Molecular dynamics simulation is commonly used method for the software, Material Studio, Lammps, Amber, etc. This visualization software, with built-in rich algorithms, a powerful interactive interface, and multi-scale and multi-functional modules, is widely used in the field of molecular property simulation. The properties of the oil–water interface [
17], the aggregation pattern of the solution [
18], and the wettability of oil droplets on the solid surface [
19] were investigated by domestic and foreign scholars under the action of chemical flooding systems. The simulation process is chosen from all-atom molecular dynamics simulation, which has the advantage of a high accuracy compared to dissipative molecular dynamics simulation [
20].
2.1. Model Construction
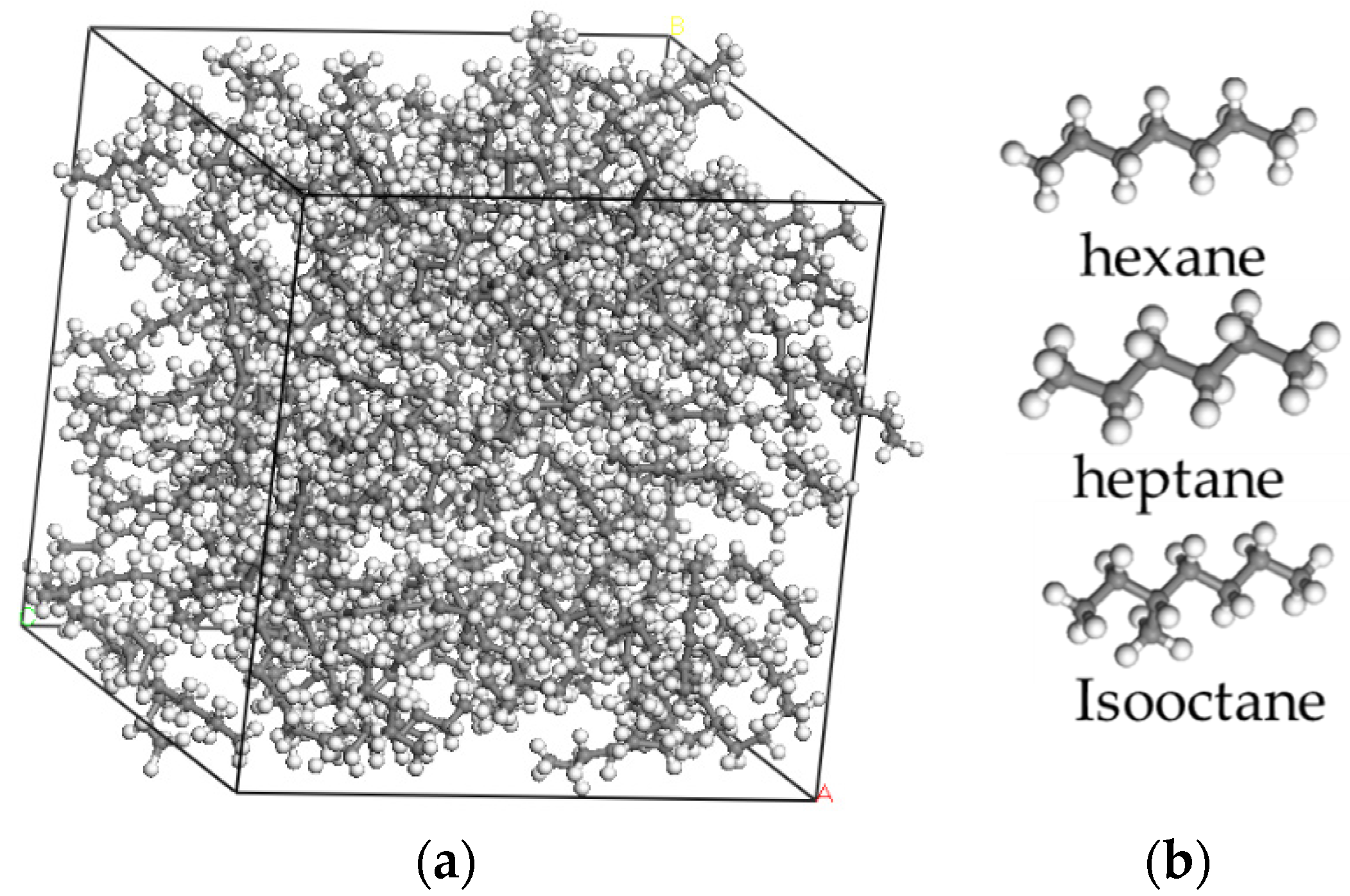
The simulations were completed with Materials Studio (MS) software, and the simulation process was carried out using the COMPASS force field [
21]. Firstly, the simulations established three crude oil systems with different molecular compositions of hexane, heptane, and isooctane, as shown in
Figure 1.
The binding energies required for the reaction of the three different coupling agents with graphene oxide were obtained according to the first principle [
22], and the parameters are shown in
Table 1. The binding energy of haloalkane with graphene oxide was +0.47, which means that the reaction of haloalkane with graphene oxide was not easy to carry out; the reaction conditions were harsh, and the resulting structures were unstable. However, the binding energies of −1.96 and −1.68, for alkylamines and silane coupling agents with graphene oxide, respectively, indicate that the reaction process of alkylamines and silane coupling agents with graphene oxide is easier; the reaction conditions are milder and the resulting products have a good structural stability.
To investigate the effect of graft length on the properties of graphene oxide, the binding energies of thirteen, sixteen, and nineteen alkylamines were separately investigated. As shown in
Table 2, the binding energies gradually decreased with increasing alkylamine carbon chain length, indicating that, as the alkylamine carbon chain length increased, the reaction proceeded with more ease, and the structure of the resulting modified graphene oxide products became more stable.
To investigate the interaction between water molecules, the reservoir surface, and modified graphene oxide, the adsorption energy of water molecules on the surface of the modified graphene oxide was simulated.
As shown in
Table 3, with 13 alkylamines, the adsorption energy of the modified water molecule is −1.06; with 16 alkylamines, the modified graphene oxide adsorption energy is −0.94; and with 19 alkyls, the modified graphene oxide adsorption energy is −0.76. The adsorption of water molecules on both types of modified graphene oxide is an exothermic process, and the surface of the modified product is strongly chemisorbed. Additionally, the octadecylamine-modified graphene oxide had the weakest interaction with water compared to the other chain lengths.
An analysis of the change in binding energy and adsorption energy shows that as the graft chain length increases, the exothermic reaction is enhanced and the binding energy increases. As the graft chain length increases, the adsorption is a spontaneous process and the adsorption energy decreases. On balance, the final choice was to choose s with a relatively smooth reaction process, a high adsorption energy of the modified molecules with water, and a better reduction in interfacial tension.
By oxidizing a thin layer of graphene, structures containing -COOH and -OH on the surface could be obtained. In this study, alkyl long-chain groups were grafted onto the surface of graphene oxide using the azide chemical reaction of cetylamine, as shown in
Figure 2.
A thin-layer, graphene oxide model with a diameter of 1.8 nm was constructed, and seven cetylamine long chains were grafted on the unilateral side of the graphene oxide model to obtain partially alkyl-modified graphene oxide nanoparticles (NGOs). The NGOs before and after modification are shown in
Figure 3.
To investigate the dispersion properties of nanoparticles in water, a water–NGO miscible system was built. A square simulation box with a size of 21.92 Å × 21.92 Å × 21.92 Å was created by “amorphous cells tools”. The mixed solution system contained 56.4% water and 42.6% NGOs. The initial constructed model is shown in
Figure 4a. To investigate the self-aggregation of nanoparticles at the oil–water interface, a columnar simulation box of 26.25 Å × 26.25 Å × 777.61 Å was constructed by the “build layer” tool. To eliminate the influence of periodic boundaries, a thickness of 10 was added above the oil model. The initial model of the vacuum layer is shown in
Figure 4b, the molecular dynamics simulation was performed using the “forcite tools”, and the simulation parameters are shown in
Table 4.
2.2. Mathematical Model Construction
This simulation used an all-atom molecular dynamics simulation approach, and the Newton’s Equation of motion (1) was used during the simulation:
where:
is combined force on the molecule,
is relative molecular mass,
is the acceleration of the molecule. The force on the atom can be obtained from the potential energy concerning the position in the coordinate system as Equation of motion (2):
Because there are no charged molecules in the system, the potential energy function [
23] discards electrostatic interactions and constructs a potential energy function that includes inter-bonding atomic interactions and van der Waals forces, with the specific functional relationship shown in Equation (3):
where:
is force constant,
is equilibrium bond length,
is equilibrium bond angle,
is van der Waals force constant between atoms,
is the minimum distance between atoms, and
is the distance between atoms at equilibrium.
The integration process uses the Verlet (1967) integral equation of motion algorithm. The advantages of the Verlet integrator are that it has only one energy evaluation per step, requires only a modest amount of memory, and allows relatively large time steps to be used. The Verlet velocity algorithm overcomes the disadvantages of the Verlet step-over method, which is not synchronous. The Verlet velocity algorithm is provided in Equations (4)–(6):
where:
is relative position,
is relative velocity, and
is the acceleration of the atom.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}