
Scale Effect on Producing Gaseous and Liquid Chemical Fuels via CO2 Reduction


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
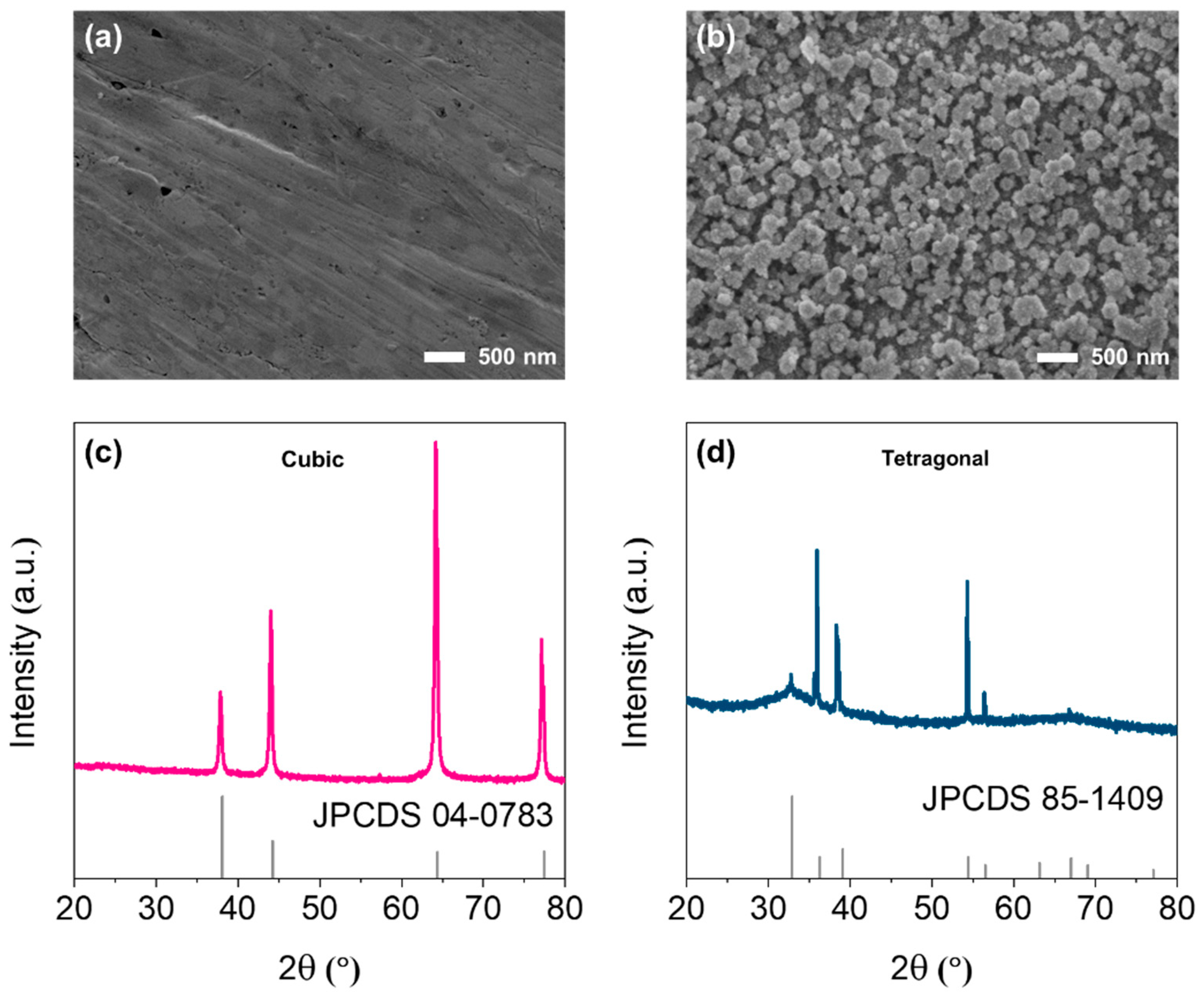
2.1. Surface Preparation
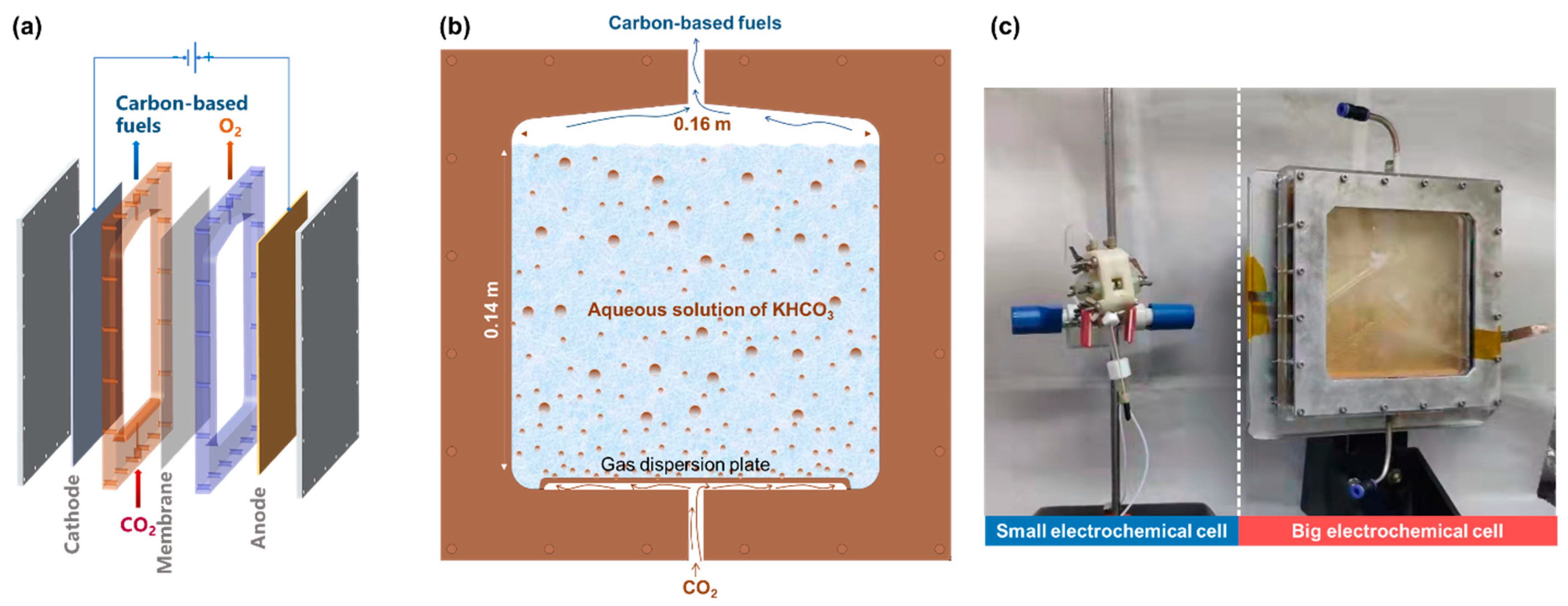
2.2. Electrochemical Cell
2.3. Product Analysis
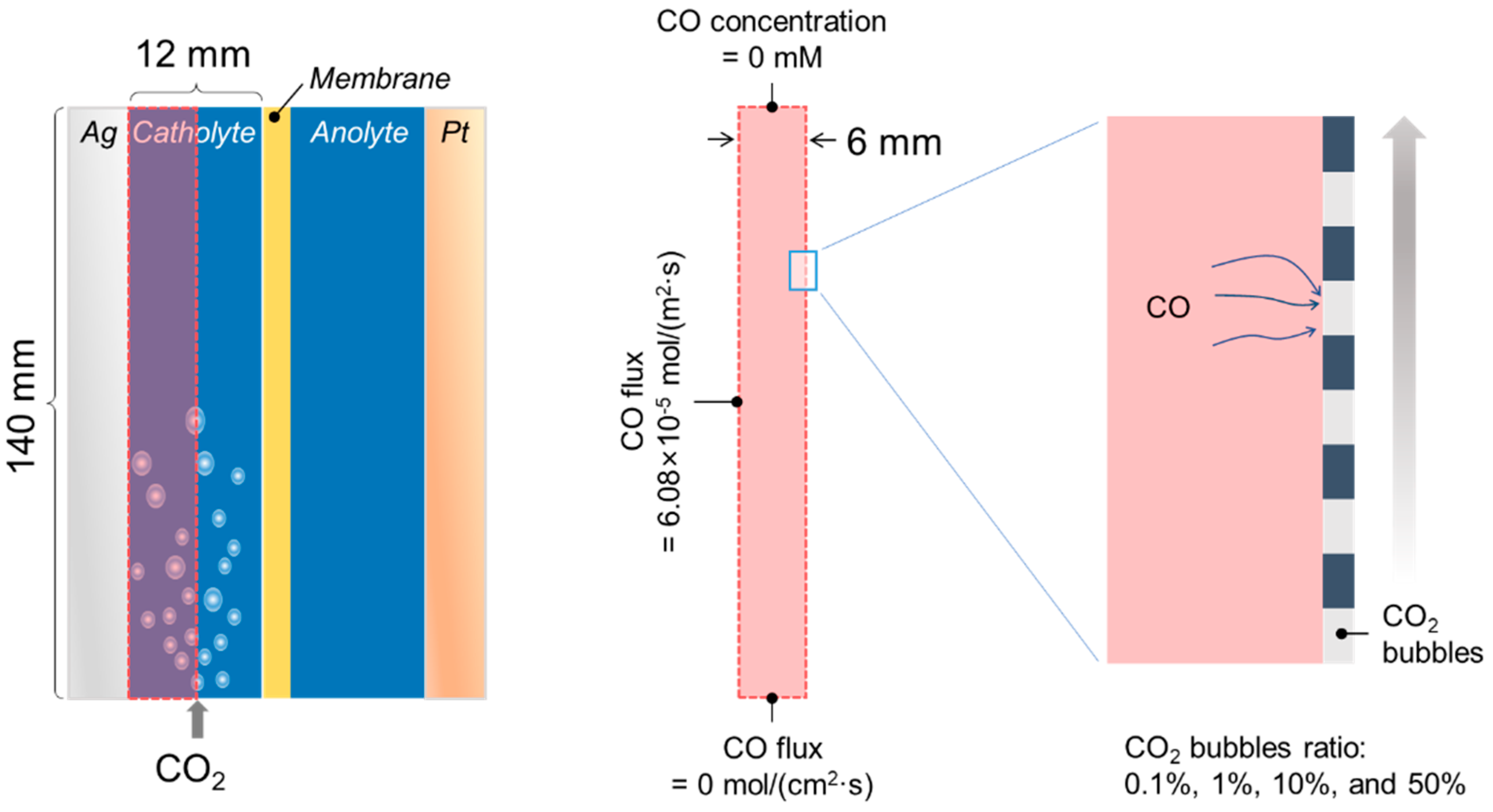
2.4. Mass Transport Simulations
3. Results and Discussion
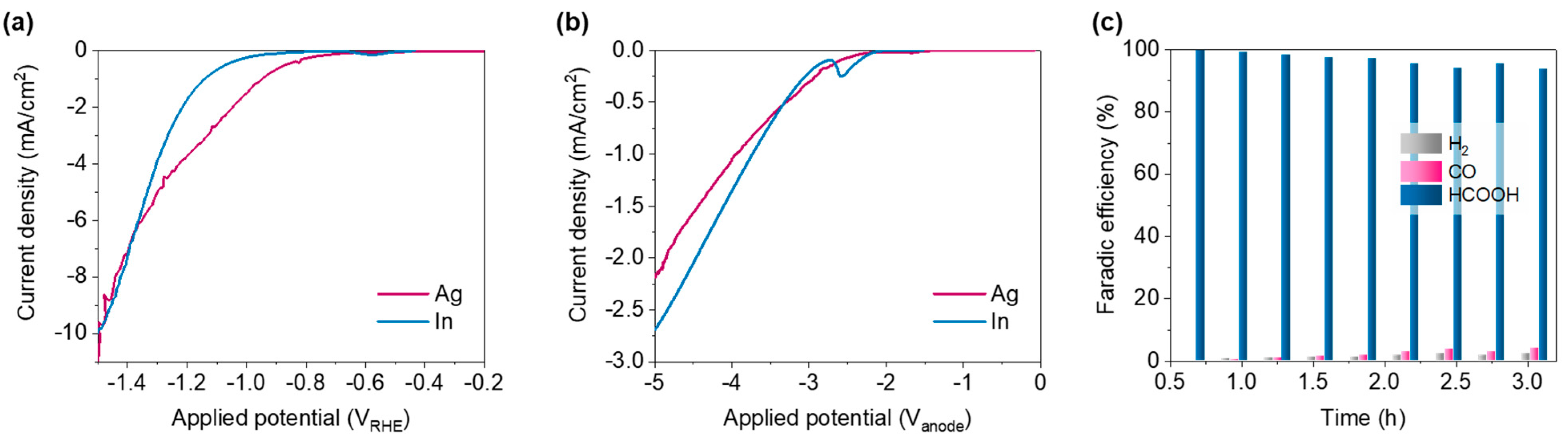
3.1. Ag and In
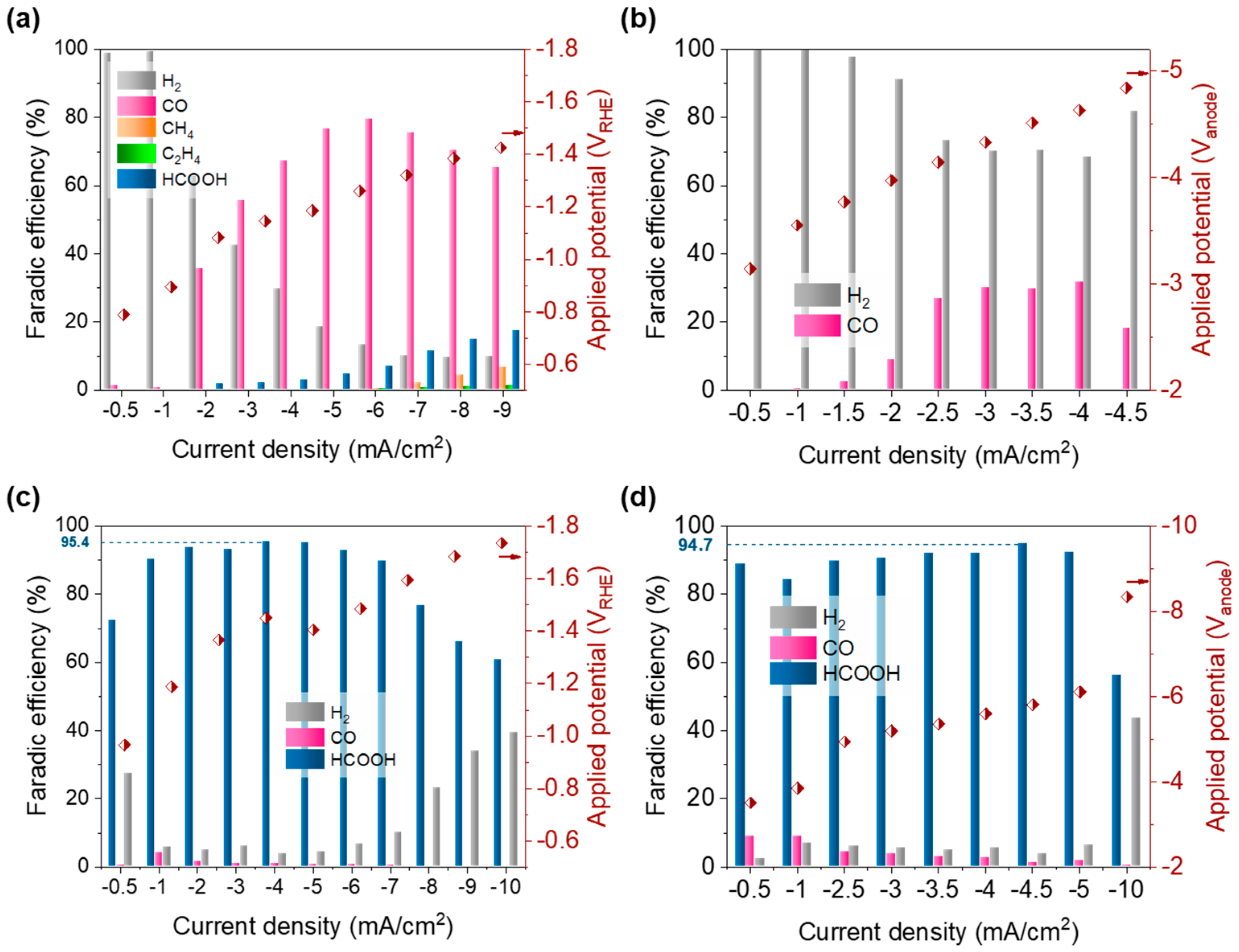
3.2. Producing Gaseous and Liquid Products
3.3. CO Blocks the Transport of CO2
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Martinez, J.; Marti Herrero, J.; Villacis, S.; Riofrio, A.J.; Vaca, D. Analysis of energy, CO2 emissions and economy of the technological migration for clean cooking in Ecuador. Energy Policy 2017, 107, 182–187. [Google Scholar] [CrossRef]
- Cheng, C.; Dong, C.-L.; Shi, J.; Mao, L.; Huang, Y.-C.; Kang, X.; Zong, S.; Shen, S. Regulation on polymerization degree and surface feature in graphitic carbon nitride towards efficient photocatalytic H2 evolution under visible-light irradiation. J. Mater. Sci. Technol. 2022, 98, 160–168. [Google Scholar] [CrossRef]
- Zhang, Y.; Shi, J.; Huang, Z.; Guan, X.; Zong, S.; Cheng, C.; Zheng, B.; Guo, L. Synchronous construction of CoS2 in-situ loading and S doping for g-C3N4: Enhanced photocatalytic H2-evolution activity and mechanism insight. Chem. Eng. J. 2020, 401, 126135. [Google Scholar] [CrossRef]
- Jia, J.; Seitz, L.C.; Benck, J.D.; Huo, Y.; Chen, Y.; Ng, J.W.D.; Bilir, T.; Harris, J.S.; Jaramillo, T.F. Solar water splitting by photovoltaic-electrolysis with a solar-to-hydrogen efficiency over 30%. Nat. Commun. 2016, 7, 13237. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Mishra, B. Photoelectrochemical Reduction of CO2 and Electrochemical Oxidation of CO. In Electrochemical Energy Conversion and Storage Systems for Future Sustainability, 1st ed.; Apple Academic Press: Palm Bay, FL, USA, 2020; pp. 57–92. [Google Scholar]
- Liu, Y.; Bai, S.; Wang, F.; Chen, Y. Photoelectrochemical technology for solar fuel generation, from single photoelectrodes to unassisted cells: A review. Environ. Chem. Lett. 2021, in press. [Google Scholar] [CrossRef]
- Nitopi, S.; Bertheussen, E.; Scott, S.B.; Liu, X.; Engstfeld, A.K.; Horch, S.; Seger, B.; Stephens, I.E.L.; Chan, K.; Hahn, C.; et al. Progress and perspectives of electrochemical CO2 reduction on copper in aqueous electrolyte. Chem. Rev. 2019, 119, 7610–7672. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Guo, L. On factors limiting the performance of photoelectrochemical CO2 reduction. J. Chem. Phys. 2020, 152, 100901. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Qiu, H.; Li, J.; Guo, L.; Ager, J.W. Tandem electrocatalytic CO2 reduction with efficient intermediate conversion over pyramid-textured Cu–Ag catalysts. ACS Appl. Mater. Interfaces 2021, 13, 40513–40521. [Google Scholar] [CrossRef]
- Hori, Y.; Murata, A.; Takahashi, R. Formation of hydrocarbons in the electrochemical reduction of carbon dioxide at a copper electrode in aqueous solution. J. Chem. Soc. Faraday Trans. 1989, 85, 2309–2326. [Google Scholar] [CrossRef]
- Hori, Y.; Wakebe, H.; Tsukamoto, T.; Koga, O. Electrocatalytic process of CO selectivity in electrochemical reduction of CO2 at metal electrodes in aqueous media. Electrochim. Acta 1994, 39, 1833–1839. [Google Scholar] [CrossRef]
- Feaster, J.T.; Shi, C.; Cave, E.R.; Hatsukade, T.; Abram, D.N.; Kuhl, K.P.; Hahn, C.; Nørskov, J.K.; Jaramillo, T.F. Understanding selectivity for the electrochemical reduction of carbon dioxide to formic acid and carbon monoxide on metal electrodes. ACS Catal. 2017, 7, 4822–4827. [Google Scholar] [CrossRef]
- Kuhl, K.P.; Hatsukade, T.; Cave, E.R.; Abram, D.N.; Kibsgaard, J.; Jaramillo, T.F. Electrocatalytic conversion of carbon dioxide to methane and methanol on transition metal surfaces. J. Am. Chem. Soc. 2014, 136, 14107–14113. [Google Scholar] [CrossRef] [PubMed]
- Peterson, A.A.; Nørskov, J.K. Activity descriptors for CO2 electroreduction to methane on transition-metal catalysts. J. Phys. Chem. Lett. 2012, 3, 251–258. [Google Scholar] [CrossRef]
- Lum, Y.; Ager, J.W. Evidence for product-specific active sites on oxide-derived Cu catalysts for electrochemical CO2 reduction. Nat. Catal. 2019, 2, 86–93. [Google Scholar] [CrossRef]
- Zheng, T.; Liu, C.; Guo, C.; Zhang, M.; Li, X.; Jiang, Q.; Xue, W.; Li, H.; Li, A.; Pao, C.-W.; et al. Copper-catalysed exclusive CO2 to pure formic acid conversion via single-atom alloying. Nat. Nanotechnol. 2021, 16, 1386–1393. [Google Scholar] [CrossRef]
- Choi, C.; Kwon, S.; Cheng, T.; Xu, M.; Tieu, P.; Lee, C.; Cai, J.; Lee, H.M.; Pan, X.; Duan, X.; et al. Highly active and stable stepped Cu surface for enhanced electrochemical CO2 reduction to C2H4. Nat. Catal. 2020, 3, 804–812. [Google Scholar] [CrossRef]
- Duan, Y.-X.; Zhou, Y.-T.; Yu, Z.; Liu, D.-X.; Wen, Z.; Yan, J.-M.; Jiang, Q. Boosting production of HCOOH from CO2 electroreduction via Bi/CeOx. Angew. Chem. Int. Ed. 2021, 60, 8798–8802. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Zhang, X.-G.; Jiang, K.; Wu, D.-Y.; Cai, W.-B. Boosting formate production in electrocatalytic CO2 reduction over wide potential window on pd surfaces. J. Am. Chem. Soc. 2018, 140, 2880–2889. [Google Scholar] [CrossRef]
- Kumawat, A.S.; Sarkar, A. Electrochemical reduction of CO2 on Pb–Bi–Sn metal mixtures: Importance of eutectics. SN Appl. Sci. 2019, 1, 317. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Li, F. Reactor design for electrochemical CO2 conversion toward large-scale applications. Curr. Opin. Green Sustain. Chem. 2021, 27, 100419. [Google Scholar] [CrossRef]
- Kuhl, K.P.; Cave, E.R.; Abram, D.N.; Jaramillo, T.F. New insights into the electrochemical reduction of carbon dioxide on metallic copper surfaces. Energy Environ. Sci. 2012, 5, 7050–7059. [Google Scholar] [CrossRef]
- Lu, W.; Zhang, Y.; Zhang, J.; Xu, P. Reduction of gas CO2 to CO with high selectivity by Ag nanocube-based membrane cathodes in a photoelectrochemical system. Ind. Eng. Chem. Res. 2020, 59, 5536–5545. [Google Scholar] [CrossRef]
- Mahyoub, S.A.; Qaraah, F.A.; Chen, C.; Zhang, F.; Yan, S.; Cheng, Z. An overview on the recent developments of Ag-based electrodes in the electrochemical reduction of CO2 to CO. Sustain. Energy Fuels 2020, 4, 50–67. [Google Scholar] [CrossRef]
- Buckley, A.K.; Cheng, T.; Oh, M.H.; Su, G.M.; Garrison, J.; Utan, S.W.; Zhu, C.; Toste, F.D.; Goddard, W.A.; Toma, F.M. Approaching 100% selectivity at low potential on Ag for electrochemical CO2 reduction to CO using a surface additive. ACS Catal. 2021, 11, 9034–9042. [Google Scholar] [CrossRef]
- Bohlen, B.; Wastl, D.; Radomski, J.; Sieber, V.; Vieira, L. Electrochemical CO2 reduction to formate on indium catalysts prepared by electrodeposition in deep eutectic solvents. Electrochem. Commun. 2020, 110, 106597. [Google Scholar] [CrossRef]
- Guo, W.; Tan, X.; Bi, J.; Xu, L.; Yang, D.; Chen, C.; Zhu, Q.; Ma, J.; Tayal, A.; Ma, J.; et al. Atomic Indium catalysts for switching CO2 electroreduction products from formate to CO. J. Am. Chem. Soc. 2021, 143, 6877–6885. [Google Scholar] [CrossRef]
- Chen, Q.; Luo, L.; Faraji, H.; Feldberg, S.W.; White, H.S. Electrochemical measurements of single H2 nanobubble nucleation and stability at Pt nanoelectrodes. J. Phys. Chem. Lett. 2014, 5, 3539–3544. [Google Scholar] [CrossRef]
- Lubetkin, S. The motion of electrolytic gas bubbles near electrodes. Electrochim. Acta 2002, 48, 357–375. [Google Scholar] [CrossRef]
- Lum, Y.; Ager, J.W. Sequential catalysis controls selectivity in electrochemical CO2 reduction on Cu. Energy Environ. Sci. 2018, 11, 2935–2944. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Lei, D.; Guo, X.; Ma, T.; Wang, F.; Chen, Y. Scale Effect on Producing Gaseous and Liquid Chemical Fuels via CO2 Reduction. Energies 2022, 15, 335. https://doi.org/10.3390/en15010335
Liu Y, Lei D, Guo X, Ma T, Wang F, Chen Y. Scale Effect on Producing Gaseous and Liquid Chemical Fuels via CO2 Reduction. Energies. 2022; 15(1):335. https://doi.org/10.3390/en15010335
Chicago/Turabian StyleLiu, Ya, Dan Lei, Xiaoqi Guo, Tengfei Ma, Feng Wang, and Yubin Chen. 2022. "Scale Effect on Producing Gaseous and Liquid Chemical Fuels via CO2 Reduction" Energies 15, no. 1: 335. https://doi.org/10.3390/en15010335