome. Acta Theol. 2006, 26, 147–164. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Luqueno, F.; López-Valdez, F.; Gamero-Melo, P.; Luna-Suárez, S.; Aguilera-González, E.N.; Martínez, A.I.; García-Guillermo, M.; Hernández-Martínez, G.; Herrera-Mendoza, R.; Álvarez-Garza, M.A. Heavy metal pollution in drinking water-a global risk for human health: A review. Afr. J. Environ. Sci. Technol. 2013, 7, 567–584. [Google Scholar]
- Fu, F.; Wang, Q. Removal of heavy metal ions from wastewaters: A review. J. Environ. Manag. 2011, 92, 407–418. [Google Scholar] [CrossRef] [PubMed]
- Wen, T.; Zhao, Y.; Zhang, T.; Xiong, B.; Hu, H.; Zhang, Q.; Song, S. Effect of anions species on copper removal from wastewater by using mechanically activated calcium carbonate. Chemosphere 2019, 230, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Charerntanyarak, L. Heavy metals removal by chemical coagulation and precipitation. Water Sci. Technol. 1999, 39, 135–138. [Google Scholar] [CrossRef]
- Blöcher, C.; Dorda, J.; Mavrov, V.; Chmiel, H.; Lazaridis, N.; Matis, K. Hybrid flotation—Membrane filtration process for the removal of heavy metal ions from wastewater. Water Res. 2003, 37, 4018–4026. [Google Scholar] [CrossRef]
- Aldrich, C.; Feng, D. Removal of heavy metals from wastewater effluents by biosorptive flotation. Miner. Eng. 2000, 13, 1129–1138. [Google Scholar] [CrossRef]
- Al-Rashdi, B.; Johnson, D.; Hilal, N. Removal of heavy metal ions by nanofiltration. Desalination 2013, 315, 2–17. [Google Scholar] [CrossRef]
- Mukherjee, R.; Bhunia, P.; De, S. Impact of graphene oxide on removal of heavy metals using mixed matrix membrane. Chem. Eng. J. 2016, 292, 284–297. [Google Scholar] [CrossRef]
- Oehmen, A.; Viegas, R.; Velizarov, S.; Reis, M.A.; Crespo, J.G. Removal of heavy metals from drinking water supplies through the ion exchange membrane bioreactor. Desalination 2006, 199, 405–407. [Google Scholar] [CrossRef]
- Wang, A.; Zhou, K.; Zhang, X.; Zhou, D.; Peng, C.; Chen, W. Arsenic removal from highly-acidic wastewater with high arsenic content by copper-chloride synergistic reduction. Chemosphere 2020, 238, 124675. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.-K.; Chiu, K.-F.; Lin, C.-Y.; Leu, H.-J. Electrochemical treatment of wastewater: Selectivity of the heavy metals removal process. Int. J. Hydrogen Energy 2017, 42, 27741–27748. [Google Scholar] [CrossRef]
- Hunsom, M.; Pruksathorn, K.; Damronglerd, S.; Vergnes, H.; Duverneuil, P. Electrochemical treatment of heavy metals (Cu2+, Cr6+, Ni2+) from industrial effluent and modeling of copper reduction. Water Res. 2005, 39, 610–616. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Gao, B.; Yao, Y.; Inyang, M.; Zhang, M.; Zimmerman, A.R.; Ro, K.S. Hydrogen peroxide modification enhances the ability of biochar (hydrochar) produced from hydrothermal carbonization of peanut hull to remove aqueous heavy metals: Batch and column tests. Chem. Eng. J. 2012, 200, 673–680. [Google Scholar] [CrossRef]
- Ihsanullah, D.; Abbas, A.; Al-Amer, A.M.; Laoui, T.; Al-Marri, M.J.; Nasser, M.S.; Khraisheh, M.; Atieh, M.A. Heavy metal removal from aqueous solution by advanced carbon nanotubes: Critical review of adsorption applications. Sep. Purif. Technol. 2016, 157, 141. [Google Scholar] [CrossRef]
- Bakkaloglu, I.; Butter, T.; Evison, L.; Holland, F.; Hancockt, I. Screening of various types biomass for removal and recovery of heavy metals (Zn, Cu, Ni) by biosorption, sedimentation and desorption. Water Sci. Technol. 1998, 38, 269–277. [Google Scholar] [CrossRef]
- Rengaraj, S.; Yeon, J.-W.; Kim, Y.; Jung, Y.; Ha, Y.-K.; Kim, W.-H. Adsorption characteristics of Cu (II) onto ion exchange resins 252H and 1500H: Kinetics, isotherms and error analysis. J. Hazard. Mater 2007, 143, 469–477. [Google Scholar] [CrossRef]
- Lin, S.H.; Lai, S.L.; Leu, H.G. Removal of heavy metals from aqueous solution by chelating resin in a multistage adsorption process. J. Hazard. Mater 2000, 76, 139–153. [Google Scholar] [CrossRef]
- Pourhashem, G.; Adler, P.R.; Spatari, S. Time effects of climate change mitigation strategies for second generation biofuels and co-products with temporary carbon storage. J. Clean. Prod. 2016, 112 Pt 4, 2642. [Google Scholar] [CrossRef]
- Bulut, Y. Removal of heavy metals from aqueous solution by sawdust adsorption. J. Environ. Sci. 2007, 19, 160–166. [Google Scholar] [CrossRef]
- Javaid, A.; Bajwa, R.; Shafique, U.; Anwar, J. Removal of heavy metals by adsorption on Pleurotus ostreatus. Biomass Bioenergy 2011, 35, 1675–1682. [Google Scholar] [CrossRef]
- Kılıc, M.; Kırbıyık, C.; Çepelioğullar, Ö.; Pütün, A.E. Adsorption of heavy metal ions from aqueous solutions by bio-char, a by-product of pyrolysis. Appl. Surf. Sci. 2013, 283, 856–862. [Google Scholar] [CrossRef]
- Inyang, M.; Gao, B.; Yao, Y.; Xue, Y.; Zimmerman, A.R.; Pullammanappallil, P.; Cao, X. Removal of heavy metals from aqueous solution by biochars derived from anaerobically digested biomass. Bioresour. Technol. 2012, 110, 50–56. [Google Scholar] [CrossRef]
- Tripathi, M.; Sahu, J.N.; Ganesan, P. Effect of process parameters on production of biochar from biomass waste through pyrolysis: A review. Renew. Sustain. Energy Rev. 2016, 55, 467–481. [Google Scholar] [CrossRef]
- Wiedner, K.; Rumpel, C.; Steiner, C.; Pozzi, A.; Maas, R.; Glaser, B. Chemical evaluation of chars produced by thermochemical conversion (gasification, pyrolysis and hydrothermal carbonization) of agro-industrial biomass on a commercial scale. Biomass Bioenergy 2013, 59, 264–278. [Google Scholar] [CrossRef]
- Sun, Y.; Gao, B.; Yao, Y.; Fang, J.; Zhang, M.; Zhou, Y.; Chen, H.; Yang, L. Effects of feedstock type, production method, and pyrolysis temperature on biochar and hydrochar properties. Chem. Eng. J. 2014, 240, 574–578. [Google Scholar] [CrossRef]
- Sevilla, M.; Ferrero, G.A.; Fuertes, A.B. Beyond KOH activation for the synthesis of superactivated carbons from hydrochar. Carbon 2017, 114, 50–58. [Google Scholar] [CrossRef] [Green Version]
- Wahi, R.; Zuhaidi, N.; Yusof, Y.; Jamel, J.; Kanakaraju, D.; Ngaini, Z. Chemically treated microwave-derived biochar: An overview. Biomass Bioenergy 2017, 107, 411–421. [Google Scholar] [CrossRef]
- Verma, D.; Fortunati, E.; Jain, S.; Zhang, X. Biomass, Biopolymer-Based Materials, and Bioenergy: Construction, Biomedical, and Other Industrial Applications; Woodhead Publishing: Cambridge, UK, 2019. [Google Scholar]
- Zhang, J.; Zhang, X. 15-The thermochemical conversion of biomass into biofuels. In Biomass, Biopolymer-Based Materials, and Bioenergy; Verma, D., Fortunati, E., Jain, S., Zhang, X., Eds.; Woodhead Publishing: Cambridge, UK, 2019; pp. 327–368. [Google Scholar]
- Regmi, P.; Moscoso, J.L.G.; Kumar, S.; Cao, X.; Mao, J.; Schafran, G. Removal of copper and cadmium from aqueous solution using switchgrass biochar produced via hydrothermal carbonization process. J. Environ. Manag. 2012, 109, 61–69. [Google Scholar] [CrossRef]
- Marfíl, A.P.; Ocampo-Pérez, R.; Collins-Martínez, V.H.; Flores-Vélez, L.M.; Gonzalez-Garcia, R.; Medellín-Castillo, N.A.; Labrada-Delgado, G.J. Synthesis and characterization of hydrochar from industrial Capsicum annuum seeds and its application for the adsorptive removal of methylene blue from water. Environ. Res. 2020, 184, 109334. [Google Scholar] [CrossRef] [PubMed]
- Funke, A.; Ziegler, F. Hydrothermal carbonization of biomass: A summary and discussion of chemical mechanisms for process engineering. Biofuels Bioprod. Biorefining 2010, 4, 160–177. [Google Scholar] [CrossRef]
- Titirici, M.-M. Sustainable Carbon Materials from Hydrothermal Processes; John Wiley & Sons: Hoboken, NJ, USA, 2013. [Google Scholar]
- Sun, K.; Tang, J.; Gong, Y.; Zhang, H. Characterization of potassium hydroxide (KOH) modified hydrochars from different feedstocks for enhanced removal of heavy metals from water. Environ. Sci. Pollut. Res. 2015, 22, 16640–16651. [Google Scholar] [CrossRef] [PubMed]
- Poo, K.-M.; Son, E.-B.; Chang, J.-S.; Ren, X.; Choi, Y.-J.; Chae, K.-J. Biochars derived from wasted marine macro-algae (Saccharina japonica and Sargassum fusiforme) and their potential for heavy metal removal in aqueous solution. J. Environ. Manag. 2018, 206, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Karnib, M.; Kabbani, A.; Holail, H.; Olama, Z. Heavy Metals Removal Using Activated Carbon, Silica and Silica Activated Carbon Composite; Elsevier Ltd.: Amsterdam, The Netherlands, 2014; Volume 50, pp. 113–120. [Google Scholar]
- Oliveira, I.; Blöhse, D.; Ramke, H.-G. Hydrothermal carbonization of agricultural residues. Bioresour. Technol. 2013, 142, 138–146. [Google Scholar] [CrossRef]
- Kruse, A.; Funke, A.; Titirici, M.-M. Hydrothermal conversion of biomass to fuels and energetic materials. Curr. Opin. Chem. Biol. 2013, 17, 515–521. [Google Scholar] [CrossRef]
- Saqib, N.U.; Sharma, H.B.; Baroutian, S.; Dubey, B.; Sarmah, A.K. Valorisation of food waste via hydrothermal carbonisation and techno-economic feasibility assessment. Sci. Total Environ. 2019, 690, 261–276. [Google Scholar] [CrossRef]
- Yu, X.; Liu, S.; Lin, G.; Yang, Y.; Zhang, S.; Zhao, H.; Zheng, C.; Gao, X. Promotion effect of KOH surface etching on sucrose-based hydrochar for acetone adsorption. Appl. Surf. Sci. 2019, 496, 143617. [Google Scholar] [CrossRef]
- Jain, A.; Balasubramanian, R.; Srinivasan, M.P. Tuning hydrochar properties for enhanced mesopore development in activated carbon by hydrothermal carbonization. Microporous Mesoporous Mater. 2015, 203, 178–185. [Google Scholar] [CrossRef]
- Sevilla, M.; Fuertes, A.B. A green approach to high-performance supercapacitor electrodes: The chemical activation of hydrochar with potassium bicarbonate. Chem. Sus. Chem. 2016, 9, 1880–1888. [Google Scholar] [CrossRef] [Green Version]
- Guo, S.; Dong, X.; Liu, K.; Yu, H.; Zhu, C. Chemical, energetic, and structural characteristics of hydrothermal carbonization solid products for lawn grass. Bio. Resour. 2015, 10, 4613–4625. [Google Scholar] [CrossRef]
- Guo, S.; Dong, X.; Wu, T.; Shi, F.; Zhu, C. Characteristic evolution of hydrochar from hydrothermal carbonization of corn stalk. J. Anal. Appl. Pyrolysis 2015, 116, 1–9. [Google Scholar] [CrossRef]
- Guo, S.; Dong, X.; Wu, T.; Zhu, C. Influence of reaction conditions and feedstock on hydrochar properties. Energy Convers. Manag. 2016, 123, 95–103. [Google Scholar] [CrossRef]
- Baccile, N.; Falco, C.; Titirici, M.-M. Characterization of biomass and its derived char using 13 C-solid state nuclear magnetic resonance. Green Chem. 2014, 16, 4839–4869. [Google Scholar] [CrossRef] [Green Version]
- Aragón-Briceño, C.; Ross, A.B.; Camargo-Valero, M.A. Evaluation and comparison of product yields and bio-methane potential in sewage digestate following hydrothermal treatment. Appl. Energy 2017, 208, 1357–1369. [Google Scholar] [CrossRef]
- Aragón-Briceño, C.I.; Grasham, O.; Ross, A.B.; Dupont, V.; Camargo-Valero, M.A. Hydrothermal carbonization of sewage digestate at wastewater treatment works: Influence of solid loading on characteristics of hydrochar, process water and plant energetics. Renew. Energy 2020, 157, 959–973. [Google Scholar] [CrossRef]
- Jackowski, M.; Niedzwiecki, L.; Lech, M.; Wnukowski, M.; Arora, A.; Tkaczuk-Serafin, M.; Baranowski, M.; Krochmalny, K.; Veetil, V.K.; Seruga, P. HTC of Wet Residues of the Brewing Process: Comprehensive Characterization of Produced Beer, Spent Grain and Valorized Residues. Energies 2020, 13, 2058. [Google Scholar] [CrossRef] [Green Version]
- Urbanowska, A.; Kabsch-Korbutowicz, M.; Wnukowski, M.; Seruga, P.; Baranowski, M.; Pawlak-Kruczek, H.; Serafin-Tkaczuk, M.; Krochmalny, K.; Niedzwiecki, L. Treatment of Liquid By-Products of Hydrothermal Carbonization (HTC) of Agricultural Digestate Using Membrane Separation. Energies 2020, 13, 262. [Google Scholar] [CrossRef] [Green Version]
- Wilk, M.; Magdziarz, A.; Jayaraman, K.; Szymańska-Chargot, M.; Gökalp, I. Hydrothermal carbonization characteristics of sewage sludge and lignocellulosic biomass. A comparative study. Biomass Bioenergy 2019, 120, 166–175. [Google Scholar] [CrossRef]
- Liu, Z.; Quek, A.; Kent Hoekman, S.; Srinivasan, M.P.; Balasubramanian, R. Thermogravimetric investigation of hydrochar-lignite co-combustion. Bioresour. Technol. 2012, 123, 646–652. [Google Scholar] [CrossRef]
- Demir-Cakan, R.; Baccile, N.; Antonietti, M.; Titirici, M.M. Carboxylate-rich carbonaceous materials via one-step hydrothermal carbonization of glucose in the presence of acrylic acid. Chem. Mater. 2009, 21, 484–490. [Google Scholar] [CrossRef]
- Liu, K.; Zhang, S.; Hu, X.; Zhang, K.; Roy, A.; Yu, G. Understanding the adsorption of PFOA on MIL-101 (Cr)-based anionic-exchange metal–organic frameworks: Comparing DFT calculations with aqueous sorption experiments. Environ. Sci. Technol. 2015, 49, 8657–8665. [Google Scholar] [CrossRef] [PubMed]
- Vishnyakov, A.; Ravikovitch, P.I.; Neimark, A.V. Molecular Level Models for CO2 Sorption in Nanopores. Langmuir 1999, 15, 8736–8742. [Google Scholar] [CrossRef]
- Gao, J.; Liu, F.; Ling, P.; Lei, J.; Li, L.; Li, C.; Li, A. High efficient removal of Cu (II) by a chelating resin from strong acidic solutions: Complex formation and DFT certification. Chem. Eng. J. 2013, 222, 240–247. [Google Scholar] [CrossRef]
- Gupta, N.K.; Sengupta, A.; Boda, A.; Adya, V.C.; Ali, S.M. Oxidation state selective sorption behavior of plutonium using N, N -dialkylamide functionalized carbon nanotubes: Experimental study and DFT calculation. Rsc. Adv. 2016, 6, 78692–78701. [Google Scholar] [CrossRef]
- Brown, A.B.; McKeogh, B.J.; Tompsett, G.A.; Lewis, R.; Deskins, N.A.; Timko, M.T. Structural analysis of hydrothermal char and its models by density functional theory simulation of vibrational spectroscopy. Carbon 2017, 125, 614–629. [Google Scholar] [CrossRef]
- Chuntanapum, A.; Matsumura, Y. Formation of tarry material from 5-HMF in subcritical and supercritical water. Ind. Eng. Chem. Res. 2009, 48, 9837–9846. [Google Scholar] [CrossRef]
- Latham, K.G.; Simone, M.I.; Dose, W.M.; Allen, J.A.; Donne, S.W. Synchrotron based NEXAFS study on nitrogen doped hydrothermal carbon: Insights into surface functionalities and formation mechanisms. Carbon 2017, 114, 566–578. [Google Scholar] [CrossRef]
- Sevilla, M.; Fuertes, A.B. Chemical and Structural Properties of Carbonaceous Products Obtained by Hydrothermal Carbonization of Saccharides. Chem. A Eur. J. 2009, 15, 4195–4203. [Google Scholar] [CrossRef]
- Falco, C.; Perez Caballero, F.; Babonneau, F.; Gervais, C.; Laurent, G.; Titirici, M.-M.; Baccile, N. Hydrothermal Carbon from Biomass: Structural Differences between Hydrothermal and Pyrolyzed Carbons via 13C Solid State NMR. Langmuir 2011, 27, 14460–14471. [Google Scholar] [CrossRef] [Green Version]
- Baccile, N.; Laurent, G.; Coelho, C.; Babonneau, F.; Zhao, L.; Titirici, M.-M. Structural Insights on Nitrogen-Containing Hydrothermal Carbon Using Solid-State Magic Angle Spinning 13C and 15N Nuclear Magnetic Resonance. J. Phys. Chem. C 2011, 115, 8976–8982. [Google Scholar] [CrossRef] [Green Version]
- Dieguez-Alonso, A.; Funke, A.; Anca-Couce, A.; Rombolà, A.G.; Ojeda, G.; Bachmann, J.; Behrendt, F. Towards biochar and hydrochar engineering—Influence of process conditions on surface physical and chemical properties, thermal stability, nutrient availability, toxicity and wettability. Energies 2018, 11, 496. [Google Scholar] [CrossRef] [Green Version]
- Naderi, M. Chapter Fourteen-Surface Area: Brunauer–Emmett–Teller (BET). In Progress in Filtration and Separation; Tarleton, S., Ed.; Academic Press: Oxford, UK, 2015; pp. 585–608. [Google Scholar]
- Sheng, K.; Zhang, S.; Liu, J.; Shuang, E.; Jin, C.; Xu, Z.; Zhang, X. Hydrothermal carbonization of cellulose and xylan into hydrochars and application on glucose isomerization. J. Clean. Prod. 2019, 237, 117831. [Google Scholar] [CrossRef]
- Clogston, J.D.; Patri, A.K. Zeta Potential Measurement. In Characterization of Nanoparticles Intended for Drug Delivery; McNeil, S.E., Ed.; Humana Press: Totowa, NJ, USA, 2011; pp. 63–70. [Google Scholar]
- Yu, G.X.; Jin, M.; Sun, J.; Zhou, X.L.; Chen, L.F.; Wang, J.A. Oxidative modifications of rice hull-based carbons for dibenzothiophene adsorptive removal. Catal. Today 2013, 212, 31–37. [Google Scholar] [CrossRef]
- Seredych, M.; Lison, J.; Jans, U.; Bandosz, T.J. Textural and chemical factors affecting adsorption capacity of activated carbon in highly efficient desulfurization of diesel fuel. Carbon 2009, 47, 2491–2500. [Google Scholar] [CrossRef]
- Boehm, H.P. Surface oxides on carbon and their analysis: A critical assessment. Carbon 2002, 40, 145–149. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09. 2009. Available online: https://gaussian.com/g09citation/ (accessed on 1 May 2020).
- Schmidt, J.R.; Polik, W.F. WebMO Enterprise, Version 20.0; WebMO LLC: Holland, MI, USA. Available online: https://www.webmo.net (accessed on 20 May 2020).
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785. [Google Scholar] [CrossRef] [Green Version]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef]
- Kunin, R.; Meitzner, E.F.; Oline, J.A.; Fisher, S.A.; Frisch, N. Characterization of amberlyst 15 macroreticular sulfonic acid cation exchange resin. Ind. Eng. Chem. Prod. Res. Dev. 1962, 1, 140–144. [Google Scholar] [CrossRef]
- Xia, Y.; Liu, H.; Guo, Y.; Liu, Z.; Jiao, W. Immobilization of heavy metals in contaminated soils by modified hydrochar: Efficiency, risk assessment and potential mechanisms. Sci. Total Environ. 2019, 685, 1201–1208. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Sun, H.; Ro, K.S.; Sun, K.; Libra, J.A.; Xing, B. Removal of antimony (III) and cadmium (II) from aqueous solution using animal manure-derived hydrochars and pyrochars. Bioresour. Technol. 2017, 234, 77–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Zhang, T.; Ren, H.; Kruse, A.; Cui, R. Polyethylene imine modified hydrochar adsorption for chromium (VI) and nickel (II) removal from aqueous solution. Bioresour. Technol. 2018, 247, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Timko, M.T.; Wang, J.A.; Burgess, J.; Kracke, P.; Gonzalez, L.; Jaye, C.; Fischer, D.A. Roles of surface chemistry and structural defects of activated carbons in the oxidative desulfurization of benzothiophenes. Fuel 2016, 163, 223–231. [Google Scholar] [CrossRef] [Green Version]
- Petrović, J.T.; Stojanović, M.D.; Milojković, J.V.; Petrović, M.S.; Šoštarić, T.D.; Laušević, M.D.; Mihajlović, M.L. Alkali modified hydrochar of grape pomace as a perspective adsorbent of Pb2+ from aqueous solution. J. Environ. Manag. 2016, 182, 292–300. [Google Scholar] [CrossRef]
- Israelachvili, J.N. Intermolecular and Surface Forces; Academic press: Cambridge, MA, USA, 2011. [Google Scholar]
- Tran, H.; You, S.-J.; Chao, H.-P. Insight into adsorption mechanism of cationic dye onto agricultural residues-derived hydrochars: Negligible role of π-π interaction. Korean J. Chem. Eng. 2017, 34, 1708–1720. [Google Scholar] [CrossRef]
- Doan, V.; Köppe, R.; Kasai, P.H. Dimerization of Carboxylic Acids and Salts: An IR Study in Perfluoropolyether Media. J. Am. Chem. Soc. 1997, 119, 9810–9815. [Google Scholar] [CrossRef]
- Semerciöz, A.S.; Göğüş, F.; Celekli, A.; Bozkurt, H. Development of carbonaceous material from grapefruit peel with microwave implemented-low temperature hydrothermal carbonization technique for the adsorption of Cu (II). J. Clean. Prod. 2017, 165, 599–610. [Google Scholar] [CrossRef]
- Elaigwu, S.E.; Greenway, G.M. Microwave-assisted and conventional hydrothermal carbonization of lignocellulosic waste material: Comparison of the chemical and structural properties of the hydrochars. J. Anal. Appl. Pyrolysis 2016, 118, 1–8. [Google Scholar] [CrossRef]
- Liang, J.; Liu, Y.; Zhang, J. Effect of Solution pH on the Carbon Microsphere Synthesized by Hydrothermal Carbonization. Procedia Environ. Sci. 2011, 11, 1322–1327. [Google Scholar] [CrossRef] [Green Version]
- Feng, Z.; Odelius, K.; Rajarao, G.K.; Hakkarainen, M. Microwave carbonized cellulose for trace pharmaceutical adsorption. Chem. Eng. J. 2018, 346, 557–566. [Google Scholar] [CrossRef]
- Mihajlović, M.; Petrović, J.; Kragović, M.; Stojanović, M.; Milojković, J.; Lopičić, Z.; Koprivica, M. Effect of KOH activation on hydrochar surface: FT-IR analysis. RAD Assoc. J. 2017, 2, 65–67. [Google Scholar]
- Libra, J.A.; Ro, K.S.; Kammann, C.; Funke, A.; Berge, N.D.; Neubauer, Y.; Titirici, M.-M.; Fühner, C.; Bens, O.; Kern, J. Hydrothermal carbonization of biomass residuals: A comparative review of the chemistry, processes and applications of wet and dry pyrolysis. Biofuels 2011, 2, 71–106. [Google Scholar] [CrossRef] [Green Version]
- Xia, Y.; Yang, T.; Zhu, N.; Li, D.; Chen, Z.; Lang, Q.; Liu, Z.; Jiao, W. Enhanced adsorption of Pb(II) onto modified hydrochar: Modeling and mechanism analysis. Bioresour. Technol. 2019, 288, 121593. [Google Scholar] [CrossRef]
- Fornes, F.; Belda, R.M.; Lidón, A. Analysis of two biochars and one hydrochar from different feedstock: Focus set on environmental, nutritional and horticultural considerations. J. Clean. Prod. 2015, 86, 40–48. [Google Scholar] [CrossRef]
- Fang, J.; Gao, B.; Chen, J.; Zimmerman, A.R. Hydrochars derived from plant biomass under various conditions: Characterization and potential applications and impacts. Chem. Eng. J. 2015, 267, 253–259. [Google Scholar] [CrossRef]
- Dupont, AMBERLYST™ 15DRY Polymeric Catalyst, product Data Sheet. 2019. Available online: https://www.dupont.com/content/dam/dupont/amer/us/en/water-solutions/public/documents/en/45-D00927-en.pdf (accessed on 1 December 2019).
- Nandan, D.; Gupta, A. Solvent sorption isotherms, swelling pressures, and free energies of swelling of polystyrenesulfonic acid type cation exchanger in water and methanol. J. Phys. Chem. 1977, 81, 1174–1179. [Google Scholar] [CrossRef]
- Davies, C.; Yeoman, G. Swelling equilibria with some cation exchange resins. Trans. Faraday Soc. 1953, 49, 968–974. [Google Scholar] [CrossRef]
- Marcus, Y.; Naveh, J. Anion exchange of metal complexes. XVII. Selective swelling of the exchanger in mixed aqueous-organic solvents. J. Phys. Chem. 1969, 73, 591–596. [Google Scholar] [CrossRef]
- Howery, D.G.; Shore, L.; Kohn, B.H. Proton magnetic resonance studies of the structure of water in Dowex 50 W. J. Phys. Chem. 1972, 76, 578–581. [Google Scholar] [CrossRef]
- Mao, Y.; Fung, B.M. A Study of the Adsorption of Acrylic Acid and Maleic Acid from Aqueous Solutions onto Alumina. J. Colloid Interface Sci. 1997, 191, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Zhao, Y.; Wu, J.-G.; Yang, Q.-C.; Guang-Xian, X. Crystal structure and FT-IR study of cesium 4-methylbenzenesulfonate. J. Mol. Struct. 1998, 471, 63–66. [Google Scholar] [CrossRef]
- Zhang, X.; Gao, B.; Fang, J.; Zou, W.; Dong, L.; Cao, C.; Zhang, J.; Li, Y.; Wang, H. Chemically activated hydrochar as an effective adsorbent for volatile organic compounds (VOCs). Chemosphere 2019, 218, 680–686. [Google Scholar] [CrossRef] [PubMed]
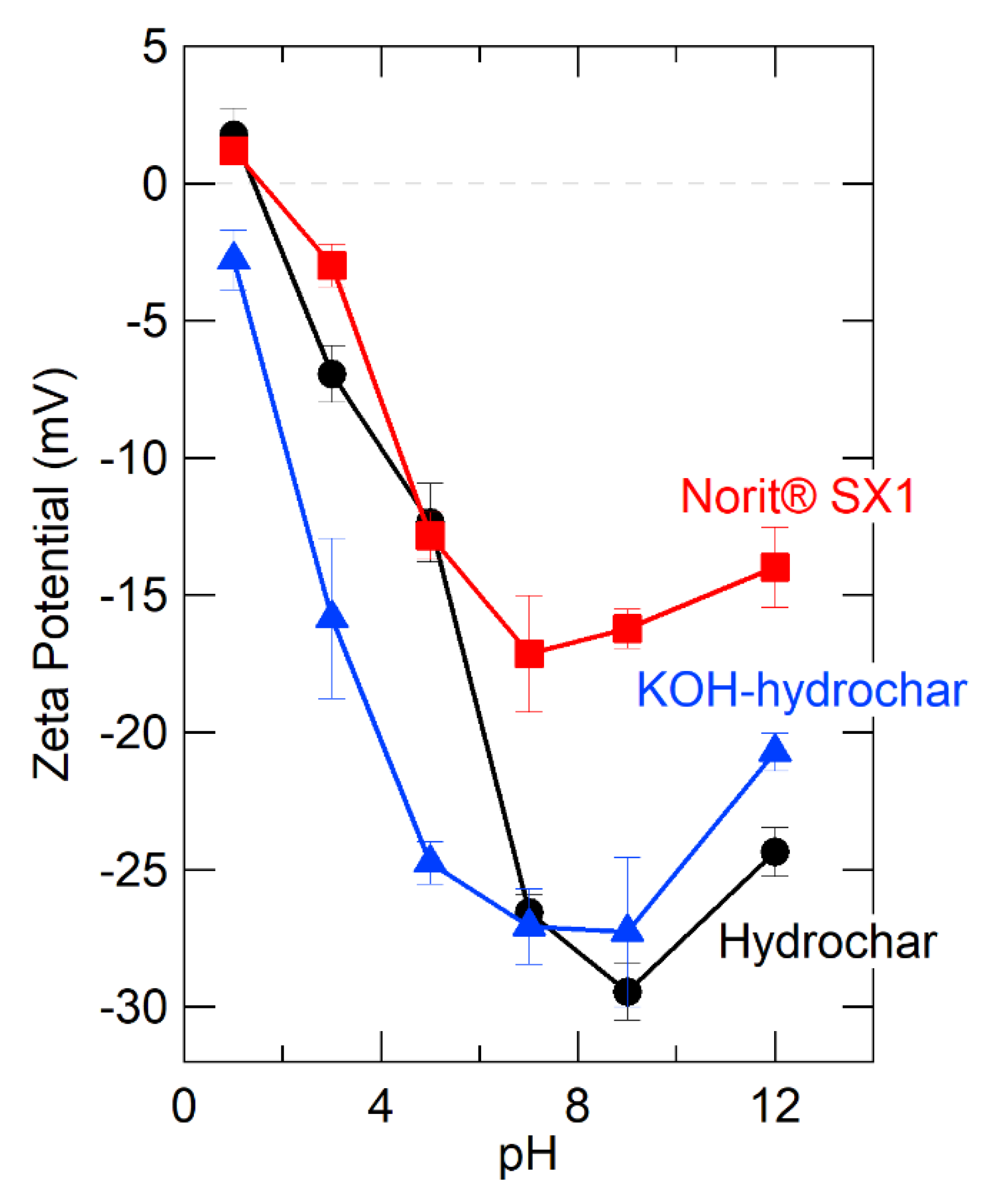

 carbon;
carbon;  hydrogen;
hydrogen;  oxygen;
oxygen;  potassium;
potassium;  sodium;
sodium;  copper.
carbon; hydrogen; oxygen; potassium; sodium; copper.
copper.
carbon; hydrogen; oxygen; potassium; sodium; copper.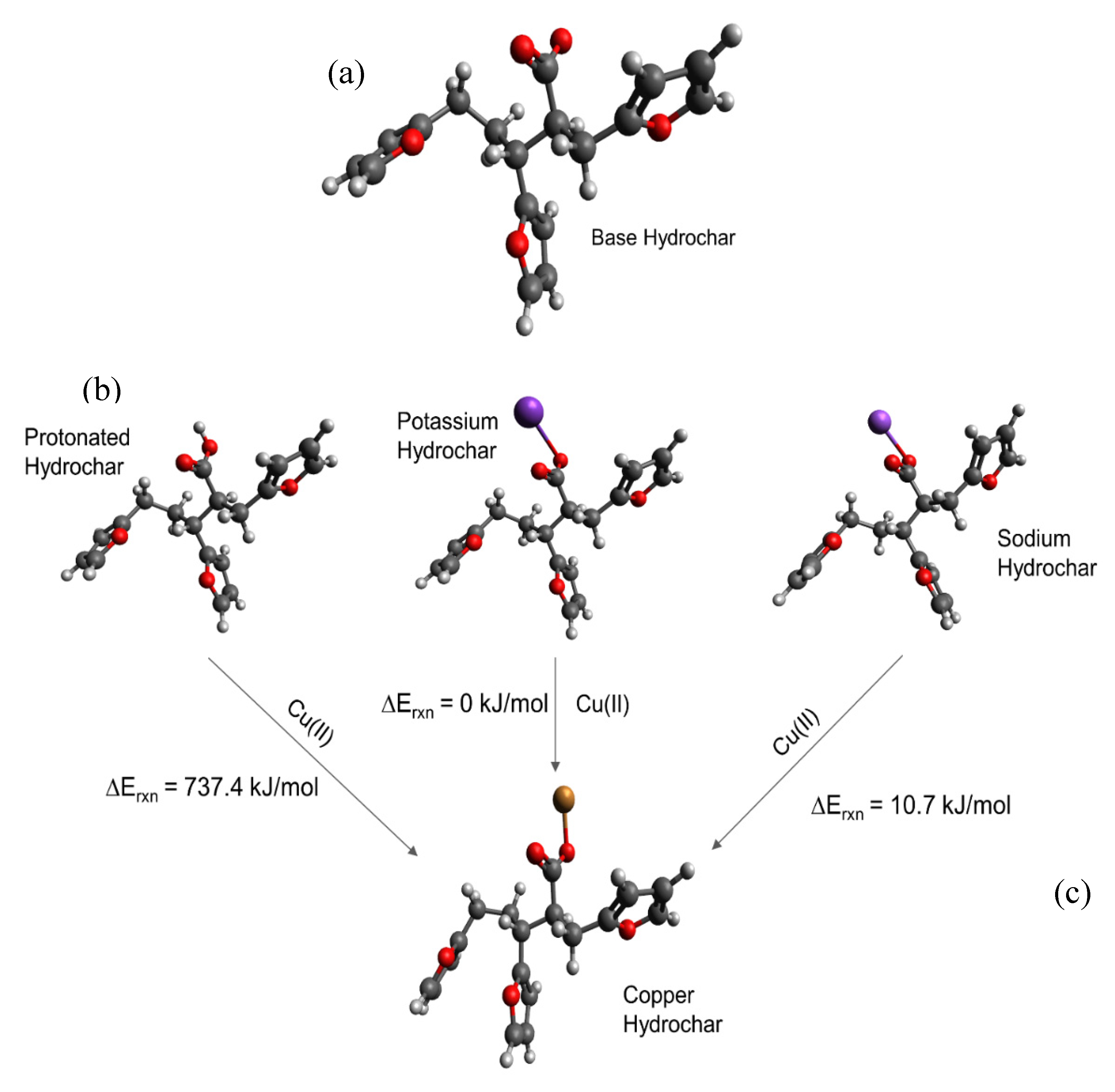
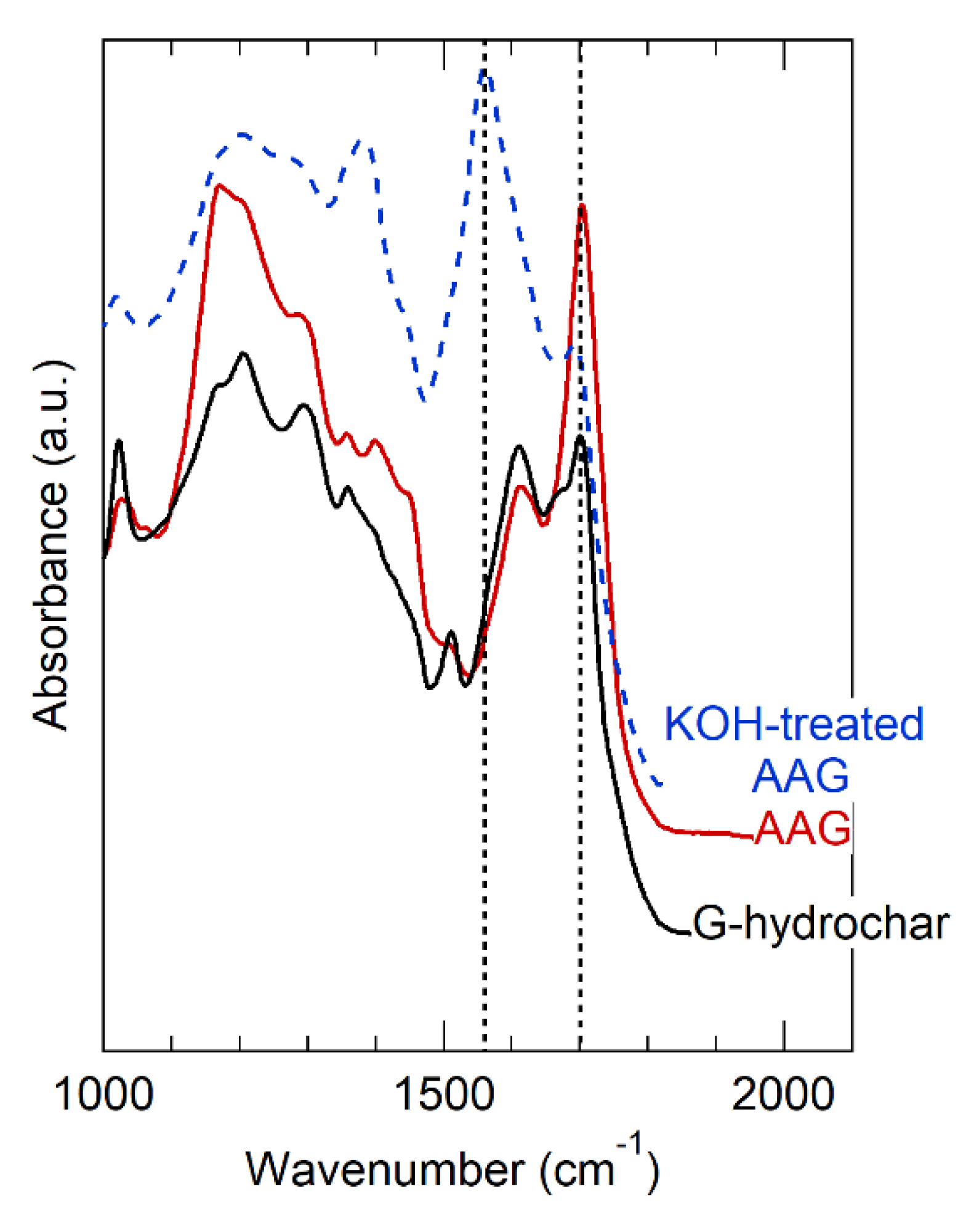
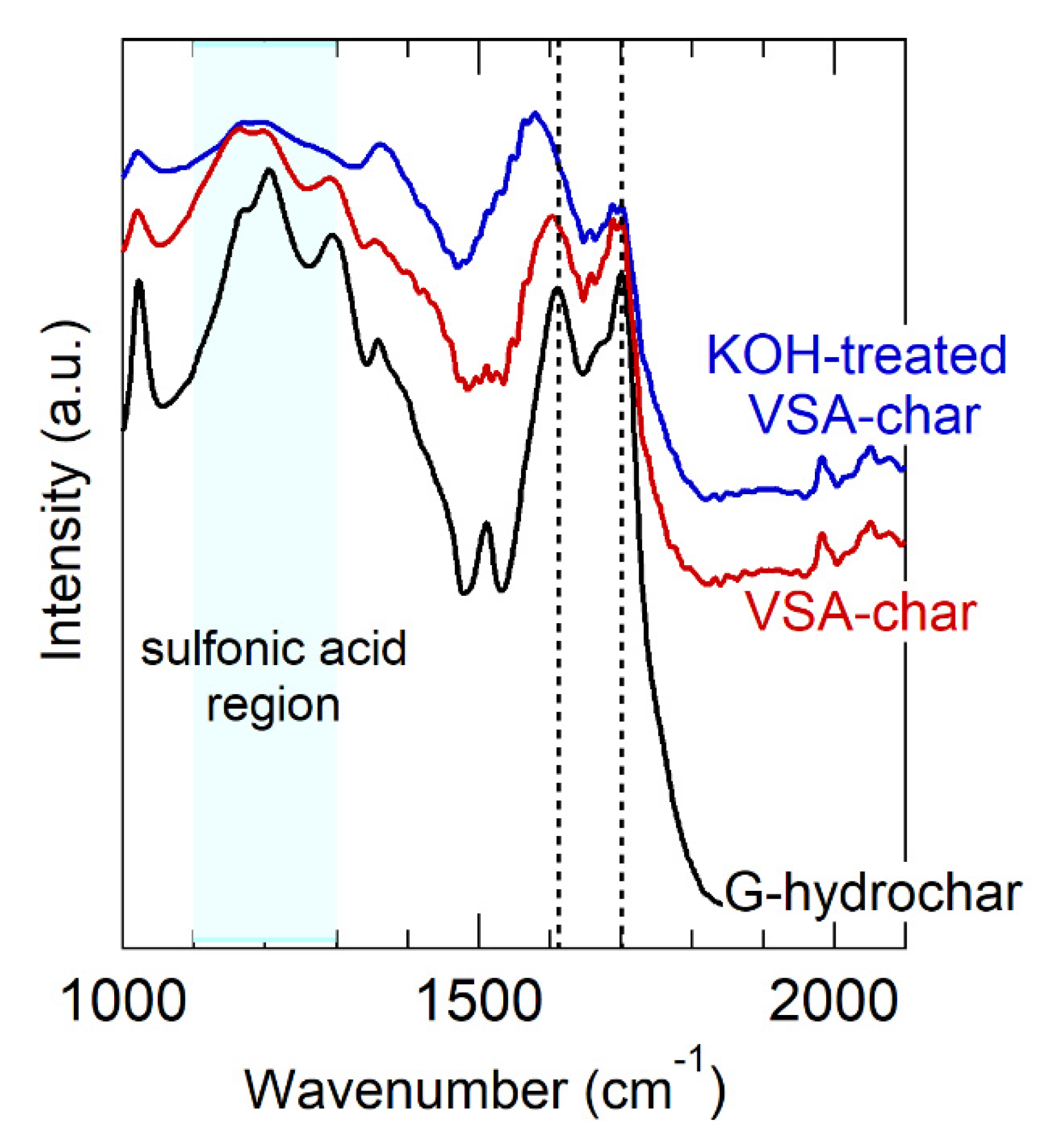 carbon; hydrogen; oxygen;
carbon; hydrogen; oxygen;  sulfur; potassium; sodium; copper.
carbon; hydrogen; oxygen; sulfur; potassium; sodium; copper.
sulfur; potassium; sodium; copper.
carbon; hydrogen; oxygen; sulfur; potassium; sodium; copper.
 depicts activated carbon,
depicts activated carbon,  ion exchange resins,
ion exchange resins,  hydrochar.
depicts activated carbon, ion exchange resins, hydrochar.
hydrochar.
depicts activated carbon, ion exchange resins, hydrochar. carbon; hydrogen; oxygen; sulfur; copper.
carbon; hydrogen; oxygen; sulfur; copper.
carbon; hydrogen; oxygen; sulfur; copper.
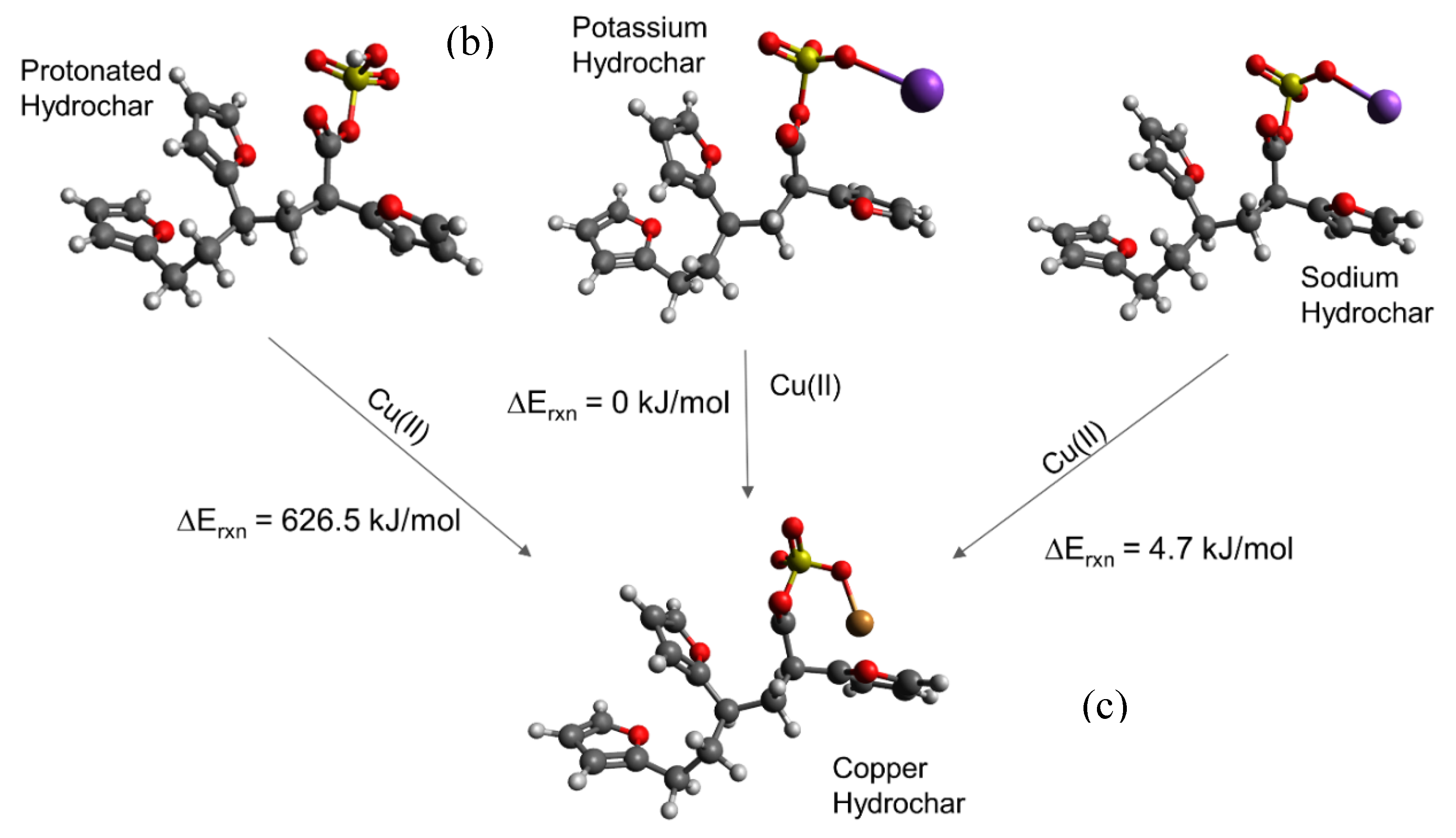
carbon; hydrogen; oxygen; sulfur; copper.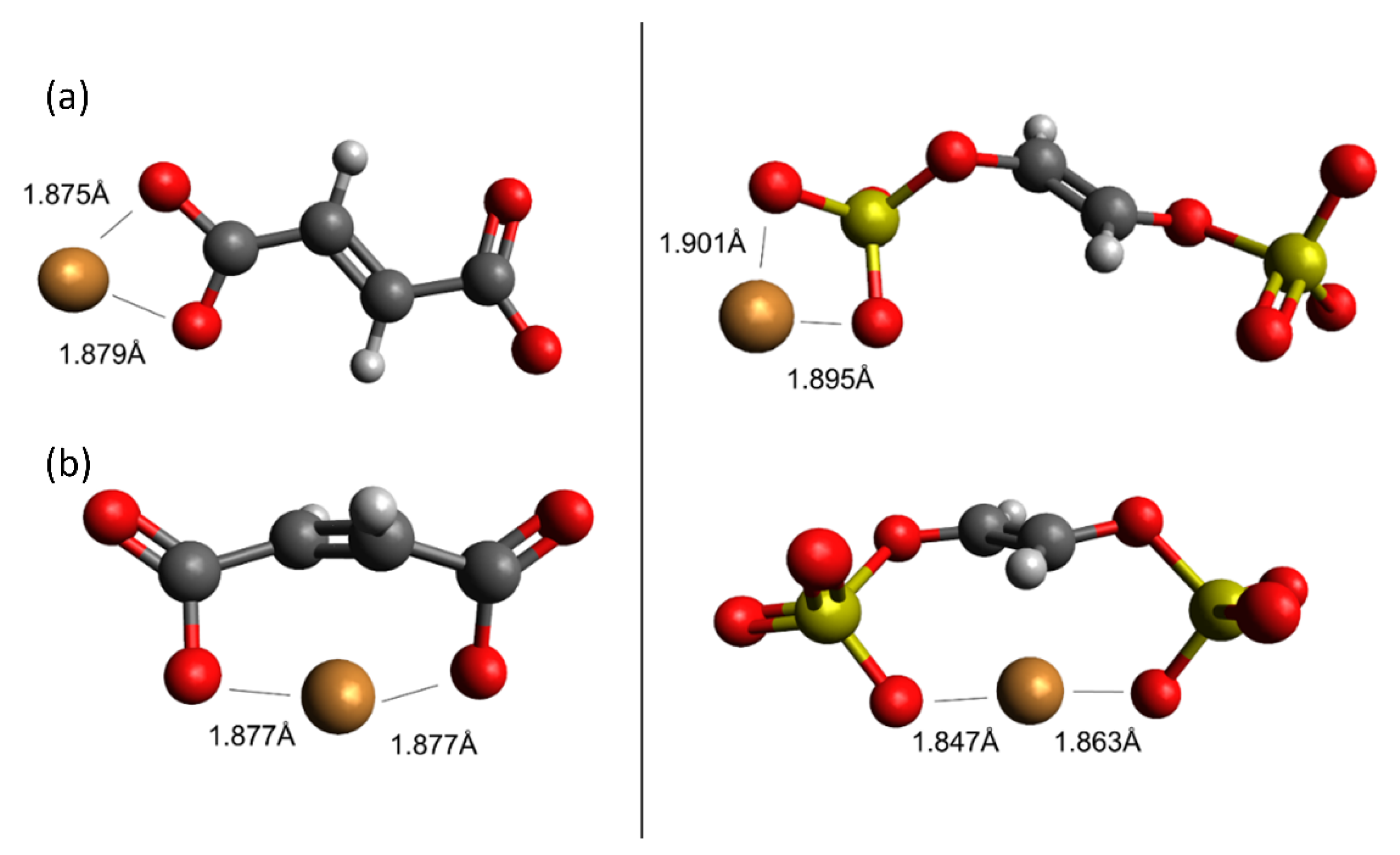 carbon; hydrogen; oxygen; sulfur; copper. Differences in energy are shown directly in the Figure.
carbon; hydrogen; oxygen; sulfur; copper. Differences in energy are shown directly in the Figure.
carbon; hydrogen; oxygen; sulfur; copper. Differences in energy are shown directly in the Figure.
carbon; hydrogen; oxygen; sulfur; copper. Differences in energy are shown directly in the Figure.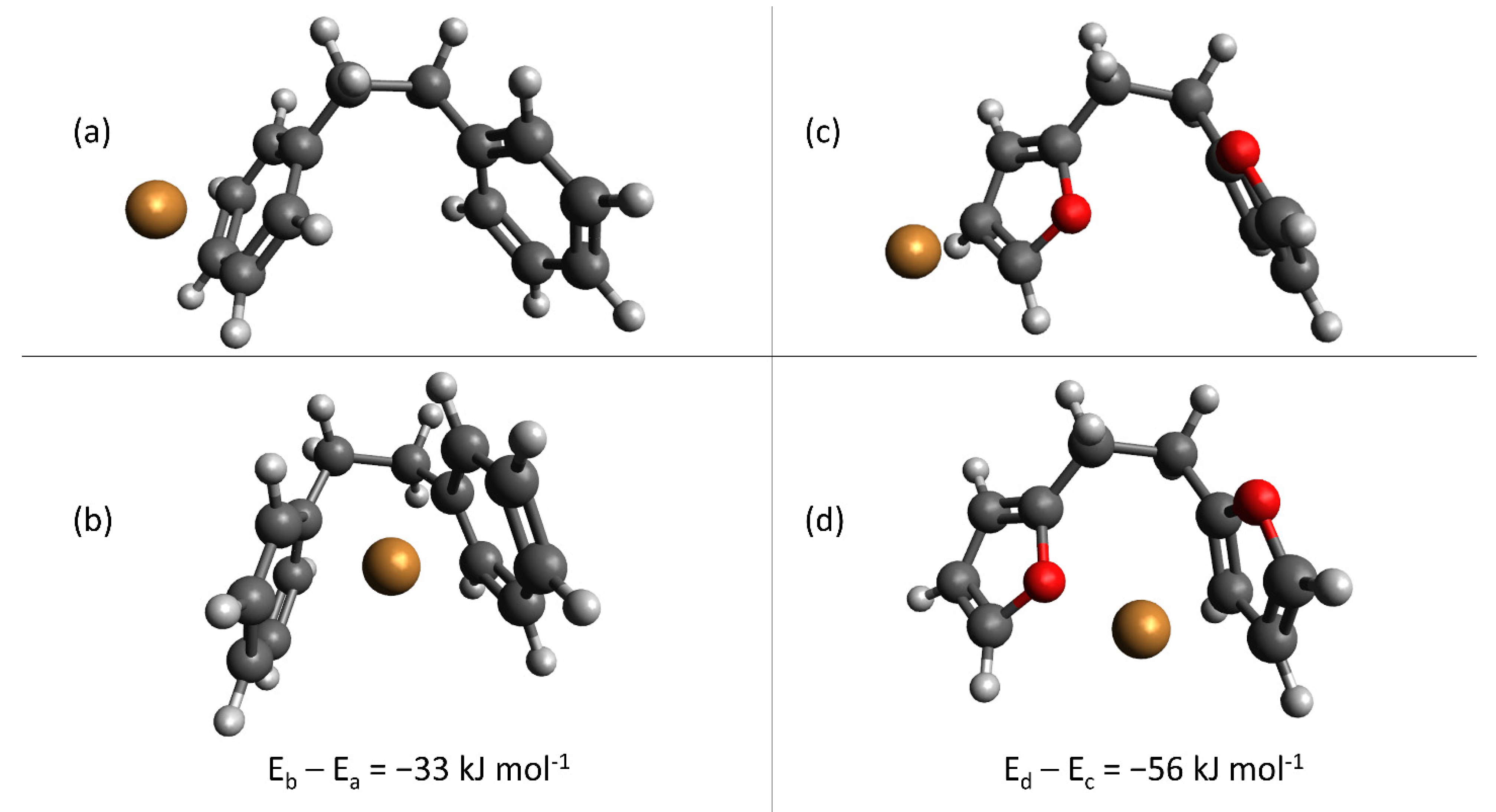

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Material | Post Treatment | Adsorption Capacity (mg Cu g−1) | BET Surface Area (m2 g−1) | ||
|---|---|---|---|---|---|
| Glucose Hydrochar | None | <3 | 4 ± 2 | ||
| Na2CO3 | 20.0 | ± 0.4 | |||
| K2CO3 | 29 | ±5 | |||
| NaOH | 35 | ±4 | |||
| KOH | 40 | ±4 | |||
| Activated Carbons | Norit® SX1 | None | 19 | ±4 | 800 ± 100 |
| KOH | 21 | ±6 | |||
| Nuchar® | None | 29 | ±3 | 1700 ± 200 | |
| KOH | 11 | ±7 | |||
| Darco® KB-G | None | 9 | ±0.1 | 1500 ± 100 | |
| KOH | 5 | ±0.8 | |||
| Darco® KB-WJ | None | 6 | ±0.2 | 1600 ± 100 | |
| KOH | 21 | ±4 | |||
| Material | Post Treatment | Adsorption Capacity (mg Cu g−1) | BET Surface Area (m2 g−1) | |||
|---|---|---|---|---|---|---|
| Hydrochars | Acrylic acid-hydrochar (AA-hydrochar) | None | <3 | 5–10 | ||
| Na2CO3 | 30 | ±7 | ||||
| K2CO3 | 26 | ±6 | ||||
| NaOH | 50 | ±4 | ||||
| KOH | 45 | ±7 | ||||
| Vinyl sulfonic acid (VSA-hydrochar) | None | <2 | <1 | |||
| NaOH | 34 | ±5 | ||||
| KOH | 51 | ±3 | ||||
| Resins | Amberlyst® | None | 128 | ±4 | 53 [97] | |
| AG® 50W-X4 | None | 109 | ±7 | <1 | ||
| Material | Carboxylic Acid Density (mmol g−1) | Carboxylic Acid Density b (mmol m−2) | |||
|---|---|---|---|---|---|
| Hydrochars | glucose-hydrochar | 1 | ±0.07 | 0.15 | ±0.02 |
| VSA-hydrochar | 0.9 | ±0.1 | 0.12 | ±0.01 | |
| AA-hydrochar | 2.8 | ±0.3 | 0.4 | ±0.1 | |
| Activated Carbons | Norit® SX1 | <0.1 a | <0.0001 | ||
| Norit® SX Ultra | <0.1 a | ||||
| Nuchar® | <0.1 a | ||||
| Resins | Amberlyst-15® | 5 | ±0.01 | 0.1 | ±0.0002 |
| AG® 50W-X4 | 1.71 | ±0.1 | 2.6 | ±0.1 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Delahaye, L.; Hobson, J.T.; Rando, M.P.; Sweeney, B.; Brown, A.B.; Tompsett, G.A.; Ates, A.; Deskins, N.A.; Timko, M.T. Experimental and Computational Evaluation of Heavy Metal Cation Adsorption for Molecular Design of Hydrothermal Char. Energies 2020, 13, 4203. https://doi.org/10.3390/en13164203
Delahaye L, Hobson JT, Rando MP, Sweeney B, Brown AB, Tompsett GA, Ates A, Deskins NA, Timko MT. Experimental and Computational Evaluation of Heavy Metal Cation Adsorption for Molecular Design of Hydrothermal Char. Energies. 2020; 13(16):4203. https://doi.org/10.3390/en13164203
Chicago/Turabian StyleDelahaye, Louise, John Thomas Hobson, Matthew Peter Rando, Brenna Sweeney, Avery Bernard Brown, Geoffrey Allen Tompsett, Ayten Ates, N. Aaron Deskins, and Michael Thomas Timko. 2020. "Experimental and Computational Evaluation of Heavy Metal Cation Adsorption for Molecular Design of Hydrothermal Char" Energies 13, no. 16: 4203. https://doi.org/10.3390/en13164203
APA StyleDelahaye, L., Hobson, J. T., Rando, M. P., Sweeney, B., Brown, A. B., Tompsett, G. A., Ates, A., Deskins, N. A., & Timko, M. T. (2020). Experimental and Computational Evaluation of Heavy Metal Cation Adsorption for Molecular Design of Hydrothermal Char. Energies, 13(16), 4203. https://doi.org/10.3390/en13164203