
The Hydration and Volume Expansion Mechanisms of Modified Expansive Cements for Sustainable In-Situ Rock Fragmentation: A Review



Abstract
:1. Introduction
2. Volume Expansion and Rock Fracturing through Expansive Materials
3. Components of SREMA
4. Hydration Mechanism of Expansive Components of SREMA
4.1. Hydration of Calcium Oxide
4.2. Hydration of Alite
4.2.1. Molecular Scale Hydration Mechanism of Alite
- Dissolution of tricalcium silicate.
- 2.
- Proton abstraction by the surface oxide groups from the surrounding water molecules.
- 3.
- Formation of hydrated silicates with further proton transfer.
- 4.
- Formation of Ca(OH)2.
- 5.
- Formation of CSH.
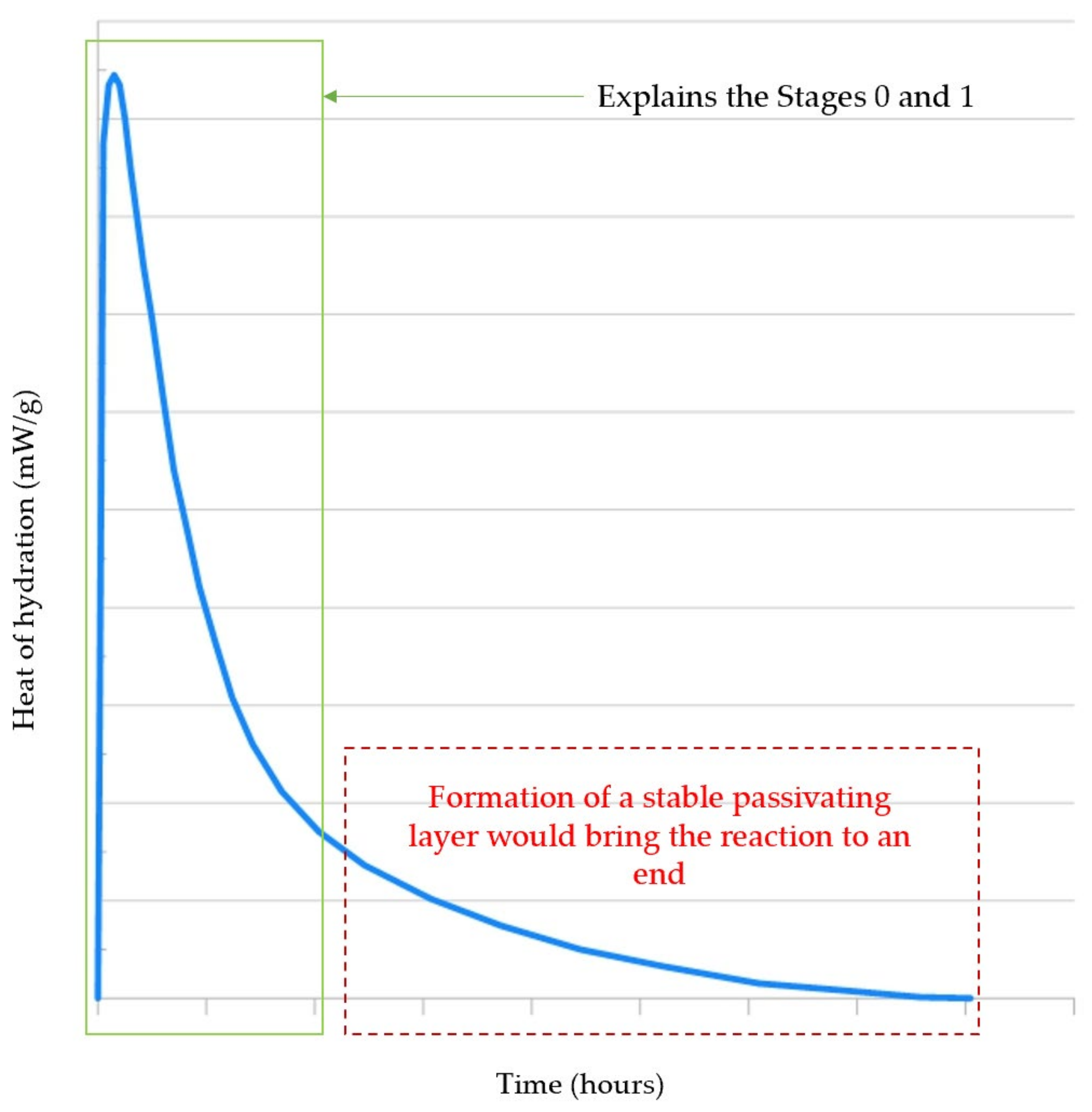
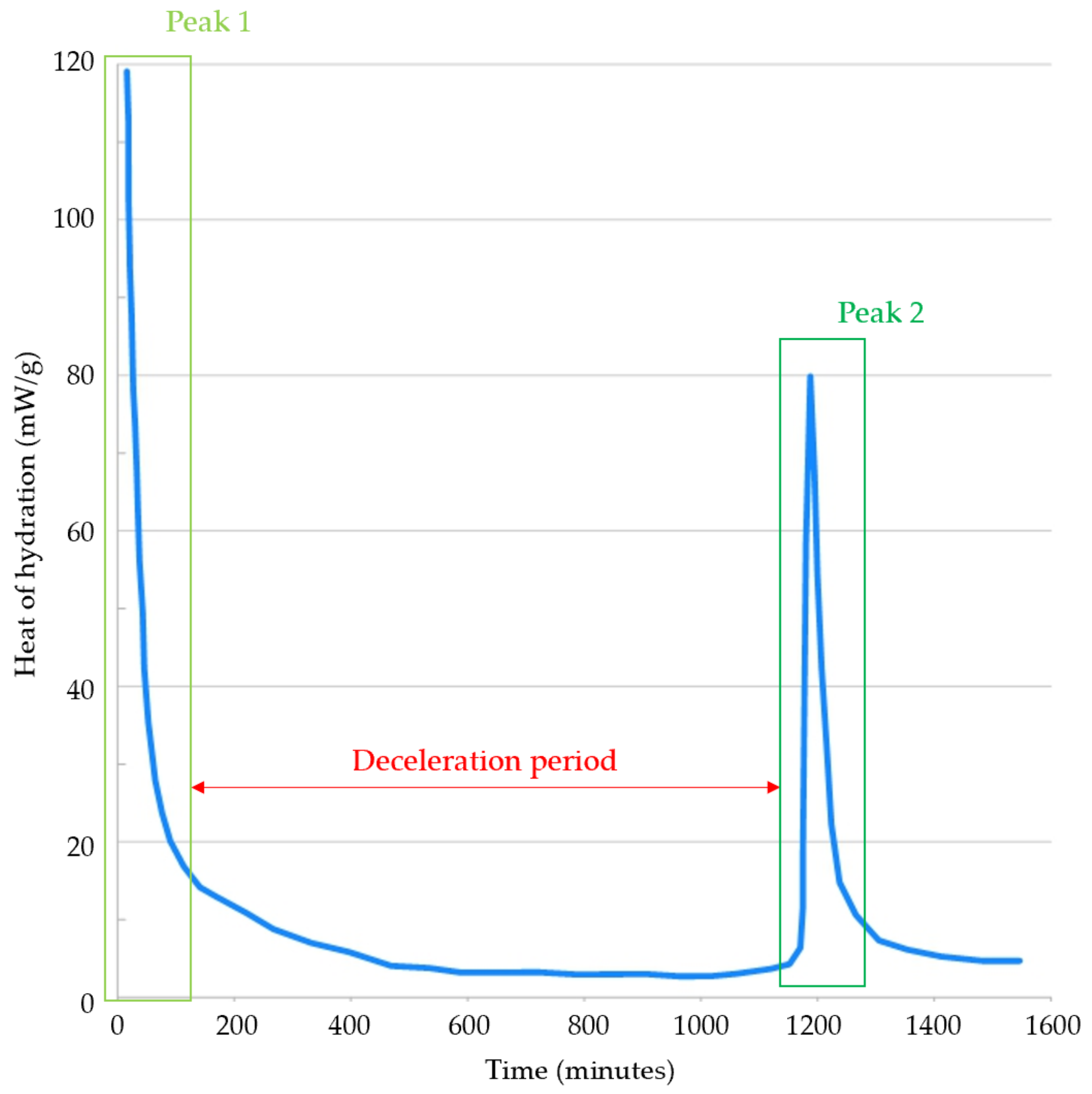
4.2.2. Time-Dependent Hydration Mechanisms of Alite
Protective Barrier Hypothesis
Slow Dissolution Hypothesis
4.3. Hydration of Tricalcium Aluminate
4.3.1. In the Absence of Sulfates
4.3.2. In the Presence of Sulfates
4.4. Combined Hydration of Alite and Tricalcium Aluminate
4.4.1. Effect of Sulfate Concentration
(CaO)3(Al2O3)(SiO2)·4H2O (s, interface) ↓
4.4.2. Effect of Alite to Tricalcium Aluminate Ratio
4.4.3. Effect of Temperature
5. Additives of SREMA and Their Mechanisms of Action
5.1. Effect of Welan Gum

5.2. Effect of SNSF

5.3. Effect of Calcium Chloride
5.4. Additives for Further Development of SREMA
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Holland, A.A. Earthquakes Triggered by Hydraulic Fracturing in South-Central Oklahoma. Bull. Seismol. Soc. Am. 2013, 103, 1784–1792. [Google Scholar] [CrossRef]
- Inoue, H.; Lisitsyn, I.V.; Akiyama, H.; Nishizawa, I. Pulsed Electric Breakdown and Destruction of Granite. Jpn. J. Appl. Phys. 1999, 38, 6502. [Google Scholar] [CrossRef]
- Winter, T.; Harvey, J.; Franke, O.; Alley, W. Ground Water and Surface Water: A Single Resource, Circular 1139; DIANE Publishing Inc.: Collingdale, PA, USA, 1998. [Google Scholar]
- Cooley, H.; Donnelly, K.; Ross, N.; Luu, P. Hydraulic Fracturing and Water Resources: Separating the Frack from the Fiction; Pacific Institute: Oakland, CA, USA, 2012. [Google Scholar]
- Levy, S.M. Calculations Relating to Concrete and Masonry. In Construction Calculations Manual; Butterworth-Heinemann: Oxford, UK, 2012; pp. 211–264. [Google Scholar]
- Bentur, A.; Ish-Shalom, M. Properties of Type K Expensive Cement of Pure Components II-Proposed Mechanism of Ettringite Formation and Expansion in Unrestrained Paste of Pure Expansive Component. Cem. Concr. Res. 1974, 4, 709–721. [Google Scholar] [CrossRef]
- Cohen, M. Modeling of Expansive Cements. Cem. Concr. Res. 1983, 13, 519–528. [Google Scholar] [CrossRef]
- Tang, S.; Huang, R.; Wang, S.; Bao, C.; Tang, C. Study of the Fracture Process in Heterogeneous Materials Around Boreholes Filled with Expansion Cement. Int. J. Solids Struct. 2017, 112, 1–15. [Google Scholar] [CrossRef]
- Hinze, J.; Brown, J. Properties of Soundless Chemical Demolition Agents. J. Constr. Eng. Manag. 1994, 120, 816–827. [Google Scholar] [CrossRef]
- Gambatese, J.A. Controlled Concrete Demolition Using Expansive Cracking Agents. J. Constr. Eng. Manag. 2003, 129, 98–104. [Google Scholar] [CrossRef]
- De Silva, V.; Ranjith, P.; Perera, M.; Wu, B.; Rathnaweera, T. Investigation of the Mechanical, Microstructural and Mineralogical Morphology of Soundless Cracking Demolition Agents during the Hydration Process. Mater. Charact. 2017, 130, 9–24. [Google Scholar] [CrossRef]
- Laefer, D.F.; Ambrozevitch-Cooper, N.; Huynh, M.; Midgette, J.; Ceribasi, S.; Wortman, J. Expansive Fracture Agent Behaviour for Concrete Cracking. Mag. Concr. Res. 2010, 62, 443–452. [Google Scholar] [CrossRef] [Green Version]
- De Silva, V.; Ranjith, P.; Perera, M.; Wu, B.; Rathnaweera, T. A Modified, Hydrophobic Soundless Cracking Demolition Agent for Non-explosive Demolition and Fracturing Applications. Process. Saf. Environ. Prot. 2018, 119, 1–13. [Google Scholar] [CrossRef]
- Gamage, R.P.; De Silva, R.S.V. Demolition Agent. U.S. Patent 10836955B2, 17 November 2020. [Google Scholar]
- Sinclair, L.; Thompson, J. In Situ Leaching of Copper: Challenges and Future Prospects. Hydrometallurgy 2015, 157, 306–324. [Google Scholar] [CrossRef]
- Zhou, Y.; Li, G.; Xu, L.; Liu, J.; Sun, Z.; Shi, W. Uranium Recovery from Sandstone-Type Uranium Deposit by Acid In-Situ Leaching-An Example from the Kujieertai. Hydrometallurgy 2020, 191, 105209. [Google Scholar] [CrossRef]
- De Rouffignac, E.P.; Vinegar, H.J.; Wellington, S.L.; Karanikas, J.M.; Berchenko, I.E.; Maher, K.A.; Zhang, E.; Fowler, T.D.; Keedy, C.R.; Ryan, R.C.; et al. In Situ Thermal Processing of a Coal Formation Using Heat Sources Positioned within Open Wellbores. U.S. Patent 6,752,210, 22 June 2004. [Google Scholar]
- Hall, A.; Scott, J.A.; Shang, H. Geothermal Energy Recovery from Underground Mines. Renew. Sustain. Energy Rev. 2011, 15, 916–924. [Google Scholar] [CrossRef]
- Sun, F.; Yao, Y.; Li, G.; Li, X. Performance of Geothermal Energy Extraction in a Horizontal Well by Using CO2 as the Working Fluid. Energy Convers. Manag. 2018, 171, 1529–1539. [Google Scholar] [CrossRef]
- Wang, K.; Yuan, B.; Ji, G.; Wu, X. A Comprehensive Review of Geothermal Energy Extraction and Utilization in Oilfields. J. Pet. Sci. Eng. 2018, 168, 465–477. [Google Scholar] [CrossRef]
- Harada, T.; Soeda, K.; Idemitsu, T.; Watanabe, A. Characteristics of Expansive Pressure of an Expansive Demolition Agent and the Development of New Pressure Transducers. Doboku Gakkai Ronbunshu 1993, 1993, 91–100. [Google Scholar] [CrossRef] [Green Version]
- Natanzi, A.S.; Laefer, D.F.; Connolly, L. Cold and Moderate Ambient Temperatures Effects on Expansive Pressure Development in Soundless Chemical Demolition Agents. Constr. Build. Mater. 2016, 110, 117–127. [Google Scholar] [CrossRef] [Green Version]
- Collepardi, M.; Borsoi, A.; Collepardi, S.; Olagot, J.J.O.; Troli, R. Effects of Shrinkage Reducing Admixture in Shrinkage Compensating Concrete under Non-Wet Curing Conditions. Cem. Concr. Compos. 2005, 27, 704–708. [Google Scholar] [CrossRef]
- Chatterji, S. Mechanism of Expansion of Concrete Due to the Presence of Dead-Burnt CaO and MgO. Cem. Concr. Res. 1995, 25, 51–56. [Google Scholar] [CrossRef]
- El Dessouki, A.; Mitri, H. Rock Breakage Using Expansive Cement. Engineering 2011, 3, 168–173. [Google Scholar] [CrossRef]
- Hirota, T.; Ishizaki, Y. Static Expansive Demolition Agent in the Three-Dimensional Form and Process for Demolishing Brittle Material Using the Same. U.S. Patent 4,600,154, 15 July 1986. [Google Scholar]
- Ishii, S.; Kubota, H.; Hida, T.; Migita, J. Expansive Demolition Agent. U.S. Patent 4807530A, 28 February 1989. [Google Scholar]
- Chang, S.-H.; Lee, C.-I.; Jeon, S. Measurement of Rock Fracture Toughness under Modes I and II and Mixed-Mode Conditions by Using Disc-Type Specimens. Eng. Geol. 2002, 66, 79–97. [Google Scholar] [CrossRef]
- Xu, Y.; Chung, D. Effect of Sand Addition on the Specific Heat and Thermal Conductivity of Cement. Cem. Concr. Res. 2000, 30, 59–61. [Google Scholar] [CrossRef]
- Arefi, M.; Javeri, M.; Mollaahmadi, E. To Study the Effect of Adding Al2O3 Nanoparticles on the Mechanical Properties and Microstructure of Cement Mortar. Life Sci. J. 2011, 8, 613–617. [Google Scholar]
- De Silva, V.; Ranjith, P.; Perera, M.; Wu, B.; Rathnaweera, T. The Influence of Admixtures on the Hydration Process of Soundless Cracking Demolition Agents (SCDA) for Fragmentation of Saturated Deep Geological Reservoir Rock Formations. Rock Mech. Rock Eng. 2019, 52, 435–454. [Google Scholar] [CrossRef]
- Campana, S.; Andrade, C.; Milas, M.; Rinaudo, M. Polyelectrolyte and Rheological Studies on the Polysaccharide Welan. Int. J. Biol. Macromol. 1990, 12, 379–384. [Google Scholar] [CrossRef]
- Sandford, P.A.; Cottrell, I.W.; Pettitt, D.J. Microbial Polysaccharides: New Products and Their Commercial Applications. Pure Appl. Chem. 1984, 56, 879–892. [Google Scholar] [CrossRef]
- Khayat, K.H.; Yahia, A. Simple Field Tests to Characterize Fluidity and Washout Resistance of Structural Cement Grout. Cem. Concr. Aggreg. 1998, 20, 145–156. [Google Scholar]
- Sakata, N.; Yanai, S.; Yokozeki, K.; Maruyama, K. Study on New Viscosity Agent for Combination Use Type of Self-Compacting Concrete. J. Adv. Concr. Technol. 2003, 1, 37–41. [Google Scholar] [CrossRef]
- Rakitsky, W.G.; Richey, D.D. Rapidly Hydrating Welan Gum. CA2063087C, 21 May 2002. [Google Scholar]
- Kaur, V.; Bera, M.B.; Panesar, P.S.; Kumar, H.; Kennedy, J. Welan Gum: Microbial Production, Characterization, and Applications. Int. J. Biol. Macromol. 2014, 65, 454–461. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Zhao, Q.; Yao, C.; Zhou, M. Impact of Welan Gum on Tricalcium Aluminate-Gypsum Hydration. Mater. Charact. 2012, 64, 88–95. [Google Scholar] [CrossRef]
- Giergiczny, Z. Effect of Some Additives on the Reactions in Fly Ash-Ca (OH) 2 System. J. Therm. Anal. Calorim. 2004, 76, 747–754. [Google Scholar] [CrossRef]
- Peterson, V.K.; Juenger, M.C.G. Hydration of Tricalcium Silicate: Effects of CaCl2 and Sucrose on Reaction Kinetics and Product Formation. Chem. Mater. 2006, 18, 5798–5804. [Google Scholar] [CrossRef]
- Arshadnejad, S.; Goshtasbi, K.; Aghazadeh, J. A Model to Determine Hole Spacing in the Rock Fracture Process by Non-explosive Expansion Material. Int. J. Miner. Metall. Mater. 2011, 18, 509–514. [Google Scholar] [CrossRef]
- Cuesta, A.; Zea-Garcia, J.D.; Londono-Zuluaga, D.; Angeles, G.; Santacruz, I.; Vallcorba, O.; Dapiaggi, M.; Sanfélix, S.G.; Aranda, M.A. Multiscale Understanding of Tricalcium Silicate Hydration Reactions. Sci. Rep. 2018, 8, 1–11. [Google Scholar]
- Bazzoni, A.; Cantoni, M.; Scrivener, K.L. Impact of Annealing on the Early Hydration of Tricalcium Silicate. J. Am. Ceram. Soc. 2014, 97, 584–591. [Google Scholar] [CrossRef]
- Pustovgar, E.; Mishra, R.K.; Palacios, M.; de Lacaillerie, J.-B.d.E.; Matschei, T.; Andreev, A.S.; Heinz, H.; Verel, R.; Flatt, R.J. Influence of Aluminates on the Hydration Kinetics of Tricalcium Silicate. Cem. Concr. Res. 2017, 100, 245–262. [Google Scholar] [CrossRef]
- Taylor, H.F. Cement chemistry; Thomas Telford Ltd.: London, UK, 1997; Volume 2. [Google Scholar]
- Ángeles, G.; De Vera, R.N.; Cuberos, A.J.; Aranda, M.A. Crystal Structure of Low Magnesium-Content Alite: Application to Rietveld Quantitative Phase Analysis. Cem. Concr. Res. 2008, 38, 1261–1269. [Google Scholar]
- Bullard, J.W.; Jennings, H.M.; Livingston, R.A.; Nonat, A.; Scherer, G.W.; Schweitzer, J.S.; Scrivener, K.L.; Thomas, J.J. Mechanisms of Cement Hydration. Cem. Concr. Res. 2011, 41, 1208–1223. [Google Scholar] [CrossRef]
- Jennings, H.M. Aqueous Solubility Relationships for Two Types of Calcium Silicate Hydrate. J. Am. Ceram. Soc. 1986, 69, 614–618. [Google Scholar] [CrossRef]
- Bullard, J.W. A Determination of Hydration Mechanisms for Tricalcium Silicate Using a Kinetic Cellular Automaton Model. J. Am. Ceram. Soc. 2008, 91, 2088–2097. [Google Scholar] [CrossRef]
- Kantro, D.L.; Brunauer, S.; Weise, C.H. Development of Surface in the Hydration of Calcium Silicates. II. Extension of Investigations to Earlier and Later Stages of Hydration. J. Phys. Chem. 1962, 66, 1804–1809. [Google Scholar] [CrossRef]
- Stein, H.; Stevels, J. Influence of Silica on the Hydration of 3 CaO, SiO2. J. Appl. Chem. 1964, 14, 338–346. [Google Scholar] [CrossRef]
- Gartner, E.M.; Jennings, H.M. Thermodynamics of Calcium Silicate Hydrates and Their Solutions. J. Am. Ceram. Soc. 1987, 70, 743–749. [Google Scholar] [CrossRef]
- Birchall, J.D.; Howard, A.; Bailey, J. On the Hydration of Portland Cement. Proc. R. Soc. London A. Math. Phys. Sci. 1978, 360, 445–453. [Google Scholar]
- Garrault, S.; Finot, E.; Lesniewska, E.; Nonat, A. Study of CSH Growth on C 3 S Surface during Its Early Hydration. Mater. Struct. 2005, 38, 435–442. [Google Scholar] [CrossRef]
- Lesko, S.; Lesniewska, E.; Nonat, A.; Mutin, J.-C.; Goudonnet, J.-P. Investigation by Atomic Force Microscopy of Forces at the Origin of Cement Cohesion. Ultramicroscopy 2001, 86, 11–21. [Google Scholar] [CrossRef]
- Juilland, P.; Gallucci, E.; Flatt, R.; Scrivener, K. Dissolution Theory Applied to the Induction Period in Alite Hydration. Cem. Concr. Res. 2010, 40, 831–844. [Google Scholar] [CrossRef]
- Marchon, D.; Flatt, R.J. Mechanisms of Cement Hydration. In Science and Technology of Concrete Admixtures; Elsevier: Amsterdam, The Netherlands, 2016; pp. 129–145. [Google Scholar]
- Tadros, M.; Skalny, J.; Kalyoncu, R. Early Hydration of Tricalcium Silicate. J. Am. Ceram. Soc. 1976, 59, 344–347. [Google Scholar] [CrossRef]
- Zheng, L.; Xuehua, C.; Mingshu, T. Hydration and Setting Time of MgO-Type Expansive Cement. Cem. Concr. Res. 1992, 22, 1–5. [Google Scholar] [CrossRef]
- Mo, L.; Deng, M.; Tang, M.; Al-Tabbaa, A. MgO Expansive Cement and Concrete in China: Past, Present and Future. Cem. Concr. Res. 2014, 57, 1–12. [Google Scholar] [CrossRef]
- Bullard, J.W.; Flatt, R.J. New Insights into the Effect of Calcium Hydroxide Precipitation on the Kinetics of Tricalcium Silicate Hydration. J. Am. Ceram. Soc. 2010, 93, 1894–1903. [Google Scholar] [CrossRef]
- Garrault, S.; Nonat, A. Hydrated Layer Formation on Tricalcium and Dicalcium Silicate Surfaces: Experimental Study and Numerical Simulations. Langmuir 2001, 17, 8131–8138. [Google Scholar] [CrossRef]
- Thomas, J.J.; Jennings, H.M.; Chen, J.J. Influence of Nucleation Seeding on the Hydration Mechanisms of Tricalcium Silicate and Cement. J. Phys. Chem. C 2009, 113, 4327–4334. [Google Scholar] [CrossRef] [Green Version]
- Garrault, S.; Behr, T.; Nonat, A. Formation of the C− S− H Layer during Early Hydration of Tricalcium Silicate Grains with Different Sizes. J. Phys. Chem. B 2006, 110, 270–275. [Google Scholar] [CrossRef]
- Meredith, P.; Donald, A.; Meller, N.; Hall, C. Tricalcium Aluminate Hydration: Microstructural Observations by In-Situ Electron Microscopy. J. Mater. Sci. 2004, 39, 997–1005. [Google Scholar] [CrossRef]
- Maier, A.-K.; Dezmirean, L.; Will, J.; Greil, P. Three-Dimensional Printing of Flash-Setting Calcium Aluminate Cement. J. Mater. Sci. 2011, 46, 2947–2954. [Google Scholar] [CrossRef]
- Bickmore, B.R.; Nagy, K.L.; Gray, A.K.; Brinkerhoff, A.R. The Effect of Al (OH) 4−on the Dissolution Rate of Quartz. Geochim. Et Cosmochim. Acta 2006, 70, 290–305. [Google Scholar] [CrossRef]
- Begarin, F.; Garrault, S.; Nonat, A.; Nicoleau, L. Hydration of Alite Containing Aluminium. Adv. Appl. Ceram. 2011, 110, 127–130. [Google Scholar] [CrossRef]
- Matschei, T.; Lothenbach, B.; Glasser, F. The AFm Phase in Portland Cement. Cem. Concr. Res. 2007, 37, 118–130. [Google Scholar] [CrossRef]
- Pourchet, S.; Regnaud, L.; Perez, J.-P.; Nonat, A. Early C3A Hydration in the Presence of Different Kinds of Calcium Sulfate. Cem. Concr. Res. 2009, 39, 989–996. [Google Scholar] [CrossRef] [Green Version]
- Skalny, J.; Tadros, M. Retardation of Tricalcium Aluminate Hydration by Sulfates. J. Am. Ceram. Soc. 1977, 60, 174–175. [Google Scholar] [CrossRef]
- Mehta, P. Morphology of Calcium Sulfoaluminate Hydrates. J. Am. Ceram. Soc. 1969, 52, 521–522. [Google Scholar] [CrossRef]
- Collepardi, M.; Baldini, G.; Pauri, M.; Corradi, M. Tricalcium Aluminate Hydration in the Presence of Lime, Gypsum or Sodium Sulfate. Cem. Concr. Res. 1978, 8, 571–580. [Google Scholar] [CrossRef]
- Scrivener, K.L.; Nonat, A. Hydration of Cementitious Materials, Present and Future. Cem. Concr. Res. 2011, 41, 651–665. [Google Scholar] [CrossRef]
- Manzano, H.; Dolado, J.S.; Ayuela, A. Structural, Mechanical, and Reactivity Properties of Tricalcium Aluminate Using First-Principles Calculations. J. Am. Ceram. Soc. 2009, 92, 897–902. [Google Scholar] [CrossRef]
- Minard, H.; Garrault, S.; Regnaud, L.; Nonat, A. Mechanisms and Parameters Controlling the Tricalcium Aluminate Reactivity in the Presence of Gypsum. Cem. Concr. Res. 2007, 37, 1418–1426. [Google Scholar] [CrossRef]
- Lerch, W. The Influence of Gypsum on the Hydration and Properties of Portland Cement Pastes. Proc. Am. Soc. Test. Mater. 2008, 46, 1252–1291. [Google Scholar]
- Quennoz, A.; Scrivener, K.L. Interactions between Alite and C3A-Gypsum Hydrations in Model Cements. Cem. Concr. Res. 2013, 44, 46–54. [Google Scholar] [CrossRef]
- Scrivener, K.L.; Pratt, P. Microstructural Studies of the Hydration of C 3 A and C 4 AF Independently and in Cement Paste. Proc. Proc. Br. Ceram. Soc. 1984, 35, 207. [Google Scholar]
- Jansson, P.-E.; Lindberg, B.; Widmalm, G.; Sandford, P.A. Structural Studies of an Extracellular Polysaccharide (S-130) Elaborated by Alcaligenes ATCC 31555. Carbohydr. Res. 1985, 139, 217–223. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, C.; Luping, Z.; Qu, C.; Zhao, Q. The impact of welan gum on C3S hydration system. In Proceedings of the 15th International Congress on the Chemistry of Cement, Budapest, Hungary, 15 July 2017; p. 353. [Google Scholar]
- Kurozumi, M.; Sugimori, D. Biodegradation of Anionic Surfactant, Sodium 2-Naphthalene Sulfonate Formaldehyde Condensates, by the Fungus Cunning-Hamella Polymorpha. Sen’i Gakkaishi 2000, 56, 109–111. [Google Scholar] [CrossRef] [Green Version]
- Peng, X.; Li, X.; Chen, D.; Ma, D. Effect of Side Chains on the Dispersing Properties of Polycarboxylate-Type Superplasticisers in Cement Systems. Mag. Concr. Res. 2013, 65, 422–429. [Google Scholar] [CrossRef]
- Winnefeld, F.; Becker, S.; Pakusch, J.; Götz, T. Effects of the Molecular Architecture of Comb-Shaped Superplasticizers on Their Performance in Cementitious Systems. Cem. Concr. Compos. 2007, 29, 251–262. [Google Scholar] [CrossRef]
- Tan, H.; Huang, J.; Ma, B.; Li, X. Effect of Superplasticiser and Sodium Tripolyphosphate on Fluidity of Cement Paste. Mag. Concr. Res. 2014, 66, 1194–1200. [Google Scholar] [CrossRef]
- Yamada, K.; Ogawa, S.; Hanehara, S. Controlling of the Adsorption and Dispersing Force of Polycarboxylate-Type Superplasticizer by Sulfate Ion Concentration in Aqueous Phase. Cem. Concr. Res. 2001, 31, 375–383. [Google Scholar] [CrossRef]
- Vovk, A.; Ushenov-Marshak, A. Physicochemical Characteristics of Hydration of Low-Water-Demand Binders. Neorg. Mater. 1993, 29, 708–716. [Google Scholar]
- Sakai, E.; Raina, K.; Asaga, K.; Goto, S.; Kondo, R. Influence of Sodium Aromatic Sulfonates on the Hydration of Tricalcium Aluminate with or without Gypsym. Cem. Concr. Res. 1980, 10, 311–319. [Google Scholar] [CrossRef]
- Hekal, E.E.; Kishar, E.A. Effect of Sodium Salt of Naphthalene-Formaldehyde Polycondensate on Ettringite Formation. Cem. Concr. Res. 1999, 29, 1535–1540. [Google Scholar] [CrossRef]
- Collepardi, M.; Monosi, S.; Moriconi, G.; Corradi, M. Combined Effect of Lignosulfonate and Carbonate on Pure Portland Clinker Compounds Hydration. I-Tetracalcium Aluminoferrite Hydration. Cem. Concr. Res. 1980, 10, 455–462. [Google Scholar] [CrossRef]
- Plank, J.; Lummer, N.R.; Dugonjić-Bilić, F. Competitive Adsorption Between an AMPS®-Based Fluid Loss Polymer and Welan Gum Biopolymer in Oil Well Cement. J. Appl. Polym. Sci. 2010, 116, 2913–2919. [Google Scholar] [CrossRef]
- Cheung, J.; Jeknavorian, A.; Roberts, L.; Silva, D. Impact of Admixtures on the Hydration Kinetics of Portland Cement. Cem. Concr. Res. 2011, 41, 1289–1309. [Google Scholar] [CrossRef]
- Kondo, H.; Asaki, Z.; Kondo, Y. Hydrolysis of Fused Calcium Chloride at High Temperature. Metall. Trans. B 1978, 9, 477–483. [Google Scholar] [CrossRef]
- Robin, V.; Wild, B.; Daval, D.; Pollet-Villard, M.; Nonat, A.; Nicoleau, L. Experimental Study and Numerical Simulation of the Dissolution Anisotropy of Tricalcium Silicate. Chem. Geol. 2018, 497, 64–73. [Google Scholar] [CrossRef]
- Juenger, M.; Monteiro, P.; Gartner, E.; Denbeaux, G. A Soft X-ray Microscope Investigation into the Effects of Calcium Chloride on Tricalcium Silicate Hydration. Cem. Concr. Res. 2005, 35, 19–25. [Google Scholar] [CrossRef]
- Onuaguluchi, O.; Ratu, R.; Banthia, N. The Effects of CaCl 2-Blended Acrylic Polymer Emulsion on the Properties of Cement Mortar. Mater. Struct. 2018, 51, 1–10. [Google Scholar] [CrossRef]
- Ramachandran, V.S.; Feldman, R.F. Time-Dependent and Intrinsic Characteristics of Portland Cement Hydrated in the Presence of Calcium Chloride; National Research Council: Ottawa, ON, Canada, 1978. [Google Scholar]
- Traetteberg, A.; Ramachandran, V.S.; Grattan-Bellew, P. A Study of the Microstructure and Hydration Characteristics of Tricalcium Silicate in the Presence of Calcium Chloride. Cem. Concr. Res. 1974, 4, 203–221. [Google Scholar] [CrossRef] [Green Version]
- Markov, I. Crystal Growth for Beginners: Fundamentals of Nucleation. In Growth and Epitaxy; World Scientific: Singapore, 2003. [Google Scholar]
- Vehmas, T.; Kronlöf, A. A Study of Early-Age Ordinary Portland Cement Hydration According to Autocatalytic Reaction Model. Nord. Concr. Res. 2011, 43, 269–272. [Google Scholar]
- Berra, M.; Carassiti, F.; Mangialardi, T.; Paolini, A.; Sebastiani, M. Effects of Nanosilica Addition on Workability and Compressive Strength of Portland Cement Pastes. Constr. Build. Mater. 2012, 35, 666–675. [Google Scholar] [CrossRef]
- Bayanak, M.; Zarinabadi, S.; Shahbazi, K.; Azimi, A. Effects of Nano Silica on Oil Well Cement Slurry Charactreistics and Control of Gas Channeling. South. Afr. J. Chem. Eng. 2020, 34, 11–25. [Google Scholar] [CrossRef]
- Khayat, K.H. Viscosity-Enhancing Admixtures for Cement-Based Materials-An Overview. Cem. Concr. Compos. 1998, 20, 171–188. [Google Scholar] [CrossRef]
- Isik, I.E.; Ozkul, M.H. Utilization of Polysaccharides as Viscosity Modifying Agent in Self-Compacting Concrete. Constr. Build. Mater. 2014, 72, 239–247. [Google Scholar] [CrossRef]
- Pourchet, S.; Comparet, C.; Nicoleau, L.; Nonat, A. Influence of PC superplasticizers on tricalcium silicate hydration. In Proceedings of the 12th International Congress on the Chemistry of Cement (ICCC 2007), Montreal, QC, Canada, 8–13 July 2007. [Google Scholar]
- Simard, M.-A.; Nkinamubanzi, P.-C.; Jolicoeur, C.; Perraton, D.; Aïtcin, P.-C. Calorimetry, Rheology and Compressive Strength of Superplasticized Cement Pastes. Cem. Concr. Res. 1993, 23, 939–950. [Google Scholar] [CrossRef]
- Vehmas, T.; Kronlöf, A.; Cwirzen, A. Calcium Chloride Acceleration in Ordinary Portland Cement. Mag. Concr. Res. 2018, 70, 856–863. [Google Scholar] [CrossRef]
- Wilding, C.; Walter, A.; Double, D. A Classification of Inorganic and Organic Admixtures by Conduction Calorimetry. Cem. Concr. Res. 1984, 14, 185–194. [Google Scholar] [CrossRef]
- Double, D. New Developments in Understanding the Chemistry of Cement Hydration. Philos. Trans. R. Soc. London. Ser. A Math. Phys. Sci. 1983, 310, 53–66. [Google Scholar]
- Fujioka, I.; Imada, K.; Nishimura, M.; Ishibashi, T. Nonexplosive Chemical Composition for Gently Breaking Rock or Concrete Mass. U.S. Patent 4,565,579, 21 January 1986. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemical Component | Molar Mass/ × 10−3 kg mol−1 | Specific Gravity/ × 103 kg m−3 | Molar Volume/ × 10−6 m3 mol−1 |
|---|---|---|---|
| CaO | 56.1 | 3.34 | 16.8 |
| Ca(OH)2 | 74.1 | 2.24 | 33.1 |
| Ettringite | 786.7 | 1.77 | 444.5 |
| Chemical Component | Percentage by Mass (%) |
|---|---|
| CaO | 81–96 |
| Al2O3 | 0.3–5.0 |
| SiO2 | 1.5–8.5 |
| Fe2O3 | 0.2–3.0 |
| MgO | 0–1.6 |
| CaSO4 | 0.6–4.0 |
| Additive | Types Commonly Used in Cement Industry | Mechanism of Action |
|---|---|---|
| VEA | Silica-based slurry | Nano/micro silica particles would easily interact with the liquid phase of the cement mixture, forming a gel-like consistency, thereby increasing the retention of water [101]. As the particles fill the voids between cement particles, it would provide additional nucleation sites for hydration and create CSH phases [102]. Thus, the resulting volume expansion may be comparatively larger. However, the workability is greatly decreased with the addition of silica-based slurries [101]. |
| Ethylenoxide derivate | These organic VEAs would retain water and decrease the mass loss, similar to welan gum as discussed in the Section 5.1, due to the multiple O atoms in the structures [103]. However, the amylopectin branch of starch facilitates relatively low viscosity. Therefore, the amount of starch-derivative necessary would be about 10 times that of a polysaccharide [104]. | |
| Other natural polysaccharides (diutan gum, xanthan gum) | ||
| Starch-based derivatives | ||
| HRWR | Polycarboxylate-type | Polycarboxylate-derivatives would behave similar to SNSF in the presence of cement, with the carbonate group interacting with water. However, these derivatives would slow down the ettringite precipitation [105]. Since the carbonate group is stronger compared to sulfonate group (in SNSF), the adsorption of the anionic head into the active sites can be seen more frequently in the presence polycarboxylate-derivatives [106]. |
| Diphosphonate-type | The phosphonate groups would interact with water similar to SNSF. However, a slight increase in Ca2+ concentration in the system can be observed, in the presence of diphosphonate-derivatives. This is due to the complex formation between the diphosphonates and Ca2+ ions leading to further dissolution of the Ca-based compounds in cement [105]. | |
| Accelerator | Other cations of a similar size and charge, that are is a similar charge density, would also show an accelerated reaction mechanism [107]. Thus, the effectiveness of cations as a chemical accelerator is as follows [108,109]: Ca2+ > Sr2+ > Ba2+ > Li+ > K+ > Na+ ≈ Cs+ > Rb+ The counter ion which would maintain the electroneutrality of the sample, should not bind to either CSH or alite during the hydration process, by replacing the silicates. As silicates are covalently bonded, highly electronegative anions such as bromides and chlorides would not take their position while anions that are less electronegative such as fluorides would replace silicates, inhibiting the acceleration [110]. Therefore, the effectiveness of anions as a chemical accelerator is as follows: Br− ≈ Cl− > SCN− > I− > NO3− > ClO4− However, considering CaBr2 and CaCl2, the chloride dissolves more efficiently and therefore is used in cement industry. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liyanage, J.B.; Gamage, R.P. The Hydration and Volume Expansion Mechanisms of Modified Expansive Cements for Sustainable In-Situ Rock Fragmentation: A Review. Energies 2021, 14, 5965. https://doi.org/10.3390/en14185965
Liyanage JB, Gamage RP. The Hydration and Volume Expansion Mechanisms of Modified Expansive Cements for Sustainable In-Situ Rock Fragmentation: A Review. Energies. 2021; 14(18):5965. https://doi.org/10.3390/en14185965
Chicago/Turabian StyleLiyanage, Janethri Buddhipraba, and Ranjith Pathegama Gamage. 2021. "The Hydration and Volume Expansion Mechanisms of Modified Expansive Cements for Sustainable In-Situ Rock Fragmentation: A Review" Energies 14, no. 18: 5965. https://doi.org/10.3390/en14185965
APA StyleLiyanage, J. B., & Gamage, R. P. (2021). The Hydration and Volume Expansion Mechanisms of Modified Expansive Cements for Sustainable In-Situ Rock Fragmentation: A Review. Energies, 14(18), 5965. https://doi.org/10.3390/en14185965