2. Analytical Dependences of Hydrate-Forming Gas Pressure Influence on Nonclathrated Water
For describing the thermodynamic properties of pore water, it is convenient to use water activity
, which depends on the water content of the sample and temperature:
where
is water vapor pressure over the soil sample with water content
(wt% water relative to the dry sample), and
is the pressure of saturated water vapor over the bulk water (in MPa or Pa).
The experimental determination of pore water activity
, depending on water content
in the soil at or close to room temperature, can be carried out by various methods. The most efficient method is to measure the dew point of air brought into equilibrium with a wet soil sample with water content
, followed by a recalculation of the dew-point temperature to water vapor pressure over wet soil and, thereby, water activity. The method for measuring pore water activity is described in detail elsewhere [
34].
The activity of pore water in a wet sample W depends on both the water content of the sample and on its temperature, i.e., . As a first approximation, the temperature dependence of pore water activity (at a fixed ) can be neglected. However, this is not the case for low water content in the soil, especially in the presence of a clay component with a sliding framework in the soil (for example, smectite).
Another task was to derive some thermodynamic dependencies connecting pore water activity in the soil (at atmospheric pressure) with the fugacity or pressure of the hydrate-forming gas at a given temperature. At the same time, it was necessary to separately describe positive and negative temperatures due to the existence of unfrozen water at negative Celsius temperatures. Unfrozen pore water in equilibrium with ice also exists at gas pressure, but gas pressure must be below pressure on the gas–ice–hydrate equilibrium line. At pressure , higher pressure (pressure at the gas–ice–hydrate equilibrium line) exists instead of ice in the gas hydrate phase (the hydrate phase becomes stabler than the ice phase). Thus, pore water at pressure should be nonclathrated water (according to the terminology considered in the introduction).
The consideration of a gas pressure effect on nonclathrated water content begins at positive Celsius temperatures. The soil sample is fixed at temperature
(when deriving thermodynamic relations, it is more convenient to set the temperature in Kelvin, while in practical examples, it is more convenient to set the temperature in Celsius). Then, experimental data of pore water activity via the water content of sample
(i.e., dependence
) were obtained. Then, the hydrate former was chosen (for example, gases such as methane, carbon dioxide, ethane, propane, nitrogen, their mixtures, and natural gas). The line of the three-phase gas–water (in bulk phase)–hydrate equilibrium was assumed either from experimental data or calculated using available software. For instance, in many books analytical approximations of three-phase equilibrium lines (gas and gas hydrates with water/ice) for pure gases were presented [
35,
36,
37].
The pressure of hydrate formation at a given temperature , gas fugacity , and gas compressibility factor were denoted assuming that the activity of pore water in sample was known from experimental measurements at atmospheric pressure. Water activity corresponds to the bulk phase of water. At pressure , there was no gas hydrate in the system. At , the amount of nonclathrated water in sample formally tended to infinity. We were interested in the thermodynamic relation between gas pressure at the three phase equilibrium gas–pore water–hydrate () and the activity of pore water , as well as the water content of the soil sample.
A preliminary remark is that from the morphological studies of hydrates [
38], the characteristic size of hydrate particles obtained in real soils, as a rule, exceeds 10 microns. It is easy to show that at a characteristic particle size of more than 1 micron, the thermodynamic properties of pore hydrate particles practically do not differ from their properties in the bulk phase. For further consideration, we excluded nanoporous media in which the thermodynamic properties of the pore hydrate could significantly differ from the properties of the bulk hydrate phase. The effect of a hydrate particle size of 0.1 microns or less on phase equilibrium requires special consideration.
According to the traditional thermodynamic model of clathrate hydrates by van der Waals–Platteeuw and Barrer–Stuart (see, for instance, [
28]), the chemical potential of water
in the hydrate phase is written as follows:
or, in an equivalent form:
where
—temperature, K;
—gas pressure, MPa;
MPa;
—molar volume of water in hydrate (
for cubic structure I and
for cubic structure II);
—chemical potential of water in an empty clathrate lattice at pressure
and temperature
;
—chemical potential of water in a clathrate lattice partially filled with guest molecules at pressure
and temperature
;
—universal gas constant,
;
,
—Langmuir constants for large and small cavities (depending only on temperature), respectively;
,
—degrees of filling small and large cavities of the structure, respectively;
and
—crystallochemical constants (
and
are for Structure I, and
,
are for Structure II).
Quantities and are related by the Langmuir isotherm (the traditional model assumes that gas molecules are sorbed by the clathrate lattice in accordance with the Langmuir isotherm). Hydrate numbers in the chemical formulas of hydrates M·nH2O, where M is a gas molecule (for example, CH4), are also used below. Hydrate number is expressed in terms of the degrees of filling cavities as follows: for hydrate Structures I and II, respectively.
The chemical potential of water
in a pore solution in a good approximation can be written as follows:
where
—chemical potential of pure bulk water at pressure
and temperature
;
—chemical potential of pore water in the soil sample at pressure
and temperature
;
—gas solubility in pore water;
—pore water activity in the soil sample measured at atmospheric pressure (pore water can also be saline);
—partial molar volume of water in pore solution, assuming that
. Gas solubility in pore water can be approximately equal to solubility in the bulk water phase. In principle, the effect of a porous medium on gas solubility can be estimated by excluding that part of pore water volume in which the gas does not dissolve (for instance, water in the interlayer space of the sliding frame clays does not dissolve the gas).
Gas solubility in bulk water under gas pressure can be determined by the Krichevsky–Kazarnovsky equation [
39] or calculated from the equations of state; experimental data can also be used. For calculations using equations of state, the cubic-plus-association (CPA) equation is recommended and is widely used in commercial software.
The Krichevsky–Kazarnovsky equation [
39] for pure gas is
where
,
, Henry’s coefficient of gas;
, partial molar volume of gas in water (
);
, gas fugacity,
(fugacity is determined by the equation of state and depends on temperature and pressure). Henry’s coefficient
only depends on temperatures up to a pressure of 20–30 MPa. By knowing Henry’s coefficient, gas fugacity
and the partial molar volume
of gas in water, it is possible to determine molar fraction
of gas in water from Equation (4). Henry’s coefficient is determined from experimental data on gas solubility in water, and the partial molar volume
may be determined from experimental data (
can also be measured in special direct volumetric experiments). Values of
and
for various gases are given in the literature [
40].
For a gas mixture, the Krichevsky–Kazarnovsky equation is generalized as follows:
where
—amount of dissolved gases,
—mole fraction of
gas in water;
—Henry coefficient of
component of gas mixture;
—fugacity of
component of gas mixture, which is determined by the gas equation of state;
—partial molar volume of
gas in water.
Phase equilibrium gas–water bulk phase–hydrate at , and a fixed temperature corresponds to the equality of the chemical potential of water in the hydrate phase according to Relation (2), and the chemical potential of water in the water bulk phase with dissolved gas according to Relation (3) at .
Equating the chemical potentials after some transformations, we obtain
or
where
—the difference between the chemical potentials of water in the hydrate phase, and liquid water at atmospheric pressure and considered temperature;
, the difference between molar volumes of water in the hydrate lattice and in bulk water;
and
for cubic Structures I and II, respectively.
Let us consider phase equilibrium gas–pore water–hydrate at a given sample water content (moisture)
, i.e., at
. In this case, the activity of pore water is equal to
Equating the chemical potentials of water in hydrate phase (2) and pore water solution (3), we obtain
or
Subtracting Relation (6) from (5) after some transformations,
Equation (7) relates pore water activity and consequently the water content W of the sample to gas fugacity and gas pressure at . In particular, from Equation (7) with , we obtain or In Equation (7), quantity is excluded, but the value of equilibrium gas pressure is included (in comparison with (6)).
Equation (7) is easily generalized to the case of a gas mixture (natural or associated petroleum gas etc.); for this, in Equation (7) one should replace with and with , where are the fugacity and Langmuir constants of the -th component of the gas mixture, respectively. For pure gases (propane, cyclopropane, and isobutane), small cavities of hydrate structure II are not filled, i.e., and . In this case, the first term disappears from the left-hand side of Equation (7). The same is true for ethane hydrate, which forms Structure I (in ethane hydrate, the small cavities are also not filled).
At a gas pressure below 6–8 MPa, as an approximation its solubility in water (except for carbon dioxide) and the influence of the Poynting effect can be neglected (i.e., value
); then, we can obtain the following simplified relationship:
or
Equations (8) and (9) can be used for methane, nitrogen, and inert gases at moderately high pressure levels (for CO2, its water solubility should not be neglected).
Let us analyze the further possibilities of simplifying Relation (7) to reduce the necessary parameters for calculating nonclathrated water. For this, let us consider the nature of cavities filling with gas molecules in hydrates of various structures. First, it should be considered that large cavities in clathrate structures are always almost completely occupied (i.e., the degree of the filling of large cavities is always close to unity, ). At temperatures close to K for Structure I, and for Structure II, . This yields the estimate of the large cavities’ degree of filling at a temperature of K: for Structure I and for Structure II. In addition, in many cases of practical interest (hydrates of methane, natural gases), the degree of the filling of small cavities of hydration structure is also close to . With increasing temperature (and gas pressure), the degrees of filling approach unity.
Let us consider separately three practically important cases of the filling of clathrate cavities guest molecules: and ; (ii) , ; and (iii), .
Let the small and large cavities be almost filled, i.e.,
,
;
,
. This situation is typical for methane and nitrogen gases and inert gases. Thus, we neglect the unit under the logarithm in expressions such as
, i.e.,
. Equation (7) is rewritten as follows:
or
For convenience, we introduce into consideration quantities
Let us rewrite (10), taking into account (11). After some transformations, we obtain
and lastly,
Relations (11) and (12) are the main result of the consideration of the thermodynamics when the filling of both types of cavities in gas hydrate structures is close to 1. Equation (12) allows for the given W and water activity a, and for the known values of and , to calculate the equilibrium gas fugacity and gas pressure . At a given gas pressure , we may determine fugacity and then the activity of pore water and . When calculating nonclathrated water content using Relation (12), no information is required on the thermodynamics of an empty clathrate lattice and the Langmuir constants of guest molecules (in contrast to Relation (10)).
If the gas is considered to be in the ideal gaseous state, then
, and Equation (12) can be rewritten as
Since, as a first approximation, , then from Equation (13), a strong nonlinear relationship between and becomes obvious. Thus, it seems from the obtained relations that, with increasing pressure, the content of nonclathrated water in a porous medium sharply decreases and is in accordance with a power law.
If the gas under consideration is weakly nonideal (for example, for methane up to pressures of about 7–8 MPa), then the approximate thermodynamic formula
should be used, where
is the gas compressibility factor. By using it, Relation (12) is rewritten as
where
are factors of gas compressibility at pressure
and
, respectively.
Let the large cavities be almost filled,
() with empty small cavities,
. This case is realized for hydrates of propane, isobutane, cyclopropane, ethane, and their mixtures. Taking
, from Relation (12) we obtain
For the hydrate of Structure I (for instance, ethane): , and for hydrates of Structure II (for propane and isobutane): .
More approximate relationships occur:
Let both cavities be filled, and the degree of the filling of large cavities is close to unity,
(
), but the degree of the filling of small cavities
can vary over a wide range (from 0 to 1). A typical example is carbon dioxide hydrate (and in its mixtures with propane, isobutane, and ethane). In such cases, Expression (7) is transformed into the following form:
Considering that
and
, after some transformations, we lastly obtain
Expression (18) is less convenient for practical use, since the Langmuir constant
of a small cavity remains. Therefore, the question arises whether it is possible to also use Equations (12)–(14) for the hydrate of carbon dioxide, substituting stoichiometric value
for the actual (or effective) hydrate numbers
. In this version, Equations (12)–(14) are rewritten as follows:
More approximately,
where
n is the actual or effective hydrate number on the three-phase gas–bulk phase of water–hydrate equilibrium line.
The simplest Approximation (20) may only be used for low pressure levels and low gas solubility in water.
A numerical comparison of approximate Relations (18) and (19) with general thermodynamic Relation (7) showed that it is practically acceptable to use the actual hydrate number (see below). Moreover, the numerical comparison of (7), (12), and (19) for methane and nitrogen hydrates (the case when both cavities are strongly filled) also showed the benefit of using Equation (20). At the same time, for hydrates in which only large cavities are strongly filled (ethane, propane, isobutane), stoichiometric hydrate numbers ( for ethane and for propane and isobutane) should be used. Thus, the above-obtained approximate Relations (12–14) can be empirically improved by using Equations (19) and (20) with an effective hydrate number (see below for the practical recommendations for choosing the values of ).
As a result, for the equilibrium of gas–pore water–gas hydrate, we obtained approximate Relations (12)–(20), which are convenient for practical applications. These relations were considered for positive temperatures in Celsius, but with some modifications they are applicable for temperatures below zero Celsius.
Let us discuss the thermodynamic correlations for calculating nonclathrated water content at temperatures below freezing Celsius. First, if at negative temperatures, the three-phase equilibrium of gas–bulk supercooled water–hydrate is used as a reference line (a continuous continuation of the line of gas–bulk water–hydrate to negative temperatures), then Relations (12)–(20) can also be applied for negative temperatures. However, the lines of the metastable three-phase equilibrium of gas–supercooled water–hydrate can be experimentally obtained only for small water droplets in especially organized experiments. Such unique data were obtained in the papers of Melnikov et al. [
41,
42] for methane, propane, and carbon dioxide gases. For other gases, up-to-date experimental information is not yet available. However, these lines (including those for ethane, isobutane, and gas mixtures) can be calculated with acceptable accuracy at least up to −15 °C using software by Istomin et al. [
43]. In other software, calculations of metastable phase equilibria of gas hydrates as a rule are not provided. Second, the pressure range from
to pressure, corresponding to the equilibrium of the gas hydrate with ice (this pressure note by
), calculated using relations of type (12), refers to the zone of nonclathrated water metastability (i.e., to a hypothetical situation, as if ice in a given system did not exist). Strictly speaking, calculations of the content of nonclathrated water at temperatures below 0 °C should be carried out only for pressures
(when there is no ice in the system because the ice is already transformed to hydrate phase).
Therefore, for thermodynamic calculations of nonclathrated water content at negative temperatures, it is preferable to use the gas–ice–hydrate line as reference. The advantage of this reference line is because cages’ filling and the hydrate number n along this line vary very slightly, with temperatures ranging from −15 to 0 °C. In a soil system, pore water as a fourth phase also exists in the equilibrium with ice, hydrate, and gas (the locus of quadrupole points). At the quadrupole point, according to the above accepted terminology, pore water may simultaneously be considered as unfrozen and nonclathrated water. At this line, the value of pore water activity describes the equilibrium between ice and pore water at atmospheric pressure. Thus, decreases with a decreasing negative temperature .
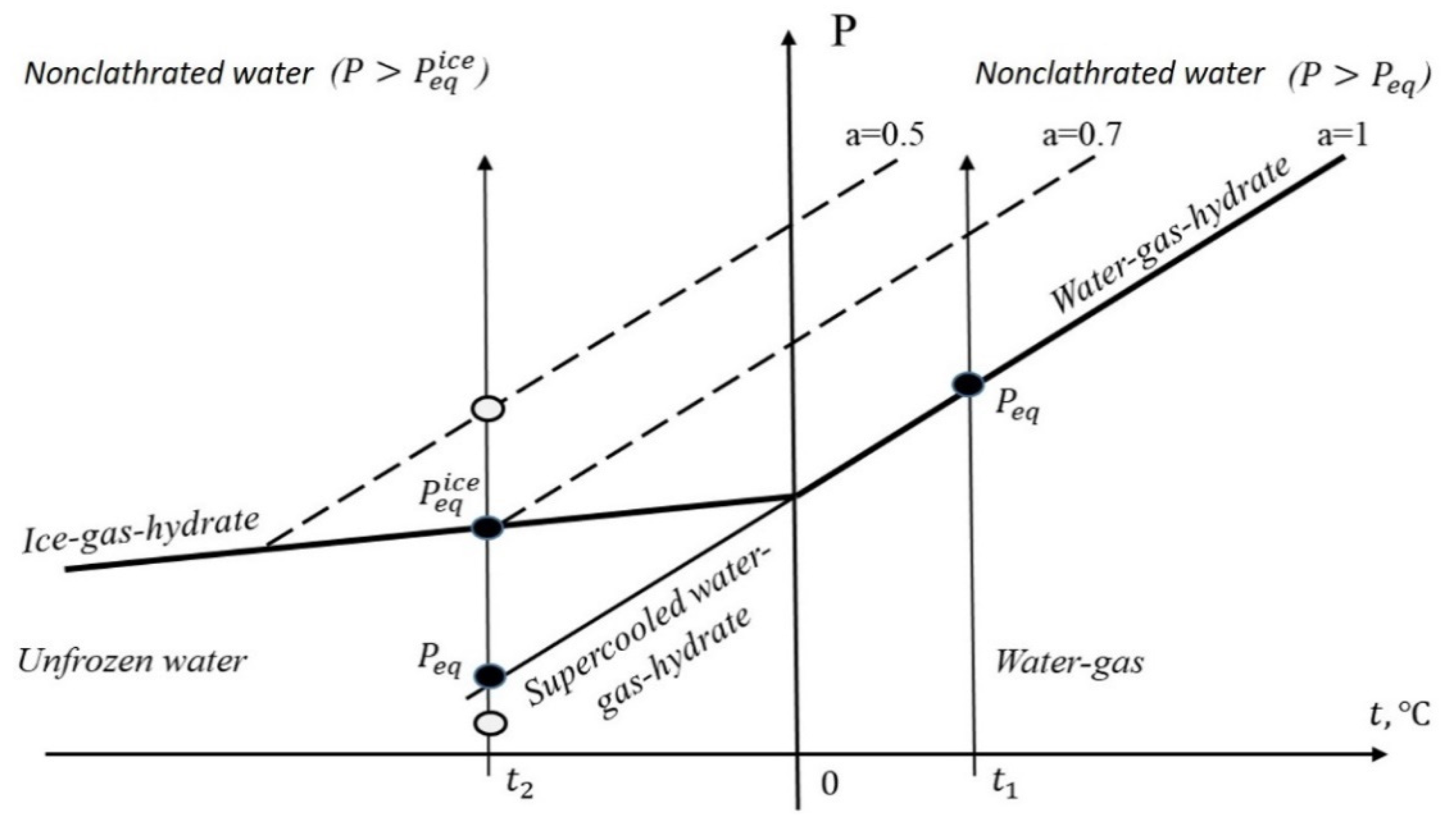
This situation is illustrated in
Figure 1. Bold lines are the lines of the three-phase equilibrium of gas–water or ice bulk phase–hydrate. Above these lines, there is a zone of nonclathrated pore water. Dotted lines are the equilibrium of gas–hydrate–nonclathrated water with given activity
of pore water (activity measured at atmospheric pressure). For positive Celsius temperatures at
, value
and the amount of nonclathrated water formally become infinite. At
,
, and when gas pressure
increases, pore water activity
and equilibrium water content
W in the sample decrease.
At a given negative Celsius temperature with gas pressure increasing, the amount of unfrozen pore water (gas–pore water–ice equilibrium) increases up to the quadrupole point (at ), but very slowly. During a further increase in pressure ( the content of nonclathrated pore water begins to sharply decrease (according to a power law as established above).
Thus, at temperatures below 0 °C, it is preferable to use the gas–ice–hydrate line as a reference line. Repeating the derivation of Relations (7)–(12), instead of Relation (12), the modified equation may be obtained. In Relation (12), should be replaced by and by ; in Relation (11), value should be replaced by , should be replaced by , and by , where is the molar volume of ice ().
As a result, using the curve gas–ice–hydrate equilibrium as a reference line, we obtain the following final equation:
where
and
and
for hydrate cubic structures I and II, respectively.
At low pressure, instead of Equation (21), the approximation may also be used
A new value
appears in the definition of
, and it is essential that
be a function of temperature, i.e.,
corresponding to the equilibrium of ice–unfrozen water in the soil under consideration (at atmospheric pressure). This means that for the practical application of Equation (21), it is necessary to determine the value of
, depending on the temperature (negative in Celsius). Such dependence was obtained [
44] for unfrozen water calculations (equilibrium of pore water and ice):
Then Equation (23) was transformed into relationship between temperature
(in degrees Celsius) and pore water activity
on the pore water–ice equilibrium line [
34]:
Equation (24) is used to calculate unfrozen water content from measured water activity when the temperature is set to Celsius. However, for our purposes,
needs to be expressed as a function of temperature
t (in degrees Celsius). By the approximation of Equation (23), we may obtain
Dependencies (23) and (25) can both be used in calculating nonclathrated water content at negative temperatures from Relation (21). Thus, the final Equation (21) is supplemented by Relation (23) or (25).
3. Nonclathrated Water Content Calculation
In the above, some thermodynamic relations were obtained (Equations (6), (7), (13) and (19)–(25)) that make it possible to calculate nonclathrated water content in a soil sample at a given temperature, depending on the pressure of hydrate-forming gas.
First, the main Equation (6) allows for the performance of thermodynamic calculations of equilibrium gas fugacity and then pressure at a given temperature, depending on pore water activity , and thereby pore water content W. However, for the application of Equation (6) in practice, we need to know (i) the structure of the hydrate, (ii) the thermodynamic properties of the empty clathrate lattice , and (iii) the Langmuir constants of the hydrate-forming gas under consideration. These values can be obtained if the hydrate-phase thermodynamic model’s parameterization is published and/or described in the software documentation. For example, such data were presented in Istomin et al. (1996), but for other software they are not documented as a rule.
Equation (7) also allows, at a given temperature , known Langmuir constants , , and the value of , for calculating water activity depending on gas pressure under consideration, and thereby determining the content of nonclathrated water W in the sample. Equation (7) excludes information about the thermodynamics of the empty hydrate lattice but contains additional information on equilibrium gas pressure However, from a practical point of view Equations (6)–(8) are not fully convenient, since the temperature dependences of the Langmuir constants for small and large cavities need to be specified for the considered hydrate-forming gas (these constants must also be consistent with the three-phase equilibrium lines). Thus, relations such as Equation (13) look more attractive from a practical point of view, but they are only a good approximation of the main thermodynamic Relations (6) and (7). Numerical analysis showed that a small additional correction of equations such as (13) can be made with the replacement of the limiting hydrate number by its effective value .
As a result, for the practical calculations of the nonclathrated water content, Relations (19) and (20) are recommended at temperatures above 0 °C, and Relations (21) and (23) (or (25)) at temperatures below 0 °C. For rough estimations, the replacement of gas fugacity by in the equation is possible. This approximation may be used for methane up to a pressure of 7–8 MPa.
Hydrate numbers
for different gases are shown in
Table 1 and
Table 2 (for positive and negative Celsius temperatures, respectively) that were calculated by using software [
43]. For C
3H
8 and i-C
4H
10, a limited hydrate number may be used.
A variant of the calculation is also possible if the unfrozen water content in frozen soils for different temperature levels () is known from the experiment. Using Equation (25), we immediately establish the dependence of pore water activity on the water content W of the sample. Then, we may calculate the nonclathrated water content as a function of for any hydrate-forming gas and any temperature (at a negative Celsius temperature according to Relations (21), (23), and (25) and at a positive Celsius temperature according to Relations (19) and (20)).
Using the proposed technique, the pressure dependence of the nonclathrated water content was calculated at a temperature of
K in a kaolinite clay and sand–clay mixture samples (sand plus 14% kaolinite clay and sand plus
kaolinite clay). This kaolinite clay was used previously to determine the effect of temperature on nonclathrated water content in porous media [
28]. Soil characteristics are presented in
Table 3.
Sand consists of quartz (more than 90%) the prevailing fraction of sand particles 0.1–0.25 mm is reach 62.9%. Kaolinite clay consists mainly of kaolinite (92%), with 95.5% silt-clay size particles, while the percentage of clay particles (<0.002 mm) reaches 42%. Kaolinite clay contains minor amounts of dissolved salts (0.04%). The specific active surface areas of sand and kaolinite clay defined by nitrogen adsorption are 0.2 and 12 m2/g, respectively.
First, experimental data of pore water activity
at atmospheric pressure via water content
were obtained (
Table 4).
Pore water activity
was determined with a WP4-T device by a method previously described in detail [
28,
34].
Thermodynamic calculations of nonclathrated water were carried out using four methods (
Figure 2 and
Table 5):
Equation (7), the most precise method, where the Langmuir constants were obtained from ratio
, and the degree of cavity filling was calculated using our software [
43].
Equations (11) and (19), where the three-phase methane–supercooled water–hydrate equilibrium line was used as a reference line, and n=.
Equation (21) and where the three-phase gas–ice–hydrate equilibrium line was used as a reference line, and .
Equations (21) and (25), where the three-phase gas–ice–hydrate equilibrium line was used as a reference line, , .
In
Figure 2, equilibrium pressure
MPa on the ice–methane–hydrate equilibrium line at a temperature of
K.
The Equation (21) approximation with
n = 6.03 gave a very similar result to that of fully correct Equation (7). This means that at negative temperatures, it is preferable to use ice–gas–hydrate as a reference line and actual hydrate numbers from
Table 2. For positive temperatures, Equation (19) and actual hydrate numbers from
Table 1 are recommended.
The data (
Table 4) were calculated using measured water activities for kaolinite clay (
Table 3). Three methods were used: squares—general (most accurate) Equation (7); crosses—approximate Equation (19), considering (11); triangles according to approximate Equation (21) using three-phase equilibrium gas–ice–hydrate as a reference line; circles—also according to Equation (21) with a hydrate number
.
A comparison of calculated data with direct experimental data obtained by the contact method (
Table 6) is shown in
Figure 2.
The contact method is a direct technique for nonclathrated water content determination in soil samples. It was proposed earlier in our papers [
45,
46]. Nonclathrated water content data has a good agreement between that calculated by thermodynamic equations and experimental data obtained by the contact method, the accuracy of which is about 0.1 wt% [
28]. The largest discrepancy of ~0.15 wt% in the data was observed in the range of 1.4–1.5 wt%.
Additional experimental data and calculations of nonclathrated water content were obtained for sand–clay mixtures, which consist of quartz sand and 14 wt% and 25 wt% of kaolinite clay, respectively (
Figure 3). These results also demonstrate a good agreement between the calculation and the experimental data. There is a regular increase in the amount of nonclathrated water in model soils with the increase in the content of clay particles. For example, the content of nonclathrated water at 4 MPa gas pressure in sand with 14 wt% kaolinite clay is 0.25%, which is two times lower than that in the sand with 25 wt% clay. The effect of gas pressure on the nonclathrated water content is weak at pressures above 6–8 MPa. However, the difference depending on the content of clay particles is also preserved under these conditions.
Thus, the proposed thermodynamic technique for nonclathrated water content calculation allows one to estimate the effect of hydrate-forming gas pressure on the equilibrium water content in hydrate-bearing soil samples. The comparison showed a sufficiently good agreement between the calculated results by the proposed technique with the direct measurements of nonclathrated water for all investigated soils.
The obtained methodological results make it possible to use the proposed technique during the efficiency estimation of methane hydrate recovery by various production methods. In contrast to the conventional approach, which takes into account only the temperature shift for the assessment of hydrate conditions in porous media, the investigated method takes into account the increase in equilibrium pore water content (non-clathrated water) due to reservoir pressure rise. The information about residual water (nonclathrated water) in hydrate-saturated reservoirs is very important for predicting the efficiency of CO2 sequestration in a hydrate form under definite temperature and pressure conditions.



 ,
, 



{kind=link}
{kind=link}
{kind=link}