
Parametric Study of Pt/C-Catalysed Hydrothermal Decarboxylation of Butyric Acid as a Potential Route for Biopropane Production



Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methodology
2.2.1. Batch Reactor Procedure
2.2.2. Product Analysis
2.2.3. Reaction Products Analyses
2.2.4. Catalyst Characterisation
3. Results
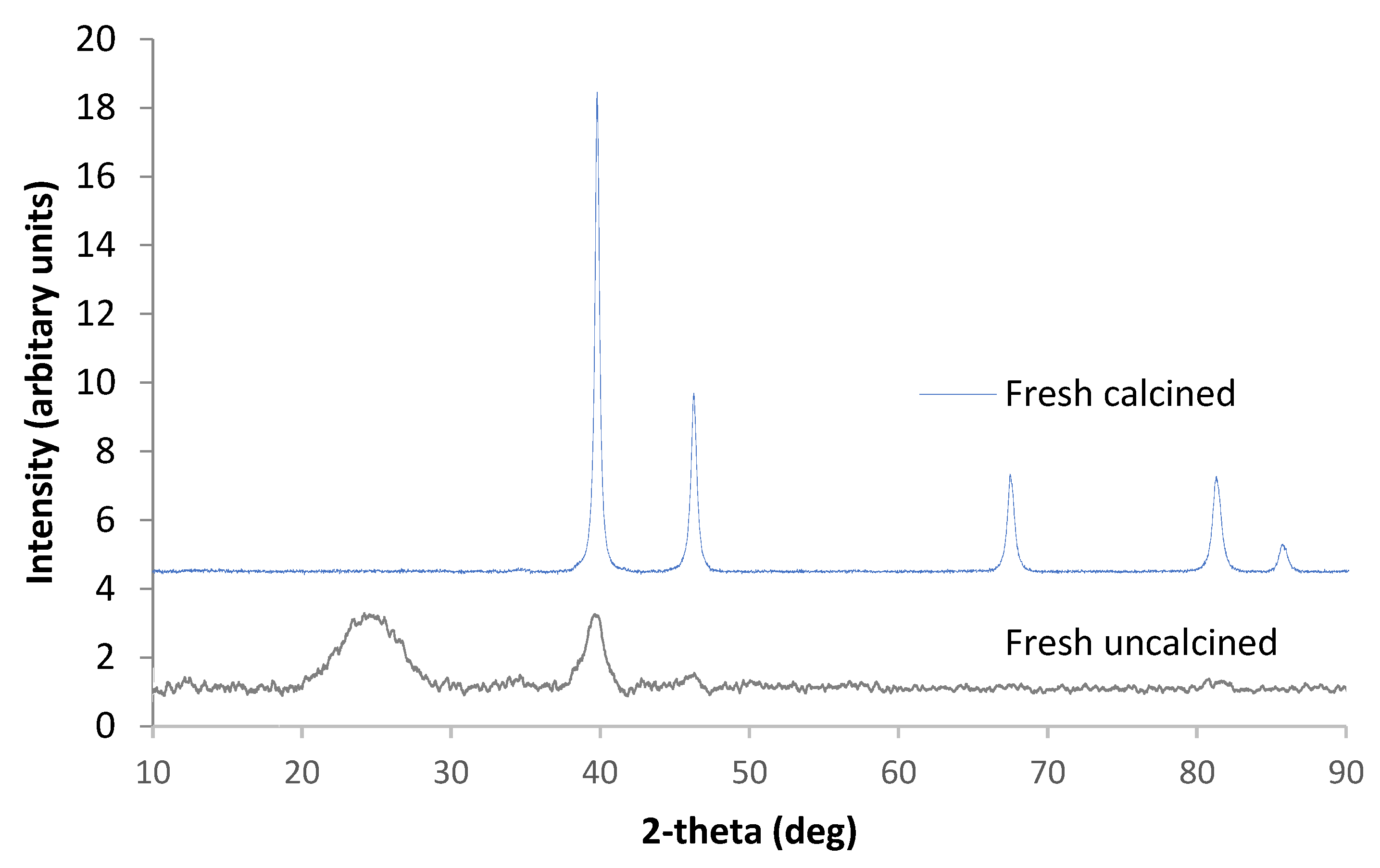
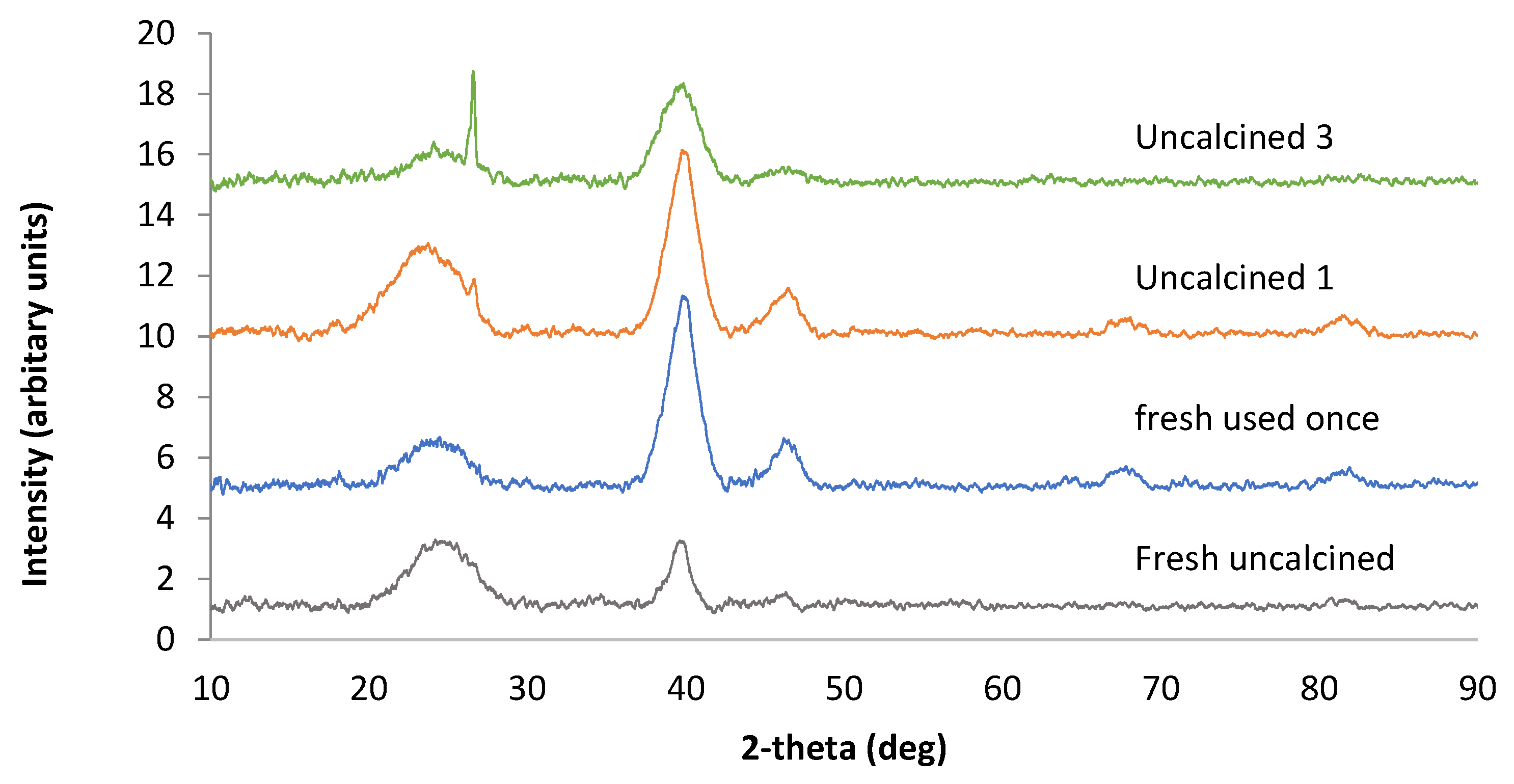
3.1. Catalyst Characterisation
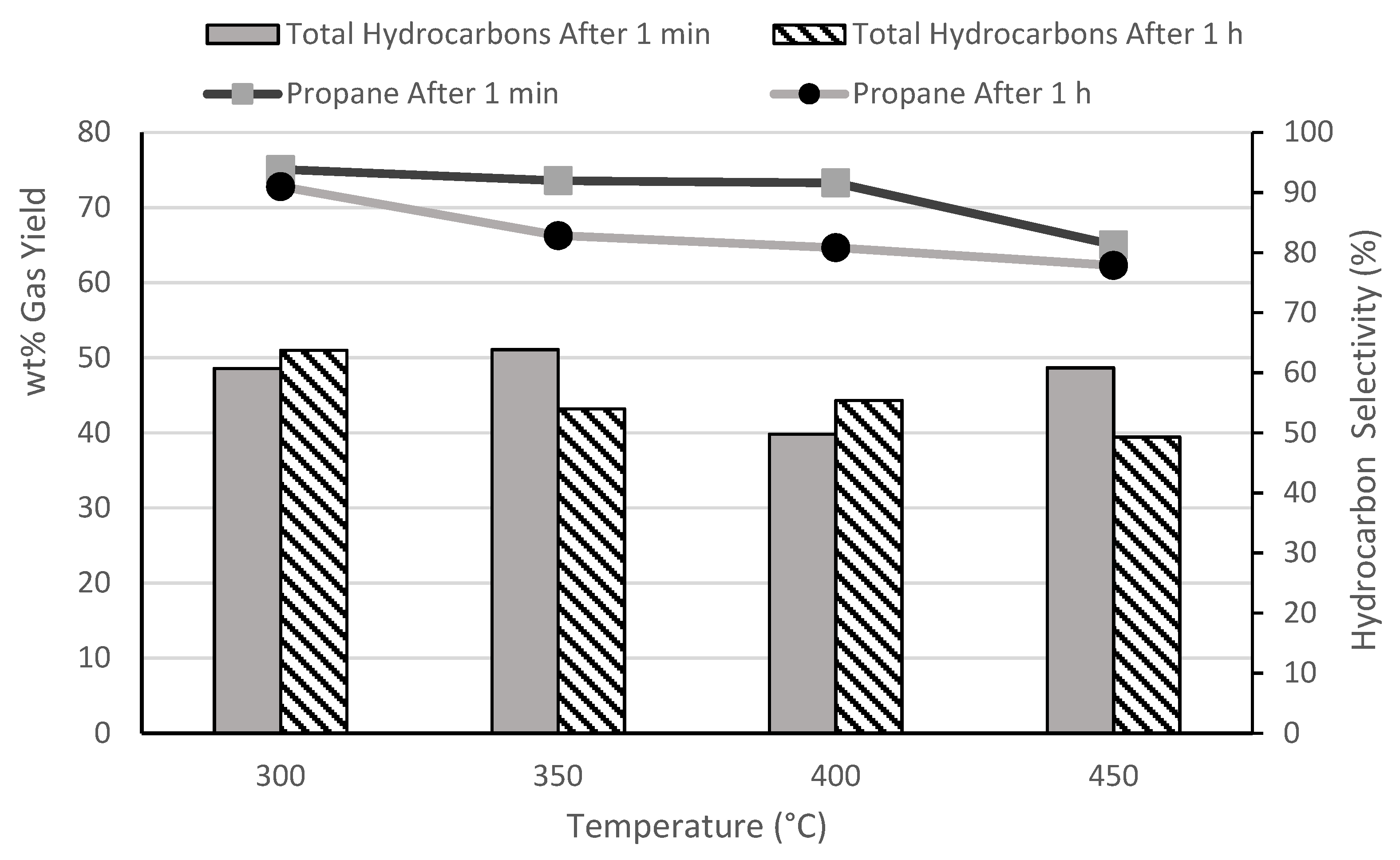
3.2. Effect of Temperature
3.3. Active Catalyst Surface
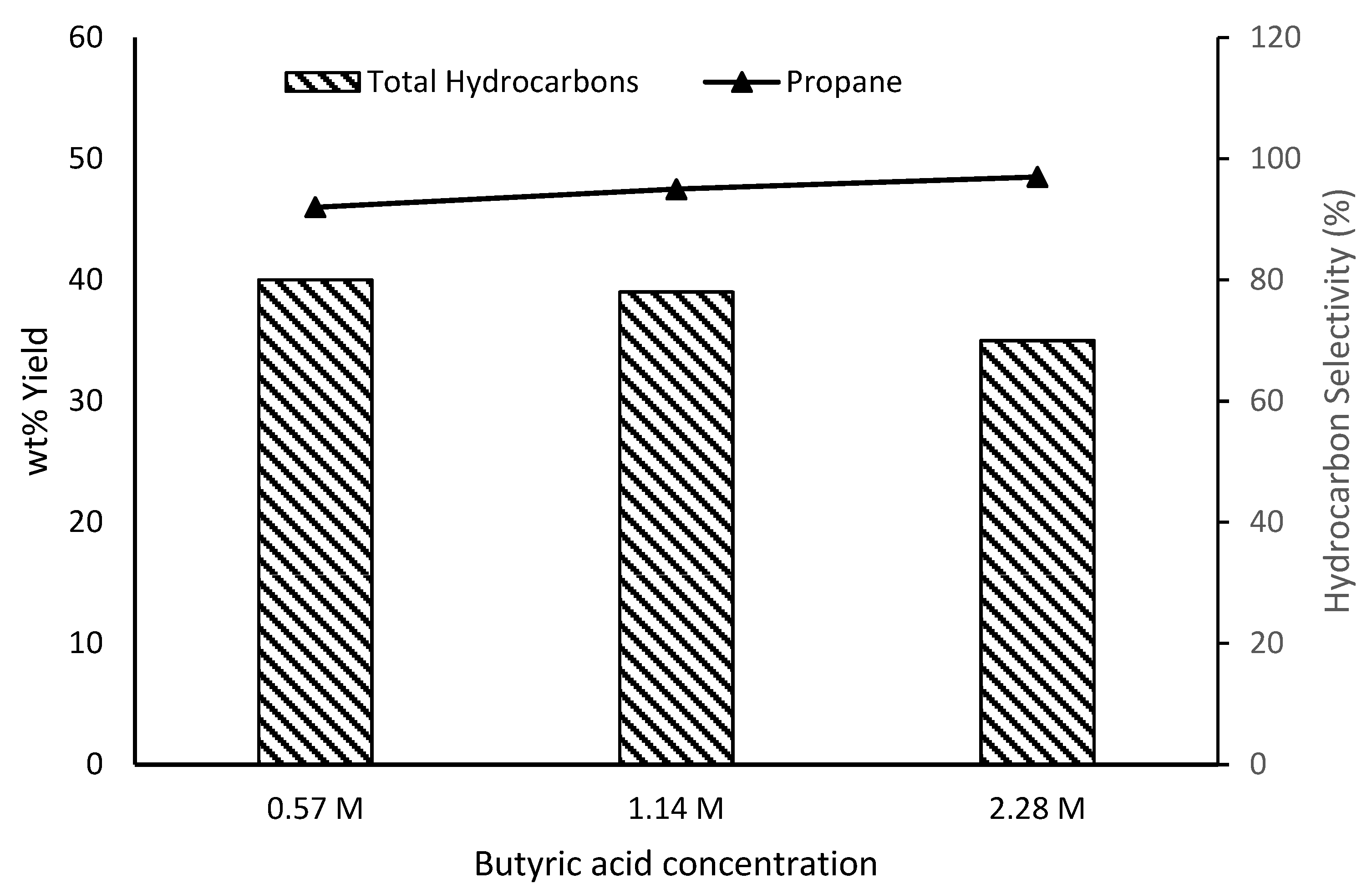
3.4. Effect of Butyric Acid Loading
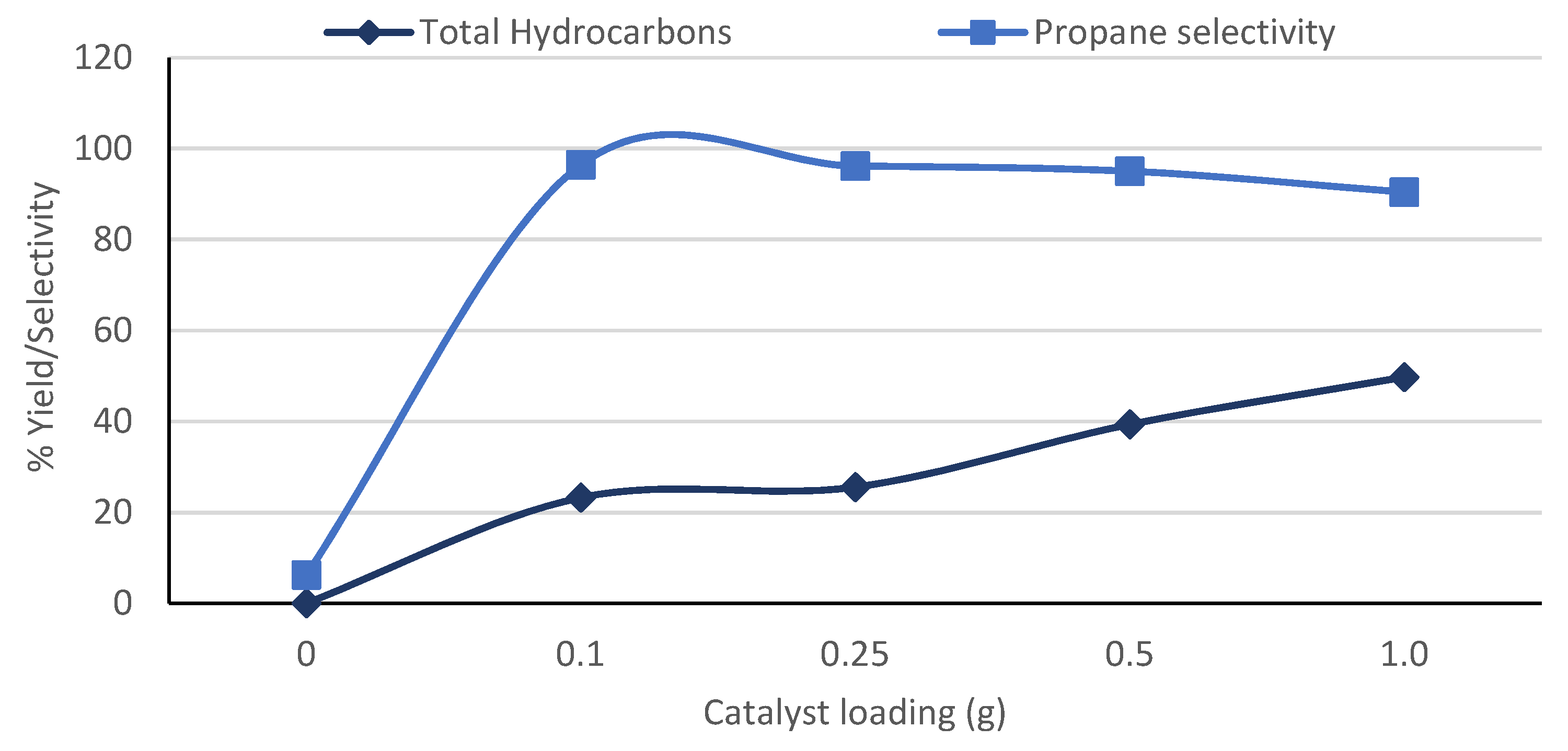
3.5. Effect of Catalyst Loading
3.6. Effect of Reaction Time with Lower Pt/C Loading
3.7. Catalyst Reuse
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Goshima, T.; Ikeda, K.; Fukudome, K.; Mizuta, K.; Mitsuyoshi, S.; Tsutsui, T. Conversion of Biomass-Derived Oxygen-Containing Intermediates into Chemical Raw Materials with Zeolite. AMM 2014, 625, 298–305. [Google Scholar] [CrossRef]
- De, S.; Saha, B.; Luque, R. Hydrodeoxygenation processes: Advances on catalytic transformations of biomass-derived platform chemicals into hydrocarbon fuels. Bioresour. Technol. 2015, 178, 108–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, T.; Santillan-Jimenez, E.; Harman-Ware, A.E.; Ji, Y.; Grubb, D.; Crocker, M. Catalytic deoxygenation of triglycerides to hydrocarbons over supported nickel catalysts. Chem. Eng. J. 2012, 189, 346−355. [Google Scholar] [CrossRef]
- Ni, L.; Xin, J.; Jiang, K.; Chen, L.; Yan, D.; Lu, X.; Zhang, S. One-step conversion of biomass-derived furanics into aromatics by Brønsted acid ionic liquids at room temperature. ACS Sustain. Chem. Eng. 2018, 6, 2541−2551. [Google Scholar] [CrossRef]
- Shi, Y.; Xing, E.; Wu, K.; Wang, J.; Yang, M.; Wu, Y. Recent progress on upgrading of bio-oil to hydrocarbons over metal/zeolite bifunctional catalysts. Catal. Sci. Technol. 2017, 7, 2385–2415. [Google Scholar] [CrossRef]
- Hollak, S.A.W.; Gosselink, R.W.; van Es, D.S.; Bitter, J.H. Comparison of tungsten and molybdenum carbide catalysts for the hydrodeoxygenation of oleic acid. ACS Catal. 2013, 3, 2837−2844. [Google Scholar] [CrossRef]
- Sotelo-Boyas, R.; Liu, Y.; Minowa, T. Renewable diesel production from the hydrotreating of rapeseed oil with Pt/Zeolite and NiMo/Al2O3 catalysts. Ind. Eng. Chem. Res. 2011, 50, 2791−2799. [Google Scholar] [CrossRef]
- Liu, Y.; Sotelo-Boyás, R.; Murata, K.; Minowa, T.; Sakanishi, K. Hydrotreatment of Vegetable Oils to Produce Bio-Hydrogenated Diesel and Liquefied Petroleum Gas Fuel over Catalysts Containing Sulfided Ni–Mo and Solid Acids. Energy Fuels 2011, 25, 4675–4685. [Google Scholar] [CrossRef]
- Johnson, E. Process technologies and projects for BioLPG. Energies 2019, 12, 250. [Google Scholar] [CrossRef] [Green Version]
- Fu, J.; Lu, X.; Savage, P.E. Hydrothermal decarboxylation and hydrogenation of fatty acids over Pt/C. ChemSusChem 2011, 4, 481–486. [Google Scholar] [CrossRef] [Green Version]
- Immer, J.G.; Lamb, H.H. Fed-Batch Catalytic deoxygenation of free fatty acids. Energy Fuels 2010, 24, 5291–5299. [Google Scholar] [CrossRef]
- López-Garzón, C.S.; Straathof, A.J. Recovery of carboxylic acids produced by fermentation. Biotechnol. Adv. 2014, 32, 873–904. [Google Scholar] [CrossRef] [PubMed]
- Peterson, E.C.; Daugulis, A.J. Demonstration of in situ product recovery of butyric acid via CO2-facilitated pH swings and medium development in two-phase partitioning bioreactors. Biotechnol. Bioeng. 2014, 111, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Chun, J.; Choi, O.; Sang, B.-I. Enhanced extraction of butyric acid under high-pressure CO2 conditions to integrate chemical catalysis for value-added chemicals and biofuels. Biotechnol. Biofuels 2018, 11, 119–126. [Google Scholar] [CrossRef]
- Hossain, M.Z.; Chowdhury, M.B.I.; Jhawar, A.K.; Xu, W.Z.; Biesinger, M.C.; Charpentier, P.A. Continuous hydrothermal decarboxylation of fatty acids and their derivatives into liquid hydrocarbons using Mo/Al2O3 catalyst. ACS Omega 2018, 3, 7046–7060. [Google Scholar] [CrossRef] [Green Version]
- Santillan-Jimenez, E.; Crocker, M. Catalytic deoxygenation of fatty acids and their derivatives to hydrocarbon fuels via decarboxylation/decarbonylation. J. Chem. Technol. Biotechnol. 2012, 87, 1041–1050. [Google Scholar] [CrossRef]
- Chen, L.; Zhu, Y.; Zheng, H.; Zhang, C.; Zhang, B.; Li, Y. Aqueous-phase hydrodeoxygenation of carboxylic acids to alcohols or alkanes over supported Ru catalysts. J. Mol. Catal. A Chem. 2011, 351, 217–227. [Google Scholar] [CrossRef]
- Fu, J.; Lu, X.; Savage, P.E. Catalytic hydrothermal deoxygenation of palmitic acid. Energy Environ. Sci. 2010, 3, 311–317. [Google Scholar] [CrossRef]
- Hossain, M.Z.; Chowdhury, M.B.I.; Jhawar, A.K.; Xu, W.Z.; Charpentier, P.A. Continuous low-pressure decarboxylation of fatty acids to fuel-range hydrocarbons with in situ hydrogen production. Fuel 2018, 212, 470–478. [Google Scholar] [CrossRef]
- Yeh, T.M.; Hockstad, R.L.; Linic, S.; Savage, P.E. Hydrothermal decarboxylation of unsaturated fatty acids over PtSnx/C catalysts. Fuel 2015, 156, 219–224. [Google Scholar] [CrossRef]
- WPLGA. Bio-LPG Charter of Benefits. 2021. Available online: https://www.wlpga.org/wp-content/uploads/2021/03/BioLPG-Charter-of-Benefits.pdf (accessed on 22 May 2021).
- Antonini, C.; Treyer, K.; Streb, A.; van der Spek, M.; Bauer, C.; Mazzotti, M. Hydrogen production from natural gas and biomethane with carbon capture and storage—A techno-environmental analysis. Sustain. Energy Fuels 2020, 4, 2967–2986. [Google Scholar] [CrossRef] [Green Version]
- Yeh, T.; Linic, S.; Savage, P.E. Deactivation of Pt catalysts during hydrothermal decarboxylation of butyric acid. ACS Sustain. Chem. Eng. 2014, 2, 2399–2406. [Google Scholar] [CrossRef]
- Borges, A.C.P.; Onwudili, J.A.; Andrade, H.M.C.; Alves, C.T.; Ingram, A.; Vieira de Melo, S.A.B.; Torres, E.A. Catlaytic supercritical water gasification of eucalyptus wood chips in a batch reactor. Fuel 2019, 255, 115804–115812. [Google Scholar] [CrossRef]
- Euopean Economic Community. Commission Regulation (EEC) No 2568/91 of 11 July 1991 on the Characteristics of Olive Oil and Olive-Residue Oil and on the Relevant Methods of Analysis; European Union: Brussels, Belgium, 1991. [Google Scholar]
- Taylor, M.J.; Beaumont, S.K.; Islam, M.J.; Tsatsos, S.; Parlett, C.A.M.; Issacs, M.A.; Kyriakou, G. Atom efficient PtCu bimetallic catalysts and ultra dilute alloys for the selective hydrogenation of furfural. Appl. Catal. B Environ. 2021, 284, 119737–119746. [Google Scholar] [CrossRef]
- ASTM. D3174-12(2018)e1. Standard Test Method for Ash in the Analysis Sample of Coal and Coke from Coal; ASTM International: West Conshohocken, PA, USA, 2018. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Temperature (°C) | Residence Time | Butyric Acid Loading (g) | Catalyst Loading (g) |
|---|---|---|---|
| 300 | 0 min | 0.5 | 0.10 |
| 350 | 1 h | 1 | 0.25 |
| 400 | 4 h | 2 | 0.50 |
| 450 | 7 h | - | 1.00 |
| Temperature (°C) | |||||
|---|---|---|---|---|---|
| Reaction Time | Conversion/ Component Yields | 300 | 350 | 400 | 450 |
| 1 min | Conversion (%) | 85 | 90 | 94 | 100 |
| Hydrogen (%) | 0.17 | 0.26 | 0.24 | 0.5 | |
| Methane (%) | 0.3 | 0.8 | 0.7 | 4.9 | |
| Ethane (%) | 2.4 | 2.9 | 2.4 | 3.5 | |
| Propane (%) | 45.6 | 47.0 | 36.5 | 39.5 | |
| Butane (%) | 0.1 | 0.1 | 0.1 | 0.1 | |
| CO2 (%) | 36.1 | 38.9 | 54.5 | 58.2 | |
| 1 h | Conversion (%) | 100 | 99 | 99 | 100 |
| Hydrogen (%) | 0.15 | 0.15 | 0.45 | 0.58 | |
| Methane (%) | 1.0 | 3.0 | 5.0 | 6.1 | |
| Ethane (%) | 3.4 | 4.2 | 2.9 | 2.0 | |
| Propane (%) | 46.4 | 35.8 | 35.8 | 30.7 | |
| Butane (%) | 0.1 | 0.1 | 0.1 | 0.1 | |
| CO2 (%) | 49.1 | 55.5 | 54.0 | 57.3 | |
| Catalyst Type | ||
|---|---|---|
| Conversion/ Component Yields | Unreduced | Reduced |
| Conversion (%) | 100 | 99 |
| Methane (%) | 0.6 | 0.4 |
| Ethane (%) | 2.3 | 1.3 |
| Propane (%) | 37.3 | 31.9 |
| Butane (%) | 0.1 | 0.1 |
| CO2 (%) | 67.5 | 69.6 |
| Total hydrocarbons | 40.4 | 39.9 |
| Propane hydrocarbon selectivity | 92.2 | 94.2 |
| Butyric Acid Loading (g) | |||
|---|---|---|---|
| Conversion/ Component Yields | 0.5 (0.57 M) | 1.0 (1.14 M) | 2.0 (2.28 M) |
| Conversion (%) | 100 | 85 | 73 |
| Methane (%) | 0.6 | 0.2 | 0.1 |
| Ethane (%) | 2.3 | 1.5 | 0.9 |
| Propane (%) | 37.3 | 37.4 | 33.9 |
| Butane (%) | 0.1 | 0.1 | 0.1 |
| CO2 (%) | 67.5 | 45.7 | 38.1 |
| Catalyst Loading (g) | |||||
|---|---|---|---|---|---|
| Conversion/ Component Yields | None | 0.1 | 0.25 | 0.5 | 1.0 |
| Conversion (%) | 2 | 33 | 66 | 85 | 87 |
| Methane (%) | 0.0 | 0.1 | 0.1 | 0.2 | 0.8 |
| Ethane (%) | 0.0 | 0.6 | 0.7 | 1.5 | 3.8 |
| Propane (%) | 0.0 | 22.4 | 24.6 | 37.4 | 45.0 |
| Butane (%) | 0.0 | 0.0 | 0.0 | 0.1 | 0.1 |
| CO2 (%) | 2.7 | 10.1 | 40.0 | 45.7 | 36.8 |
| Residence Time (h) | ||||
|---|---|---|---|---|
| Catalyst Loading | Conversion/ Components Yields | 1 | 4 | 7 |
| 0.1 g Pt/C | Conversion (%) | 33 | 53 | 55 |
| Methane (%) | 0.1 | 0.1 | 0.1 | |
| Ethane (%) | 0.6 | 0.8 | 1 | |
| Propane (%) | 22.4 | 30.9 | 32 | |
| Butane (%) | 0 | 0 | 0 | |
| CO2 (%) | 10.1 | 20.8 | 21.9 | |
| 0.25 g Pt/C | Conversion (%) | 66 | 65 | 65 |
| Methane (%) | 0.1 | 0.1 | 0.2 | |
| Ethane (%) | 0.7 | 1 | 1.2 | |
| Propane (%) | 24.6 | 32.6 | 35.2 | |
| Butane (%) | 0 | 0 | 0 | |
| CO2 (%) | 40 | 31.3 | 28.6 | |
| Catalyst Reuse | ||||
|---|---|---|---|---|
| Conversion/ Component Yields | Fresh | Uncalcined 1 | Uncalcined 2 | Uncalcined 3 |
| Conversion (%) | 78 | 86 | 92 | 95 |
| Methane (%) | 0.3 | 0.3 | 0.3 | 0.2 |
| Ethane (%) | 1.7 | 1.5 | 1.8 | 1.5 |
| Propane (%) | 41.2 | 39.5 | 39.1 | 35.4 |
| Butane (%) | 0.1 | 0.1 | 0.1 | 0.1 |
| CO2 (%) | 34.7 | 44.6 | 50.9 | 57.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Razaq, I.; Simons, K.E.; Onwudili, J.A. Parametric Study of Pt/C-Catalysed Hydrothermal Decarboxylation of Butyric Acid as a Potential Route for Biopropane Production. Energies 2021, 14, 3316. https://doi.org/10.3390/en14113316
Razaq I, Simons KE, Onwudili JA. Parametric Study of Pt/C-Catalysed Hydrothermal Decarboxylation of Butyric Acid as a Potential Route for Biopropane Production. Energies. 2021; 14(11):3316. https://doi.org/10.3390/en14113316
Chicago/Turabian StyleRazaq, Iram, Keith E. Simons, and Jude A. Onwudili. 2021. "Parametric Study of Pt/C-Catalysed Hydrothermal Decarboxylation of Butyric Acid as a Potential Route for Biopropane Production" Energies 14, no. 11: 3316. https://doi.org/10.3390/en14113316