
Solar-Driven Simultaneous Electrochemical CO2 Reduction and Water Oxidation Using Perovskite Solar Cells


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
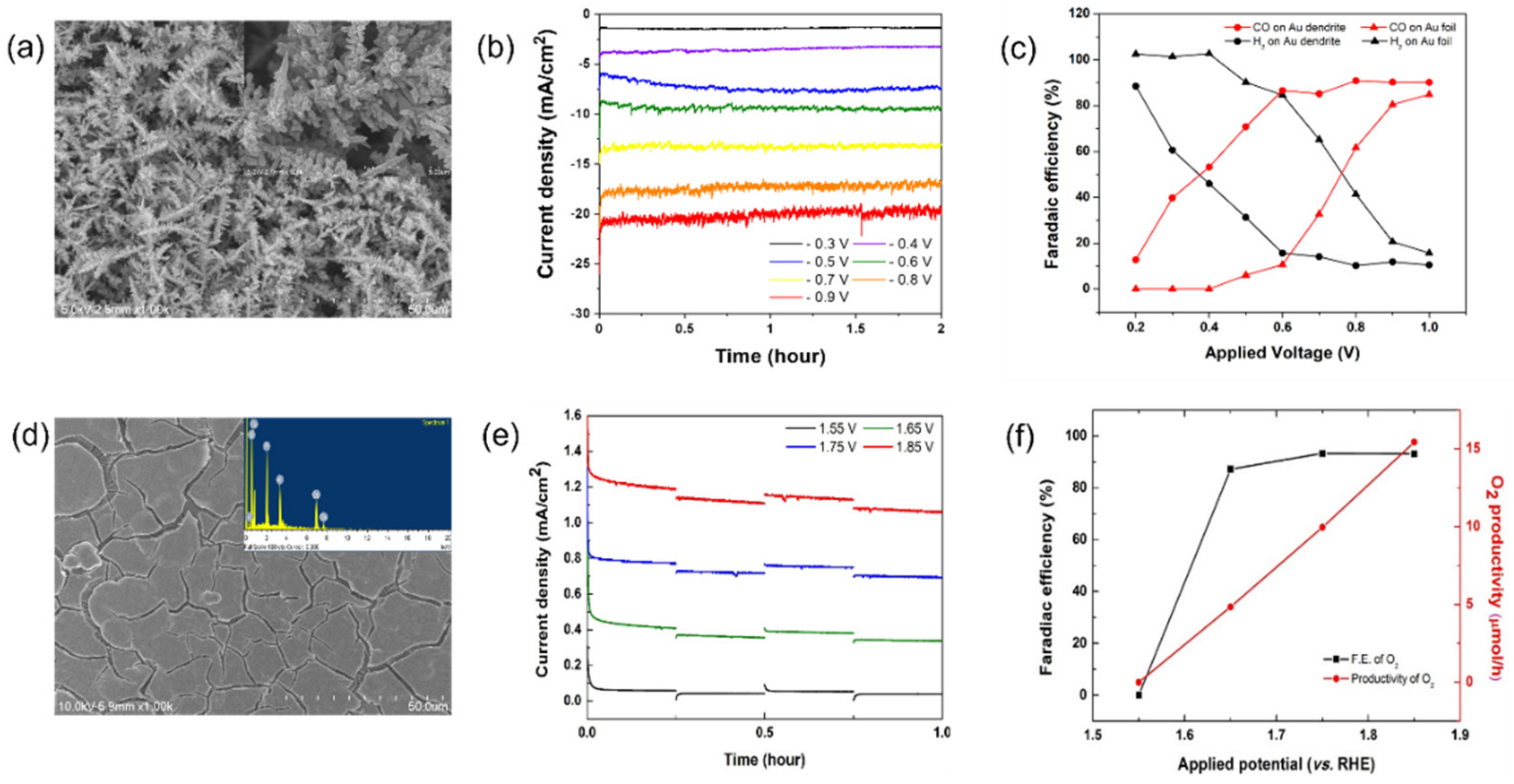
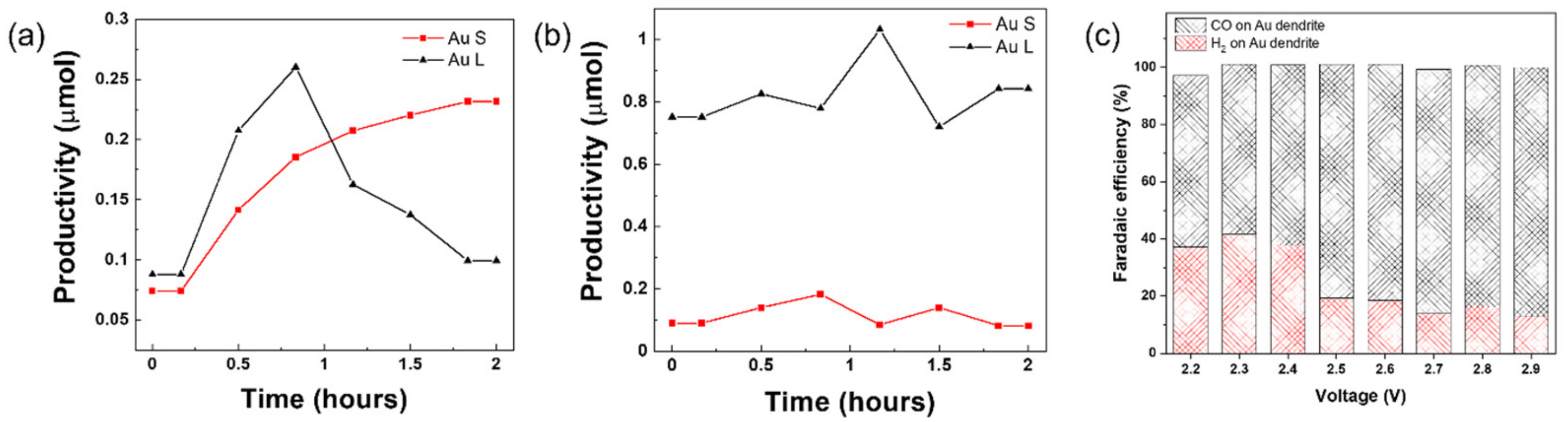
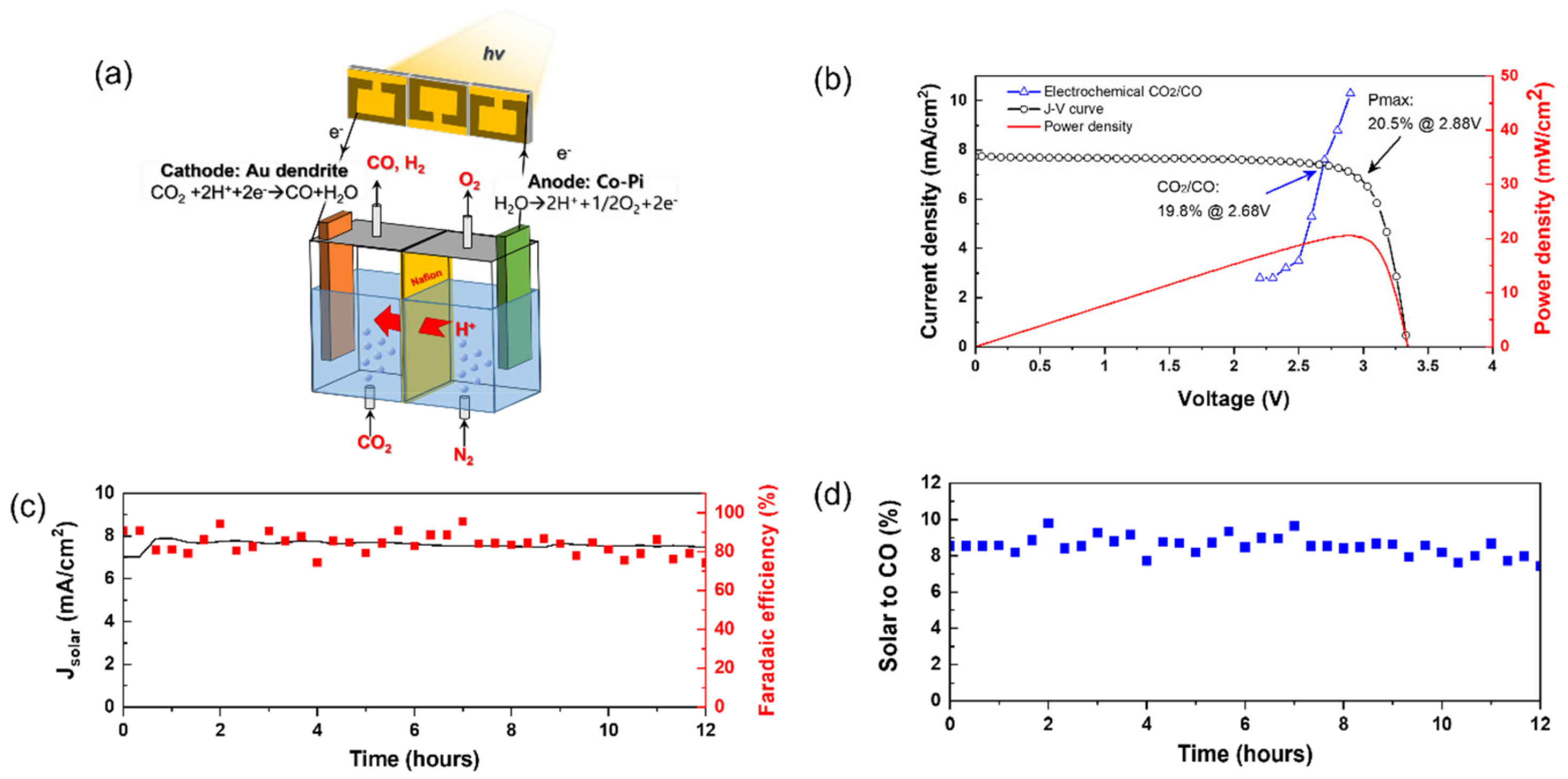
2. Results
3. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Marshall, J. Solar energy: Springtime for the artificial leaf. Nature 2014, 510, 22–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, Y.J.; Jeong, I.; Lee, J.; Lee, J.; Ko, M.J.; Lee, J.S. Unbiased sunlight-driven artificial photosynthesis of carbon monoxide from CO2 using a ZnTe-based photocathode and a perovskite solar cell in tandem. ACS Nano 2016, 10, 6980–6987. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Im, J.-H.; Mayer Matthew, T.; Schreier, M.; Nazeeruddin Mohammad, K.; Park, N.-G.; Tilley, S.D.; Fan Hong, J.; Grätzel, M. Water photolysis at 12.3% efficiency via perovskite photovoltaics and earth-abundant catalysts. Science 2014, 345, 1593–1596. [Google Scholar] [CrossRef]
- Gurudayal; Sabba, D.; Kumar, M.H.; Wong, L.H.; Barber, J.; Grätzel, M.; Mathews, N. Perovskite–hematite tandem cells for efficient overall solar driven water splitting. Nano Lett. 2015, 15, 3833–3839. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, Y.; Wang, H.; Wang, H.; Ma, W.; Zong, X.; Li, C. Carbon encapsulation of organic–inorganic hybrid perovskite toward efficient and stable photo-electrochemical carbon dioxide reduction. Adv. Energy Mater. 2020, 10, 2002105. [Google Scholar] [CrossRef]
- Chen, J.; Dong, C.; Idriss, H.; Mohammed, O.F.; Bakr, O.M. Metal halide perovskites for solar-to-chemical fuel conversion. Adv. Energy Mater. 2020, 10, 1902433. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Vermaas, D.A.; Bi, D.; Hagfeldt, A.; Smith, W.A.; Grätzel, M. Bipolar membrane-assisted solar water splitting in optimal pH. Adv. Energy Mater. 2016, 6, 1600100. [Google Scholar] [CrossRef]
- Chen, H.; Zhang, M.; Tran-Phu, T.; Bo, R.; Shi, L.; Di Bernardo, I.; Bing, J.; Pan, J.; Singh, S.; Lipton-Duffin, J.; et al. Integrating low-cost earth-abundant co-catalysts with encapsulated perovskite solar cells for efficient and stable overall solar water splitting. Adv. Funct. Mater. 2021, 31, 2008245. [Google Scholar] [CrossRef]
- Koo, B.; Kim, D.; Boonmongkolras, P.; Pae, S.R.; Byun, S.; Kim, J.; Lee, J.H.; Kim, D.H.; Kim, S.; Ahn, B.T.; et al. Unassisted water splitting exceeding 9% solar-to-hydrogen conversion efficiency by Cu(In, Ga)(S, Se)2 photocathode with modified surface band structure and halide perovskite solar cell. ACS Appl. Energy Mater. 2020, 3, 2296–2303. [Google Scholar] [CrossRef]
- Schreier, M.; Curvat, L.; Giordano, F.; Steier, L.; Abate, A.; Zakeeruddin, S.M.; Luo, J.; Mayer, M.T.; Grätzel, M. Efficient photosynthesis of carbon monoxide from CO2 using perovskite photovoltaics. Nat. Commun. 2015, 6, 7326. [Google Scholar] [CrossRef] [Green Version]
- Mikkelsen, M.; Jørgensen, M.; Krebs, F.C. The teraton challenge. A review of fixation and transformation of carbon dioxide. Energy Environ. Sci. 2010, 3, 43–81. [Google Scholar] [CrossRef]
- Schreier, M.; Gao, P.; Mayer, M.T.; Luo, J.; Moehl, T.; Nazeeruddin, M.K.; Tilley, S.D.; Grätzel, M. Efficient and selective carbon dioxide reduction on low cost protected Cu2O photocathodes using a molecular catalyst. Energy Environ. Sci. 2015, 8, 855–861. [Google Scholar] [CrossRef]
- Kang, P.; Chen, Z.; Nayak, A.; Zhang, S.; Meyer, T.J. Single catalyst electrocatalytic reduction of CO2 in water to H2+CO syngas mixtures with water oxidation to O2. Energy Environ. Sci. 2014, 7, 4007–4012. [Google Scholar] [CrossRef]
- Hori, Y. Electrochemical CO2 Reduction on metal electrodes. In Modern Aspects of Electrochemistry; Vayenas, C.G., White, R.E., Gamboa-Aldeco, M.E., Eds.; Springer: New York, NY, USA, 2008; pp. 89–189. [Google Scholar]
- Gattrell, M.; Gupta, N.; Co, A. A review of the aqueous electrochemical reduction of CO2 to hydrocarbons at copper. J. Electroanal. Chem. 2006, 594, 1–19. [Google Scholar] [CrossRef]
- Jiang, T.; Mowbray, D.J.; Dobrin, S.; Falsig, H.; Hvolbæk, B.; Bligaard, T.; Nørskov, J.K. Trends in CO oxidation rates for metal nanoparticles and close-packed, stepped, and kinked surfaces. J. Phys. Chem. C 2009, 113, 10548–10553. [Google Scholar] [CrossRef]
- Falsig, H.; Hvolbaek, B.; Kristensen, I.S.; Jiang, T.; Bligaard, T.; Christensen, C.H.; Norskov, J.K. Trends in the catalytic CO oxidation activity of nanoparticles. Angew. Chem. Int. Ed. Engl. 2008, 47, 4835–4839. [Google Scholar] [CrossRef]
- Gong, M.; Li, Y.; Wang, H.; Liang, Y.; Wu, J.Z.; Zhou, J.; Wang, J.; Regier, T.; Wei, F.; Dai, H. An advanced Ni-Fe layered double hydroxide electrocatalyst for water oxidation. J. Am. Chem. Soc. 2013, 135, 8452–8455. [Google Scholar] [CrossRef]
- McDonald, K.J.; Choi, K.-S. Synthesis and photoelectrochemical properties of Fe2O3/ZnFe2O4 composite photoanodes for use in solar water oxidation. Chem. Mater. 2011, 23, 4863–4869. [Google Scholar] [CrossRef]
- Masayuki Yagi, E.T.; Sakita, S.; Kuwabara, T.; Nagai, K. Self-Assembly of active IrO2 colloid catalyst on an ITO electrode for efficient electrochemical water oxidation. J. Phys. Chem. B 2005, 109, 21489–21491. [Google Scholar] [CrossRef]
- Lee, Y.; Suntivich, J.; May, K.J.; Perry, E.E.; Shao-Horn, Y. Synthesis and activities of rutile IrO2 and RuO2 nanoparticles for oxygen evolution in acid and alkaline solutions. J. Phys. Chem. Lett. 2012, 3, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Zeng, K.; Zhang, D. Recent progress in alkaline water electrolysis for hydrogen production and applications. Prog. Energy Combust. Sci. 2010, 36, 307–326. [Google Scholar] [CrossRef]
- Jeon, N.J.; Noh, J.H.; Yang, W.S.; Kim, Y.C.; Ryu, S.; Seo, J.; Seok, S.I. Compositional engineering of perovskite materials for high-performance solar cells. Nature 2015, 517, 476–480. [Google Scholar] [CrossRef]
- Yang Woon, S.; Noh Jun, H.; Jeon Nam, J.; Kim Young, C.; Ryu, S.; Seo, J.; Seok Sang, I. High-performance photovoltaic perovskite layers fabricated through intramolecular exchange. Science 2015, 348, 1234–1237. [Google Scholar] [CrossRef] [PubMed]
- Jung, E.H.; Jeon, N.J.; Park, E.Y.; Moon, C.S.; Shin, T.J.; Yang, T.-Y.; Noh, J.H.; Seo, J. Efficient, stable and scalable perovskite solar cells using poly (3-hexylthiophene). Nature 2019, 567, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Min, H.; Lee, D.Y.; Kim, J.; Kim, G.; Lee, K.S.; Kim, J.; Paik, M.J.; Kim, Y.K.; Kim, K.S.; Kim, M.G.; et al. Perovskite solar cells with atomically coherent interlayers on SnO2 electrodes. Nature 2021, 598, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Fu, Y.; Meng, F.; Wu, X.; Gong, Z.; Ding, Q.; Gustafsson, M.V.; Trinh, M.T.; Jin, S.; Zhu, X.Y. Lead halide perovskite nanowire lasers with low lasing thresholds and high quality factors. Nat. Mater. 2015, 14, 636–642. [Google Scholar] [CrossRef]
- Luo, J.; Wang, X.; Li, S.; Liu, J.; Guo, Y.; Niu, G.; Yao, L.; Fu, Y.; Gao, L.; Dong, Q.; et al. Efficient and stable emission of warm-white light from lead-free halide double perovskites. Nature 2018, 563, 541–545. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.; Moon, C.S.; Yang, T.-Y.; Kim, Y.Y.; Chung, J.; Jung, E.H.; Shin, T.J.; Jeon, N.J.; Park, H.H.; Seo, J. A thermally induced perovskite crystal control strategy for efficient and photostable wide-bandgap perovskite solar cells. Sol. RRL 2020, 4, 2000033. [Google Scholar] [CrossRef]
- Gharibzadeh, S.; Abdollahi Nejand, B.; Jakoby, M.; Abzieher, T.; Hauschild, D.; Moghadamzadeh, S.; Schwenzer, J.A.; Brenner, P.; Schmager, R.; Haghighirad, A.A.; et al. Record open-circuit voltage wide-bandgap perovskite solar cells utilizing 2D/3D perovskite heterostructure. Adv. Energy Mater. 2019, 9, 1803699. [Google Scholar] [CrossRef]
- Huan, T.N.; Dalla Corte, D.A.; Lamaison, S.; Karapinar, D.; Lutz, L.; Menguy, N.; Foldyna, M.; Turren-Cruz, S.-H.; Hagfeldt, A.; Bella, F.; et al. Low-cost high-efficiency system for solar-driven conversion of CO2 to hydrocarbons. Proc. Natl. Acad. Sci. USA 2019, 116, 9735. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Yin, J.; Zheng, X.; Ait Ahsaine, H.; Zhou, Y.; Dong, C.; Mohammed, O.F.; Takanabe, K.; Bakr, O.M. Compositionally screened eutectic catalytic coatings on halide perovskite photocathodes for photoassisted selective CO2 reduction. ACS Energy Lett. 2019, 4, 1279–1286. [Google Scholar] [CrossRef] [Green Version]
- Nesbitt, N.T.; Ma, M.; Trześniewski, B.J.; Jaszewski, S.; Tafti, F.; Burns, M.J.; Smith, W.A.; Naughton, M.J. Au dendrite electrocatalysts for CO2 electrolysis. J. Phys. Chem. C 2018, 122, 10006–10016. [Google Scholar] [CrossRef]
- Chen, Y.; Li, C.W.; Kanan, M.W. Aqueous CO2 reduction at very low overpotential on oxide-derived Au nanoparticles. J. Am. Chem. Soc. 2012, 134, 19969–19972. [Google Scholar] [CrossRef]
- Chung, J.; Won, D.H.; Koh, J.; Kim, E.-H.; Woo, S.I. Hierarchical Cu pillar electrodes for electrochemical CO2 reduction to formic acid with low overpotential. Phys. Chem. Chem. Phys. 2016, 18, 6252–6258. [Google Scholar] [CrossRef] [PubMed]
- Won da, H.; Choi, C.H.; Chung, J.; Chung, M.W.; Kim, E.H.; Woo, S.I. Rational design of a hierarchical tin dendrite electrode for efficient electrochemical reduction of CO2. ChemSusChem 2015, 8, 3092–3098. [Google Scholar] [CrossRef]
- Klahr, B.; Gimenez, S.; Fabregat-Santiago, F.; Bisquert, J.; Hamann, T.W. Photoelectrochemical and impedance spectroscopic investigation of water oxidation with “Co–Pi”-coated hematite electrodes. J. Am. Chem. Soc. 2012, 134, 16693–16700. [Google Scholar] [CrossRef] [Green Version]
- Kanan, M.W.; Nocera, D.G. In situ formation of an oxygen-evolving catalyst in neutral water containing phosphate and Co2+. Science 2008, 321, 1072–1075. [Google Scholar] [CrossRef] [Green Version]
- Weekes, D.M.; Salvatore, D.A.; Reyes, A.; Huang, A.; Berlinguette, C.P. Electrolytic CO2 reduction in a flow cell. Acc. Chem. Res. 2018, 51, 910–918. [Google Scholar] [CrossRef] [PubMed]
- Endrődi, B.; Bencsik, G.; Darvas, F.; Jones, R.; Rajeshwar, K.; Janáky, C. Continuous-flow electroreduction of carbon dioxide. Prog. Energy Combust. Sci. 2017, 62, 133–154. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chung, J.; Jeon, N.J.; Noh, J.H. Solar-Driven Simultaneous Electrochemical CO2 Reduction and Water Oxidation Using Perovskite Solar Cells. Energies 2022, 15, 270. https://doi.org/10.3390/en15010270
Chung J, Jeon NJ, Noh JH. Solar-Driven Simultaneous Electrochemical CO2 Reduction and Water Oxidation Using Perovskite Solar Cells. Energies. 2022; 15(1):270. https://doi.org/10.3390/en15010270
Chicago/Turabian StyleChung, Jaehoon, Nam Joong Jeon, and Jun Hong Noh. 2022. "Solar-Driven Simultaneous Electrochemical CO2 Reduction and Water Oxidation Using Perovskite Solar Cells" Energies 15, no. 1: 270. https://doi.org/10.3390/en15010270