1. Introduction
Supersonic flight put forward a significant challenge to cooling aeroengines. Higher flight speeds and compressor pressure ratios increase the compressor exit temperature. When flight speed reaches Mach 2.4, the temperature of cooling air from the compressor outlet is as high as 890 °C [
1]. At this temperature, it is difficult to meet the stringent cooling requirements of hot components. An innovative and effective way to accomplish this is to fully use the fuel’s heat sink to precool the cooling air; this is known as cooled cooling air (CCA) technology [
1]. In a CCA system, the bleed air is removed at the compressor exit and then cooled as it passes through an air-to-fuel heat exchanger. One principal barrier to using CCA technology is the coking of high temperature fuel; carbonaceous surface deposits in the heat exchange passages reduce the performance of the heat transfer, and even endanger the fuel system.
Heated hydrocarbon fuels can produce surface deposits through autoxidation and pyrolysis reactions. The former reaction, called thermal oxidative deposition, is dominant at bulk temperatures between 150 and 450 °C [
2]. When fuel temperature is greater than 480 °C, the dissolved oxygen is depleted, and pyrolytic deposition reactions become dominant [
3]. Thermal oxidative deposition is the main concern in air-to-fuel heat exchange due to the low fuel operating temperature (generally no more than 500 °C). In recent decades, more research has focused on thermal oxidative deposition. It was found that many physical and chemical factors such as temperature [
4], dissolved oxygen concentration [
5,
6], system pressure [
7], surface material [
8,
9] and fuel composition [
2,
10] can greatly affect the formation of thermal oxidative depositions. Unfortunately, due to the complexity of aviation fuel composition and differences in experimental conditions, many conclusions from different researchers are inconsistent or even contradictory, especially regarding pressure. Most experimental results regarding pyrolytic deposition showed that elevated pressure promotes surface deposition because elevated pressure is conducive to the thermal cracking reaction [
3]. Ervin et al. [
11] observed that amorphous pyrolytic deposits increased several times when the pressure increased from 3.89 to 6.31 MPa. Jin et al. [
12] also found that elevating pressure from 0.7 to 3.5 MPa promoted a 4.4 times increase in the formation of pyrolytic deposits. In the fuel cracking reaction, elevated pressure increases the formation rate of aromatic products, which are the key precursors of pyrolytic coke. In contrast, the influence of pressure on thermal oxidative deposition is complex and varies with different fuels. Watt et al. [
13] studied the effect of pressures from 0.017 to 4.24 MPa for three different fuels. The peak deposit amount for the first fuel was observed at 2.17 MPa, and was approximately twice that at 1.13 MPa. The deposit amount for the second fuel at 2.17 MPa was more than those at 1.13 and 4.24 MPa. The deposit amount for the third fuel decreased with an increase in pressure. Tao et al. [
14] also tested the thermal oxidation stability of two RP-3 fuels, one mainly containing C
7−C
12 and the other mainly containing C
9−C
12. Elevated pressure inhibited deposition of the fuel with a lower distillation range (C
7−C
12) but had no obvious effect on the other fuel. Some studies have also reported that pressure has little impact on thermal oxidative deposition [
11,
15,
16]. Chin et al. [
15] observed that the deposition rate above a minimum pressure was independent of fuel pressure. This minimum pressure was considered to be related to fuel temperature and volatility. Ervin et al. [
11] also showed that elevated pressures essentially had no influence on thermal oxidative deposition but promoted pyrolytic deposition. Taylor [
16] found that the deposit amount of deoxygenated JP-5 fuel did not change as pressure increased from 1.8 MPa to 7 MPa, whereas elevated pressure inhibited the deposition of another deoxygenated fuel. Although many studies have investigated the effect of pressure on oxidative deposition, conclusions from different researchers were inconsistent or even contradictory; only the apparent laws, based on experimental phenomena, were found in the open literature. The effect and mechanism of pressure on the deposit formation of aviation fuel are confusing because there is a lack of insight into their internal mechanisms. It is necessary to analyze the effects of pressure on the formation of surface deposits further.
The formation of oxidative deposits involves complex chemical and physical processes. There are two pathways to form surface deposits [
17,
18,
19,
20,
21]. In the first pathway, hydrocarbon fuels react with dissolved oxygen in the bulk liquid. Oxidation products further react with heteroatom species to form soluble macromolecular oxidatively reactive species (SMORS) [
21]. These highly polar SMORS with a large molecular weight have limited solubility in hydrocarbon fuel. SMORS can precipitate out from bulk fuel via further polymerization or clustering reactions. The conversion between SMORS and insoluble gums depends on the solubility of SMORS in hydrocarbon fuel. Finally, insoluble gums will be deposited on the surface via a physical deposition and adhesion process. In another pathway, a portion of SMORS or reactant precursors in bulk fuel can be transported to the hot wall surface and then form deposits through a series of surface reactions. Generally, the chemical process of oxidative deposition is independent of pressure, because oxidation reactions occur in the liquid phase with weak degrees of reaction. The physical process of oxidative deposition can be affected by pressure, but its intrinsic mechanism is still poorly understood.
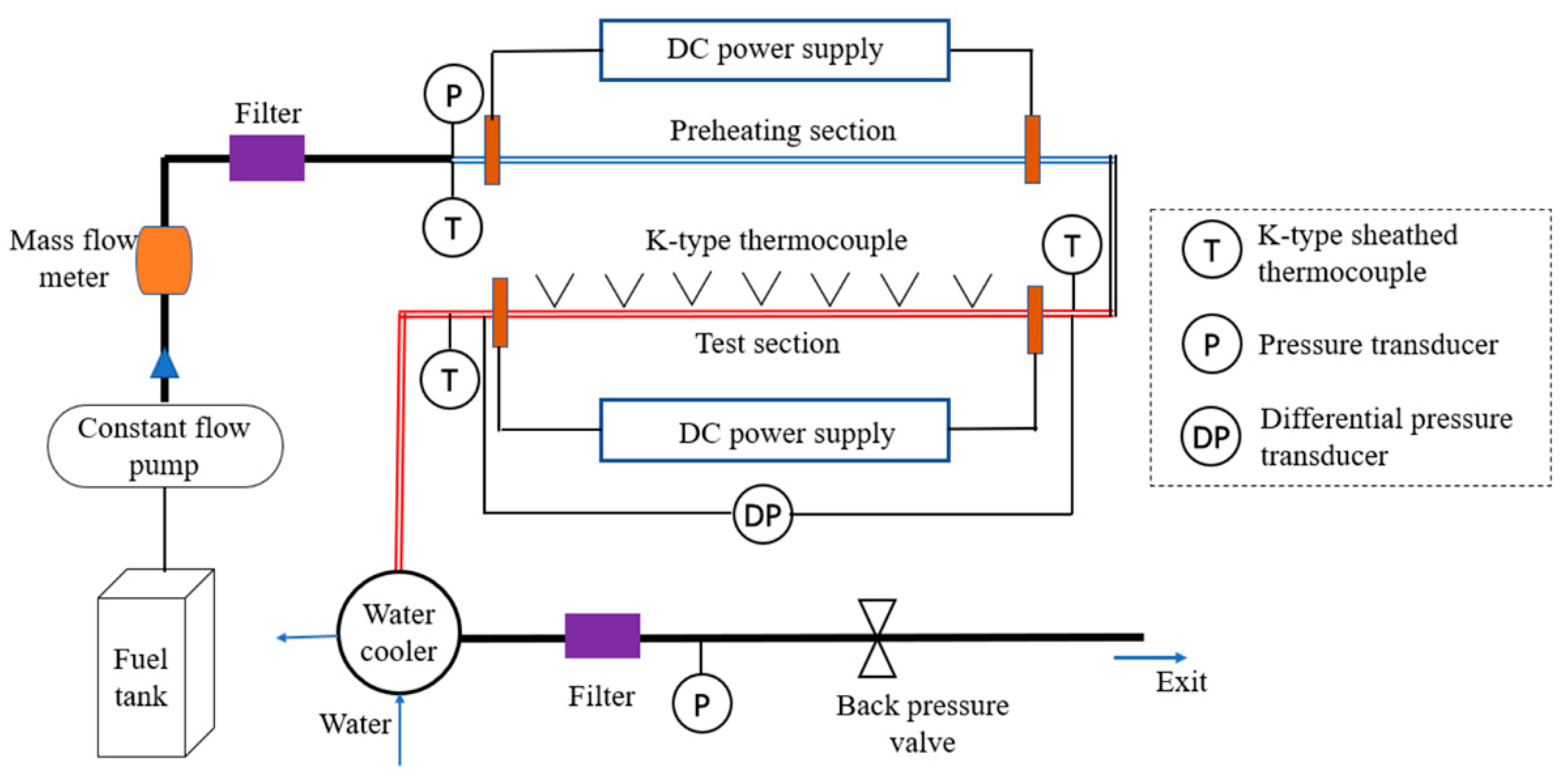
To gain additional insight regarding the influence of pressure on the deposit formation process, this study carried out aviation fuel surface deposition experiments under different pressures. Fuel in an aero-engine fuel system usually works at supercritical pressure. Therefore, deposition behaviors of fuel in the supercritical pressure range of 2.5–5.5 MPa were studied. In addition, a subcritical pressure experiment (1.5 MPa) was also included in this study to help understand the effect mechanism of pressure on deposition. A wide heat flux range (0.08–0.4 MW/m2) was applied to cover possible thermal conditions that fuel experiences in an air-to-fuel heat exchanger. In addition, some experimental cases were numerically simulated to provide more detailed information regarding the flow and heat transfer inside the tube.
3. Numerical Method
To obtain the detailed flow field and heat transfer, steady single-phase and multi-phase flow simulations were carried out for experimental cases 2–6 and case 1, respectively. The conservation equations of mass, momentum, and energy of steady single-phase incompressible flow are given below:
where
and
are velocity vectors,
is the density,
is the static pressure,
is the stress tensor,
is the gravitational volume force,
is the effective thermal conductivity, and
and
are internal energy and temperature, respectively. The first two terms on the right side in Equation (4) represent energy transport by heat conduction, and viscous dissipation, respectively.
is the heat source term.
The volume of fluid (VOF) model [
25] was used to simulate steady multi-phase flow by solving a single set of momentum equations and tracking the volume fraction of each of the fluids throughout the domain. A continuity equation for the volume fraction of one of the phases was solved to track the interface between the phases. For the
phase, this equation is:
where
is the mass transfer from phase
to phase
and
is the mass transfer from phase
to phase
.
is the volume of phase
. The Lee model [
26] was used to model the fuel evaporation process. The constraint on the volume fractions of all phases is:
A single momentum equation is identical to Equation (3) throughout the domain, and the resulting velocity field is shared among the phases, depending on the volume fractions of all phases. The energy equation is shown as Equation (4), also shared among the phases. Energy,
, as mass-averaged variables, is expressed as:
where
for each phase is based on the specific heat of that phase and the shared temperature. The present study selected the renormalization group (RNG)
turbulence model [
25] with an enhanced wall treatment to close the Reynolds-averaging momentum equations.
In the solid domain, only the heat conduction process needs to be solved:
where
is the solid thermal conductivity. The temperatures and heat fluxes on either side of the solid–fluid interface must be equal.
A commercial software, Fluent [
25], was used to simulate the coupled fluid–thermal–solid interactions. A pressure-based solver using the SIMPLE pressure–velocity coupling scheme was employed to solve steady-state governing equations. The diffusion and convection terms of governing equations were discretized using second-order central-difference and second-order upwind schemes, respectively. The temperature-dependent thermal physical properties of the aviation fuel were determined using the four-component model described in the previous section. A grid independence verification was carried out before starting simulations. Grid schemes with 58,000, 120,000 and 240,000 mesh points were compared; the mesh with 120,000 points was selected. The numerical results of Case 5 were compared with experimental results to verify the rationality of the numerical calculation method. As shown in
Figure 4, simulated wall temperatures were in good agreement with measured results. The simulation method above was applicable when the fuel temperature was less than 550 °C, because the endothermic effect of fuel cracking should be considered at higher fuel temperatures. Multi-phase flow simulation was adopted when the pressure was lower than the critical pressure of aviation fuel (2.319 MPa), whereas single-phase flow simulation was adopted when the pressure was higher than the critical pressure.
4. Results and Discussion
Deposition experiments under subcritical pressures of 1.5 MPa and 3.0 MPa were carried out to study the effect of phase transition on surface deposition; profiles of temperature and deposition rates are compared in
Figure 5. There is only a single peak in the deposition rate profile under supercritical pressure, indicating that the surface deposition rates increased as temperature increased, but decreased with the depletion of dissolved oxygen and the consumption of precursors. However, there are two deposition peaks under subcritical pressure. The first peak appears in the region where the wall temperature approached the phase transition temperature; the second peak appears in the region where the bulk temperature approached the phase transition temperature. Detailed distributions of temperature, density and liquid phase volume fraction under the subcritical pressure of 1.5 MPa are shown in
Figure 6. Under subcritical pressure, wall temperatures were maintained near the phase transition temperature as bulk temperatures rose from 180 to 332 °C (the phase transition temperature). Fuel liquid and vapor coexisted in this region due to the occurrence of the phase transition. Interestingly, high levels of surface deposition also occurred in this region, which indicates that phase transition played an important role in the formation of surface deposits. The surface deposition level at the end of the channel, where only fuel vapor exists, was very low. Obviously, the vapor phase was not the reason for the significant increase in the surface deposition. The first peak appears in the region where the bulk fuel was still in a liquid phase but changed to a vapor phase near the wall. The second peak appeared in the region where the bulk liquid completely changed to vapor. The change in fuel properties caused by phase transition played a key role in the formation of surface deposits.
Velocity, vector and density distributions on the cross-section at the first deposition peak position (X = 325 mm) are shown in
Figure 7. The radial density gradient at 1.5 MPa was significantly greater than that at 3.0 MPa due to the phase transition at the wall. In the gravity field, the secondary flow induced by the density gradient greatly enhanced mass transport between the wall and the bulk fuel. In addition, the sharp change in solubility caused by the density gradient was a key factor affecting surface deposition. A schematic diagram of the deposition acceleration mechanism is shown in
Figure 8. The density of the vapor layer near the wall was much lower than that of the bulk liquid in the tube’s center. Buoyancy force and turbulence drove the mass transfer between the vapor layer and the bulk liquid. The coking precursors produced from the liquid reaction were transported to the wall through the mass transfer mechanism. The solubility of coking precursors in the vapor phase was much lower than that in the liquid phase because solubility was strongly positively correlated with density [
27,
28,
29,
30]. Therefore, the precursors precipitated in the vapor to form insoluble species which were deposited on the wall. The precursors in the bulk fuel were continuously carried to the wall by the strong secondary flow and then further reacted to form insoluble species. This process greatly accelerated the formation of surface deposits. Consequently, the maximum surface deposition rate under subcritical pressure (1.5 MPa) was more than 7 times greater than it was under supercritical pressure (3.0 MPa).
In an actual aero-engine, heat flux conditions of the air-to-fuel heat exchanger vary with flight conditions. In this study, the effects of supercritical pressure on surface deposition at two different heat flux levels were investigated. The temperature and deposition rate profiles at a heat flux of 0.2 MW/m
2 are shown in
Figure 9. All outlet fuel temperatures were controlled to 500 °C, but wall temperatures were different under different pressures due to pressure’s influence on the fuel’s physical properties. The wall temperature difference occurred in the low deposition rate region; therefore, it had little impact on the deposition. The deposition peak appeared at the bulk temperature of approximately 150 °C. The magnitude of the deposition rate peak shows obvious pressure dependence. The deposition level at the pressure of 2.5 MPa was the highest. The deposition rate profile at 5.5 MPa was identical to that at 4.5 MPa, as the bulk temperature was below 425 °C. Variation in the deposition rate at different pressures was attributed to differences in fuel’s physical properties (especially density) at different pressures. The density gradient near the pseudocritical temperature increased as the pressure approached the critical pressure. Temperature and density distributions at the deposition peak position under 2.5 MPa are shown in
Figure 10. The temperature of the fluid near the wall was higher than that of the fluid in the center. A significant radial density gradient appeared near the wall when wall temperatures approach the pseudocritical temperature. This significant radial density gradient accelerated the formation of surface deposits through the mechanism illustrated in
Figure 8. The distributions of
at the cross-section Z = 125 mm under different pressures are shown in
Figure 11. The parameter
represents the deviation between the local density and the average density of the cross-section. The larger the parameter was, the stronger the deposition acceleration mechanism was. The
near the tube wall decreased as the pressure increased. The minimum
at 2.5 MPa was only 0.16, indicating that there was a strong deposition acceleration effect. The distributions of
at 2.5 MPa and 5.5 MPa were similar. Variation in deposition rate with pressure was highly consistent with that of the density deviation with pressure, which shows the rationality of the deposition acceleration mechanism described above.
Deposition rates at 5.5 MPa rose rapidly as the bulk temperature exceeded 425 °C. A small number of gaseous products were collected at the tube outlet under 5.5. These gaseous products were mainly composed of low molecular weight gases, such as hydrogen and methane, which are typical products of the hydrocarbon fuel pyrolysis reaction. Therefore, the high deposition level at the end of the tube under 5.5 MPa resulted from the contribution of pyrolytic deposition. Deposit morphologies at the peak position (L = 150 mm) and the high temperature section (L = 550 mm) under different pressures are shown in
Figure 12. Deposit morphologies at the deposition peaks of all pressures were similarly amorphous, which is a typical oxidative deposit morphology. In the high temperature section, granular crystallites were observed at 2.5 MPa. Granular crystallite coke was only observed on the high temperature wall, and was produced by wall catalytic pathways [
31]. The granular coke was gradually covered by another type of coke with an increase in pressure. The wall was entirely covered by condensed spherical coke at 5.5 MPa, which is the typical pyrolytic deposit [
12]; this indicated that elevated pressure promotes the formation of pyrolytic deposits.
The temperature and deposition rate profiles at a heat flux of 0.4 MW/m
2 are shown in
Figure 13. The heat flux and mass flow rate were doubled to 0.4 MW/m
2 and 1 g/s, respectively. The outlet temperatures were still maintained at 500 °C. The difference in heat transfer performance between different pressures was remarkable at the higher heat flux. The wall temperatures in the first half of the tube remained constant at 2.5 MPa and 3.5 MPa, which was similar to the wall temperature distribution in the boiling heat transfer tube. This was caused by the drastic change in the physical properties of aviation fuel (especially the specific heat and density) near the pseudocritical temperature. The wall temperature decreased as pressure approached critical. Unexpectedly, a low wall temperature did not reduce the surface deposition. The maximum deposition level still appeared at 2.5 MPa. Obviously, the deposition acceleration mechanism caused by the radial density gradient played an important role. The distribution tendencies of the deposition rate varied with pressure at a heat flux of 0.4 MW/m
2, which differed from that at a heat flux of 0.2 MW/m
2. Excessively high wall temperature led to the decomposition of oxidative deposit precursors and inhibited the formation of oxidative deposits. With the exception of a pressure of 5.5 MPa, the deposition rate decreased with increasing pressure at a heat flux of 0.4 MW/m
2. The highest deposition level appeared in the middle section of the tube under 5.5 MPa. Moreover, deposition rates at 5.5 MPa were significantly higher than those at other pressures in the high temperature section (T
bulk > 350 °C) were. The morphologies of deposits at a heat flux of 0.4 MW/m
2 are shown in
Figure 14. Condensed spherical deposit morphologies were observed in the deposition peak position (L = 250 mm) at 5.5 MPa. Condensed spherical coke is produced by homogeneous noncatalytic pyrolysis reactions [
12]. Obviously, the deposition peak at 5.5 MPa originated in the pyrolysis deposition. In addition, the gaseous products formed in the fuel pyrolysis reactions were observed at the tube outlet under 5.5 MPa. Elevated pressure promotes the pyrolysis reaction of fuel. An increase in pressure leads to pyrolysis deposition in advance. There were amorphous oxidative deposits at the inlet section (L = 50 mm) of the tube under pressures of 2.5 and 4.5 MPa, whereas the formation of oxidative deposits at the same position under a pressure of 5.5 MPa was greatly inhibited due to the excessively high wall temperature. At the end of the tube (L = 550 mm), the oxidative deposition disappeared, and only granular deposits were observed.
The maximum deposition rates for all supercritical experiment cases are summarized in
Figure 15. Except for the case with a pressure of 5.5 MPa and heat flux of 0.4 MW/m
2, the maximum deposition rate of experiment cases came from the oxidative deposition pathway. The maximum oxidative deposition rate increased as the pressure approached critical; this can be attributed to the deposition acceleration mechanism caused by the radial density gradient. The drastic change in fuel density on the tube’s cross-section led to the enhancement of mass transfer and the precipitation of insoluble substances on the wall; this promoting effect increased as pressure decreased because the fuel density gradient near the pseudocritical temperature increased as the pressure approached critical. Interestingly, maximum oxidative deposition rates in the high heat flux tube were lower than those in the lower heat flux tube; this was because excessive wall temperature (more than 400 °C) led to the decomposition of the deposit precursors [
4,
7]. However, a higher heat flux may increase the formation of pyrolytic deposits if the pressure is high enough (more than 5.5 MPa). The proper selection of heat flux and pressure can maintain the heat exchange channel at a low surface deposition level.
The dependence of the surface deposition rate on bulk and wall temperatures for all experiment cases is shown in
Figure 16. High oxidative deposition rates were concentrated in the region with wall temperatures from 300 to 400 °C, whereas pyrolytic deposition occurred only where the wall temperature was above 480 °C. However, the relationship between the oxidative deposition level and bulk temperature was weaker. The bulk temperature in the high oxidative deposition region varied from 50 to 350 °C, but the initial temperature of the oxidation reaction was approximately 150 °C [
19]. Pyrolytic deposition also occurred at a bulk temperature of 250 °C, which was far from the initial temperature of the fuel pyrolysis reaction. Obviously, the dependence of surface deposition on wall temperature was stronger than its dependence on bulk temperature; this was because the formation of coking precursors and the precipitation and deposition of insoluble substances occurred in the fluid region near the wall.





















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}