
The Relationship between Structure and Catalytic Activity-Stability of Non-Precious Metal-Based Catalysts towards Levulinic Acid Hydrogenation to γ-Valerolactone: A Review

Abstract
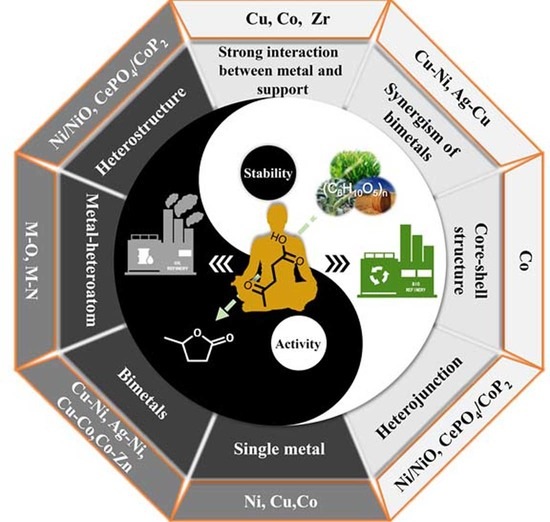
:
1. Introduction
2. Reaction Mechanism
3. Structure–Activity Relationship
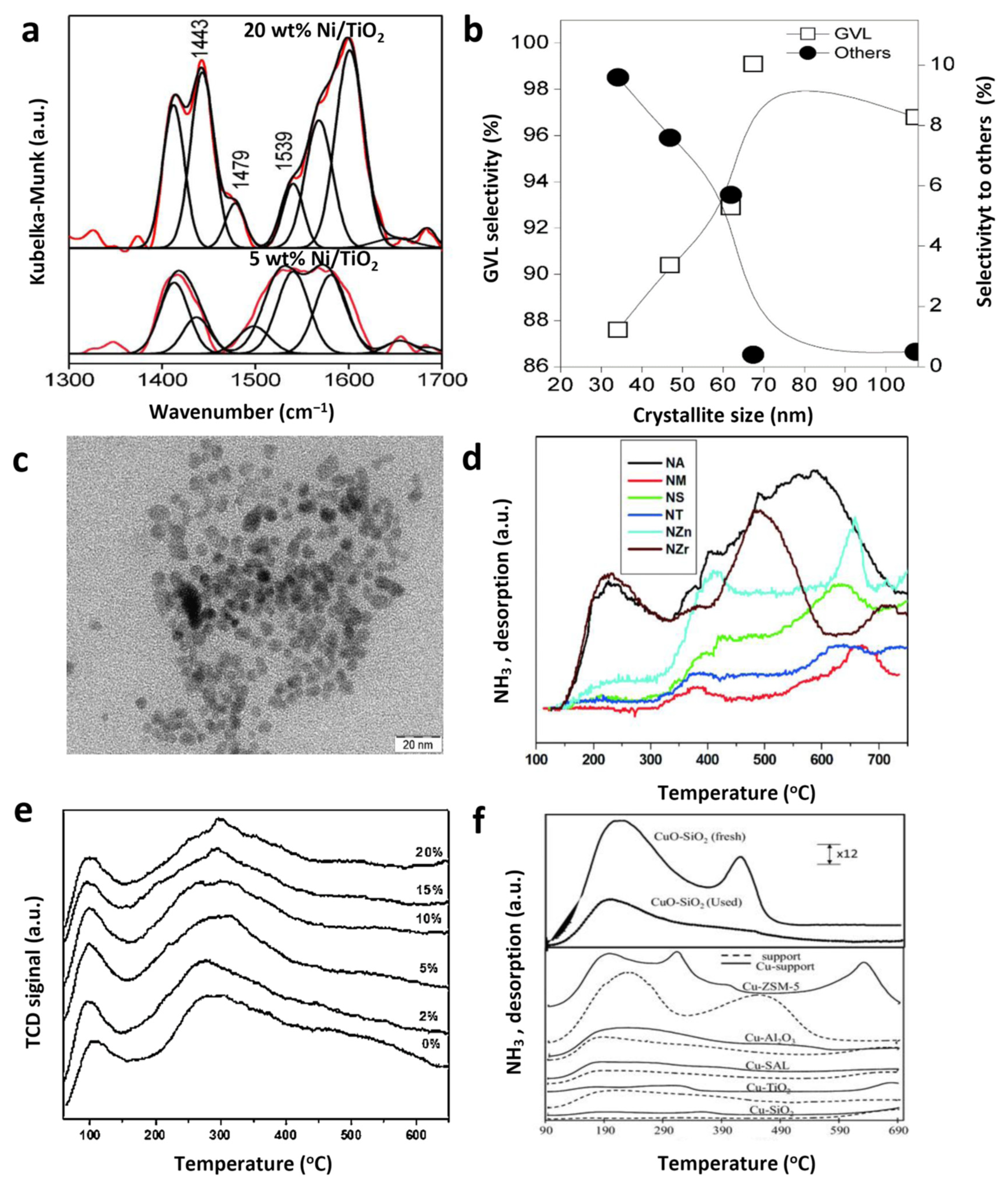
3.1. Single Metal Catalyst

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Catalyst | H Donor | Solvent | Catalyst Amount (mg) | Metal Loading (wt.%) | LA Amount (mol) | Temperature (°C) | Time (h) | H2 Pressure (MPa) | LA Conversion (%) | GVL Selectivity (%) | References |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 5 wt.%Ni/TiO2 | H2 | H2O | - | 5 | 10 [a] | 270 | - | - | 26 | 88 | [52] |
| 2 | 10 wt.%Ni/TiO2 | H2 | H2O | - | 10 | 10 [a] | 270 | - | - | 55 | 90 | |
| 3 | 15 wt.%Ni/TiO2 | H2 | H2O | - | 15 | 10 [a] | 270 | - | - | 80 | 93 | |
| 4 | 20 wt.%Ni/TiO2 | H2 | H2O | - | 20 | 10 [a] | 270 | - | - | 100 | 99 | |
| 5 | 25 wt.%Ni/TiO2 | H2 | H2O | - | 25 | 10 [a] | 270 | - | - | 92 | 97 | |
| 6 | 40%Ni/MgO | H2 | 1,4-dioxane | 100 | 40 | 0.0086 | 160 | 1 | 3 | 43 | 97.3 | [30] |
| 7 | 40%Ni/MgAl0.5O1.75 | H2 | 1,4-dioxane | 100 | 40 | 0.0086 | 160 | 1 | 3 | 83 | 93.4 | |
| 8 | 40%Ni/MgAlO2.5 | H2 | 1,4-dioxane | 100 | 40 | 0.0086 | 160 | 1 | 3 | 100 | 99.7 | |
| 9 | 40%Ni/MgAl2O4 | H2 | 1,4-dioxane | 100 | 40 | 0.0086 | 160 | 1 | 3 | 100 | 94.9 | |
| 10 | 40%Ni/Al2O3 | H2 | 1,4-dioxane | 100 | 40 | 0.0086 | 160 | 1 | 3 | 22 | 95.6 | |
| 11 | Cu/γ-Al2O3 [c] | H2 | H2O | 300 | 5 | 10 [a] | 265 | - | 98 | 87 | [54] | |
| 12 | Cu/γ-Al2O3 [c] | H2 | H2O | 300 | 2 | 10 [a] | 265 | - | 82 | 76 | ||
| 13 | Cu/γ-Al2O3 [c] | H2 | H2O | 300 | 10 | 10 [a] | 265 | - | 76 | 71 | ||
| 14 | Cu/γ-Al2O3 [c] | H2 | H2O | 300 | 15 | 10 [a] | 265 | - | 69 | 66 | ||
| 15 | Cu/γ-Al2O3 [c] | H2 | H2O | 300 | 20 | 10 [a] | 265 | - | 41 | 56 | ||
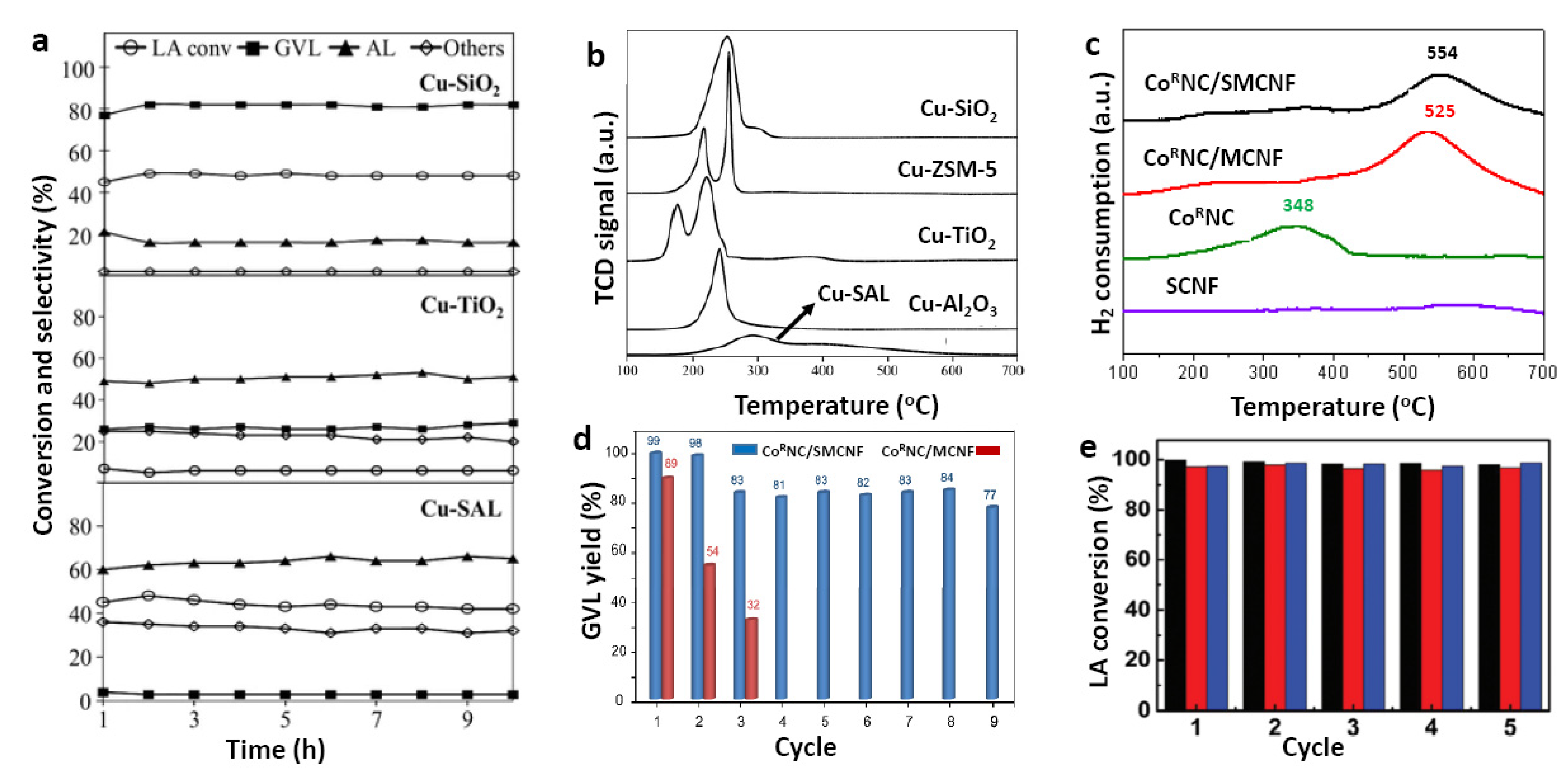
| 16 | Cu/SiO2 | FA | 1000 | 6 | 1 [b] | 250 | 10 [d] | 0.1 | 48 | 17 | [55] | |
| 17 | Cu/TiO2 | FA | 1000 | 6 | 1 [b] | 250 | 10 [d] | 0.1 | 8 | 55 | ||
| 18 | Cu/ZSM-5 | FA | 1000 | 6 | 1 [b] | 250 | 10 [d] | 0.1 | 38 | 82 | ||
| 19 | Cu/Al2O3 | FA | 1000 | 6 | 1 [b] | 250 | 10 [d] | 0.1 | 24 | 15 | ||
| 20 | Cu/SAL | FA | 1000 | 6 | 1 [b] | 250 | 10 [d] | 0.1 | 48 | 64 | ||
| 21 | 1 Co/Al2O3 | H2 | 1,4-dioxane | 33 | 54 | 0.02 | 180 | 3 | 5 | 43 | 99 | [58] |
| 22 | 2 Co/Al2O3 | H2 | 1,4-dioxane | 25 | 70 | 0.02 | 180 | 3 | 5 | 46 | 99 | |
| 23 | 3 Co/Al2O3 | H2 | 1,4-dioxane | 23 | 78 | 0.02 | 180 | 3 | 5 | 43 | 99 | |
| 24 | 4 Co/Al2O3 | H2 | 1,4-dioxane | 22 | 82 | 0.02 | 180 | 3 | 5 | 100 | 99 | |
| 25 | 5 Co/Al2O3 | H2 | 1,4-dioxane | 21 | 85 | 0.02 | 180 | 3 | 5 | 93 | 99 |
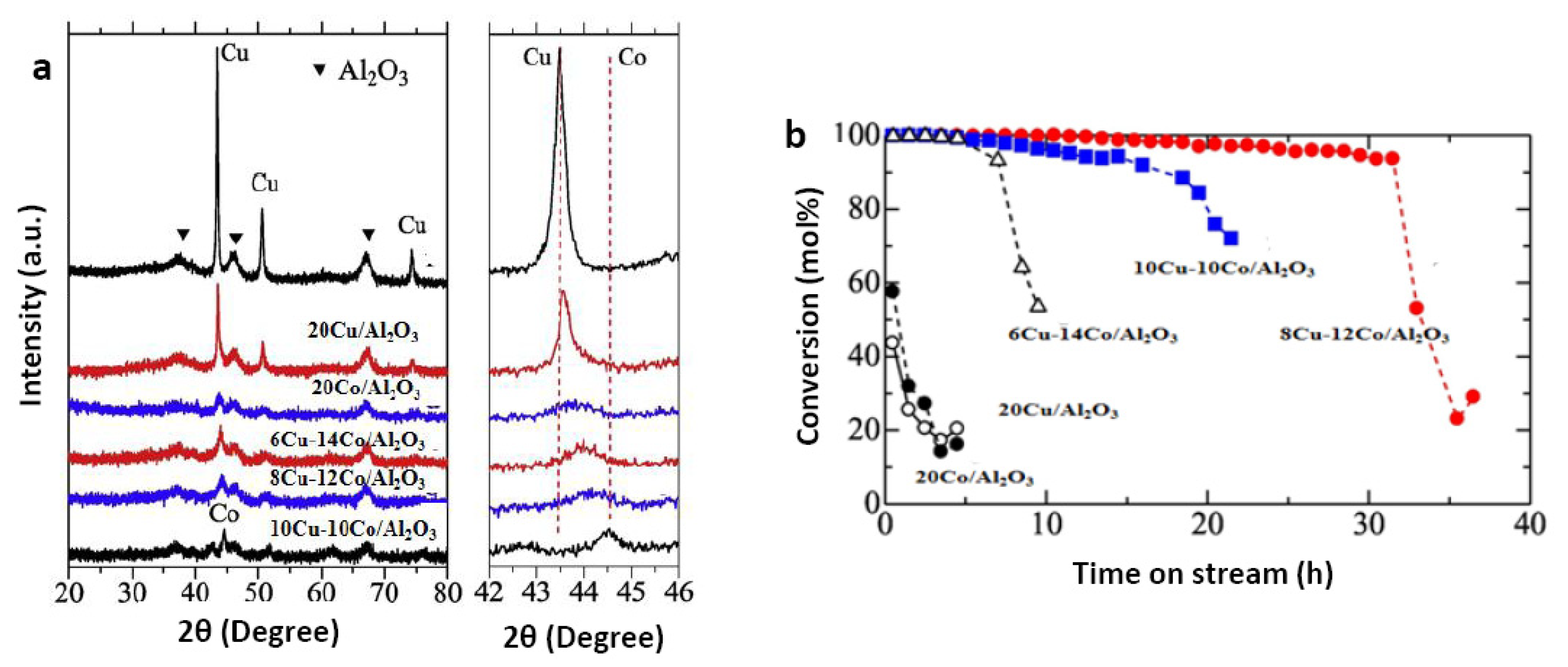
3.2. Bimetallic Catalyst
| Entry | Catalyst | H Donor | Solvent | Catalyst Amount (mg) | Metal Loading (wt.%) | LA Amount (mmol) | Temperature (°C) | Time (h) | H2 Pressure (MPa) | LA Conversion (%) | GVL Selectivity (%) | References |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Ni/Al2O3 (WI) [a] | H2 | H2O | 1000 | 36 (Ni) | 200 [c] | 250 | 6 | 6.5 | 100 | 91–92 | [59] |
| 2 | Cu/Al2O3 (WI) [a] | H2 | H2O | 1000 | 30 (Cu) | 200 [c] | 250 | 6 | 6.5 | 75 | 66 | |
| 3 | Ni–Cu/Al2O3 (WI) [a] | H2 | H2O | 1000 | 20 (Ni) 13 (Cu) | 200 [c] | 250 | 2 | 6.5 | 100 | 91–92 | |
| 4 | Ni–Cu/Al2O3 (SG 300) [b] | H2 | H2O | 1000 | 13 (Ni) 9 (Cu) | 200 [c] | 250 | 2 | 6.5 | 100 | 96 | |
| 5 | Ni–Cu/Al2O3 (SG 450) [b] | H2 | H2O | 1000 | 9 (Ni) 6 (Cu) | 200 [c] | 250 | 2 | 6.5 | 100 | 92 | |
| 6 | Cu/Al | H2 | 1,4-dioxane | 100 | 70 (Cu) | 4.3 | 140 | 3 | 3 | 76 | 58 | [20] |
| 7 | Ni/Al | H2 | 1,4-dioxane | 100 | 54 (Ni) | 4.3 | 140 | 3 | 3 | 8 | 8 | |
| 8 | Mg/Al | H2 | 1,4-dioxane | 100 | - | 4.3 | 140 | 3 | 3 | 5 | 3 | |
| 9 | Cu/Mg/Al | H2 | 1,4-dioxane | 100 | 41 (Cu) | 4.3 | 140 | 3 | 3 | 100 | 82 | |
| 10 | Ni/Mg/Al | H2 | 1,4-dioxane | 100 | 39 (Ni) | 4.3 | 140 | 3 | 3 | 15 | 15 | |
| 11 | Cu/Ni/Mg/Al | H2 | 1,4-dioxane | 100 | 30 (Cu) 28 (Ni) | 4.3 | 140 | 3 | 3 | 100 | 100 | |
| 12 | Cu/Ni/Mg/Al | H2 | 1,4-dioxane | 100 | 24 (Cu) 30 (Ni) | 4.3 | 140 | 3 | 3 | 100 | 100 | |
| 13 | Cu/Ni/Mg/Al | H2 | 1,4-dioxane | 100 | 18 (Cu) 32 (Ni) | 4.3 | 140 | 3 | 3 | 80 | 78 | |
| 14 | Cu/Ni/Mg/Al | H2 | 1,4-dioxane | 100 | 28 (Cu) 18 (Ni) | 4.3 | 140 | 3 | 3 | 100 | 100 | |
| 15 | Cu/Ni/Mg/Al | H2 | 1,4-dioxane | 100 | 33 (Cu) 10 (Ni) | 4.3 | 140 | 3 | 3 | 100 | 85 | |
| 16 | 10% Ag/ZrO2 | FA | H2O | 500 | 10 (Ag) | 43 | 220 | 5 | - | 22 | 22 | [32] |
| 17 | 20% Ni/ZrO2 | FA | H2O | 500 | 20 (Ni) | 43 | 220 | 5 | - | 34 | 34 | |
| 18 | 10% Ag-20% Ni/ZrO2 | FA | H2O | 500 | 10 (Ag) 20 (Ni) | 43 | 220 | 5 | - | 100 | 99 | |
| 19 | 10% Ag-20% Ni/ZrO2 | FA | H2O | 500 | 10 (Ag) 20 (Ni) | 43 | 200 | 7 | - | 99 | 98 | |
| 20 | 10% Ag-20% Ni/ZrO2 | FA | H2O | 500 | 10 (Ag) 20 (Ni) | 43 | 150 | 7 | - | 21 | 21 | |
| 21 | 10% Ag-20% Ni/ZrO2 | FA | H2O | 500 | 10 (Ag) 20 (Ni) | 43 | 220 | 1 | - | 34 | 34 | |
| 22 | 5% Ag-20% Ni/ZrO2 | FA | H2O | 500 | 5 (Ag) 20 (Ni) | 43 | 220 | 5 | - | 53 | 52 | |
| 23 | 10% Ag-10% Ni/ZrO2 | FA | H2O | 500 | 10 (Ag) 10 (Ni) | 43 | 200 | 5 | - | 61 | 60 | |
| 24 | 10% Ag-20% Ni/ZrO2 | FA | H2O | 500 | 10 (Ag) 20 (Ni) | 86 | 220 | 5 | - | 79 | 78 | |
| 25 | 10% Ag/ZrO2 + 20% Ni/ZrO2 | FA | H2O | 500 | 10 (Ag) 20 (Ni) | 43 | 220 | 5 | - | 41 | 41 | |
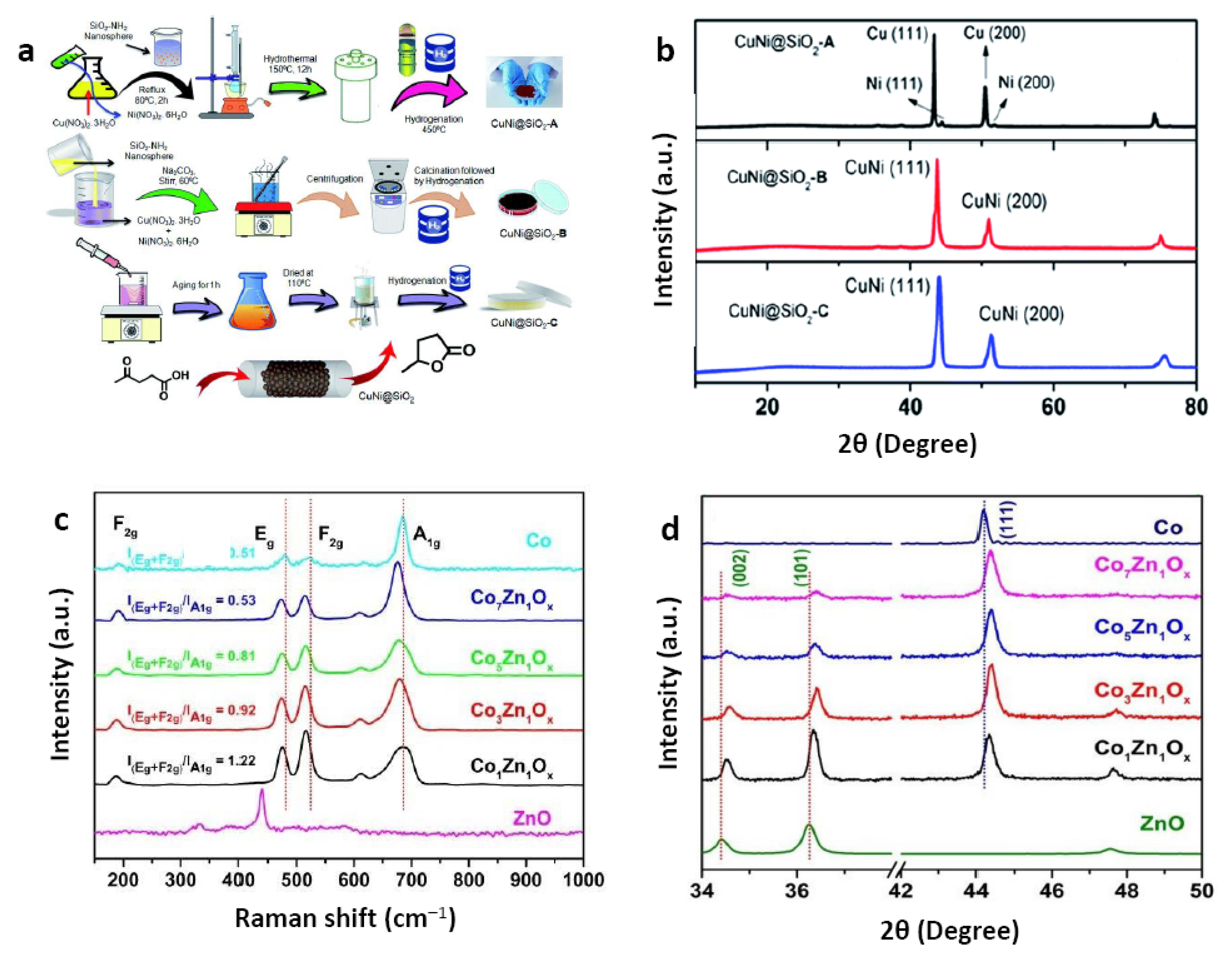
| 26 | CuNi@SiO2-A | H2 | 2-PrOH [d] | 65 | - | 2 | 120 | 13 | 4 | 68.9 | 75.3 | [61] |
| 27 | CuNi@SiO2-B | H2 | 2-PrOH | 65 | 22 (Cu) 11 (Ni) | 2 | 120 | 13 | 4 | 99.3 | 96.8 | |
| 28 | CuNi@SiO2-C | H2 | 2-PrOH | 65 | - | 2 | 120 | 13 | 4 | 34.6 | 27.6 | |
| 29 | Cu@SiO2 | H2 | 2-PrOH | 65 | - | 2 | 120 | 13 | 4 | 55.8 | 61.2 | |
| 30 | Ni@SiO2 | H2 | 2-PrOH | 65 | - | 2 | 120 | 13 | 4 | 31.2 | 26.7 | |
| 31 | Cu-SiO2 + Ni-SiO2 | H2 | 2-PrOH | 65 | - | 2 | 120 | 13 | 4 | 75.6 | 65.3 | |
| 32 | CuCo@SiO2 | H2 | 2-PrOH | 65 | - | 2 | 120 | 13 | 4 | 42.8 | 61.3 | |
| 33 | CuFe@SiO2 | H2 | 2-PrOH | 65 | - | 2 | 120 | 13 | 4 | 23.6 | 52.3 | |
| 34 | Co5Zn1Ox | H2 | MeOH [e] | 20 | 19 (Co) 4 (Zn) | 1 | 150 | 3 | 4 | 79 | 77 | [62] |
| 35 | Co | H2 | MeOH | 20 | - | 1 | 150 | 3 | 4 | 42 | 55 |
3.3. Metal–Heteroatom (M-H) Active Sites
3.3.1. M-O Active Sites
3.3.2. M-N Active Sites
3.4. Heterostructural Catalysts
| Entry | Catalyst | H Donor | Solvent | Catalyst Amount (g) | LA Loading (mmol) | Temperature (°C) | Time(h) | H2 Pressure (MPa) | LA Conversion (%) | GVL Selectivity (%) | References |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | UiO-66-SO3H | 2-PrOH | 2-PrOH | 0.1 | 1 | 140 | 24 | 0.5 [a] | 99 | 86 | [67] |
| 2 | Zr-CA | 2-PrOH | 2-PrOH | 0.2 | 1 | 130 | 4 | 100 | 97 | [68] | |
| 3 | ZrF | 2-PrOH | H2O | 0.1 | 0.5 | 200 | 2 | 1 [b] | 96 | 98 | [69] |
| 4 | Co-900 | H2 | H2O | 0.02 | 1 | 25 | 24 | 2 | 83 | 96 | [34] |
| 5 | Co-440 | H2 | H2O | 0.02 | 1 | 60 | 6 | 2 | 14 | 97 | |
| 6 | Co-900-B | H2 | H2O | 0.02 | 1 | 60 | 6 | 2 | 15 | 97 | |
| 7 | Co/AC | H2 | H2O | 0.02 | 1 | 60 | 6 | 2 | 2 | 95 | |
| 8 | CoRNC/SMCNF | H2 | H2O | 0.065 | 2 | 180 | 4 | 4.5 | 100 | 99 | [72] |
| 9 | CoR-SMCNF | H2 | H2O | 0.065 | 2 | 180 | 2 | 4.5 | 59 | 44 | |
| 10 | CoR/SMCNF | H2 | H2O | 0.065 | 2 | 180 | 2 | 4.5 | 47 | 47 | |
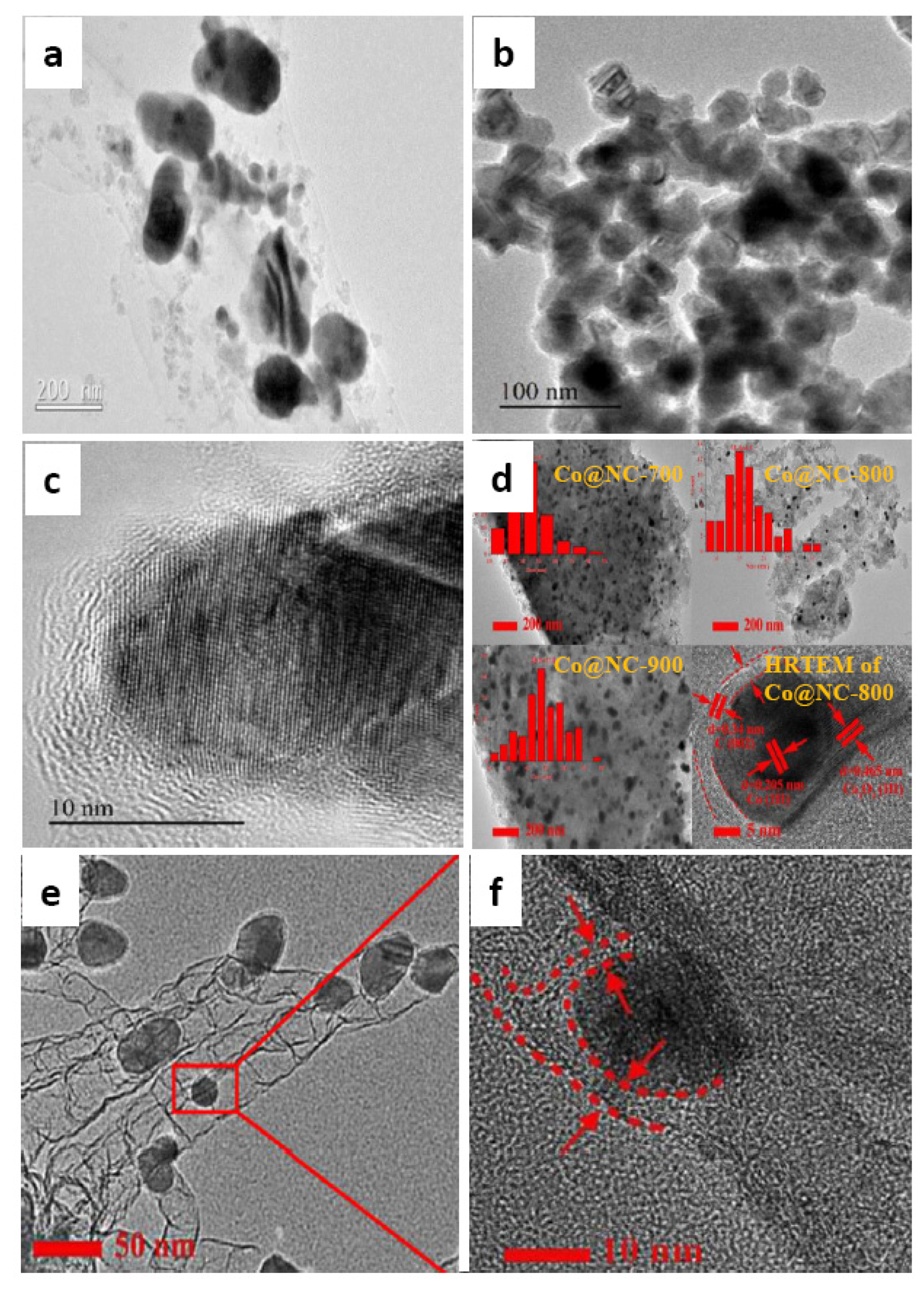
| 11 | Co@NC-800 | H2 | H2O | 0.1 | 8.6 | 220 | 5 | 2 | 100 | 100 | [33] |
| 12 | Co@NC-600 | H2 | H2O | 0.1 | 8.6 | 220 | 5 | 2 | 11 | 83 | |
| 13 | Co@NC-700 | H2 | H2O | 0.1 | 8.6 | 220 | 5 | 2 | 96 | 98 | |
| 14 | Co@NC-900 | H2 | H2O | 0.1 | 8.6 | 220 | 5 | 2 | 93 | 99 | |
| 15 | Co@N-800 | H2 | H2O | 0.1 | 8.6 | 220 | 5 | 2 | 45 | 95 | |
| 16 | Co@C-800 | H2 | H2O | 0.1 | 8.6 | 220 | 5 | 2 | 16 | 64 | |
| 17 | Co/NC-800 | H2 | H2O | 0.1 | 8.6 | 220 | 5 | 2 | 72 | 96 |
| Entry | Catalyst | H Donor | Solvent | Catalyst Amount (g) | LA Loading (mmol) | Temperature (°C) | Time (h) | H2 Pressure (MPa) | LA Conversion (%) | GVL Selectivity (%) | Mass Activity (mmol g−1 h−1) | References |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | (CePO4)0.16/Co2P | H2 | H2O | 0.1 | 2 | 90 | 1.5 | 4 | 100 | 97 | - | [42] |
| 2 | Co2P | H2 | H2O | 0.1 | 2 | 90 | 1.5 | 4 | - | 26 | - | |
| 3 | CePO4 | H2 | H2O | 0.1 | 2 | 90 | 1.5 | 4 | - | - | - | |
| 4 | (CePO4)0.08/Co2P | H2 | H2O | 0.1 | 2 | 90 | 1.5 | 4 | - | 72 | - | |
| 5 | (CePO4)0.34/Co2P | H2 | H2O | 0.1 | 2 | 90 | 1.5 | 4 | - | 61 | - | |
| 6 | NiO-523 | H2 | 1,4-dioxane | 0.2 | 10 | 120 | 24 | 2 | 100 | 100 | 14.1 | [35] |
| 7 | NiO | H2 | 1,4-dioxane | 0.2 | 10 | 120 | - | 2 | - | 100 | 0.8 | |
| 8 | Ni foil | H2 | 1,4-dioxane | 0.2 | 10 | 120 | - | 2 | - | 100 | 0.3 | |
| 9 | NiO-473 | H2 | 1,4-dioxane | 0.2 | 10 | 120 | - | 2 | - | 100 | 6.7 | |
| 10 | NiO-573 | H2 | 1,4-dioxane | 0.2 | 10 | 120 | - | 2 | - | 100 | 11.5 | |
| 11 | NiO-673 | H2 | 1,4-dioxane | 0.2 | 10 | 120 | - | 2 | - | 100 | 1.3 | |
| 12 | NiO-773 | H2 | 1,4-dioxane | 0.2 | 10 | 120 | - | 2 | - | 100 | 1.2 |
4. Structure-Stability Relationship
4.1. Strong Interaction between Active Site and Support
4.2. Synergism of Bimetallic Catalysts
4.3. Core–Shell Structure

4.4. Heterojunction
5. Summary and Perspective
- (1)
- High metal dispersion, tiny particles and optimum total acidity contribute significantly to LA hydrogenation. However, it is challenging to develop such non-precious metal-based catalysts because of their easy aggregation upon heat treatment. The characteristics of selected supports and catalyst preparation methods need to be considered comprehensively. This calls for new strategies to be explored in order to render preferable metal dispersion and loading. In addition, the non-precious metal single-atom catalysts with the highest metal dispersion may be promising for LA hydrogenation to GVL, though they are not referred to.
- (2)
- The hydrogen source is crucial for LA hydrogenation to GVL. Compared with conventional hydrogenation routes, the management of liquid alcohols used in the CTH route is more convenient and promising. The CTH route via MPV reduction shows excellent selectivity to the carbonyl group in LA, avoiding the over-hydrogenated byproducts derived from GVL. Alcohol used as both solvent and internal hydrogen source decreases the process cost. LA hydrogenation to GVL via the CTH route can also overcome the drawbacks of the FA/LA catalytic system that require harsh temperatures or long reaction times and the higher requirements for metal active sites that are resistant to FA-poisoning. Therefore, it is highly desirable to develop high-performance non-precious metal-based catalysts with the goal of LA hydrogenation via the CTH route at a lower temperature, especially at room temperature.
- (3)
- At present, M–N plays a positive role in the hydrogenation of LA to GVL in terms of activity and stability. However, the current M–N catalysts are limited to Co–N catalysts because of their facile synthesis from Co-containing MOFs. M–P catalysts can activate the carbonyl of LA and have been used in heterostructured catalysts which can be added into the library of the existing non-precious metal-based catalysts. In addition, M–S (metal–sulfur) catalysts may be feasible for LA hydrogenation, though they have not be investigated, and they are widely used in hydrodesulfurization, hydrodenitrogenation and aromatics hydrogenation reactions.
- (4)
- Coking deposition, metal sintering and leaching are important factors in the deactivation of catalysts. The deactivation of catalyst caused by coke deposition is considered to be reversible, and it can be reduced by a combination of calcination and reduction. However, repeated calcination may damage the structure of the catalyst. It is a promising strategy to design an active structure for the hydrogenation of LA to GVL at a mild temperature. New immobilization strategies, such as heteroatom doping and increasing the interaction between supports and active sites, are feasible and need to be further explored.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hayes, D.J. An examination of biorefining processes, catalysts and challenges. Catal. Today 2009, 145, 138–151. [Google Scholar] [CrossRef]
- Rose, M.; Palkovits, R. Cellulose-based sustainable polymers: State of the art and future trends, Macromol. Rapid Comm. 2011, 32, 1299–1311. [Google Scholar] [CrossRef] [PubMed]
- Cherubini, F. The biorefinery concept: Using biomass instead of oil for producing energy and chemicals. Energ. Convers. Manag. 2010, 51, 1412–1421. [Google Scholar] [CrossRef]
- Serrano-Ruiz, J.C.; Luque, R.; Sepulveda-Escribano, A. Transformations of biomass-derived platform molecules: From high added-value chemicals to fuels via aqueous-phase processing. Chem. Soc. Rev. 2011, 40, 5266–5281. [Google Scholar] [CrossRef]
- Kan, T.; Strezov, V.; Evans, T.J. Lignocellulosic biomass pyrolysis: A review of product properties and effects of pyrolysis parameters. Renew. Sust. Energ. Rev. 2016, 57, 1126–1140. [Google Scholar] [CrossRef]
- Yu, Z.H.; Lu, X.B.; Bai, H.; Xiong, J.; Feng, W.L.; Ji, N. Effects of solid acid supports on the bifunctional catalysis of levulinic acid to γ-valerolactone: Catalytic activity and stability. Chem. Asian J. 2020, 15, 1182–1201. [Google Scholar] [CrossRef]
- Ármin, S.; Márk, M.; Dibó, G.; Mika, L.T. Microwave-assisted conversion of carbohydrates to levulinic acid: An essential step in biomass conversion. Green Chem. 2013, 15, 439–445. [Google Scholar]
- Wright, W.R.H.; Palkovits, R. Development of heterogeneous catalysts for the conversion of levulinic acid to γ-valerolactone. ChemSusChem 2012, 5, 1657–1667. [Google Scholar] [CrossRef]
- Horváth, I.T.; Mehdi, H.; Fábos, V.; Boda, L.; Mika, L.T. γ-Valerolactone-a sustainable liquid for energy and carbon-based chemicals. Green Chem. 2008, 10, 238–242. [Google Scholar] [CrossRef]
- Manzer, L.E. Catalytic synthesis of α-methylene-γ-valerolactone: A biomass-derived acrylic monomer. Appl. Catal. A Gen. 2004, 272, 249–256. [Google Scholar] [CrossRef]
- Du, X.L.; He, L.; Zhao, S.; Liu, Y.M.; Cao, Y. Hydrogen-independent reductive transformation of carbohydrate biomass into gamma-valerolactone and pyrrolidone derivatives with supported gold catalysts. Angew. Chem. Int. Ed. 2011, 50, 7815–7819. [Google Scholar] [CrossRef] [PubMed]
- Bond, J.Q.; Alonso, D.M.; Wang, D.; West, R.M.; Dumesic, J.A. Integrated catalytic conversion of gamma-valerolactone to liquid alkenes for transportation fuels. Science 2010, 327, 1110–1114. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Qiao, B.; Liu, F.; Zhang, L.; Su, Y.; Wang, A.; Zhang, T. Correction: Catalytic production of 1,4-pentanediol from furfural in a fixed-bed system under mild conditions. Green Chem. 2020, 22, 3532–3538. [Google Scholar] [CrossRef]
- Al-Shaal, M.G.; Dzierbinski, A.; Palkovits, R. Solvent-free γ-valerolactone hydrogenation to 2-methyltetrahydrofuran catalysed by Ru/C: A reaction network analysis. Green Chem. 2014, 16, 1358–1364. [Google Scholar] [CrossRef]
- Carine, C.; Marcello, M.; Rinaldo, P.; Ravasio, N.; Zaccheriaa, A.F. New generation biofuels: γ-valerolactone into valeric esters in one pot. RSC Adv. 2013, 3, 1302–1306. [Google Scholar]
- Yan, K.; Lafleur, T.; Wu, X.; Chai, J.; Wu, G.; Xie, X. Cascade upgrading of γ-valerolactone to biofuels. Chem. Commun. 2015, 51, 6984–6987. [Google Scholar] [CrossRef]
- Molleti, J.; Tiwari, M.S.; Yadav, G.D. Novel synthesis of Ru/OMS catalyst by solvent-free method: Selective hydrogenation of levulinic acid to γ-valerolactone in aqueous medium and kinetic modelling. Chem. Eng. J. 2018, 334, 2488–2499. [Google Scholar] [CrossRef]
- Li, W.; Xie, J.; Lin, H.; Zhou, Q. Highly efficient hydrogenation of biomass-derived levulinic acid to γ-valerolactone catalyzed by iridium pincer complexes. Green Chem. 2012, 14, 2388–2390. [Google Scholar] [CrossRef]
- Yan, K.; Lafleur, T.; Jarvis, C.; Wu, G. Clean and selective production of γ-valerolactone from biomass-derived levulinic acid catalyzed by recyclable Pd nanoparticle catalyst. J. Clean. Prod. 2014, 72, 230–232. [Google Scholar] [CrossRef]
- Gupta, S.S.R.; Kantam, M.L. Selective hydrogenation of levulinic acid into γ-valerolactone over Cu/Ni hydrotalcite-derived catalyst. Catal. Today 2018, 309, 189–194. [Google Scholar] [CrossRef]
- Liu, D.; Zhang, L.; Han, W.; Tang, M.; Zhou, L.; Zhang, Y.; Li, X.; Qin, Z.; Yang, H. One-step fabrication of Ni-embedded hierarchically-porous carbon microspheres for levulinic acid hydrogenation. Chem. Eng. J. 2019, 369, 386–393. [Google Scholar] [CrossRef]
- Dutta, S.; Yu, I.K.; Tsang, D.C.; Ng, Y.H.; Ok, Y.S.; Sherwood, J.; Clark, J.H. Green synthesis of gamma-valerolactone (GVL) through hydrogenation of biomass-derived levulinic acid using non-noble metal catalysts: A critical review. Chem. Eng. J. 2019, 372, 992–1006. [Google Scholar] [CrossRef]
- Chia, M.; Dumesic, J.A. Liquid-phase catalytic transfer hydrogenation and cyclization of levulinic acid and its esters to γ-valerolactone over metal oxide catalysts. Chem. Commun. 2011, 47, 12233–12235. [Google Scholar] [CrossRef]
- Yu, Z.H.; Lu, X.B.; Xiong, J.; Ji, N. Transformation of levulinic acid to valeric biofuels: A review on heterogeneous bifunctional catalytic systems. ChemSusChem 2019, 12, 3915–3930. [Google Scholar] [CrossRef] [PubMed]
- Upare, P.P.; Lee, J.; Hwang, Y.K.; Hwang, D.W.; Lee, J.; Halligudi, S.B.; Hwang, J.; Chang, J. Direct hydrocyclization of biomass-derived levulinic acid to 2-methyltetrahydrofuran over nanocomposite copper/silica catalysts. ChemSusChem 2011, 4, 1749–1752. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; Li, Q.; Liu, J.; Yao, G.; Wang, J.; Zeng, X.; Huo, Z.; Jin, F. New method for highly efficient conversion of biomass-derived levulinic acid to γ-valerolactone in water without precious metal catalysts. ACS Sustain. Chem. Eng. 2017, 5, 6517–6523. [Google Scholar] [CrossRef]
- Grilc, M.; Likozar, B. Levulinic acid hydrodeoxygenation, decarboxylation and oligmerization over NiMo/Al2O3 catalyst to bio-based value-added chemicals: Modelling of mass transfer, thermodynamics and micro-kinetics. Chem. Eng. J. 2017, 330, 383–397. [Google Scholar] [CrossRef]
- Kumar, V.V.; Naresh, G.; Sudhakar, M.; Anjaneyulu, C.; Bhargava, S.K.; Tardio, J.; Reddy, V.K.; Padmasri, A.H.; Venugopal, A. An investigation on the influence of support type for Ni catalyzed vapor phase hydrogenation of aqueous levulinic acid to γ-valerolactone. RSC Adv. 2016, 6, 9872–9879. [Google Scholar] [CrossRef]
- Sudhakar, M.; Kumar, V.V.; Naresh, G.; Kantam, M.L.; Bhargava, S.K.; Venugopal, A. Vapor phase hydrogenation of aqueous levulinic acid over hydroxyapatite supported metal (M = Pd, Pt, Ru, Cu, Ni) catalysts. Appl. Catal. B Environ. 2016, 180, 113–120. [Google Scholar] [CrossRef]
- Jiang, K.; Sheng, D.; Zhang, Z.; Fu, J.; Hou, Z.; Lu, X. Hydrogenation of levulinic acid to γ-valerolactone in dioxane over mixed MgO–Al2O3 supported Ni catalyst. Catal. Today 2016, 274, 55–59. [Google Scholar] [CrossRef]
- Hengst, K.; Ligthart, D.M.; Doronkin, D.E.; Walter, K.M.; Kleist, W.; Hensen, E.J.M.; Grunwaldt, J. Continuous synthesis of γ-valerolactone in a trickle-bed reactor over supported nickel catalysts. Ind. Eng. Chem. Res. 2017, 56, 2680–2689. [Google Scholar] [CrossRef]
- Hengne, A.M.; Malawadkar, A.V.; Biradar, N.S.; Rode, C.V. Surface synergism of an Ag–Ni/ZrO2 nanocomposite for the catalytic transfer hydrogenation of bio-derived platform molecules. RSC Adv. 2014, 4, 9730–9736. [Google Scholar] [CrossRef]
- Li, W.; Geng, W.; Liu, L.; Shang, Q.; Liu, L.; Kong, X. In situ-generated Co embedded in N-doped carbon hybrids as robust catalyst for the upgrading of levulinic acid in aqueous phase. Sustain. Energy Fuels 2020, 4, 2043–2054. [Google Scholar] [CrossRef]
- Gong, W.; Lin, Y.; Chen, C.; Al-Mamun, M.; Lu, H.S.; Wang, G.; Zhang, H.; Zhao, H. Nitrogen-doped carbon nanotube confined Co–Nx sites for selective hydrogenation of biomass-derived compounds. Adv. Mater. 2019, 31, 1808341. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Yao, S.; Cao, J.; Guan, N.; Li, L. Heterostructured Ni/NiO composite as a robust catalyst for the hydrogenation of levulinic acid to γ-valerolactone. Appl. Catal. B Environ. 2017, 217, 115–124. [Google Scholar] [CrossRef]
- Ye, L.; Han, Y.; Feng, J.; Lu, X. A review about GVL production from lignocellulose: Focusing on the full components utilization. Ind. Crop. Prod. 2020, 144, 112031–112047. [Google Scholar] [CrossRef]
- Xu, Q.; Li, X.; Pan, T.; Yu, C.; Deng, J.; Guo, Q.; Fu, Y. Supported copper catalysts for highly efficient hydrogenation of biomass-derived levulinic acid and γ-valerolactone. Green Chem. 2015, 4, 2043–2054. [Google Scholar] [CrossRef]
- Zhang, L.; Mao, J.; Li, S.; Zhou, J.; Jiang, C. Hydrogenation of levulinic acid into gamma-valerolactone over in situ reduced CuAg bimetallic catalyst: Strategy and mechanism of preventing Cu leaching. Appl. Catal. B Environ. 2010, 232, 1–10. [Google Scholar] [CrossRef]
- Wang, J.; Jaenicke, S.; Chuah, G. Zirconium-beta zeolite as a robust catalyst for the transformation of levulinic acid to γ-valerolactone via meerwein–ponndorf–verley reduction. RSC Adv. 2014, 4, 13481–13489. [Google Scholar] [CrossRef]
- Yang, Z.; Huang, Y.B.; Guo, Q.X.; Fu, Y. Ni catalyzed transfer hydrogenation of levulinate esters to γ-valerolactone at room temperature. Chem Commun. 2013, 49, 5328–5330. [Google Scholar] [CrossRef]
- Abdelrahman, O.A.; Heyden, A.; Bond, J.Q. Analysis of kinetics and reaction pathways in the aqueous-phase hydrogenation of levulinic acid to form γ-valerolactone over Ru/C. ACS Catal. 2014, 4, 1171–1181. [Google Scholar] [CrossRef]
- Feng, H.; Li, X.; Qian, H.; Zhang, Y.; Zhang, D.; Zhao, D.; Hong, S.G.; Zhang, N. Efficient and sustainable hydrogenation of levulinic-acid to gamma-valerolactone in aqueous solution over acid-resistant CePO4/Co2P catalysts. Green Chem. 2019, 21, 1743–1756. [Google Scholar] [CrossRef]
- Sun, D.; Takahashi, Y.; Yamada, Y.; Sato, S. Efficient formation of angelica lactones in a vapor-phase conversion of levulinic acid. Appl. Catal. A Gen. 2016, 526, 62–69. [Google Scholar] [CrossRef] [Green Version]
- Yan, Z.; Lin, L.; Liu, S. Synthesis of γ-valerolactone by hydrogenation of biomass-derived levulinic acid over Ru/C catalyst. Energy Fuel 2009, 23, 3853–3858. [Google Scholar] [CrossRef]
- Upare, P.P.; Lee, J.; Hwang, D.W.; Halligudi, S.B.; Hwang, Y.K.; Chang, J. Selective hydrogenation of levulinic acid to γ-valerolactone over carbon-supported noble metal catalysts. J. Ind. Eng. Chem. 2011, 17, 287–292. [Google Scholar] [CrossRef]
- Mamun, O.; Walker, E.; Faheem, M.; Bond, J.Q.; Heyden, A. Theoretical investigation of the hydrodeoxygenation of levulinic acid to γ-valerolactone over Ru (0001). ACS Catal. 2016, 7, 215–228. [Google Scholar] [CrossRef]
- Mamun, O.; Saleheen, M.; Bond, J.Q.; Heyden, A. Investigation of solvent effects in the hydrodeoxygenation of levulinic acid to γ-valerolactone over Ru catalysts. J. Catal. 2019, 379, 164–179. [Google Scholar] [CrossRef]
- Yu, Z.; Lu, X.; Xiong, J.; Li, X.; Bai, H.; Ji, N. Heterogeneous catalytic hydrogenation of levulinic acid to γ- valerolactone with formic acid as internal hydrogen source: Key issues and their effects. ChemSusChem 2020, 13, 2916–2930. [Google Scholar] [CrossRef]
- Kuwahara, Y.; Kaburagi, W.; Osada, Y.; Fujitani, T.; Yamashita, H. Catalytic transfer hydrogenation of biomass-derived levulinic acid and its esters to γ-valerolactone over ZrO2 catalyst supported on SBA-15 silica. Catal. Today 2017, 281, 418–428. [Google Scholar] [CrossRef]
- Yu, J.; Savage, P.E. Decomposition of formic acid under hydrothermal conditions. Ind. Eng. Chem. Res. 1998, 37, 2–10. [Google Scholar] [CrossRef]
- Assary, R.S.; Curtiss, L.A.; Dumesic, J.A. Exploring meerwein–ponndorf–verley reduction chemistry for biomass catalysis using a first-principles approach. ACS Catal. 2013, 3, 2694–2704. [Google Scholar] [CrossRef]
- Kumar, V.V.; Naresh, G.; Sudhakar, M.; Tardio, J.; Bhargava, S.K.; Venugopal, A. Role of brønsted and lewis acid sites on Ni/TiO2 catalyst for vapour phase hydrogenation of levulinic acid: Kinetic and mechanistic study. Appl. Catal. A Gen. 2015, 505, 217–223. [Google Scholar] [CrossRef]
- Mohan, V.; Venkateshwarlu, V.; Pramod, C.V.; Raju, B.D.; Rao, K.S.R. Vapour phase hydrocyclisation of levulinic acid to γ-valerolactone over supported Ni catalysts. Catal. Sci. Technol. 2014, 4, 1253–1259. [Google Scholar] [CrossRef]
- Putrakumar, B.; Nagaraju, N.; Kumar, V.P.; Chary, K.V.R. Hydrogenation of levulinic acid to γ-valerolactone over copper catalysts supported on γ-Al2O3. Catal. Today 2015, 250, 209–217. [Google Scholar] [CrossRef]
- Lomate, S.; Sultana, A.; Fujitani, T. Vapor phase catalytic transfer hydrogenation (CTH) of levulinic acid to γ-valerolactone over copper supported catalysts using formic acid as hydrogen source. Catal. Lett. 2018, 148, 348–358. [Google Scholar] [CrossRef]
- Murugesan, K.; Alshammari, A.S.; Sohail, M.; Jagadeesh, R.V. Levulinic acid derived reusable cobalt-nanoparticles-catalyzed sustainable synthesis of γ-valerolactone. ACS Sustain. Chem. Eng. 2019, 7, 14756–14764. [Google Scholar] [CrossRef]
- Novodárszki, G.; Solt, H.E.; Valyon, J.; Lónyi, F.; Hancsók, J.; Deka, D.; Tuba, R.; Mihályi, M.R. Selective hydroconversion of levulinic acid to γ-valerolactone or 2-methyltetrahydrofuran over silica-supported cobalt catalysts. Catal. Sci. Technol. 2019, 9, 2291–2304. [Google Scholar] [CrossRef]
- Long, X.; Sun, P.; Li, Z.; Lang, R.; Xia, C.; Li, F. Magnetic Co/Al2O3 catalyst derived from hydrotalcite for hydrogenation of levulinic acid to γ-valerolactone. Chin. J. Catal. 2015, 36, 1512–1518. [Google Scholar] [CrossRef]
- Obregón, I.; Corro, E.; Izquierdo, U.; Requies, J.; Arias, P.L. Levulinic acid hydrogenolysis on Al2O3-based Ni-Cu bimetallic catalysts. Chin. J. Catal. 2014, 35, 656–662. [Google Scholar] [CrossRef]
- Yanase, D.; Yoshida, R.; Kanazawa, S.; Yamada, Y.; Sato, S. Efficient formation of γ-valerolactone in the vapor-phase hydrogenation of levulinic acid over Cu-Co/alumina catalyst. Catal. Commun. 2020, 139, 105967–105971. [Google Scholar] [CrossRef]
- Pendem, S.; Mondal, I.; Shrotri, A.; Rao, B.S.; Lingaiah, N.; Mondal, J. Unraveling the structural properties and reactivity trends of Cu–Ni bimetallic nanoalloy catalysts for biomass-derived levulinic acid hydrogenation. Sustain. Energy Fuels 2018, 2, 1516–1529. [Google Scholar] [CrossRef]
- He, Z.; Jiang, C.; Wang, Z.; Wang, K.; Sun, Y.; Yao, M.; Li, Z.; Liu, Z. Catalytic hydrodeoxygenation of biomass-derived oxygenates to bio-fuels over Co-based bimetallic catalysts. Sustain. Energy Fuels 2020, 4, 4558–4569. [Google Scholar] [CrossRef]
- Yepez, A.; De, S.; Climent, M.; Romero, A.; Luque, R. Microwave-assisted conversion of levulinic acid to γ-valerolactone using low-loaded supported iron oxide nanoparticles on porous silicates. Appl. Sci. 2015, 5, 532–543. [Google Scholar] [CrossRef] [Green Version]
- Hussain, S.; Velisoju, V.K.; Rajan, N.P.; Kumar, B.P.; Chary, K.V.R. Synthesis of γ-valerolactone from levulinic acid and formic acid over Mg-Al hydrotalcite like compound. ChemistrySelect 2018, 3, 6186–6194. [Google Scholar] [CrossRef]
- Xu, S.; Yu, D.; Ye, T.; Tian, P. Catalytic transfer hydrogenation of levulinic acid to γ-valerolactone over a bifunctional tin catalyst. RSC Adv. 2017, 7, 1026–1031. [Google Scholar] [CrossRef] [Green Version]
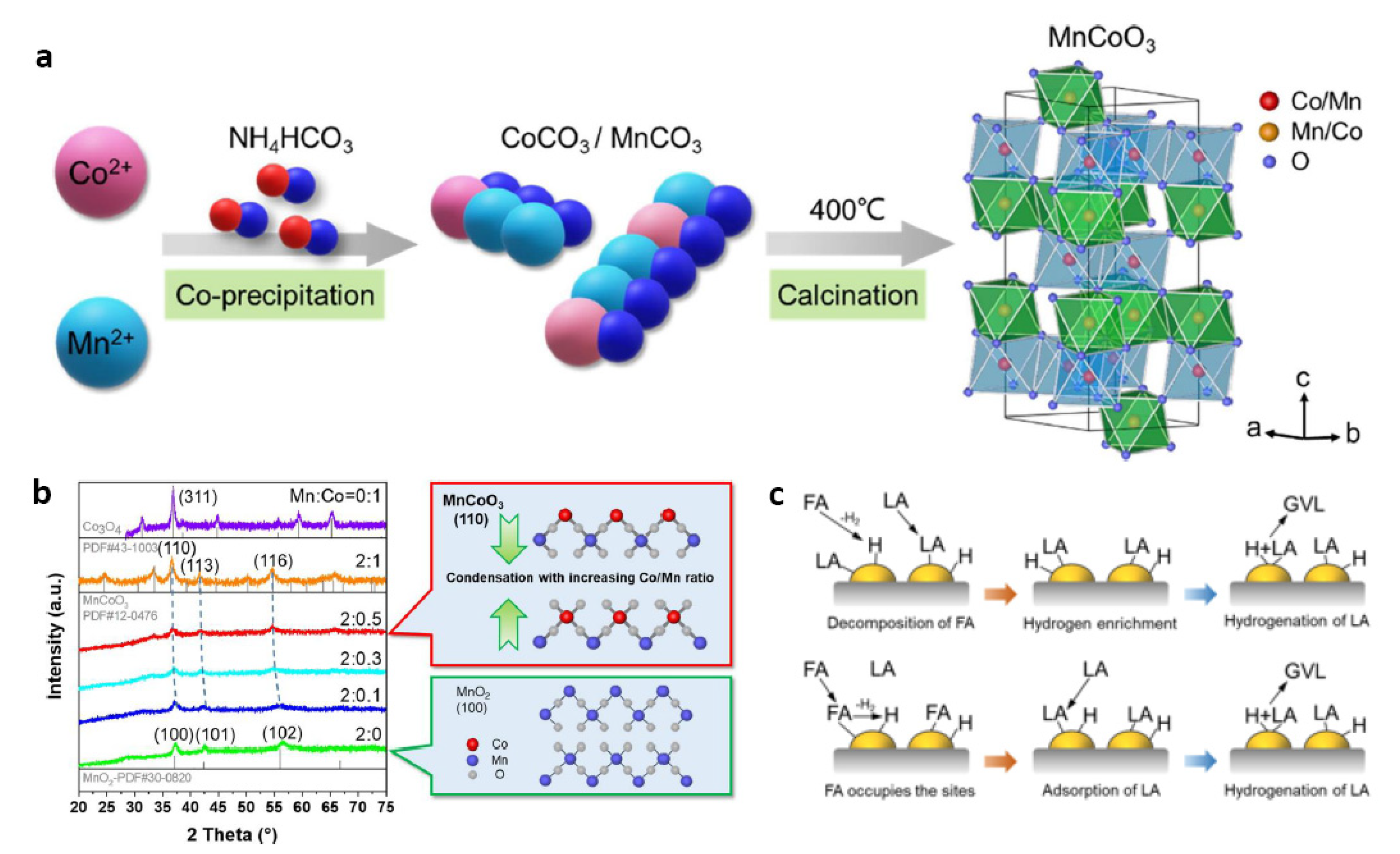
- Wang, J.; Zhang, G.; Liu, M.; Xia, Q.; Yu, X.; Zhang, W.; Shen, J.; Yang, C.; Jin, X. Lattice distorted MnCo oxide materials as efficient catalysts for transfer hydrogenation of levulinic acid using formic acid as H-donor. Chem. Eng. Sci. 2020, 222, 115721. [Google Scholar] [CrossRef]
- Kuwahara, Y.; Kango, H.; Yamashita, H. Catalytic transfer hydrogenation of biomass-derived levulinic acid and its esters to γ-valerolactone over sulfonic acid-functionalized UiO-66. ACS Sustain. Chem. Eng. 2016, 5, 1141–1152. [Google Scholar] [CrossRef]
- Xue, Z.; Jiang, J.; Li, G.; Zhao, W.; Wang, J.; Mu, T. Zirconium–cyanuric acid coordination polymer: Highly efficient catalyst for conversion of levulinic acid to γ-valerolactone. Catal. Sci. Technol. 2016, 6, 5374–5379. [Google Scholar] [CrossRef]
- Yun, W.C.; Yang, M.T.; Lin, K.A. Water-born zirconium-based metal organic frameworks as green and effective catalysts for catalytic transfer hydrogenation of levulinic acid to gamma-valerolactone: Critical roles of modulators. J. Colloid. Interface Sci. 2019, 543, 52–63. [Google Scholar] [CrossRef]
- Rai, P.K.; Kumar, V.; Lee, S.; Raza, N.; Kim, K.H.; Ok, Y.S.; Tsang, D. Nanoparticle-plant interaction: Implications in energy, environment, and agriculture. Green Energy Environ. 2018, 119, 1–19. [Google Scholar] [CrossRef]
- Yang, Y.; Gu, L.; Guo, S.; Shao, S.; Li, Z.; Sun, Y.; Hao, S. N-doped mesoporous carbons: From synthesis to applications as metal-free reduction catalysts and energy storage materials. Front. Chem. 2019, 7, 761. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Gao, F.; Concepción, P.; Corma, A. A new strategy to transform mono and bimetallic non-noble metal nanoparticles into highly active and chemo selective hydrogenation catalysts. J. Catal. 2017, 350, 218–225. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Y.; Sun, Y.; Luo, X. The Relationship between Structure and Catalytic Activity-Stability of Non-Precious Metal-Based Catalysts towards Levulinic Acid Hydrogenation to γ-Valerolactone: A Review. Energies 2022, 15, 8093. https://doi.org/10.3390/en15218093
Yang Y, Sun Y, Luo X. The Relationship between Structure and Catalytic Activity-Stability of Non-Precious Metal-Based Catalysts towards Levulinic Acid Hydrogenation to γ-Valerolactone: A Review. Energies. 2022; 15(21):8093. https://doi.org/10.3390/en15218093
Chicago/Turabian StyleYang, Ying, Yuhang Sun, and Xinruo Luo. 2022. "The Relationship between Structure and Catalytic Activity-Stability of Non-Precious Metal-Based Catalysts towards Levulinic Acid Hydrogenation to γ-Valerolactone: A Review" Energies 15, no. 21: 8093. https://doi.org/10.3390/en15218093