
Molecular Simulation Study and Analytical Model for Oil–Water Two-Phase Fluid Transport in Shale Inorganic Nanopores

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Molecular Simulation
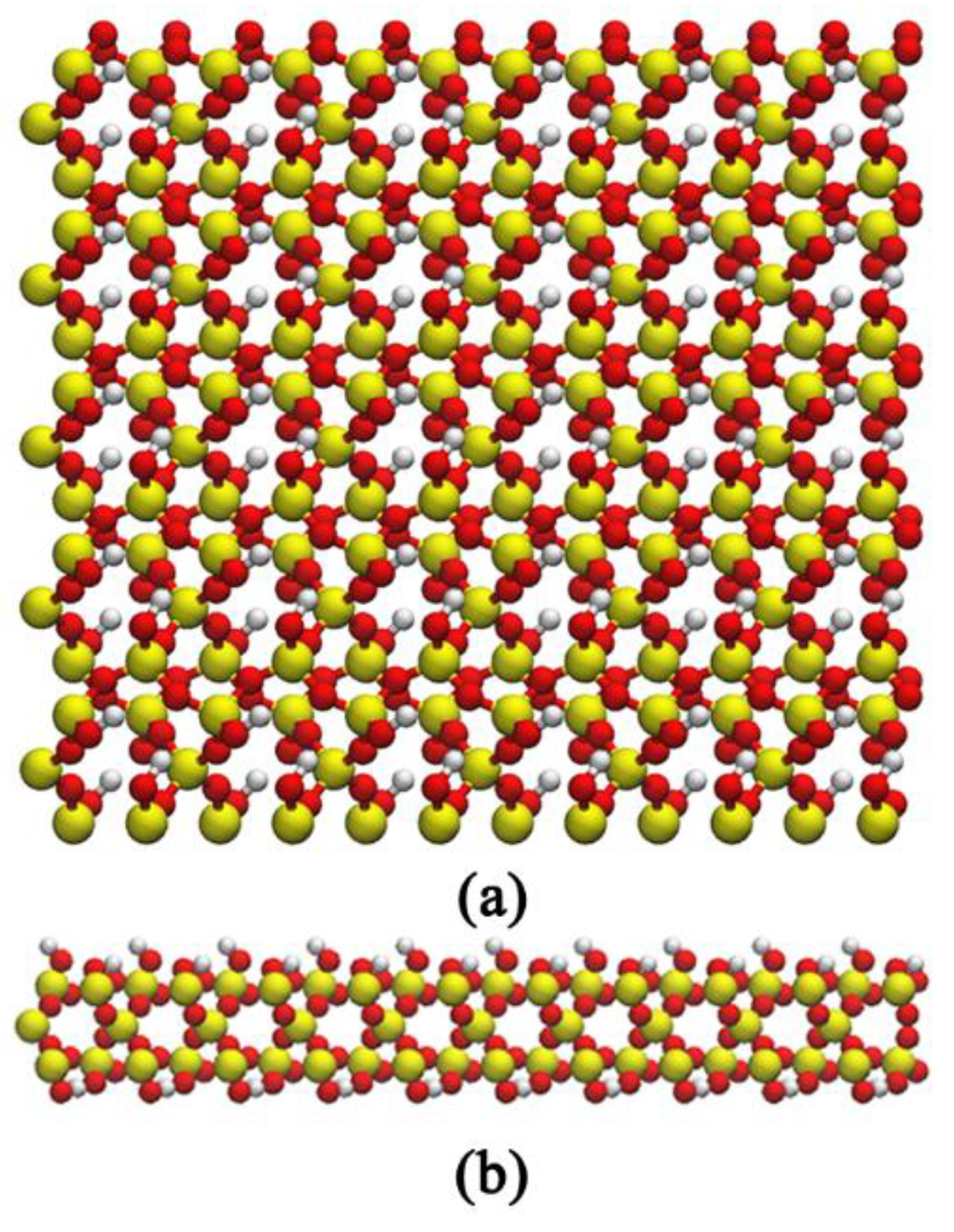
2.1. Model Construction and Simulation
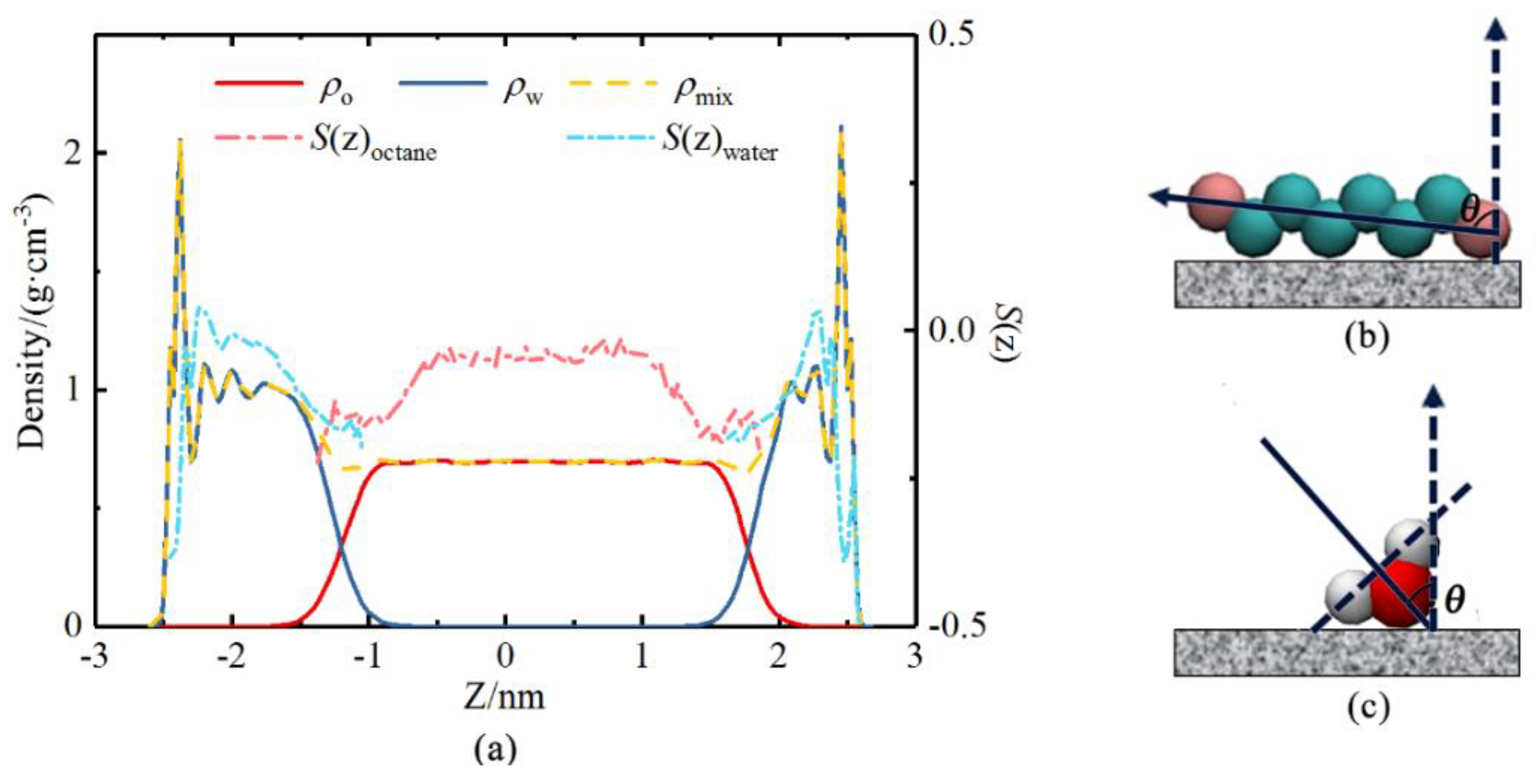
2.2. Results of Molecular Simulation
3. Mathematical Model
3.1. Liquid Slip Model
3.2. Apparent Viscosity Model
4. Model Validation and Discussion
4.1. Model Validation of Liquid–Liquid Slip Model
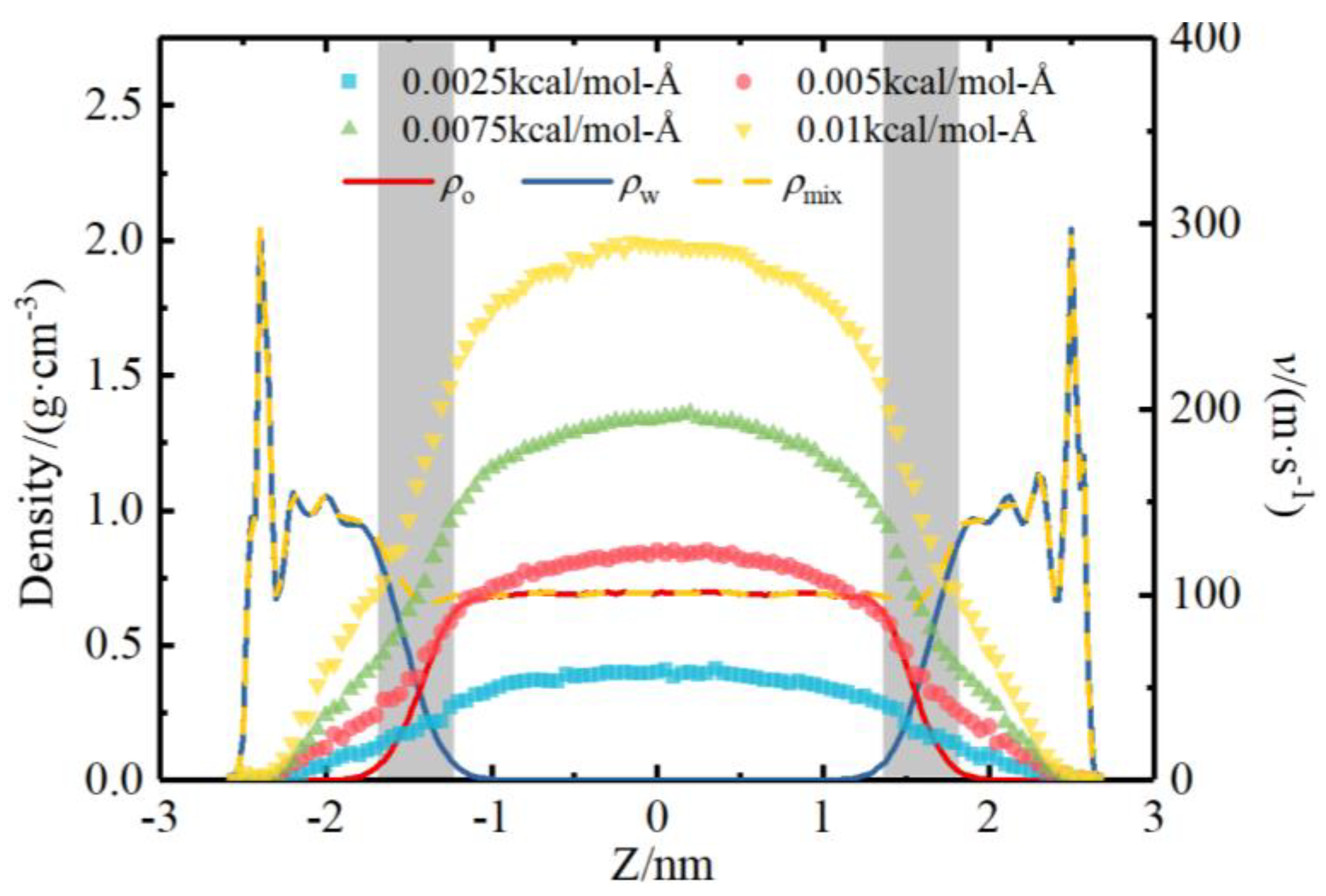
4.2. Model Validation of Apparent Viscosity Model
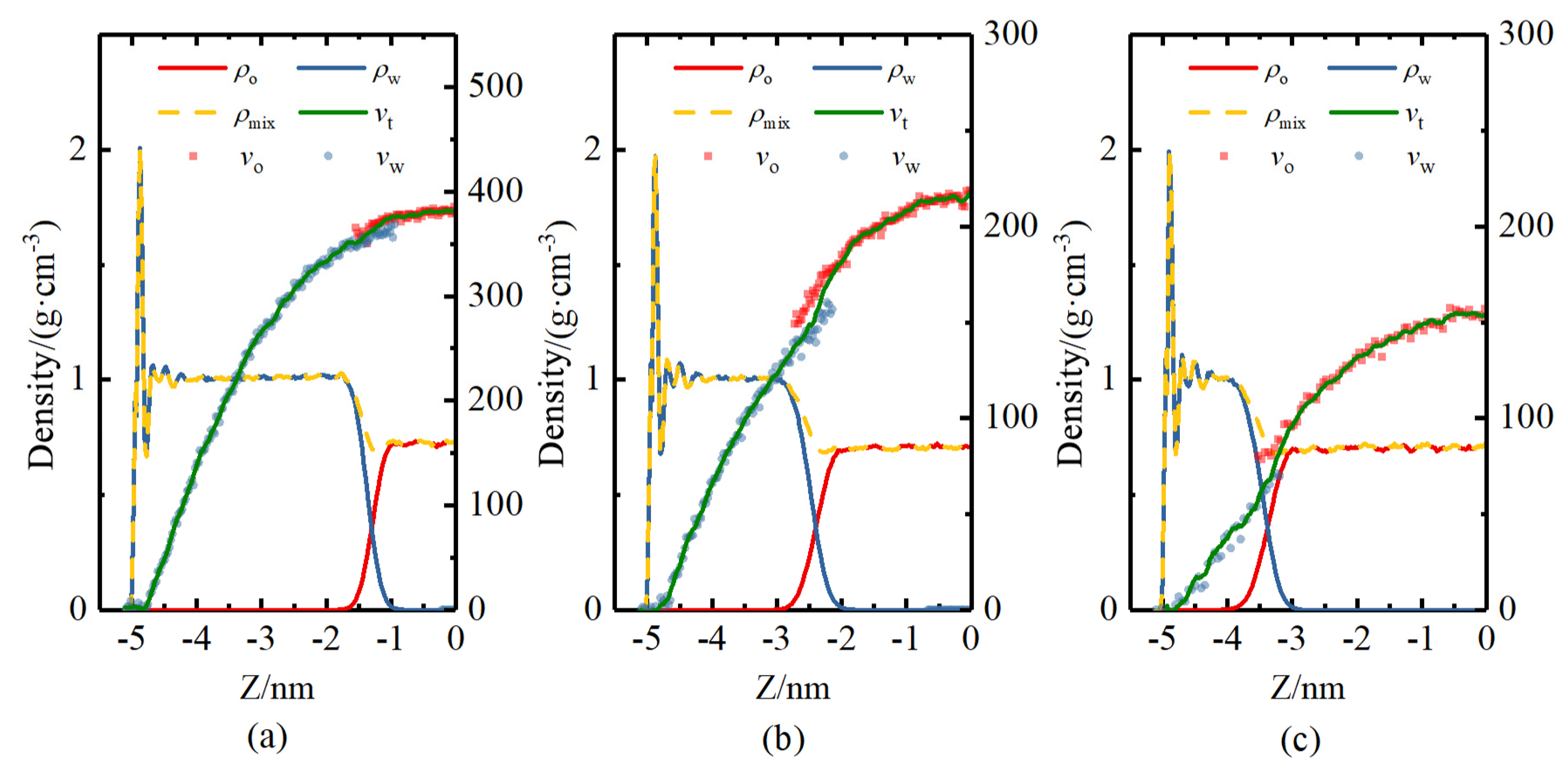
4.3. Flow Rate
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Suyun, H.; Wenzhi, Z.; Lianhua, H.; Zhi, Y.; Rukai, Z.; Songtao, W.; Bin, B.; Xu, J. Development potential and technical strategy of continental shale oil in China. Pet. Explor. Dev. 2020, 47, 877–887. [Google Scholar]
- US EIA. Drilling Productivity Report for Key Tight Oil and Shale Gas Regions; US Energy Information Administration: Washington, DC, USA, 2015.
- Petroleum, B. Statistical Review of World Energy 2009; BP: London, UK, 2009. [Google Scholar]
- Nelson, P.H. Pore-throat sizes in sandstones, tight sandstones, and shales. AAPG Bull. 2009, 93, 329–340. [Google Scholar] [CrossRef]
- Yang, C.; Zhang, J.; Tang, X.; Ding, J.; Zhao, Q.; Dang, W.; Chen, H.; Su, Y.; Li, B.; Lu, D. Comparative study on micro-pore structure of marine, terrestrial, and transitional shales in key areas, China. Int. J. Coal Geol. 2017, 171, 76–92. [Google Scholar] [CrossRef]
- Xing, X.; Feng, Q.; Zhang, W.; Wang, S. Vapor-liquid equilibrium and criticality of CO2 and n-heptane in shale organic pores by the Monte Carlo simulation. Fuel 2021, 299, 120909. [Google Scholar] [CrossRef]
- Vishnyakov, A.; Neimark, A.V. Studies of Liquid—Vapor equilibria, criticality, and spinodal transitions in nanopores by the gauge cell Monte Carlo simulation method. J. Phys. Chem. B 2001, 105, 7009–7020. [Google Scholar] [CrossRef]
- Riewchotisakul, S.; Akkutlu, I.Y. Adsorption-Enhanced Transport of Hydrocarbons in Organic Nanopores. SPE J. 2016, 21, 1960–1969. [Google Scholar] [CrossRef]
- Wu, H.; Chen, J.; Liu, H. Molecular Dynamics Simulations about Adsorption and Displacement of Methane in Carbon Nanochannels. J. Phys. Chem. C 2015, 119, 13652–13657. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, J.; Yao, J.; Kou, J.; Li, Z.; Wu, T.; Zhang, K.; Zhang, L.; Sun, H. Adsorption behaviors of shale oil in kerogen slit by molecular simulation. Chem. Eng. J. 2020, 387, 124054. [Google Scholar] [CrossRef]
- Bao, B.; Zhao, S. A review of experimental nanofluidic studies on shale fluid phase and transport behaviors. J. Nat. Gas. Sci. Eng. 2020, 86, 103745. [Google Scholar] [CrossRef]
- Nojabaei, B.; Johns, R.T.; Chu, L. Effect of Capillary Pressure on Phase Behavior in Tight Rocks and Shales. SPE Reserv. Eval. Eng. 2013, 16, 281–289. [Google Scholar] [CrossRef]
- Nan, Y.; Li, W.; Jin, Z. Slip length of methane flow under shale reservoir conditions: Effect of pore size and pressure. Fuel 2020, 259, 116237. [Google Scholar] [CrossRef]
- Heinbuch, U.; Fischer, J. Liquid flow in pores: Slip, no-slip, or multilayer sticking. Phys. Rev. A 1989, 40, 1144–1146. [Google Scholar] [CrossRef] [PubMed]
- Frenkel, D.; Smit, B. Understanding Molecular Simulation: From Algorithms to Applications, 2nd ed.; Academic Press: San Diego, CA, USA, 2001; ISBN 0122673514. [Google Scholar]
- Wilson, M.R. Molecular simulation of liquid crystals: Progress towards a better understanding of bulk structure and the prediction of material properties. Chem. Soc. Rev. 2007, 36, 1881–1888. [Google Scholar] [CrossRef] [Green Version]
- Martini, A.; Eder, S.J.; Dörr, N. Tribochemistry: A Review of Reactive Molecular Dynamics Simulations. Lubricants 2020, 8, 44. [Google Scholar] [CrossRef] [Green Version]
- Scheraga, H.A.; Khalili, M.; Liwo, A. Protein-Folding Dynamics: Overview of Molecular Simulation Techniques. Annu. Rev. Phys. Chem. 2007, 58, 57–83. [Google Scholar] [CrossRef] [Green Version]
- Ho, T.A.; Wang, Y.; Ilgen, A.; Criscenti, L.J.; Tenney, C.M. Supercritical CO2-induced atomistic lubrication for water flow in a rough hydrophilic nanochannel. Nanoscale 2018, 10, 19957–19963. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Qu, Z.; Yin, Y.; Bai, J.; Yu, B. Review of Molecular Simulation Method for Gas Adsorption/desorption and Diffusion in Shale Matrix. J. Therm. Sci. 2019, 28, 1–16. [Google Scholar] [CrossRef]
- Nagayama, G.; Cheng, P. Effects of interface wettability on microscale flow by molecular dynamics simulation. Int. J. Heat Mass Transf. 2004, 47, 501–513. [Google Scholar] [CrossRef]
- Cao, B.-Y.; Chen, M.; Guo, Z.-Y. Liquid flow in surface-nanostructured channels studied by molecular dynamics simulation. Phys. Rev. E 2006, 74, 066311. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Fan, J.; Xia, J.; Liu, H.; Wu, H. Multiscale gas transport behavior in heterogeneous shale matrix consisting of organic and inorganic nanopores. J. Nat. Gas. Sci. Eng. 2020, 75, 103139. [Google Scholar] [CrossRef]
- Wang, S.; Feng, Q.; Javadpour, F.; Zha, M.; Cui, R. Multiscale Modeling of Gas Transport in Shale Matrix: An Integrated Study of Molecular Dynamics and Rigid-Pore-Network Model. SPE J. 2020, 25, 1416–1442. [Google Scholar] [CrossRef]
- Wang, S.; Yao, X.; Feng, Q.; Javadpour, F.; Yang, Y.; Xue, Q.; Li, X. Molecular insights into carbon dioxide enhanced multi-component shale gas recovery and its sequestration in realistic kerogen. Chem. Eng. J. 2021, 425, 130292. [Google Scholar] [CrossRef]
- Ho, T.A. Water and Methane in Shale Rocks: Flow Pattern Effects on Fluid Transport and Pore Structure. In Springer Theses; Springer Science and Business Media LLC: Berlin, Germany, 2017; pp. 53–64. [Google Scholar]
- Ho, T.A.; Wang, Y. Molecular Origin of Wettability Alteration of Subsurface Porous Media upon Gas Pressure Variations. J. ACS Appl. Mater. Interfaces 2021, 13, 41330–41338. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Zhao, J.; Liu, Z.; Wang, C.; Zhang, H. Study on effect of different surface roughness on nanofluid flow in nanochannel by using molecular dynamics simulation. J. Mol. Liq. 2022, 346, 117148. [Google Scholar] [CrossRef]
- Xiong, H.; Devegowda, D.; Huang, L. Water Bridges in Clay Nanopores: Mechanisms of Formation and Impact on Hydrocarbon Transport. Langmuir 2020, 36, 723–733. [Google Scholar] [CrossRef]
- Zhan, S.; Su, Y.; Jin, Z.; Zhang, M.; Wang, W.; Hao, Y.; Li, L. Study of liquid-liquid two-phase flow in hydrophilic nanochannels by molecular simulations and theoretical modeling. Chem. Eng. J. 2020, 395, 125053. [Google Scholar] [CrossRef]
- Zhang, W.; Feng, Q.; Jin, Z.; Xing, X.; Wang, S. Molecular simulation study of oil-water two-phase fluid transport in shale inorganic nanopores. Chem. Eng. Sci. 2021, 245, 116948. [Google Scholar] [CrossRef]
- Zhang, W.; Feng, Q.; Wang, S.; Xing, X. Oil diffusion in shale nanopores: Insight of molecular dynamics simulation. J. Mol. Liq. 2019, 290, 111183. [Google Scholar] [CrossRef]
- Zhang, T.; Li, X.; Yin, Y.; He, M.; Liu, Q.; Huang, L.; Shi, J. The transport behaviors of oil in nanopores and nanoporous media of shale. Fuel 2019, 242, 305–315. [Google Scholar] [CrossRef]
- Cui, R.; Feng, Q.; Chen, H.; Zhang, W.; Wang, S. Multiscale random pore network modeling of oil-water two-phase slip flow in shale matrix. J. Pet. Sci. Eng. 2019, 175, 46–59. [Google Scholar] [CrossRef]
- Wang, H.; Su, Y.; Wang, W.; Li, L.; Sheng, G.; Zhan, S. Relative permeability model of oil-water flow in nanoporous media considering multi-mechanisms. J. Pet. Sci. Eng. 2019, 183, 106361. [Google Scholar] [CrossRef]
- Yang, Y.; Cai, S.; Yao, J.; Zhong, J.; Zhang, K.; Song, W.; Zhang, L.; Sun, H.; Lisitsa, V. Pore-scale simulation of remaining oil distribution in 3D porous media affected by wettability and capillarity based on volume of fluid method. Int. J. Multiph. Flow 2021, 143, 103746. [Google Scholar] [CrossRef]
- Liu, C.; Zhang, L.; Li, Y.; Liu, F.; Martyushev, D.A.; Yang, Y. Effects of microfractures on permeability in carbonate rocks based on digital core technology. Adv. Geo.-Energy Res. 2022, 6, 86–90. [Google Scholar] [CrossRef]
- Yu, X.; Wang, Y.; Yang, Y.; Wang, K.; Yao, J.; Zhang, K.; Sun, H.; Zhang, L.; Song, W.; Lisitsa, V. Effect of particle content on relative permeabilities in water flooding. J. Pet. Sci. Eng. 2021, 205, 108856. [Google Scholar] [CrossRef]
- Prokhorov, D.; Lisitsa, V.; Khachkova, T.; Bazaikin, Y.; Yang, Y. Topology-based characterization of chemically-induced pore space changes using reduction of 3D digital images. J. Comput. Sci. 2021, 58, 101550. [Google Scholar] [CrossRef]
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Skelton, A.; Wesolowski, D.J.; Cummings, P.T. Investigating the quartz (1010)/water interface using classical and ab initio molecular dynamics. Langmuir 2011, 27, 8700–8709. [Google Scholar] [CrossRef]
- Skelton, A.; Fenter, P.; Kubicki, J.D.; Wesolowski, D.J.; Cummings, P.T. Simulations of the quartz (1011)/water interface: A comparison of classical force fields, ab initio molecular dynamics, and X-ray reflectivity experiments. J. Phys. Chem. C 2011, 115, 2076–2088. [Google Scholar] [CrossRef]
- Cygan, R.T.; Liang, J.-J.; Kalinichev, A. Molecular Models of Hydroxide, Oxyhydroxide, and Clay Phases and the Development of a General Force Field. J. Phys. Chem. B 2004, 108, 1255–1266. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Zielkiewicz, J. Structural properties of water: Comparison of the SPC, SPCE, TIP4P, and TIP5P models of water. J. Chem. Phys. 2005, 123, 104501. [Google Scholar] [CrossRef] [PubMed]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Lorentz, H.A. Ueber die Anwendung des Satzes vom Virial in der kinetischen Theorie der Gase. Ann. Phys. 1881, 248, 127–136. [Google Scholar] [CrossRef] [Green Version]
- Evans, D.J.; Holian, B.L. The Nose—Hoover thermostat. J. Chem. Phys. 1985, 83, 4069–4074. [Google Scholar] [CrossRef]
- Hockney, R.W.; Eastwood, J.W. Computer Simulation Using Particles; CRC Press: Boca Raton, FL, USA, 1988. [Google Scholar]
- Jin, Z.; Firoozabadi, A. Methane and carbon dioxide adsorption in clay-like slit pores by Monte Carlo simulations. Fluid Phase Equilibria 2013, 360, 456–465. [Google Scholar] [CrossRef]
- Zhang, W.; Feng, Q.; Wang, S.; Xing, X.; Jin, Z. CO2-regulated octane flow in calcite nanopores from molecular perspectives. Fuel 2021, 286, 119299. [Google Scholar] [CrossRef]
- Galliero, G. Lennard-Jones fluid-fluid interfaces under shear. Phys. Rev. E 2010, 81, 056306. [Google Scholar] [CrossRef]
- Wang, X.; Binder, K.; Chen, C.; Koop, T.; Pöschl, U.; Su, H.; Cheng, Y. Second inflection point of water surface tension in the deeply supercooled regime revealed by entropy anomaly and surface structure using molecular dynamics simulations. Phys. Chem. Chem. Phys. 2019, 21, 3360–3369. [Google Scholar] [CrossRef]
- Li, W.; Jin, Z. Effect of ion concentration and multivalence on methane-brine interfacial tension and phenomena from molecular perspectives. Fuel 2019, 254, 115657. [Google Scholar] [CrossRef]
- Koplik, J.; Banavar, J.R. Slip, Immiscibility, and Boundary Conditions at the Liquid-Liquid Interface. Phys. Rev. Lett. 2006, 96, 044505. [Google Scholar] [CrossRef] [Green Version]
- Boţan, A.; Rotenberg, B.; Marry, V.; Turq, P.; Noetinger, B. Hydrodynamics in Clay Nanopores. J. Phys. Chem. C 2011, 115, 16109–16115. [Google Scholar] [CrossRef]
- Burgess, D. Thermochemical Data, NIST Chemistry WebBook; NIST Standard Reference Database Number 69; Linstrom, P.J., Mallard, W.G., Eds.; National Institute of Standards and Technology: Gaithersburg, MD, USA, 1998; p. 20899.
- Zhao, R.; Macosko, C.W. Slip at polymer–polymer interfaces: Rheological measurements on coextruded multilayers. J. Rheol. 2002, 46, 145–167. [Google Scholar] [CrossRef]
- Poesio, P.; Damone, A.; Matar, O.K. Slip at liquid-liquid interfaces. Phys. Rev. Fluids 2017, 2, 044004. [Google Scholar] [CrossRef] [Green Version]
- Barsky, S.; Robbins, M.O. Molecular dynamics study of slip at the interface between immiscible polymers. Phys. Rev. E 2001, 63, 021801. [Google Scholar] [CrossRef] [PubMed]
- Razavi, S.; Koplik, J.; Kretzschmar, I. Molecular Dynamics Simulations: Insight into Molecular Phenomena at Interfaces. Langmuir 2014, 30, 11272–11283. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Sun, S.; Qiao, R. Structure, thermodynamics, and dynamics of thin brine films in oil–brine–rock systems. Langmuir 2019, 35, 10341–10353. [Google Scholar] [CrossRef]
- Wu, K.; Chen, Z.; Li, J.; Li, X.; Xu, J.; Dong, X. Wettability effect on nanoconfined water flow. Proc. Natl. Acad. Sci. USA 2017, 114, 3358–3363. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, W.; Feng, Q.; Wang, S.; Zhang, X.; Zhang, J.; Cao, X. Molecular Simulation Study and Analytical Model for Oil–Water Two-Phase Fluid Transport in Shale Inorganic Nanopores. Energies 2022, 15, 2521. https://doi.org/10.3390/en15072521
Zhang W, Feng Q, Wang S, Zhang X, Zhang J, Cao X. Molecular Simulation Study and Analytical Model for Oil–Water Two-Phase Fluid Transport in Shale Inorganic Nanopores. Energies. 2022; 15(7):2521. https://doi.org/10.3390/en15072521
Chicago/Turabian StyleZhang, Wei, Qihong Feng, Sen Wang, Xianmin Zhang, Jiyuan Zhang, and Xiaopeng Cao. 2022. "Molecular Simulation Study and Analytical Model for Oil–Water Two-Phase Fluid Transport in Shale Inorganic Nanopores" Energies 15, no. 7: 2521. https://doi.org/10.3390/en15072521