1. Introduction
Dibenzothiophene and its derivatives are major polycyclic aromatic sulfur heterocyclics (PASHs) in coals, crude oils and sedimentary organic matter [
1,
2,
3,
4]. Due to the excellent micro-biological degradation resistance and a high thermal stability of DBTs [
5,
6,
7,
8,
9], their geochemical parameters such as distribution and concentration have been widely applied to evaluate the maturity of coals and suggested as the depositional environment indicator of sedimentary organic matter as well as the tracer of hydrocarbon migration pathways [
10,
11,
12,
13,
14,
15,
16]. In view of their valuable applications as geochemical markers, great efforts have been devoted to elucidating the origin of DBTs [
17,
18,
19,
20].
Generally, it has been assumed that aromatic sulfur compounds in nature are formed from the reactions of hydrocarbons such as alkenes, benzenes, and biphenyls with pyrite, sulfides or elemental sulfur during the coalification process [
17,
18,
19,
20]. Particularly, DBTs have been shown to be promptly generated through direct insertion of heterosulfur bridges into biaryls [
18]. The early studies on the insertion reaction of biaryls have considerably increased our understanding on the chemistry associated with the formation of DBTs. However, the reactivity of elemental sulfur is rather high and hence not all ranks of coals have elemental sulfur needed in the direct insertion reaction [
15]. What is more, DBTs generally have oxygen counterparts such as dibenzofurans (DBFs) [
4,
17,
21,
22]. Although DBF can be generated from the reaction of biphenyl with oxygen [
23], the formation of alkyl substituted DBFs requires alkyl substituted biaryls, to which the insertion of oxygen bridge would be much more difficult than that of sulfur bridge since benzylic hydrogen is much more reactive than benzene ring toward oxygen, especially at a relatively low temperature to which coals have been exposed. Thus, the chemistry involved in the formation of substituted DBTs might not be applicable to that of substituted DBFs. This casts doubt on the biaryls’ route to DBT(s) and DBF(s).
Considering that sulfur and oxygen are congeners, it would be much desirable that similar chemistry can be constructed for the formation of DBTs and DBFs. In this aspect, the thermal degradation of phenol or thiophenol might be a good alternative for the generation of DBTs or DBFs. It has been demonstrated that the gas-phase thermolysis of phenol produces DBF at 665–865 °C [
24]. Although this pyrolysis temperature may be too high for coals, the temperature can be reduced to 300–500 °C in the presence of activated charcoal [
25]. Thus, DBF from phenol is a feasible process in nature.
Recently, we discovered that the thermal degradation of phenols is also catalyzed by alkalis and generates 2,2′-biphenol and 2-phenylphenol [
26,
27], which can cyclize to yield DBF [
28]. On the basis of this result, it is speculated that thiophenol can produce DBT too under mild conditions in the presence of alkalis. Since this route is based on the catalyzed thermal degradation of phenol or thiophenol, it does not require external agents such as oxygen and elemental sulfur except the presence of inorganic salts which are ubiquitous or readily available in coals and oils [
29,
30,
31]. Therefore, the chemistry is essentially the same for the generations of DBF and DBT.
In this paper, we report the thermal degradation of sodium thiophenolate at various temperatures. Under conditions used in this study, DBT and a series of related compounds have indeed been detected among the degradation products of thiophenolate. Detailed chemistry involved in the formation of DBT and its derivatives is constructed. Our work will provide insights into the origin of DBTs in coals and crude oils.
2. Experimental
2.1. Materials
Sodium thiophenolate, sodium sulfide and diphenyl disulfide were bought from Aladdin Co. (Shanghai, China). Dibenzothiophene, 2,2′-biphenol and diphenyl sulfide were purchased from Sigma-Aldrich Co. Ltd. (Shanghai, China). Acetonitrile was obtained from Sinopharm Chemical Reagent Co. Ltd. (Shanghai, China). All materials were used without further purification.
2.2. Thermal Treatments
Thermal treatment in TGA: About 15 mg of sodium thiophenolate was loaded in a platinum pan and heated in a TA Q500 thermogravimetric analyzer (TA Instrument, New Castle, DE, USA) up to 600 °C at a heating rate of 10 °C/min under nitrogen with a flow rate of 60 mL/min. Residues at different temperatures (400, 440, 470, 500 °C) collected at different temperatures (400, 440, 470, 500 °C) during the major degradation step were subjected to further analysis.
Thermal treatment in alcohol flame: samples (sodium thiophenolate, sodium sulfide/2,2′-biphenol, diphenyl disulfide or dibenzothiophene) of about 20 mg was each loaded in a long nuclear magnetic resonance (NMR) glass tube filled with nitrogen. The tubes were subsequently capped and directly heated in alcohol flame for a predetermined duration (up to 40 s). The residues were subject to quadra pole-time of flight mass spectrograph (Q-TOF) analysis or gas chromatography/mass spectrograph (GC-MS) analysis to quantify the yield of DBT. The inside temperature of NMR tubes was estimated to be lower than 600 °C on the basis of which the majority of bisphenol A bis(diphenyl phosphate) (BDP) kept intact in NMR tubes for such short-duration thermal treatment although BDP completely degrades at 600 °C [
32].
Thermal degradation at the low temperature: sodium thiophenolate of about 40 mg was loaded in a long glass NMR tube. It was heated in an oil bath of 220–225 °C under nitrogen flow. The residue was collected for Q-TOF and GC-MS analysis to determine if products obtained at high temperatures are generated at a low temperature.
2.3. Characterizations
Thermogravimetric analysis/infrared spectra/mass spectra (TGA-FTIR-MS) of sodium thiophenolate were recorded on a Perkin Elmer TGA 8000 thermogravimetric instrument (Perkin Elmer, Columbus, OH, USA) coupled with a Spectrum Two FTIR spectrophotometer (Perkin Elmer) and a Clarus SQ 8 T mass analyzer (Perkin Elmer). The transferring line between the equipment was kept at 280 °C. Approximate 8 mg sample was heated from 50 to 600 °C under nitrogen (50 mL/min). The heating rate was 10 °C/min. The spectra were recorded every 40 s for 85 min with a resolution of 4 cm−1. The range for the mass analysis was from m/z = 2 to 300 Da.
Gas chromatography/mass spectrograph (GC-MS) of thermally treated sodium thiophenolate, diphenyl sulfide, diphenyl disulfide, dibenzothiophene and 2,2′-biphenol/sodium sulfide was recorded on an Agilent 7890B gas chromatograph interfaced to a 5977A mass-selective detector (Agilent Technologies, Santa Clara, CA, USA). 1 μL of the extracts was injected into a HP-5MS capillary column (30 m × 250 μm × 0.25 μm) using a PTV-injection. The temperature was initially set at 40 °C and then raised up to 320 °C at 15 °C/min and held at this temperature for 15 min. The range of the mass investigation using electron ionization was selected from m/z = 2 to 500 Da. The solvent delay was set to 3 min.
Fourier transform infrared spectra (FTIR) of the solids left from the TGA thermal treatment were performed on an Agilent Cary 660 FTIR analyzer (Agilent Technologies) interfaced to a Pike GladiATR (Pike Technologies, Fitchburg, WI, USA) at a resolution of 4 cm−1. A diamond crystal was used as the sample loading platform.
Quadra pole-time of flight mass spectrograph (Q-TOF) of thermally treated sodium thiophenolate was performed on an AB Sciex TripleTOF 4600 mass analyzer (AB Sciex, Framingham, MA, USA). Both the negative ion and positive ion modes were performed. The atmospheric pressure chemical ionization (APCI) mode was used to detect the fragment ions. A declustering potential of 80 V and an ion spray voltage of 4500 V were used. The vaporizer was heated to 500 °C. The samples were injected at a rate of 5 μL/min. The range of the measurement was selected from m/z = 50 to 1000 Da.
3. Results and Discussion
3.1. TGA-FTIR-MS of Sodium Thiophenolate
Since thiophenolate is structurally related to phenolate and also because phenolate can be thermally transformed to DBFs, the thermal degradation of thiophenolate is first compared with that of phenolate.
Figure 1 shows their thermogravimetric analyses. It can be clearly seen that the temperature at the maximum mass loss of thiophenolate is more than 50 °C lower than that of phenolate. Thus, DBTs might be more easily formed from thiophenolate than DBFs from phenolate.
The notably reduced thermal stability of thiophenolate, compared with that of phenolate, likely stems from the great electronic donating ability of the sulfur anion. According to our earlier work, the oxygen anion activates the homolysis of the NaOPh‒H bond through facilitating tautomerization of NaOPh to its keto forms followed by the regain of aromaticity [
26], sulfur anion should weaken the NaSPh‒H bond more thanks to its great electron donating ability. Consequently, the much weakened NaSPh‒H bond expedites the generation of NaSPh radical and hydrogen radical. The homolysis of the NaSPh‒H bond is strongly supported by the presence of benzene in the gaseous products. There is a clear absorption at 673 cm
−1, which is due to the characteristic C‒H deformation vibration of benzene [
33], in the FTIR spectra of gaseous products as shown in
Figure 2. Benzene is also positively identified in the MS spectrum as illustrated in the
Figure S1. Since the formation of benzene requires hydrogenolysis, its early presence suggests the yield of hydrogen radical during the initial thermal degradation of thiophenolate. Therefore, thiophenolate follows the similar degradation chemistry as phenolate with the bond cleavage of Ar‒H taking place first [
26,
27].
The thermal degradation of thiophenolate beginning with the homolysis of Ar‒H has important consequences. In the case of phenolate, the homolysis leads to hydroarylation which produces phenylphenolate or self-combination of aryl radicals to biphenolates [
27]. Likewise, thiophenolate is expected to form phenylthiophenolate or bithiophenolate, the cyclization of which should yield DBT [
34,
35]. Unfortunately, the IR absorptions of DBT overlap significantly with those of diphenyl sulfide, which is identified by its notable absorptions at 1584 and 1479 cm
−1 as well as a series of small peaks attributed to vibrational overtones of benzene ring as shown in
Figure 2. As a result, the FTIR spectra of off-gases from TGA experiments are not suitable to positively pinpoint DBT. However, the generation of DBT is clearly suggested by the MS spectrum with a peak at MW = 184 as illustrated in
Figure S1. This implies that DBT can be formed from the thermal degradation of thiophenolate. To firmly establish the presence of DBT and quantify its yield, thiophenolate was subjected to thermal treatment in NMR tubes on alcohol flame.
3.2. GC-MS of Sodium Thiophenolate Thermally Treated on Alcohol Flame
Sodium thiophenolate in NMR tubes was directly heated on alcohol flame to accelerate the investigation. The collected residues were subjected to the GC-MS analysis. Detectable degradation products are identified and listed in
Table 1. The chief volatile products are diphenyl sulfide and DBT with a small amount of diphenyl disulfide as shown in
Figure 3. This result solidly supports that the generation of DBT from the thermal degradation of thiophenolate is chemically feasible.
To quantify the yield of DBT, thiophenolate was heated for different durations in glass NMR tubes filled with nitrogen. To verify that DBT was collected instead of escaping from the tube, the same thermal treatment was carried out on standard DBT samples for comparison.
Figure 4 illustrates the recovery rates of standard DBT samples and yields of DBT from thermally treated samples. It can be seen that all the standard DBT samples were essentially recovered, indicating that DBT barely decomposes or escapes from the glass tubes. This lends confidence to the measurement of yields of DBT from the thermal degradation of thiophenolate. Noticeably, the yield of DBT increases first then stays about 6–7% during 20–40 s, suggesting that the thermal degradation of thiophenolate is complete within 20 s. Considering that the mass loss of thiophenolate is about 55% as shown in
Figure 1, the value of 6–7% represents a significant quantity among volatile species and validates the chemical route of DBT from thiophenolate.
After establishing the feasibility of thiophenolate as a source of DBT, one thing that remains to be clarified is the reaction pathway to DBT from thiophenolate. It has been known that diphenyl sulfide, which is another major degradation product, can transform to DBT [
36,
37,
38]. To check whether DBT is formed from diphenyl sulfide or diphenyl disulfide, both of them were subjected to flame tests. No DBT can be clearly spotted in thermally treated diphenyl disulfide in
Figure 3. However, DBT is indeed found among the degradation products of diphenyl sulfide with a yield of 3.3% at 30 s. This yield is about 50% lower than that from thiophenolate. Thus, a considerable amount of DBT must come from other sources.
As pointed out above, a likely precursor of DBT is 2,2′-bithiophenolate. Unfortunately, an authentic 2,2′-bithiophenolate sample was not available. To probe whether it is possible for 2,2′-bithiophenolate to cyclize to DBT, 2,2′-biphenol was instead mixed with sodium sulfide and the mixture was heated in alcohol flame for 30 s. It has been shown that phenol can be readily converted to thiophenol by sulfides [
39,
40].
Figure 3 shows the gas chromatogram of degradation products. Evidently, there is a peak at 16.2 min, which is attributed to be DBT by its retention time and MS spectrum (to see in
Figure S2). This result indicates the formation of DBT from 2,2′-bithiophenolate or similar sulfur substituted biaryl compounds which are generated from 2,2′-biphenol and sulfide. In addition, this transformation is backed by the presence of hydroxyl-DBT, which appears at 16.9 min and is identified by its MS spectrum (to see in
Figure S3). The generation of hydroxyl-DBT suggests that the cyclization of 2-phenylthiophenolate to DBT can occur too. The overall conversion of 2,2′-biphenol to DBT and hydroxyl-DBT is shown in
Scheme 1.
3.3. FTIR and Q-TOF Analyses of Residues
To examine the chemical structures of species in the condensed phase, residues were collected at pre-determined temperatures in TGA experiments and subjected to a detail examination.
Figure 5 shows the FTIR spectra of the residues from the thermal degradation of thiophenolate at different temperatures. Notably, the intensity of the absorption of Ph−S at 1089 cm
−1 is reduced with the increase of the temperature [
33]. Simultaneously, new peaks at 968 cm
−1, 641 cm
−1 and 500 cm
−1 appear at 400 °C and grow with the temperature. These peaks are assigned to Na
2SO
3 [
41], which is formed by the oxidation of Na
2S in the air before and during the FTIR experiment, similar to the formation of Na
2CO
3 from the thermal degradation of phenolate [
26]. The presence of Na
2SO
3 implies the disconnect of the Ph‒S bond, which is consistent with the presence of benzene in the gaseous phase.
More information about residues is obtained from the Q-TOF analyses. As shown in
Figure 6, two strong peaks appear at 440 °C. They are respectively assigned to thiophenolate (MW = 109) and phenylthiophenolate (MW = 185) that differ by one phenylene group (MW = 76). At higher temperatures, major peaks can be characterized by a pattern of MW = 109 + 32 m + 76 k (
m = 0, 1, 2;
k = 0, 1, 2, 3), suggesting the continuous buildup of NaS• and phenylene group to thiophenolate. Additionally, a series of peaks with MW = 215 + 76 n (
n = 0, 1, 2) emerge at 440 °C and grow with the temperature. A species matching MW = 183 + 32 m + 76 k (
m = 1;
k = 0) is NaS‒substituted DBT. Apparently, the NaS‒substituted DBT and its phenylene insertion products are related to species with MW = 109 + 32 m +76 k (
m = 1;
k = 1, 2, 3). The former can be viewed as the cyclization products of the latter by losing NaH. Similarly, species with MW = 185 + 76 n (
n = 0, 1, 2) which observed in the positive mode (to see in
Figure 7), is linked to those with MW = 109 + 76 k (
k = 1, 2, 3) in
Figure 6. The extensive one-to-one correspondence suggests that the cyclization of 2-aryl-thiophenolate by losing NaH should be common.
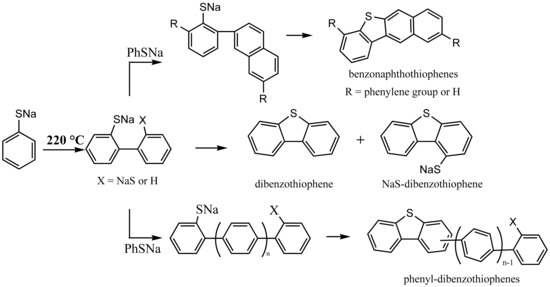
3.4. Thermal Degradation Chemistry of Thiophenolate
On the basis of analysis of gaseous products and residues, the thermal degradation of thiophenolate has very similar chemistry as that of phenolate.
Scheme 2 lists typical steps that account for the results obtained in the TGA-FTIR, GC-MS, FTIR, and Q-TOF.
First, the homolysis of the NaSPh‒H bond generates an aryl radical anion (I) and a H• as shown in Reaction (1). The H• attacks the ipso-carbon bonding to –SNa yielding benzene as observed in the TGA-FTIR. Hydroarylation of thiophenolate by the aryl radical and the H radical followed by the elimination of NaSH produces a phenylthiophenolate (II), which subsequently undergoes more hydroarylation reactions to generate polyaromatics with MW = 109 + 76 k (k = 0, 1, 2, 3) as detected in the Q-TOF. Phenylthiophenolate can also cyclize to DBT which is discovered in the gas phase while a cyclization of phenyl substituted phenylthiophenolate readily generates phenyl substituted DBT (phenyl-DBT), which appears at MW = 185 + 76 n (n = 0, 1, 2) in the Q-TOF positive mode.
In addition, the radical (I) can self-combine to a bithiophenolate (III), whose hydroarylation produces species (IV) with MW = 109 + 32 m +76 k (
m = 1;
k = 0, 1, 2, 3). With –SNa in an ortho- position of the benzene ring, (IV) cyclize to DBT or NaS-substituted DBT or those with MW = 215 + 76 n (
n = 0, 1, 2) in
Figure 7 as shown in Reaction (9). On the other hand, the sulfur radical couples with the radical (I) to form a benzenedithiolate and subsequently benzenetrithiolate, accounting for species with MW = 109 + 32 m (
m = 0, 1, 2).
The above schemes illustrate not only the formation of DBT but also the generation of some unusual species such as phenyl-DBT. The latter has been proposed as an alternative molecular marker of maturity of coals but its origin has remained elusive [
15,
42]. Further, benzonaphthothiophenes, which have been found in crude oils, coals, and sediment extracts [
15], can also be readily elucidated. Referencing the formation of polyaromatic hydrocarbons during the thermal degradation of phenol [
27], an exemplified pathway to benzonaphthothiophenes is shown in
Scheme 3. In addition, the presence of benzonaphthothiophenes is consistent with MW = 235 + 76 n (
n = 0, 1, 2) observed in the APCI positive mode.
The thermal degradation of thiophenolate has been so far investigated at relatively high temperatures. To confirm that it can also take place at coalification temperatures which are typically below 300 °C, sodium thiophenolate was further subjected to thermal treatment at a reduced temperature.
3.5. Thermal Degradation of Thiophenolate at Low Temperature
Sodium thiophenolate was thermally treated at 220–225 °C from 30 to 120 h. The GC spectra of the collected residues are shown in
Figure 8. It can be seen that a new peak albeit with a weak absorption emerges at 16.2 min after 30 h, suggesting the presence of trace DBT. The slow generation of DBT from the thermolysis of thiophenolate at the relatively low temperature should come without surprise since the formation of DBTs in nature takes many years. However, the peak of DBT grows significantly with time and becomes one of major species in the residue obtained after 120 h. This species is further confirmed to be DBT by its MS spectrum with a molecular weight of 184.
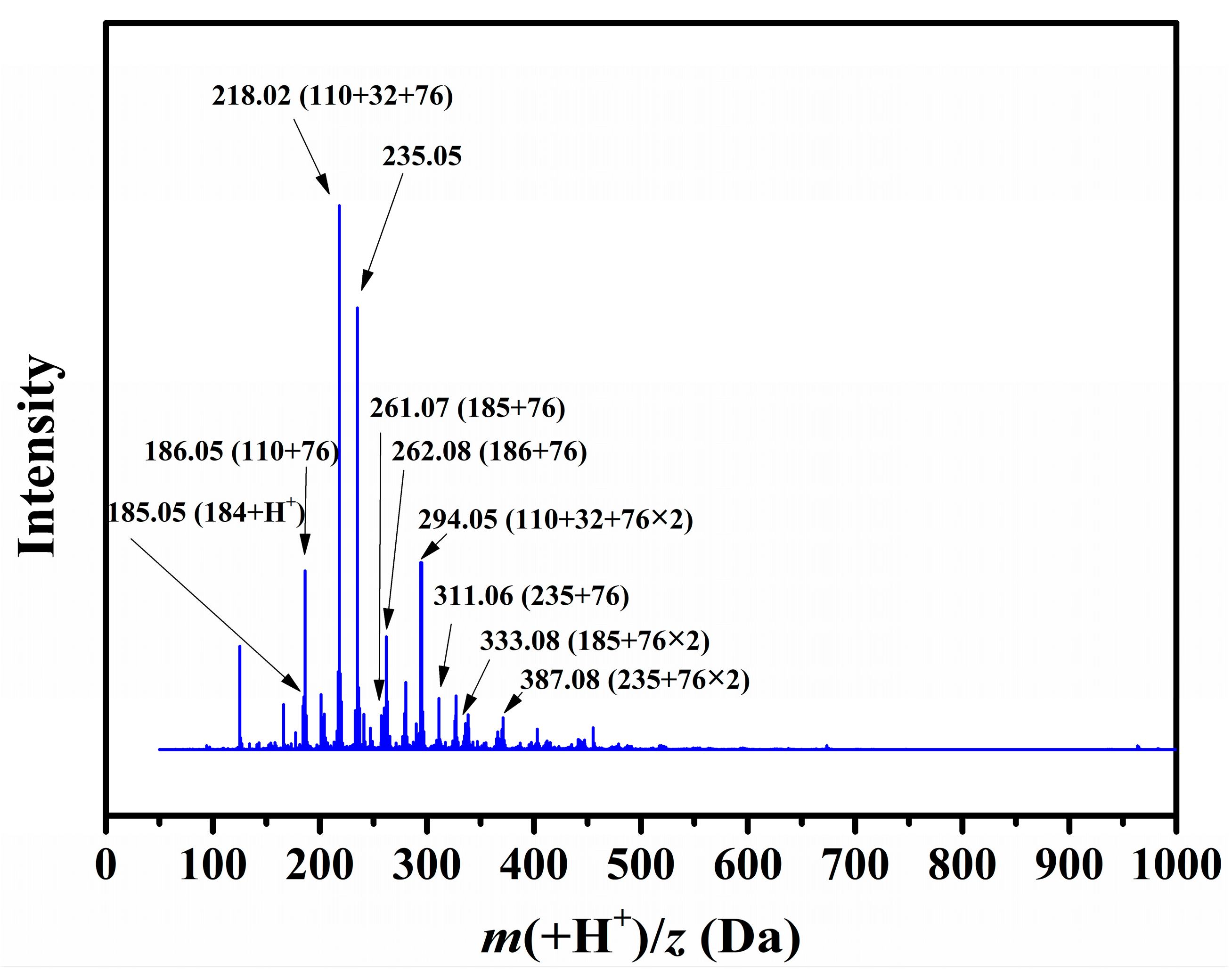
What is more, the Q-TOF results of the residues clearly indicates the presence of polyaromatics with MW = 109 + 76 k (
k = 0, 1, 2) (in the negative mode) and 110 + 76 k (
k = 0, 1, 2, 3) (in the positive mode), NaS‒substituted DBT (MW = 215) (in the negative mode), phenyl‒DBT with MW = 185 + 76 n (
n = 0, 1) (in the positive mode) and benzonaphthothiophenes with MW = 235 + 76 n (
n = 0, 1, 2) (in the positive mode) as shown in
Figure 9 and
Figure 10. The presence of these species is strong evidence that the chemistry leading to the generation of DBTs at high temperature also takes place at coalification temperatures well below 300 °C.
3.6. Geochemical Feasibility of DBTs from Thiophenols in Coals and Oils
The above investigations clearly demonstrate that thiophenolate can be transformed to DBTs. Although this transformation is realized in a lab environment, the conditions used in this work are actually readily achievable in nature. As a matter of fact, the chemistry leading to the generation of DBTs is so simple that it only requires the presence of thiophenolate and a mild temperature.
For thiophenolate, it is simply yielded from a neutralization reaction of thiophenol with alkalis or inorganic salts. The existing evidence points out that thiophenol is one of the major organic sulfur-containing components in certain coals and oils [
6,
10,
43]. For other types of coals or sedimentary matter containing limited quantities of thiophenol, it can be generated from phenol and inorganic sulfide [
39,
40]. In this aspect, thiophenol works as an intermediate for the generation of DBT so a large quantity of thiophenol is not required. On the other hand, alkalis or inorganic salts are abundantly distributed in sedimentary organic matter [
29,
31,
44]. As a matter of fact,
Scheme 2 clearly shows the ion, Na
+, is regenerated so only a catalytic amount of salts is needed to catalyze the thermal degradation of thiophenol. Thus, neither thiophenolate or salts are exotic compounds in nature, nor are they necessitated in large quantities.
For the condition of the mild reaction temperature, it can be easily achieved by the geothermal energy that also provides heat for the coalification process [
45,
46,
47]. What is more, the current investigation shows that the higher the temperature of the thermal degradation of thiophenolate is, the faster the generation of DBTs is. Thus, the thermal degradation of thiophenolate can undoubtedly occur at a much lower temperature albeit at a slower rate. Consequently, both the reactants and the thermal condition for the generation of DBTs from thiophenol are clearly accessible in a real coalification scenario.
Further, the chemistry of thiophenolate to DBTs is hardly affected by external substances surrounding thiophenolate since thiophenolate is a solid even at 220 °C. The degradation reactions occurring inside the solid shall be the same as those taking place in the lab environment. Although DBT is only one of the major sulfur-containing products in the lab simulation, other major products such as diphenyl sulfide can transform into DBT albeit with a slower rate. Consequently, DBT with a much higher yield will be generated from thiophenolate in nature. Overall, on the basis of the availability of raw materials and reaction conditions that are attainable in nature, the formation of DBTs from thiophenols is highly geochemically feasible.
4. Conclusions
The thermal degradation of sodium thiophenolate has been studied at various temperatures. Similar to phenolate, the thiophenolate begins the degradation with an initial cleavage of the Ar−H bond which generates aryl radicals and hydrogen radicals. The subsequent hydrogenolysis and hydroarylation followed by the elimination of NaSH or the cyclization reaction largely account for the formation of benzene, multi-sulfuretted benzenes, DBT, phenyl or –SNa substituted DBTs, benzonapthothiophenes as well as sulfur-containing oligomers.
The generation of DBTs from thiophenolate is chemically simple and highly geochemically feasible. This new transformation represents a completely different approach from those based on the insertion of sulfur into biphenyl. It has a merit to easily unify the chemical pathways to DBTs from thiophenolate and DBFs from phenolate. Further, it clearly shows that a variety of PASHs come from a common starting material, thiophenolate. This suggests that a ratio of these PASHs, for example, the ratio of phenyl-DBT/DBT or benzonapthothiophene/DBT, might be a good indicator for the evaluation of maturity of coals and oils since they are formed in a sequential manner. Since thiophenol can be readily generated from phenol, an important source of DBTs in coals, crude oils and organic sedimentary matter might very well be inorganic sulfides and phenols.















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





