Abstract
Palladium is among the most versatile noble-metal atoms that, when dispersed on solid supports, can be stabilized in 0, +1, +2, +3 redox states. Moreover, despite its noble-metal character, Pd shows a considerable degree of chemical reactivity. In Pd Nanoparticles (NPs), atomic {Pdn+-X} states, where n = 0, 1, 2, 3, and X = atom or hydride, can play key roles in catalytic processes. Pd-oxygen moieties can be stabilized at nanointerfaces of Pd in contact with metal-oxides. These {Pdn+-X}s can be either isolated Pd atoms dispersed on the support, or, more interestingly, atomic states of Pd occurring on the Pd NPs. The present review focuses on the role of such {Pdn+-X} states in catalytic processes related to energy storage or energy conversion, with specific focus on photocatalysis, H2 production reaction (HRR), oxygen reduction reaction (ORR), and water-splitting. Synthesis of atomic {Pdn+-X} states and their detection methodology is among the current challenges. Herein, the chemistry of {Pdn+-X} states on Pd- [metal oxide] interfaces, methods of detection, and identification are discussed. The implication of {Pdn+-X} in transient catalytic intermediates is reviewed. Finally, the role of {Pdn+-X} in photo electrocatalytic processes is critically discussed.
Keywords:
palladium; single-atom; nanoclusters; subnanoclusters; catalysis; HER; ORR; photocatalysis 1. Introduction
Palladium nanoparticles (NPs), Pd-clusters or single-Pd-atoms find wide used in many research and technology fields thanks to their chemical reactivity, large surface-to-volume ratio, and size-dependent optical properties [1]. In parallel, in the last decades, a wide variety of Pd-based organometallic complexes have been used as effective catalysts in diverse catalytic reactions. These molecular-Pd catalysts generally contain individual Pd centres i.e., not particles, thus, conceptually, one may argue that these compounds represent the early examples of ‘single atom catalysts’ [2]. In the nanomaterials’ context, it is well known that several parameters i.e., such as size, electronic and geometric structure of Pd species [1], can play crucial roles in the catalytic behavior of the final material [2,3], see Figure 1. However, in the case of Pd-clusters and nanoparticles, due to the orbital overlapping between Pd metal atoms and the support, new properties may emerge [1]. As the metal particle-size increases, the bandgap between the highest-occupied-molecular orbital (HOMO) and the lowest-unoccupied-molecular orbital (LUMO) becomes smaller than those in subnanometric Pd clusters. In the case of larger Pd-nanoparticles (>2 nm), quasi-periodic lattice structures start to form, with energy states that tend to have band-structure, with the upper occupied level defining its Fermi state, see Figure 1A.
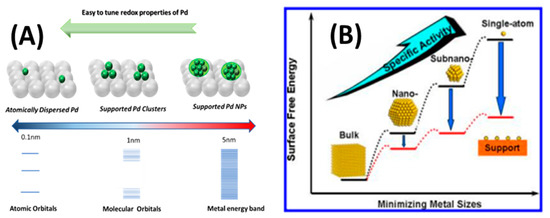
Figure 1.
(A) Pd species with different size (single atom, clusters, and nanoparticles) supported on metal oxide matrix, and the electronic structure on different species is also presented (B); schematic illustration of the surface free energy vs. supported Pd species size.
Therefore, most research on Pd NPs has been based on ensemble-averaged properties of many NPs [3,4]. However, the study of the atomic states of Pd occurring on Pd NPs may offer several advantages, including reactive intermediates or Pd active species, as well as the discovery of unconventional physicochemical events that cannot be observed in bigger nanoparticles. Identification of such active species and the rate determining step are crucial for optimizing the catalytic performance.
With regard to catalysis, it is commonly assumed that Pd NPs or clusters supported on oxides are the active components for several types of catalytic reactions [4,5,6]. However, there is increasing evidence that single atoms, or very small clusters, can offer district advances in catalytic efficiency, especially in the case of the supported metal [7] see Figure 1. In these cases, the interfacing of the Pd-atoms with the support metal oxide can easily supply oxygen atoms [8]. Single atoms, on the same support, can have different coordination environments that will translate into different electronic and catalytic behaviours.
It is, therefore, highly important to precisely determine the coordination of the single Pd-atoms on the support. According to the literature, the coordination environment is strongly related to the specific synthesis process applied each time, as well as the post-synthesis protocols [9]. The metal-oxide type can also act synergistically with the Pd-species towards a specific catalytic process. So far, supports e.g., as metal oxides [8], carbon nanostructures [5], polymeric-resins [7], zeolites [7], and mesoporous materials [7], have been studied, among others, as supports of Pd-nanoparticles, see Figure 2.

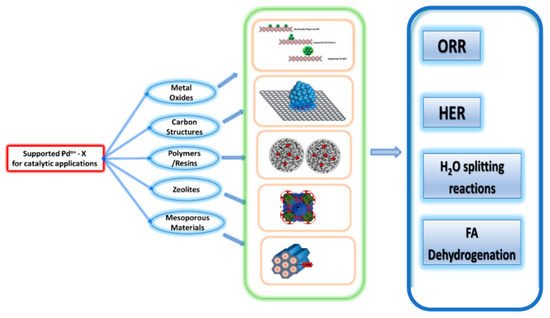
Figure 2.
Supported Pd species on different matrixes and possible catalytic applications.
Aim of the Review
The present review focuses on the role of atomically distinct Pd-states formed by Pd-atoms that, despite that fact they may belong to a Pd-nanoparticle lattice, their redox/electronic configuration retains some atomic character. Herein, we use the term “Pdn+” to describe these Pd-states. ‘n+’ signifies that the oxidation state of the Pd-atom is not “0”, i.e., it can be oxidized to its +1, +2, +3 or +4 state. Moreover, the role of neighboring atoms to the Pd-atom, herein described by the term “-X”, is reviewed concerning the effect of “X” on the Pdn+ state. In this context, herein the term “Pdn+-X” refers to a functional unit that contains atomic character Pd-atoms linked to a moiety X in such a way that the functional unit has a distinct effect on a catalytic process. The structure of the present review is as follows: in Section 1 and Section 2, we review the type of solid support and the type of Pdn+-X states that each support stabilizes. In Section 3, we review the methods of detection/identification of Pdn+-X states. In Section 4, we review the synthesis methods in relation to the stabilization of Pdn+-X states. In Section 5, we discuss pertinent examples of implicathe tions of Pdn+-X states in catalytic applications. These include dehydrogenation of HCOOH, photocatalytic H2-production from H2O, oxygen reduction reaction (ORR), and electrocatalytic H2-evoluton reaction (HER).
2. Definition of {Pdn+-X} Species at Supported Pd-Catalysts
There is ample evidence that oxidized states of Pd-atoms formed on Pd-nanoparticles can play a key role in specific catalytic processes. In principle, Pd can adopt a wide range of oxidation states, Pd+1, Pd+2, Pd+3, and Pd+4, as well as the metallic Pd0 state [10,11,12], see Figure 3. In addition, Pd is known for its high tendency for concerted processes and high affinity toward π- and n-donors. Furthermore, Pd is relatively electronegative and, hence, has a low reactivity toward reactants with polar functional groups but is, on the other hand, highly chemoselective [13]. Typically, in heterogeneous catalysts where Pd-particles are supported on metal-oxides, metallic Pd0 particles are usually predominant, while oxidized atomic states Pdn+ occur at the interface with the oxide support [14,15]. Interaction of the Pd with the support is an important factor controlling characteristics, e.g., dispersion, size, distribution, redox state, and coordination environments [9,15].
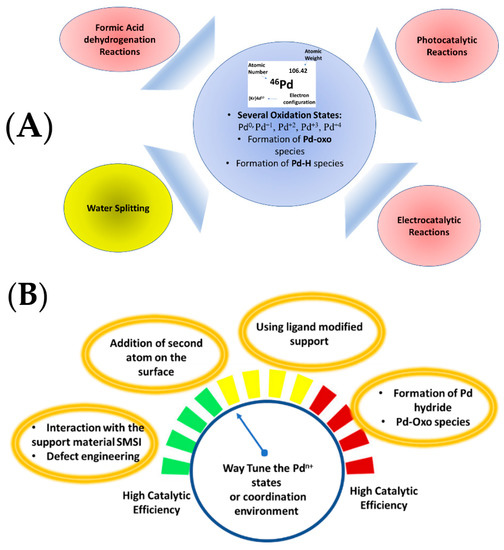
Figure 3.
(A) Chemical properties of the palladium and possible catalytic applications. (B) Possible ways to tune the oxidation state and/or coordination environment of supported Pd species.
2.1. Palladium-Support Interactions
In many catalytic processes, in situ formation of oxidized atomic Pd-states can occur during catalysis. In Pd-{metal oxide} interfaces, strong metal support interactions (SMSI) can be evidenced by the presence of oxidized atomic states Pdn+ [8,16]. These interactions may tune the charge density and the d-orbital states of the metal active sites, enabling, in this way, optimization of the adsorption energy for the intermediates on the metal surface. As the metal active sites decrease in size, i.e., towards single atom, their sensitivity to the support becomes maximal. Dipole–dipole interactions via metal-oxygen bonding {Pdn+-O}, see Figure 4, or electrostatic interactions induced by oxygen vacancies in reducible metal oxides [17,18] can promote the formation of active {Pdn+-X} species, see Figure 4.
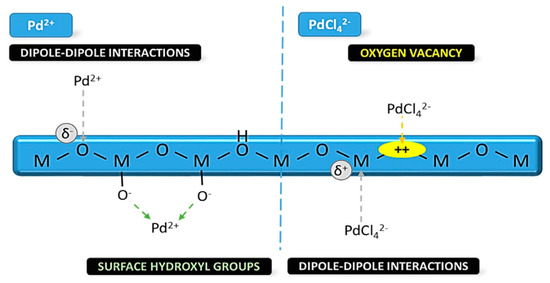
Figure 4.
Schematic illustration of Pd interactions with surface sites or the oxygen vacancies formed on reducible metal oxide. Adapted with permission from [9].
2.2. Τhe {Pdn+-O} Species on Reducible and Non-Reducible Metal Oxides
Metal oxides (MOn) can be divided into [i] reducible or [ii] non-reducible. Reducibility is an essential characteristic of a given metal oxide, strongly affected by the reversible oxidation state of the metal from Mn+ state to M(m−1)+. Transition-metal oxides, such as TiO2, WO3, NiO, Fe2O3, and CeO2, are typical reducible metal-oxides [17,19]. Non-reducible oxides include, among others, ZrO2, ZnO, MgO, and Al2O3. In reducible oxides, usually, the lowest empty states available (conduction band states) on the oxide material consist of cation d-orbitals, which lie at energies higher than the valence band states [17,18,19]. Reducible oxides can be also viewed as being prone to lose O atoms from their structure. Such removal of oxygen results in excess electrons left on the solid matrix, which are redistributed on the cation empty levels [19]. Especially, oxygen vacancies are easily available on the surfaces of oxides and can play an important role in the catalytic process: they can be used as either reducing active centers for reactant species or sites for anchoring the catalytic nanoparticles and single atoms [18]. The energy requirements for the formation of vacancies in reducible oxides is relatively low [19] compared to that of other materials and is thermodynamically favorable at high temperatures and low partial pressures of oxygen [19].
The general physicochemical concept is as follows: the reversible cycling between the reduced/oxidized states renders these materials potent for e.g., oxygen storage and release, as well as, for a various catalytic processes, see Figure 5. Thus, a key property of reducible metal oxides is the presence of oxygen defects on the metal-oxide surface [20,21,22]. This property promotes rich surface-chemistry on reducible metal oxides. As a result, interaction of Pd with this type of oxides can generate several Pd redox species, with different oxidation states and coordination environment. Understanding the interaction mechanism of oxygen with reducible oxide surfaces is crucial for understanding the properties of oxide catalysts. As we discuss hereafter, often, lattice vacancies can interact with O2 molecules forming peroxo [MOO, M = metal] species on the surface [19], which can be extremely reactive, strong oxidants. Due to the increase in bonding distances relative to O2 and also the corresponding displacement of the O–O, superoxo, and peroxo stretching frequency [18], surface MOO species can be experimentally identified from the vibrational (infrared or Raman) spectroscopy. Hence, the adsorption energy of the different oxygen species on the surface of the metal, alters the dynamics of oxygen donation and its reincorporation [18].
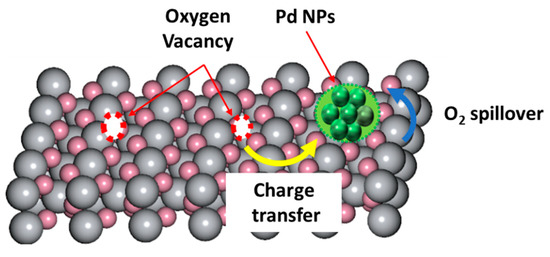
Figure 5.
Electron transfer between Pd and reducible metal oxide.
Examples of supported {Pdn+-O} species on metal-oxide supports are presented below. In 2011, Tonbul et al. [23] had studied CeO2 impregnated with Pd nanoparticles, with sizes between 2-6 nm. Metallic Pd0 and Pdn+-Oy species were detected using XPS. Several reducing agents were studied, such as methanol and H2(g) (weak reducing capacity) [24], or NaBH4 (strong reducing capacity). Satsuma and co-workers [14] combined XPS and STEM for the identification of {Pdn+-O} species supported on different metal oxide surfaces, i.e., such as θ-Al2O3, γ-Al2O3, ZrO2, Ce2O. They proposed that the ΔfHM-O0 value, i.e., the standard formation enthalpy of metal–oxygen bond on the oxide, plays key role on the character of the formed {Pdn+-O} species. A relationship between the standard formation enthalpy (ΔfHM-O0) vs. the Pd0 fraction stabilized on different substrate is presented in Figure 6. Based on ΔfHM-O0 value, we can observe trends: (i) Al2O3 and ZrO2 promoted [Pd0-core-PdO-shell] structures, see Figure 6; and (ii) TiO2, Nb2O5, and SnO2 promoted the stabilization of fully reduced Pd species Pd0. Finally, metal oxides with higher ΔfHM-O0, i.e., such as MgO and La2O3, drive to stabilization of fully oxidized PdOx species, see Figure 6, which is difficult to reduce during catalytic reaction.
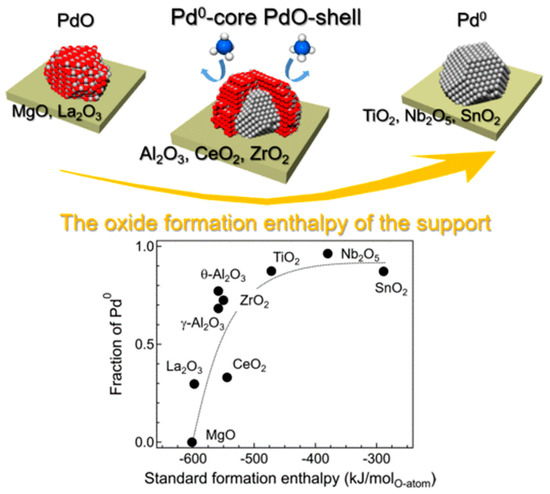
Figure 6.
Effect of ΔfHM-O0 value i.e., the standard formation enthalpy of metal-oxygen bond on the oxide on the stabilization of {Pdn+-O} species supported on different metal oxides. Adapted from [14] with permission.
Recently, we have reported that a complex landscape may occur in the case of Pd-TiO2 materials, prepared using flame spray pyrolysis (FSP) technology [25]. We have observed that, depending on the oxygen abundance and the local temperature at the Pd-TiO2 interface, interfacial Pd atoms can be stabilized at Pd1+ or Pd3+ states, see Figure 7A. More precisely, using electron paramagnetic resonance (EPR) spectroscopy, we identified the formation of four distinct Pd states i.e., Pd0 Pd1+, {Pd2+-O2−}, Pd3+ at Pd/TiO2 interface [25]. Pd0 species were present under O2 Lean preparation conditions or highly reducing conditions (treatment with H2 without presence of H2O). Interestingly, formation of two uncommon Pd1+ and Pd3+ states was shown to be correlated with oxygen abundancy [25]. A non-conventional {Pd2+-OO−} state was found to be stabilized under certain conditions e.g., H2/H2O treatment or catalytic HCOOH dehydrogenation [25]. The orbital-coupling process for the formation of {Pd2+-OO−} was described in detail in reference [25]. This state can be identified by its EPR fingerprint, see Figure 7C. As analyzed in [25], it can be viewed as a weakly coupled OO− anion on a Pd2+ atom.
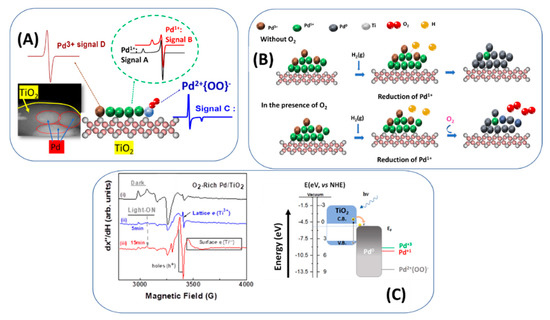
Figure 7.
(A) Schematic depiction of structural assignment of the detected Pdn+ species. (B) Proposed mechanism of formation of {Pd2+-O2−} species (C) Schematic photoinduced electron transfer mechanism under photocatalytic conditions. (i–iii) materials with different treatment. Adapted from [25] with permission.
Zheng and co-workers [26] had studied the critical role of the oxidation state and coordination environment of atomically-dispersed Pd on a Cu2O surface. According to their results, formation of interfacial {Pd-(O-Cu)2} structures was promoted, which contains the oxidation state Pd+1, see Figure 8.
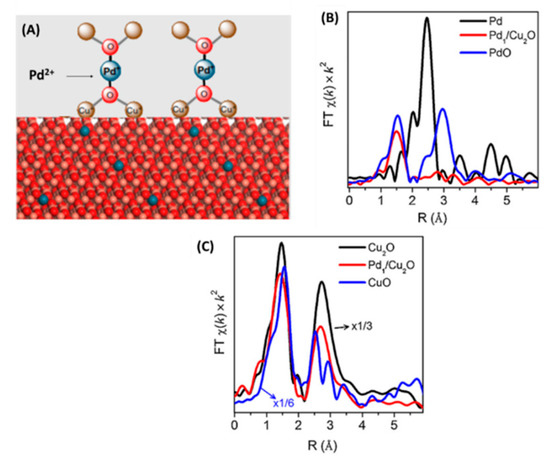
Figure 8.
(A) state and coordination environment of Pd atomically dispersed on Cu2O surface. (B) a FT EXAFS of different Pd species (C) a FT EXAFS of different Cu species. Adapted from [26] with permission.
Guo et al. [27] had studied the effect of metal–support interaction between Pd and hierarchical Nb2O5 tuned via oxygen defects. XPS data, as well as in situ EPR and DFT calculations, confirmed that the Pd species were prone to occupy the oxygen defects and interact with Lewis acids [27]. Additionally, enhanced metal–support interactions between the interfacial {Pd-Nb2O5} decreased the electron density of the Pd species associated with the generation of interfacial Pd-NbOx sites [28]. Notably, the lower Pd electron-density, i.e., resulting from the electron withdrawing effect of Nb on H-Nb2O5, could facilitate the activation of the adsorbed catalytic intermediates, leading to a much higher catalytic activity. Moussa et al. [28] had prepared graphene-stabilized Pd-NPs without using any strong reducing agent or stabilizing agents. Mixture of a Pd precursor with graphene oxide suspension were treated using pulsed laser irradiation (PLI). As shown, photoexcitation of the sp2 domains in the graphite oxide using the lasers’ light energy, which exceeded their band gaps, generated {electron-hole} pairs [28]. This initiated a redox reaction in which the photogenerated electrons reduced Pd2+ to Pd0. On the other hand, the photogenerated holes reacted with H2O, leading to the formation of gaseous CO and CO2, which introduced structural defects in the carbon support [28]. Thus, after PLI, the final material contained several {Pdn+-O} species. Partial oxidation of the Pd on the surface, as well as the partial reduction of the graphene oxide, seem to play a crucial role on the catalytic efficiency of the system. This was most likely due to a competition between the sp2 domains of the graphite oxide and the Pd2+ species for the photogenerated electrons [28].
In some reports, possible existence of {Pdn+-O} species was implied, however, with no direct experimental detection. Park et al. [21] reported that a pronounced enhancement in activity for the water-gas-shift reaction occurred when Pd was supported on CeO2 or TiO2 surfaces [21]. They considered that interaction of Pd/TiO2 occurred via charge transfer from the Pd to the Op bonding state of TiO2, mediated through Pd-O-Ti at the material interface. In a theoretical study of CO2 hydrogenation and formation of methanol over Pd/TiO2 [21], it was suggested that bidentate adsorption of CO2 on a Pdx cluster could be achieved via charge transfer from Pd to antibonding orbitals of the CO2 molecule [21]. Pd on a CeO2 support has been shown to exhibit strong metal support interactions, which are implicated in enhanced catalytic activity [24]. The ability of ceria to shift easily between the oxidized and reduced state (Ce3+↔Ce4+), under operating conditions, is considered to be of importance for the formation of different Pdn+ species at the Pd/CeO2 interface [21].
2.3. The {Pdn+-H} Species
Metallic Pd has the well established property of absorbing H2 to form {Pd-hydrides}, even at low H2-pressures and room temperature [3,4,29,30]. Understanding the formation of the {Pdn+-H} species is of immediate pertinence for its implications in the catalytic activity [29,30]. From these studies, it is evidenced that the tendency to form the Pd-hydride state is enhanced with increasing dispersion of Pd over the supporting solid phase. This indicates that the deposition of atomic-Pd is a route to pursue towards maximal {Pd-hydride} states. In this context, a close relation between the efficiency of HCOOH-dehydrogenation and the Pd-hydride formation tendency has been previously reported at increasing Pd-dispersion over supported Pd catalysts [25,26,27,28]. Other researchers found a similar effect with a Pd-SiO2 catalyst, and, in this case, the main pathway via the dehydrogenation reaction was the hydride formation [7]. Other works found that, for Pd/silica catalysts, the energy barrier for hydride formation decreases with decreasing Pd particle size and catalysis [29,30,31].
3. Detection Methods of the {Pdn+-X} Species Stabilized on Oxide-Supported Pd-Catalysts
Detection, determination of Pd-redox state, and coordination environment of the {Pdn+-X} species is crucial for understanding their role of this sites in catalysis [29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47]. Since these {Pdn+-X} species are not crystalline, and are formed at rather minor concentrations, their detection remains challenging, and thus they relies on advanced characterization techniques. Hereafter, we highlight some pertinent data to emphasize the complexity and challenges of reliable detection of {Pdn+-X} species. For instance, advanced high-resolution scanning transmission electron microscopy (STEM) can visualize atomically dispersed Pd atoms, see Figure 9, and probably distinguish them from metal–particle aggregates. STEM offers useful information to study the particle size distribution, as well as the coordination configuration of the Pd active sites on the supports, due to the obvious difference in contrast between metal atoms and light elements. Such in situ STEM technology has been used to observe the evolution process of the Pd from bulk metals, see Figure 9. It should be underlined, however, that the detection of single Pd atoms by STEM remains a highly challenging task.

Figure 9.
STEM detection of atomically dispersed Pd atoms on TiO2 metal oxide. (a–d) STEM images of different Pd/TiO2 samples. Adapted from [31] with permission.
To gather information about electronic interactions between Pd-centers and their coordination environment, spectroscopic tools may be employed. X-ray absorption spectroscopy (XAS) [31], including the extended X-ray absorption fine structure (EXAFS), and X-ray absorption near-edge structure (XANES) spectroscopies, are important tools to reveal the electronic state, local geometric structure, and charge transfer trend of Pd [31]. XANES can provide information on the oxidation state and coordination of Pd-atom, i.e., due to its sensitivity to the charge-states of the metals and surrounding coordination atoms. EXAFS is sensitive to the local structure without the need of long-range order [31], thus it can provide information about the coordination number and bond distance of the Pd in Figure 10a.
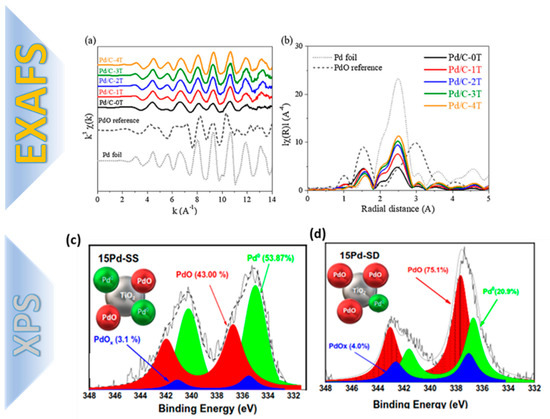
Figure 10.
(a) EXAFS data on Pd/Al2O3 catalyst, indicating the distinguishing fingerprint of Pd0 (i.e., in a reference metal-Pd foil) vs. the PdO reference. The oxidation state of Pd was followed by recording spectra either in the QEXAFS mode (during changes of reaction conditions) or taking EXAFS spectra in the step scanning mode (after reaching steady state). Adapted from [31] with permission. (b) Evolution of the oxidized PdO in this sample is also reflected by the coordination number and the distance to the nearest neighbors. (c,d) XPS data for Pd on Pd/TiO2 nanocatalysts prepared with different FSP protocols by in situ deposition of Pd on TiO2 by flame spray pyrolysis technology. Enhancement of the PdO species in (d) is controlled by oxygen-rich FSP process. Adapted from [25] with permission.
X-ray photoelectron spectroscopy (XPS) can also detect the oxidation states, chemical environments, and bonding information of the Pd centers and its surrounding heteroatoms (e.g., such as N,S,O) [32,33]. In our recent work [25], we have used XPS as a diagnosis tool for controlled formation of PdO states, i.e., up to 75% of total Pd content of Pd/TiO2 nano catalysts [25] by flame spray pyrolysis technology.
Electron paramagnetic resonance (EPR) spectroscopy [25] is eminently suited for detection, quantification, and characterization of the chemical state and coordination environment of atomic Pd-species. Among the various Pdn+ oxidation states, Pd1+ and Pd3+ are paramagnetic, thus detectable by EPR, with configurations 4d9(S = ½) and 4d7(S = 1/2), respectively [25].
The most common, stable oxidation state Pd2+(S = 0) is EPR silent. So far, most EPR studies of Pd on particles and materials referred to Pd incorporated in zeolites as a host matrix [34,35,36,37,38,39,40,41,42,43]. In these works, Pd1+, and Pd3+ states were stabilized due to the interaction with the zeolite matrix. Additionally, zeolites have been shown to promote interaction of O2 with Pd1+, resulting in stabilization of unconventional Pd2+-O2− or Pd2+-O33− states [43]. Recently, we have demonstrated that EPR offers a versatile, sensitive tool to distinguish and quantify Pd1+, Pd3+ species formed at the interface with TiO2 particles [25], see Figure 11. Moreover, we have shown that EPR allows detection on non-conventional {Pd2+-OO−} species [25]. Such spectroscopic data can be rationalized by combination with theoretical calculations, i.e., such as density functional theory (DFT) calculations [7].
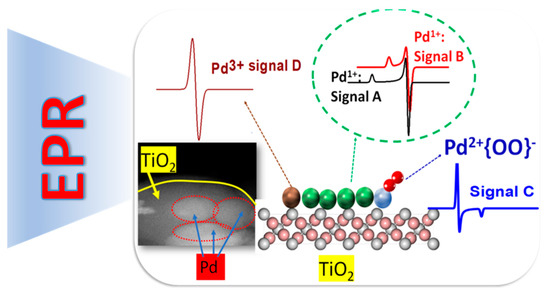
Figure 11.
Detection techniques of different Pd species. Adapted from [25] with permission.
4. Methods for {Pdn+-X} Synthesis on Supported Pd-Catalysts
Several methods for controlled synthesis of atomically dispersing Pd on catalyst supports have been elaborated during the last decade [48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83]. Common strategies include minimizing the metal-loading quantity, increasing metal–support interactions, and using voids and/or vacancy defects on catalytic supports. The majority of reported works indicate that the supports for atomically distributed-catalysts should be selected purposefully [48], i.e., since this may determine the feasibility/or not of fine-dispersion. In the case of Pd-species in addition to particle size, shape and chemical composition, the Pd-redox state, i.e., typically Pd2+ or Pd0, is of pertinence [9]. It is anticipated that decreasing the Pd loading to a minimal amount can theoretically at least result in highly desirable single-atom-catalysts (SAC) that can be considered as the ultimate limit of maximum catalytic efficiency per Pd atom. In this context, {Pdn+-X} species can be considered as a more general description of SACs. In general, the literature categorizes the different synthesis’ strategies to ex situ and in situ bottom-up methods [49]. In in situ methods, SACs are developed immediately on the surface of a support. Ex situ, bottom-up synthesis, on the other hand, impregnates the support with a colloidal dispersion of pre-synthesized SACs [9]. In a more general context, in this review we choose to categorize the synthetic routes according to physical, chemical, and electrochemical methods.
4.1. Physical Synthesis-Methods
Physical synthesis approaches entail the use of metal precursors that are not chemically altered, but instead undergo molecular rearrangements, while neither new substances, nor chemical bonds, are generated [50]. Nanoscale films, nanoparticles, nanowires, and nanorods are among Pd-based nanomaterials created using this approach [51]. A wide range of such synthetic routes include sputtering, ion- οr electron-beam deposition, and laser ablation.
Methods using sputtering enable the creation of nanostructures with precisely regulated cluster size and dispersion. As indicated by Razaee and Ghobadi [51], direct current (dc) magnetron sputtering deposition allowed the development of Pd–alloy thin films with excellent stability. Ion- and electron-beam-induced deposition (IBID and EBID) techniques are appealing due to their ability to produce three-dimensional structures in a single step. IBID and EBID permit the modification of morphologies, dimensions, and interparticle distances using high-energy focused beams, ions, and electrons, respectively [53,54]. The laser ablation (LA) method includes the controlled deposition of a vaporized plasma onto a target by laser radiation with a certain energy flow [55], see Figure 12. Nevertheless, for LA synthesis in liquids, the nanoparticle dimensions and morphologies are largely dependent on a number of experimental factors, such as the kind of solvent and the laser-related fluency, wavelength, and ablation durations.
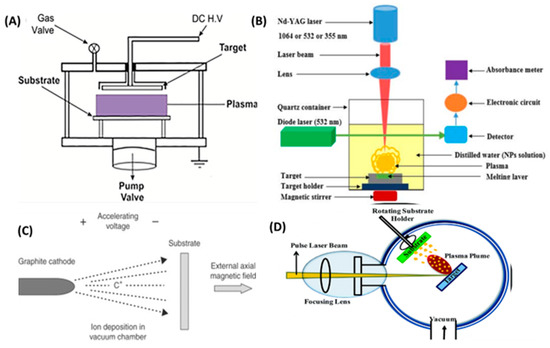
Figure 12.
(A) Schematic illustration of DC–magnetron sputtering system. (B) Experimental setup of laser ablation technique. (C) Ion Beam Deposition technique. (D) electron beam evaporation. Adapted from [59] with permission.
4.2. Chemical Synthesis-Methods
Chemical synthesis methods can be divided into two sub-categories: dry and wet methods.
4.2.1. Dry Methods
Atomic layer deposition (ALD): in dry-methods, a very attractive synthesis way for {Pdn+-X} species is Atomic layer deposition (ALD) [56]. ALD is a cyclic process based on successive self-limiting molecular-level surface interactions between gas phase chemical precursors and the surface of a solid substrate. It includes four basic steps, depicted in Figure 13 [56]. In brief, the catalyst support is located in a vacuum chamber into which the pulse stream of precursor, containing the atomically distributed active component, diffuses and interacts with the functional groups on the support surface. The precursor stream ceases when the reaction between the precursor and substrate is completed. The leftover precursor vapor and byproducts are evacuated or purged with an inert gas. These two actions constitute the first half-reaction. After the first half-reaction, one cycle of ALD is complete. If numerous ALD cycles are necessary, a second precursor stream is pulsed into the chamber to renew the ligands or functional groups on the surface of the support. After purging to eliminate residuals and byproducts, the substrate is prepared for the subsequent ALD cycle. ALD methods create catalysts with monodisperse size, homogenous composition, and exceptional repeatability, in contrast to traditional catalyst production techniques. The self-limiting feature of ALD processes enables homogenous deposition over supports, resulting in catalysts with enhanced activity, selectivity, and durability.
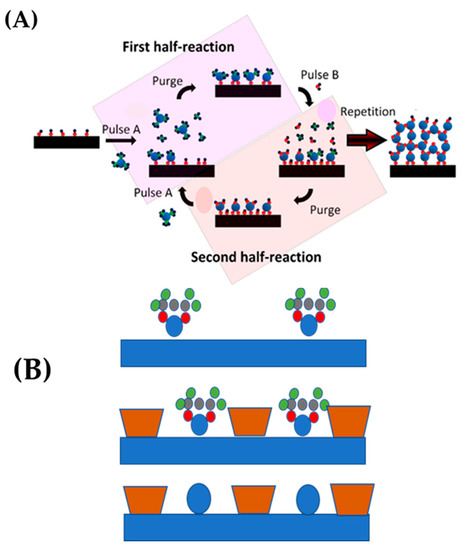
Figure 13.
(A) Schematic representation of the ALD technique using a binary (A,B) precursor system. (B) Exposure to a first precursor vapor. Adapted from [57] with permission.
Piernavieja-Hermida et al. [57] described a method for enhancing the thermal stability of homogeneously distributed Pd“1”, i.e., single-atom catalysts, by using ultrathin metal oxide protective coatings of TiO2 created by ALD method. After 300 °C calcination, the single-atom Pd1 catalysts, shielded by 14 cycles of ALD, demonstrated considerably increased thermal stability with no discernible agglomeration. L3 edge XANES and XANES characterizations revealed that the 14TiPd1 catalysts have drastically different chemical behavior, i.e., being much more efficient than Pd NPs [57].
Flame spray pyrolysis (FSP): FSP is a gas-phase combustion technology [18] where the metal ions are forming particles via a droplet-to-particle process that occurs at high temperatures, 2000–3000 K [18]. The deposition of {Pdn+-X} species can be accomplished by use of the so-called single-nozzle FSP or the double-nozzle-FSP [17]. In these FSP-based methods, Pd dispersion, e.g., over metal-oxide such as TiO2, is controlled via the Pd concentration in the Pd precursor, or via the flame properties, e.g., allowing more diffuse flames [17]. Recently, we have presented the concept of sequential-deposition FSP (SD-FSP), where the Pd is deposited on the metal-oxide support in a second step, after the formation of the metal oxide [17,25], see Figure 14.
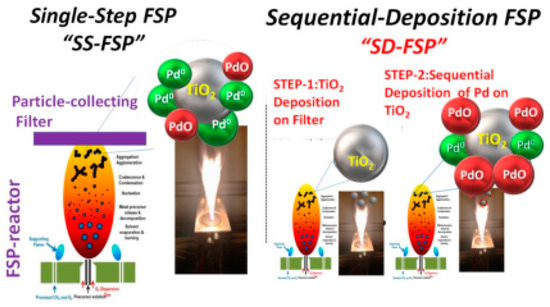
Figure 14.
Schematic representation of FSP prosses used for the deposition of Pd on TiO2 surface. In the sequential-deposition FSP (SD-FSP) method, Pd atoms are deposited on the metal-oxide support, TiO2, in a second-step, after the formation of the metal oxide. Adapted from [25] with permission.
The importance of this sequential-deposition FSP approach is that is allows quantitative control of the (Pdn+-O} species deposited on the metal-oxide support [26]. So far, FSP has been extensively used for the {Pdn+-X} species production, dedicated to catalytic applications. Fujiwara et al. [17] have used FSP-made Pd/TiO2 for catalytic oxidation of NO. Muravev et al. [58] compared the efficiency of impregnated and FSP-made Pd1/CeO2 SACs in a CO oxidation process. Under CO oxidation conditions, the flame spray pyrolysis-prepared Pd/CeO2 catalyst demonstrated a remarkable resistance to sintering [58].
As a mechanochemical approach, ball milling utilizes mechanical interactions to transform reactants into products throughout the reaction process. It plays a crucial function in organic synthesis. It has several advantages, such as scalability and broad application range, without the need of solvents.
4.2.2. Wet Methods
In wet processes, the following methods can be included: hydrothermal/solvothermal, wet impregnation, and photochemical methods [49,60,61,62]. One of the most important steps during the synthetic route in a chemical method is the stabilization of the nanoparticle, which, in wet-chemistry methods, is usually accomplished using a protective stabilizer, see Figure 15. Stabilization is accomplished by electrostatic, steric, or a mixture of the two forces, i.e., electrosteric forces, [49,63]. Typically, the stabilizer is added during the formation of nanoparticles, which is followed by the chemical or electrochemical reduction or thermal degradation of metallic precursor [63], see Figure 15. Among the different supports, zeolites have been extensively used [64,65]. The ensuing interaction between the stabilizer and the nanoparticle’s surface is very dynamic, with its strength and nature often dictating long-term stability of {Pdn+-X} species [62]. Stabilizers include surfactants, polymers, dendrimers, and different types of ligands [62]. The hydrothermal/solvothermal process, see Figure 15, are typically performed in confined sealed containers, where the temperature of the solvent may be increased to near its critical point by heating in conjunction with autogenous pressures [63]. When water is employed as a solvent, the process is referred to as “hydrothermal”, in contrast with “solvothermal”, where an organic solvent is used [63]. The hydrothermal/solvothermal synthesis of {Pdn+-X} species is a crystallization process that typically involves two steps: crystal nucleation and subsequent growth. By adjusting processing factors such as temperature, pH, reactant concentrations, and additives, the particle size and morphology of the final product can be tailored. The phenomena underpinning the regulation of size and morphology, except from nucleation and growth state, are highly dependent on saturation [63].
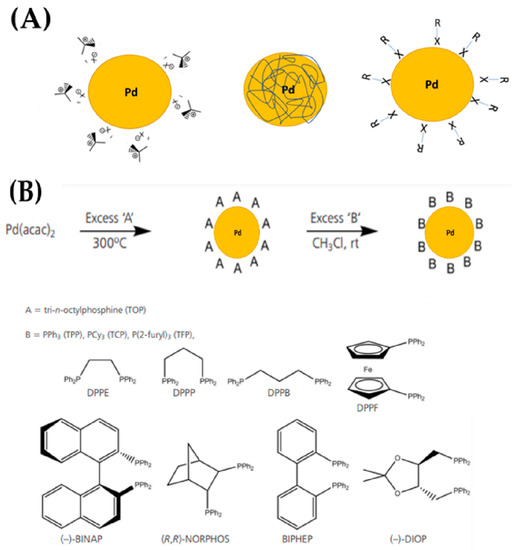
Figure 15.
(A) Schematic representing the stabilization of palladium nanoparticles using different protecting groups: surfactants; polymers; and ligands. (B) Formation of phosphine-stabilized palladium nanoparticles via direct thermolysis and subsequent ligand exchange reactions.
In many cases, PdCl2 can serve as the main precursor, while ammonium formate (AF) [62], formaldehyde [63], poly(vinylpyrrolidone) (PVP) [68], alcohols, and ethylene glycol (EG) [65] were typical reducing agents employed in the hydrothermal/solvothermal production of {Pdn+-X} species. Guo et al. [69] have presented a hydrothermal synthesis of highly dispersive and ultrafine Pd clusters within the crystals of beta-zeolite (Pd@Beta) by employing disodium tetrachloropalladate (Na2PdCl4) as the noble metal precursor and using acidic co-hydrolysis procedure for initial gelation, as well as by using tetraethyl orthosilicate and concentrated hydrochloric acid [69].
Wet impregnation employs a surplus of precursor solution, resulting in a diffusion-driven process, and, in most cases, it is combined with chemical reduction. In a typical procedure, the supporting matrices, e.g., ZrO2, Al2O3, TiO2, are added to an aqueous Pd2+ precursor solution, which is typically composed of PdCl2 [24] or Pd(NO3)2 [70] or Na2PdCl4 [71]. Notably, the choice of precursor will affect the interactions of the Pd ions with the metal oxide, i.e., dipole–dipole interactions via metal–oxygen bondings (Pd2+, PdCl42−) and electrostatic interactions generated by oxygen vacancies (PdCl42−) [72].
Frequently, the impregnation process is followed immediately by a wet Pd reduction. Prior to wet reduction, however, the Pd2+ impregnated metal oxides may be isolated from the precursor solution and purified using (multiple) washing procedures. Utilizing an appropriate reducing agent, typically an aqueous NaBH4 solution or, to a lesser extent, hydrazine, is a common method to produce Pd-NPs on metal oxides, i.e., since these strong reducing agents are known to promote formation of small NPs with narrow size distribution. For example, NaBH4-reduction of Pd2+-impregnated CeO2 (1.18 wt % Pd, water 10 mL, 5 mM NaBH4) at room temperature for 1 h resulted in Pd-NPs with diameters ranging from 2 to 6 nm. Weakly reducing agents, i.e., methanol or H2-gas, on the other hand, result in a lower super-saturation compared to strongly reducing agents; hence, fewer and bigger Pd-NPs are anticipated for the weakly reducing agents. In addition to the strength of the reducing agent, its concentration, the type and concentration of the metal precursor, the reduction temperature/time, and the support properties are the most influential parameters.
For example, hydrazine in water (strong reducing agent), compared to methanol (weak reducing agent) results in higher supersaturation, thus bigger Pd-NPs (12.1 nm) were grown [75,76,77,78,79,80,81]. Differently stated, this method disfavors the formation of atomic {Pd-X} species. To conduct a dry reduction, the Pd2+-loaded metal oxides were isolated from the precursor solution, purified, freeze-dried, or calcined, and then reduced without the addition of a solvent, for example, under a hydrogen gas flow. This is the so called “wetness impregnation”, see Figure 16, where the catalyst is impregnated with an aqueous or organic metal salt solution, with the metal salt strongly absorbed to the support surface. The leftover solvent is evaporated to produce the salt-impregnated catalyst. The impregnation procedure is highly dependent on the surface’s adsorption ability for organometallic complexes or inorganic salts, with the metal salt–support interaction to be crucial and considerably affecting the quantity of metal loading and the metal’s dispersion on the support surface. The essential criteria for regulating metal dispersion are: (i) the type and concentration of the metal salt, (ii) the pH value of the aqueous solution, and (iii) the kind of catalyst support and the surface functional groups. In this context, Kim et al. have used the wetness impregnation to create {Pdn+-X} species supported on C@gC3N4, used for electrochemical reactions [80].
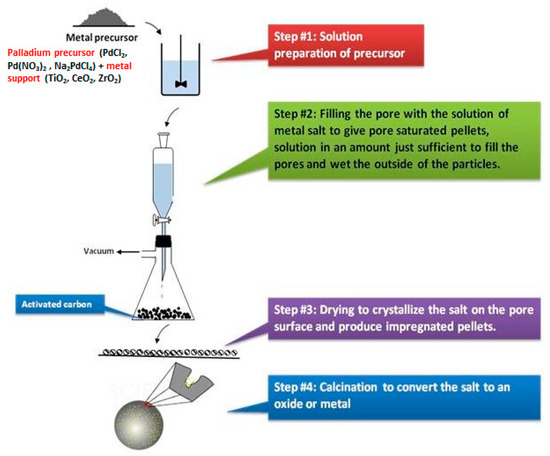
Figure 16.
The wetness impregnation procedure.
A more advanced synthesis procedure that combine the benefits of wet chemistry and conventional wet impregnation methods is the generic wet impregnation method [82]. In contrast to wet chemistry approaches, the originality of this new method is that it omits the use of organic solvent and capping reagent, which simplifies the synthetic operation and eliminates surface contamination of the produced nanoparticles. See Figure 17.
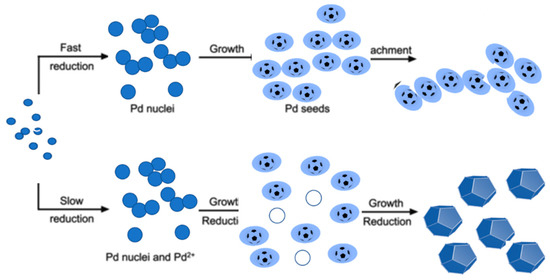
Figure 17.
Schematic illustration of the effect of reaction kinetics affects the nucleation, growth, and final morphology of Pd nanostructures in a polyol synthesis.
In another approach, photochemical deposition can be used for the production of ultrathin films and nanoclusters. In this concept, Tian et al. [60] used photo-assisted deposition for PdAu nanoparticles supported on TiO2-nanotubes. This starts with immersing of a pre-grown TiO2 nanotube plate in a solution of 0.04 M Pd and Au precursors [Pd(NO3)2 and AuCl3] and 5 mL of 50% (v/v) methanol [61]. The solution was then subjected to UV light for 30 min, which promoted formation of 20 nm stable alloyed PdAu nanoparticles. Atomically distributed noble metal catalysts often display strong catalytic capabilities, but the metal loading density must be maintained low (typically below 0.5% w/w) to prevent creation of metal nanoparticles by sintering. In this context, Zhang et al. [73] used the photochemical method to produce atomically dispersed palladium–titanium oxide catalyst (Pd1/TiO2) on ethylene glycole (EG)-stabilized ultrathin TiO2 nanosheets containing up to 1.5% Pd [32]. The catalytic activity of the Pd1/TiO2 catalyst in the hydrogenation of C=C bonds exceeded by a factor of ~9 that of Pd atoms on the surface of commercial Pd catalysts [32].
4.3. Electrochemical Synthesis Methods
Electrodeposition is a straightforward, cost-effective approach for creating nanoparticles, nanowires, and nanorods [83]. Typically, electrodeposition method utilizes a two- or three-electrode setup, with the electrolyte serving as both the Pd source and the conductive medium. By manipulating the potential or current density of an electrochemical cell, an oxidized form in solution is converted to a metallic state at the surface. For the synthesis of Pd-based nanomaterials, several electrodeposition methods are available, such as cyclic voltammetry, square-wave voltammetry, chronoamperometry, and chronopotentiometry. Frequently, cyclic voltammetry (CV) [83] is used as a supplementary technique for the creation of nanomaterials. Zhao et al. [84] deposited dendritic Pd nanoarchitectures on single-crystal n-GaN(0001) through CV electrochemically. nGaN was employed as the working electrode, Pt wire (=1 mm) as the counter electrode, and Ag/AgCl as the reference electrode in a typical three-electrode cell. Pd catalysts were formed on n-GaN by cycling, one, three, five, and seven times [81]. Chronopotentiometry (CP) utilizes a constant current [35], i.e., galvanostatic procedures. Zhao et al. generated porous tubular Pd nanostructures with an applied current density of 0.4 mAcm-2 for 10 h, using a three-electrode cell in combination with an electrolyte consisting of 1.5 gL−1 H2PdCl4 and 25 gL−1 H2BO3, with a pH of 6. Utilizing a CdS-modified porous anodic alumina (PAA) membrane as a template, the shape of Pd nanostructures was achieved.
Overall, during the last decades specific, highly efficient synthesis methodologies have been devoted for the production of {Pdn+-X} species supported solid matrices. In all cases, the challenging steps are related: (i) to avoid growth of large Pd-particles, i.e., to favor small Pd-clusters or atomically dispersed {Pdn+-X} species, as well as (ii) to avoid complete reduction to the Pd0-state, i.e., to retain some of the {Pdn+} oxidation states with n = +1, +2 or +3.
5. The Role of the {Pdn+-X} Species on Catalysis
5.1. Dehydrogenation of HCOOH
H2 production via dehydrogenation of formic acid (HCOOH, FA), sodium formate (HCOONa, SF), or their mixtures, at near-ambient conditions, T < 100 °C, P = 1 bar, is intensively pursued in the context of the most economically and environmentally eligible technologies. Generally, catalytic decomposition of FA can proceed via two pathways, i.e., dehydrogenation to generate H2 and CO2 (reaction (1)) and undesirable dehydration (reaction (2)), which produces poisonous CO provoking deactivation of the catalysts:
HCOOH (l) → H2 (g) + CO2 (g) ΔG (298 K) = −35 kJ mol−1
HCOOH (l) → H2O (l) + CO (g) ΔG (298 K) = −14.9 kJ mol−1
In aqueous solvent, sodium formate (SF, HCOONa) is added during FA dehydrogenation promoting catalytic activity and H2 generation efficiency [1]. In H2O, SF generates H2 via hydrolysis (reaction (3)) with sodium bicarbonate as secondary product:
HCOONa + H2O (l) → H2 (g) + NaHCO3
Among the various noble metal catalysts explored for FA dehydrogenation, palladium is optimal for FA dehydrogenation compared to other metals such as Pt, Au, and Ag. Several studies for Pd-NPs, supported on various supports, provide convincing evidence that the support plays also a role in the catalytic activity. Matrices, such as carbon materials or metal oxides, i.e., TiO2, SiO2 ZnO, and CeO2, have been studied. Among them, TiO2 is of notable interest due to its abundance, stability, and excellent optical properties [18]. Moreover, it exhibits strong metal–support interactions (SMSIs) with Pd [17]. SMSIs can substantially affect the catalytic performance by altering particle properties (e.g., size, morphology, and electronic state) [82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104].
The catalytic cycle of FA–dehydrogenation is also a matter of investigation. A schematic illustration of the catalytic cycle proposed, which comes from our group in the case of a {Pd@SiO2-Gallic-Acid} hybrid material. which is presented in Figure 18.
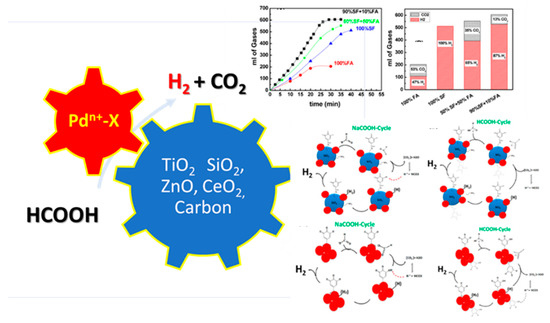
Figure 18.
Schematic illustration of FA dehydrogenation reaction. Catalytic results from catalytic dehydrogenation of FA over the Pd@SiO2GA material. Adapted from [82] with permission.
More specifically, in 2016, our group [83] had demonstrated that the ternary nanohybrid [Pd/SiO2-Gallic acid], i.e., combining [metal] with [oxide support] with [organic cofactor], achieves a high H2 production 19 mL min−1(Ea = 42.2 kJmol−1), see Table 1. Recently, H2 production by dehydrogenation of aqueous ammonium formate at a rate of ~10 mL min−1, at RT, was achieved by TiO2-supported Pd nanoparticles Ea = 37 kJ mol−1. Highly efficient FA dehydrogenation at RT was also reported in 2017 by Akbayrak et al. [23] for a Pd/CeO2 nanocatalyst achieving 10 mL H2 min−1. All these studies dictate that the efficiency of H2 production by FA decomposition can be decisively affected by the intrinsic electronic and structural properties of the Palladium on the surface such as composition, size, dispersion, and oxidation state.

Table 1.
Pd-based materials used for H2-production via dehydrogenation of HCOOH.
In this dehydrogenation process, oxidation state of the Pd-catalyst, i.e., the occurrence of {Pd-O} states on the Pd0-nanparticles, can be of key importance. Recently, Li et al. [102] have noticed that Pd NPs supported on carbon show H2 generation activity in FA decomposition that was substantially enhanced in the presence of a larger proportion of positively charged Pd species (Pd+2 and Pd+4) due to electron transfer from Pd to the carbon support through metal–support interactions. This is corroborated by the work of Lv et al. [102], who suggest that, in carbon-supported Pd catalysts, the Pd0/PdO interface can be a quite efficient active site in H2 production by FA decomposition, i.e., PdO pulls H2 from reaction intermediates facilitating the dehydrogenation process while eliminating self-poisoning dehydration route towards CO formation. Thus, all recent data, including our research on FSP-made Pd/TiO2 [25], evidence that the Pd0/PdO interface should be considered as an optimizable parameter, i.e., in tandem with Pd particle-size when catalytic efficiency is pursued. So far, however, a systematic decoupling of these two parameters (Pd-particle size vs. Pd2+/Pd0 ratio) remains challenging.
5.2. Photocatalytic H2-Production
Another aspect of hydrogen production—besides electrocatalysis—is the photocatalytic production of H2 from H2O using nanocomposite catalysts [105,106]. Most recently [107], single atom catalysts (SACs) attained considerable attention in photocatalysis in view of using single atoms of noble metal as co-catalysts in H2 production and as a substitute of noble metal nanoparticles. Palladium (Pd) co-catalyst attracts much attention in the photocatalytic H2 production due to the ability to boost charge transfer/separation and provides active sites for H2 evolution owing to its suitable work function (Φ = 5.12 eV vs. vacuum) for trapping the photogenerated electrons. Despite their high efficiencies, single atom catalysts require further optimization to overcome the tend to aggregate due to high surface energy [106]. A summary of some Pd-based single atom photocatalytic systems is presented in Table 2.
Table 2.
Summary of Pd-based single atom photocatalytic systems used for H2 production from H2 O and their respective hydrogen evolution rates.
In general, in photo catalysis, a semiconductor is illuminated with irradiation equal/larger than its bandgap that generates mobile charge carriers in both valence and conduction band of the semiconductor [109,110,111]. To facilitate this step, noble metals are widely used as co-catalysts due to their work function they can rapidly trap electrons from the conduction band and prevent them from the undesired recombination, and they also serve as active sites for H2 evolution. Single atom noble metal catalysts can maximize the efficiency of noble metal and have a large proportion of active sites due to their high number of under-coordinated metal atoms.
Wang et al. [105] designed a TiO2 decorated with single Pd atoms (SA) (codenamed PdSA/TiO2-VO) by a pyrolysis process. The dispersion states of Pd species were controlled by thermal reduction of PdSA/TiO2-VO in H2/Ar mixed gas at 200 °C (Figure 19A). Noticeably, the coexistence of {Pd SAs + clusters + VOs} (PdSA+C/TiO2-VO material) exhibited superior H2 production (Figure 19B). DFT calculations (Figure 19C,D) indicated that the introduction of VO created a band tailing effect near the CB minimum of pristine TiO2, which resulted in a decrease in band gap, which agrees with the DRS data [105]. The surface configurations PdSA/TiO2-VO and PdSA+C/TiO2-VO have been analysed by DFT and showed that the introduction of Vo and Pd species influence the total DOS of both VB and CB. The key finding of this research lies in the electron transfer from Pd SAs to TiO2 substrate, which contributes to the stabilization of VO and Ti3+ defects through the Pd-O-Ti3+ interface [104]. Meanwhile, the presence of Pd clusters promotes the photogenerated charge separation and provides active sites for H2 evolution.
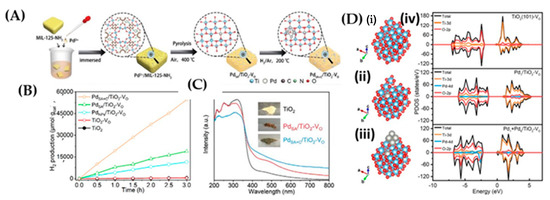
Figure 19.
(A) Schematic representation of synthesis of PdSA/TiO2-VO and PdSA+C/TiO2-VO; (B) Photocatalytic H2 production from MeOH aqueous solutions (20 vol%) where PdSA+C/TiO2-VO exhibits the highest H2 production rate; (C) UV-Vis DRS spectra and the photographs of the samples (inset); (D) Optimized structures of (i) TiO2(101)-VO, (ii) PdSA/TiO2-VO, (iii) PdSA+C/TiO2-VO and (iv) the corresponding PDOS diagrams. Adapted from [106] with permission.
In other studies, [107,108], Pd/g-CN photocatalysts have been designed where the Pd atoms occupy the space among the adjacent layers promoting the charge transfer from the bulk to the surface (Figure 20A). As suggested, [108], electrons can be transferred either via the vertical Pd bridge or the directional in-plane migration to the surface Pd atoms, which serve as active sites for sequential reduction. Moreover, DFT calculations [108] suggest that the work function of Pd (≈4.98 eV) is larger than that of g-CN (≈4.18 eV), denoting that the surface Pd atoms serve as electron collectors [108], and, additionally, the Pd incorporation greatly improves the adsorption of H2 O molecules due to the more negative values of adsorption energy for H2O on the surface of Pd/g-CN (−1.93 eV) than that of pristine g-CN (−0.68 eV). This remarkable activity of Pd/g-CN is attributed to the unique bridge-like structure and the surface anchor of Pd atoms [108] (Figure 20B). The interlayered Pd atoms chemically bonded with C and N atoms and can enhance the π-electron delocalization and subsequently improve charge mobility. Comparing, Pd/g-CN with both Pd/g-CN-R1 (0.11 wt% Pd same as Pd/g-CN but different configuration) and Pd/g-CN-R2 (0.33 wt% Pd), it becomes clear that the Pd single atom modification boosts the photocatalytic performance while increasing the Pd loading the appearance of clusters/nanoparticles (Figure 20D) affecting the photocatalytic performance (Figure 20C).
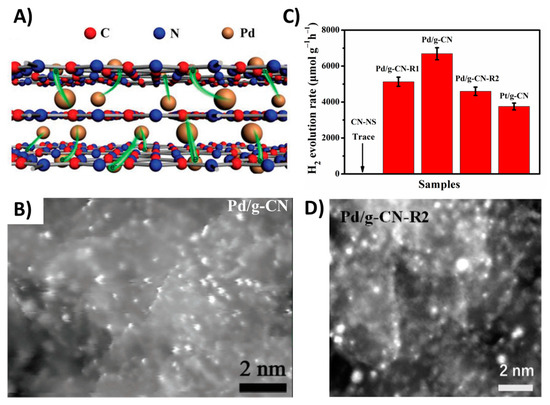
Figure 20.
(A) Schematic illustration of Pd/g-CN hybrid with the Pd single atom bridges and the surface anchor of Pd atoms; (B) Aberration-corrected HAADF-STEM image of Pd/g-CN where with bright dots we can see the Pd single atoms; (C) Comparison of photocatalytic H2 evolution activities of the different Pd-based single atom g-CN catalysts and the pristine CN-NS material; (D) HAADF-STEM image of the reference sample Pd/g-CN-R2 where we can see the formation of Pd clusters/nanoparticles. Adapted from [108] with permission.
5.3. Involvement of {Pdn+-X} Species in Electrocatalytic Processes
A number of modern energy technologies, e.g., such as fuel cells, water electrolysis, metal–air batteries, and CO2 to fuel conversion, have at their core a series of electrochemical processes. Hereafter, for conciseness, we reiterate the basics of these processes. Electrocatalytic hydrogen oxidation reaction (HOR) and oxygen reduction reaction (ORR) occur on the anode and on the cathode of a hydrogen–oxygen fuel cell, respectively. Hydrogen evolution reaction (HER) and oxygen evolution reaction (OER) at the cathode and the anode of an electrolytic cell producing gaseous molecular hydrogen and oxygen, respectively. These (four half-cell) electrochemical reactions are reversible, but with some differences: HER and OER obey the Butler-Volmer model [112,113,114,115,116,117,118,119,120,121,122,123,124,125,126,127,128,129,130,131,132,133,134,135,136,137,138,139,140], even with very high overpotentials. On the other hand, in HOR and ORR under high overpotentials, the apparent current approaches a constant value i.e., due to mass-transfer rate-limitations. Generally, the two-electron transfer in half-cell HER and HOR is fast, while ORR and OER are sluggish, therefore for these reactions noble metals are used [112,113,114,115,116,117,118,119,120,121,122,123,124,125,126,127,128,129,130,131,132,133,134,135,136,137,138,139,140], such as platinum (Pt), palladium (Pt), iridium (IR), and ruthenium (Ru) catalysts, to achieve beneficial kinetics.
5.3.1. Oxygen Reduction Reaction (ORR)
Oxygen reduction reaction is one of the most important electrochemical reactions that serve many processes, such as life processes and energy conversation [113,114,115,116,117,118,119,120,121,122,123,124,125,126,127,128]. ORR in aqueous solutions occurs mainly by two pathways [117]. The direct (four-electron) pathway from O2 to H2O and the (two-electron) pathway from O2 to H2O2 (hydrogen peroxide). In nonaqueous and in alkaline solutions, the one electron transfer pathway from O2 to O2− can occur as shown in Figure 21.
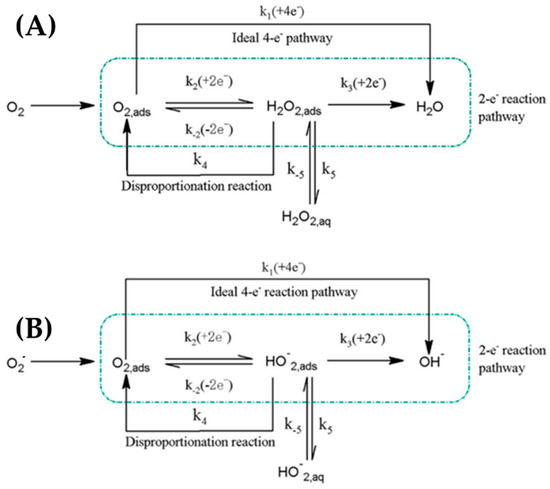
Figure 21.
Simplified ORR mechanism through direct 4e− reduction or two steps. (A) peroxide pathways on acidic and neutral dielectrics and (B) intermediate pathways in alkaline dielectric. Adapted from [114] with permission.
ORR involves either a four-electron (4e−) or two electron steps (2e−) transfer the reactants [114] and products differ for each case of electrolyte solution, as shown in Table 3. In the case of a fuel cell—no matter its type—a direct four electron pathway is highly preferred in order to obtain high efficiency [114], while the two-electron pathway is mainly used for H2O2 production.
Table 3.
Overall reaction equations of ORR in acidic and alkaline solutions.
There are two possible mechanisms for the 4e- pathway, i.e., associative and dissociative [115]. On most metal surfaces, ORR follows the dissociative pathway, i.e., due to strong adsorption of O2 [117]. The dissociative pathway in an alkaline medium, see Figure 22, can be described as:
where * denotes one catalytic active site on a certain surface. The mechanism can be described as O2 adsorption, followed by an O–O bond breaking and a new formation of two adsorbed atomic O* species, which further gain two protons and two electrons to directly form OH− without OOH− generation.
O2 + 2* → 2O*
2O* + 2e− + 2H2O → 2OH* + 2OH−
2OH* + 2e−→ 2OH− + 2*
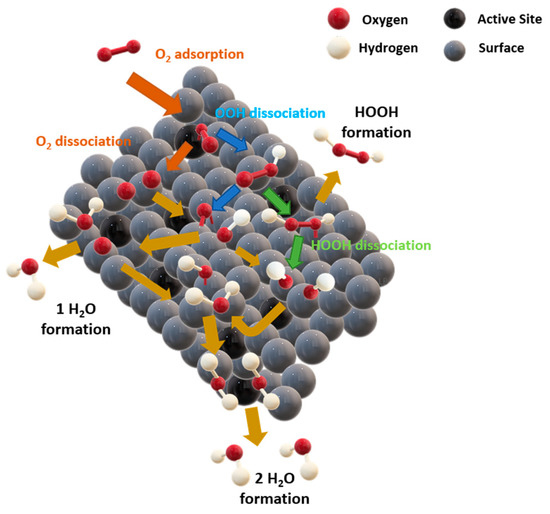
Figure 22.
Schematic representation of dissociative ORR mechanism.
The so-called volcano plot, Figure 23A, is useful to correlate the intrinsic surface adsorption properties and electronic structure (electron orbital levels or wok-functions) of a catalyst, with its apparent catalytic activity [118]. Volcano plots can be used as a guide for the development of highly active catalysts. All these volcano plots reflect the Sabatier principle [116], which states that the optimal catalytic activity can be achieved by a catalytic surface with appropriate binding energies for reactive intermediates. When the intermediates bind too weakly, their activation on the surface is difficult. However, when they bind strongly, all available surface-active sites are occupied, which drives to the poisoning effect. In ORR, the oxygen adsorption strength ΔΕO* can serve as an atomic level descriptor through which the activity obeys the volcano plot. For OER, the activity has been related to a single descriptor (ΔGO*-ΔGOH*) [115,116], where ΔGO* is oxygen adsorption free energy, as shown in Figure 23B. Density functional theory (DFT) can provide predictive tools for selection of ORR and OER catalysts, as shown in Figure 23C,D.
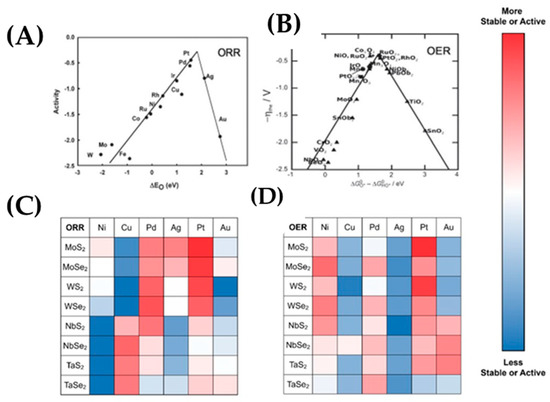
Figure 23.
Volcano plots, free energy diagram for different materials for both oxygen reactions (ORR, OER), (A) ORR activity for a range of pure metals plotted against O* adsorption energy. (B) Activity trends for OER as a function of ΔGO*-ΔGOH*. (C) DFT calculations for ORR and (D) for OER for pure metals on different substrates versus activity and stability. Figure adapted with permission from [115,116].
Adsorbed oxygen is known as a key intermediate in the ORR, and so the binding energy of oxygen is often used to evaluate the catalyst, i.e., in Figure 24B the adsorbed intermediates for the two most active electrocatalysts (Pt, Pd) are shown. Because the binding energy of O2 on Pd is weaker than on Pt [115,116], it is expected for Pd to be more selective to H2O2 species, while Pt is expected to produce H2O through dissociation. This is confirmed by the DOS profile, as shown in Figure 24C, where the d-band center in a Pd SAC is lower than a Pt SAC, which leads to a weaker interaction with an adsorbate.
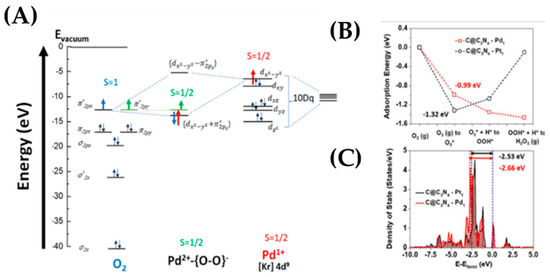
Figure 24.
(A) Suggested coupling Figure for the Pd2+{O-O}- state for Pd on TiO2. Taken from 25 (Β) Energy profile of intermediates on atomic Pd and Pt along ORR pathways at UV for the Pd/C@C3N4 material. Adapted from Reference 25, with permission. (C) Calculated density of states of d-band of Pd and Pt. for the for the Pd/C@C3N4 material. Adapted from [80] with permission.
In Figure 24C, the calculation of density of d-states for Pd and Pt and the surface charge polarization on the substrate@C3N4 reveals the reason of enhancement of ORR activity. It is clear that the density of Pd is shifted down toward the Fermi level in comparison to that of Pt. The calculated d-band center of Pd is located further to Fermi level (−2.66 eV) than Pt (−2.53 eV). The downshift of Pd SAC leads to a weaker interaction with an absorbate, hence higher ORR activity. Noble metal nanoparticles, such as Pt and Pd, show extremely good activity, but their high costs and low natural abundance hamper the commercialization process [80]. An intuitive strategy is to decrease the size of the nanoparticles and hence to reduce the cost of those state of art catalysts, since bulk atoms are not directly involved in the reactions and single atom catalysts (SACs), which represent an ideal solution [119]. For SACs, the strong metal–support interaction is maximal [118], where the active metal sites may be enhanced by two factors, firstly by the substrate (support) and, secondly, by contributions for neighboring atoms of the surface [122]. This has been exemplified for Pd on [MnO2 nanowire-carbon networks], see Figure 25. Pd atoms can interact with nanotubes via their weak adsorption on the carbon nanotube surface, forming a strong chemical bond with O or N containing functional groups or binding on defect sizes. XAS measurements showed that Pd SAs were charged positively with a valence of +2 and coordinated with four oxygen atoms [122]. Two main structures, one with Pd-O4 on carbon networks and the other with Pd-O4 on the surface of the MnO2 nanowires, were detected [122], as shown in Figure 25A,B. Through that study, it was shown that, with these structures, high mass activity towards ORR was achieved, 20 times better than that of commercial Pd/C in alkaline medium-electrolyte and a high number of electrons 3.84, which indicates an almost 4e− pathway, as shown in Figure 25. In another study, Li et al. [123] synthesized Pd SAs on MoO3 nanoparticles. XPS analysis of Pd@MoO3 compared to Pd nanoparticles on MoO3 support revealed Pd2+ states, probably due to the atomic Pd–O bonds formed in the MoO3 lattice [121]. In another study by Liu et al. [124], Pd SAs on a hollow nitrogen doped carbon cages (HNC) matrix was observed. The atomic ratio Pd2+:Pd0 was 2.3 for the best sample (Pd-HNC10) versus Pd2+:Pd0 = 0.77 for Pd nanoparticles in the same matrix, Figure 25A,B. In the same work [123], it was shown that the performance increases with the increment of Pd loading, but the Eonset and E1/2 for Pd-HNC10 was +1.00 and 0.898 V, respectively, while Eonset and E1/2 for Pd-HNC was +0.975 and 0.853 V, respectively, in alkaline medium electrolyte, as shown in Figure 25A,B. This suggested that atomic Pd species in the HNC matrix made dominant contributions to the ORR activity with minor contributions from the nanoparticles. Atomically dispersed Pd atoms on the nanoparticles significantly increased the number of active sites.
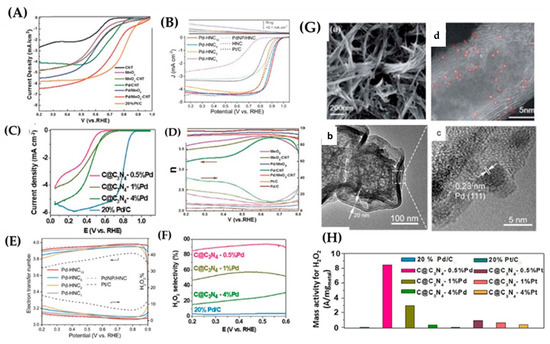
Figure 25.
ORR linear sweep voltammetry curves on RDE in oxygen saturated cell of (A) MnO2-CNT substrate and Pd/MnO2-CNT 0.1 M KOH (B) Pd-HNC series in 0.1 M KOH and (C) single atom to nanoparticles Pd series at C@C3N4 substrate in 0.1 M HClO4. Corresponding electron transfer, peroxide yield on the potential (%H2O2) on RRDE of (D) MnO2-CNT substrate and Pd/MnO2-CNT and (E) Pd-HNC series. (F) the peroxide selectivity of different Pd species on C@C3N4. (G) Representative TEM images of Pd SAs on MnO2-CNT and Pd SAs species on HNC. (a–d) TEM images of different nanomaterials. (H) percentage of active Mass of peroxides from commercial Pd down to SA Pd. Figures adapted with permission from [120,122,123].
Liu et al. prepared Pd SAs supported on graphitic carbon nitride with carbon black (C@C3N4), where the Pd SAs were bonded to C or N atoms, in order to produce stable SACs, with a total coordination number around 3.5, as shown in Figure 25A,B. The Pd SACs were active towards ORR by catalyzing the 2e− pathway, which can occur at single atomic sites that preserve the O–O bonds [126], of O2. The selectivity seemed to decrease with the increment of Pd content, probably because the Pd catalyzed four electron pathway producing H2O [126]. The Pd SACs on the C@C3N4 substrate had similar behavior to that of Pt SACs [126], and a probable reason might be the binding energies of O2 on C@C3N4 Pd1, which is weaker than C@C3N4 Pt1, and the O–O bond length is similar [126]. As shown in Figure 25, it is important to mention that the commercial 20%Pd/C has a much lower H2O2 selectivity than that of SA Pd species. Table 4 provides a summary of pertinent PD-SACs from the literature. It seems that, in most cases, when Pd is dispersed to single atoms it performs a 2e− pathway, and, when the mass of Pd is increasing, it proceeds to a 4e− pathway.
Table 4.
Summary of ORR activity of some Pd-based Single Atom Catalysts in acidic/alkaline solutions.
5.3.2. Hydrogen Evolution Reaction (HER) at Pd Single Atom Catalysts
Hydrogen evolution reaction (HER) holds a special place in electrochemistry, i.e., since it is the most direct way to produce hydrogen of high purity considered to be the fuel of the future [129,130,131,132,133,134,135,136,137,138,139,140]. Despite the great research efforts, a cheap and abundant HER electrocatalyst is missing today. The basic HER mechanism is summarized in Table 5 and Figure 26A. The first step of HER is the adsorption of one hydrogen atom (H) on the catalyst surface by transferring a proton from the acid electrolyte, which combines with an electron transferred from the catalyst surface to form an adsorber hydrogen atom . This process is called the Volmer step [129] (Figure 26A). Then, this combines with an electron and a to form a molecule, and this step is named the Heyrovsky step (Figure 26A) [129,130,131,132,133]. Alternatively, there is a second possibility to form a molecule through the Tafel step i.e., the combination of two (Figure 27A). Regarding the HER in alkaline media, due to the lack of , we begin from molecules dissociation. This additional step of proton generation from H2O in alkaline media introduces an additional energy barrier and affects the whole reaction kinetics.
Table 5.
Overall reaction pathways for HER in acidic/alkaline solutions.
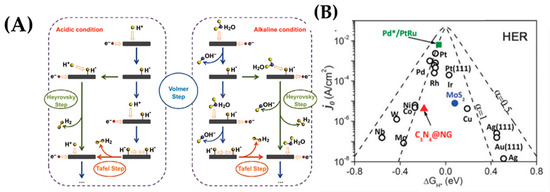
Figure 26.
(A) Schematic representation of HER pathways in acidic and alkaline solutions, adapted with permission from [129]; (B) Volcano plot for HER j0 vs. hydrogen adsorption free energy for various metals, alloys, and non-metallic materials. Adapted from [115] with permission.
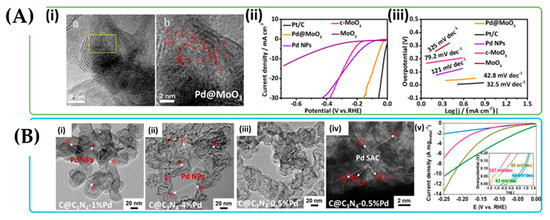
Figure 27.
(A) (i) HRTEM images of Pd@MoO3 where the Pd atoms are marked with red circles dispersed in the MoO3 sheets; (ii,iii) LSV curves and Tafel plots respectively for Pd@MoO3, c-MoO3, MoO3, Pd NPs and Pt/C catalysts (a) and (b) TEM images in different scale. (B) (i,ii) TEM images of C@C3N4-1%Pd (left) and C@C3N4-4%Pd (right). With red circles, we mark the Pd NPs, which are formed at these relatively high concentrations of noble metal; (iii,iv) TEM and HAADF-STEM images of C@C3N4-0.5%Pd where no particles were observed in the TEM images whilst many bright dots were observed in the HAADF-STEM image; (v) Hydrogen evolution reaction results performed by linear sweep voltammetry in Ar-saturated 0.1 M HClO4 solution on 20% Pd/C (blue), C@C3N4-4%Pd (green), C@C3N4-1%Pd (mustard), and C@C3 N4-0.5%Pd (magenta). The inset shows HER Tafel slope of the catalysis adοpted with permission from [121].
In general, the relationship between the electrode material and the activation energy is expressed through the volcano plot by plotting versus (Figure 27B). The most common volcano plot of the HER rate description is based on the Langmuir type of adsorption where the maximum is located near the region where the hydrogen adsorption free energy zero is [129]. is the rate-determining parameter for HER, and the highly active materials (e.g., Pt, Pd, Rh) are located near the top of the volcano plot and possess an optimal value (Figure 27B).
From the volcano plot in Figure 27B, it is obvious that, for a material to be an ideal catalyst for HER, it should be able to bind the reaction intermediates neither too strongly nor too weakly, i.e., a material that can minimize subscript value can be considered as a possible candidate. Noble metal electrocatalysts, such as Pt, Pd, Rh, and Ir, are notorious for their high activity, despite their high cost and low abundance [132,133,134,135]. Single atom catalysts (SACs) have attracted considerable attention and have expanded the potential pool of nanoscale candidates, even if HER is not considered a bottleneck in electrochemical applications. Compared with nanoscale catalysts, single-atom catalysts are characterized by isolated active sites and high dispersion leading to maximum atom utilization efficiency. Herein, we focus on the Pd SACs deposited on a variety of substrate materials (metal sulfide, carbon material, metal oxides etc.) (Table 6).
Table 6.
Summary of the HER activity of SACs in acidic/alkaline solutions.
A relatively small Tafel slope is highly desirable, and we can achieve that by exposing more active sites for reaction. Thus, we increase the catalytic current density and decrease overpotentials, resulting in a decrease in the Tafel slopes. Lau et al. [134] designed an uncommon Pd-1 T-sMoS2 nanosheet where 1 T phase is found to be metallic and thermally metastable by exfoliation of bulk 2 H-sMoS2 using BuLi intercalation. The incorporation of single Pd atoms at 3 wt% can successfully increase the HER performance compared to undoped 1 T-sMoS2. The reference catalyst 20% Pt/C gave a slope of 33 mV dec−1, which is in good accordance with previous results [131]. A Tafel slope of ~30 mV dec−1 is generally accepted and indicates that the HER proceeds through the Tafel pathway. On the other hand, the 2 H- sMoS2 material gave a slope of 103 mV dec−1, which indicates that the kinetics for H2G.
H2 formation is limited by the first Volmer proton reduction step, and the material follows the Volmer-Tafel mechanism. The Tafel slope of Pd-1 T-sMoS2 is relatively smaller (50 mV dec−1) compared to the pristine 1 T-sMoS2 (61 mV dec−1), as shown in Table 6.
In another study, [133] designed a two-dimensional (2D) MoO3 with petal-shaped nanosheets confining Pd nanoparticles (NPs) and atomically dispersed Pd substituted sites [132]. The high surface area of Pd@MoO3, the interface between MoO3 and Pd NPs, and the atomically dispersed Pd sites in the MoO3 lattice significantly enlarge the number of active sites and facilitate electron transfer. As a result, Pd@MoO3 material exhibits high HER activity with a small overpotential of 71 mV and an extremely low Tafel slope of 42.8 mV dec−1 in acidic solution compared with the reference catalyst 20% Pt/C.
Besides metal oxides and metal sulfides, carbon materials are another attractive substrate. Zhang et al. [135] reported the use of thin layers of graphitic carbon nitride with carbon black (C@C3N4) as a substrate material to immobilize Pd single atoms (Figure 27B(i–iv)). The use of carbon in electrochemical applications is rather usual due to its high surface area and conductivity. The addition of dopants, such as N or B, provides anchoring sites, and specifically N-doped carbon can anchor metal atoms and prevent metal agglomeration, and also nitrogen doping enhances the electron-donor capacity of the carbon matrix, resulting in an improvement of the interaction of carbon support and guest molecules [121]. The abundance of N-anchoring sites of C@C3N4 support facilitate the synthesis of single atom Pd catalysts without forming metal nanoparticles. The Pd loading on C@C3N4 was 0.5, 1, and 4%, with C@C3N4-0.5%Pd being slightly more active than conventional 20% Pd/C, but less active than Pd nanoparticles supported on C@C3N4. However, C@C3N4-0.5%Pd shows a higher Tafel slope of 127 mV dec−1 compared to 20% Pd/C with a slope of 49 mV dec−1 (Figure 27B(v)).
Downsizing metal nanoparticles to atomic level boosts electrocatalytic performance even if SACs suffer from one major drawback. Due to the high activity of isolated metal atoms and the rough conditions under which HER occurs (acidic or alkaline solution) SACs, besides Pt-based catalysts, tend to aggregate and dissolve as they suffer from low stability, thus affecting their catalytic performance. The design of a stable structure with the ability to improve the stability of SACs is essential. The role of support becomes crucial and determines the coordination of the catalytic metal centre and its properties. Compared with nanoparticles, the active sites of SACs are directly fixed to the support by chemical bonds. By controlling the chemical structure of the support, one can control the properties of SACs and optimize them. Carbon nitride is a good support matrix for stabilizing metal single atoms due to its unique structure, the large surface area, and the many nitrogen species that it possesses [134]. For instance, Co SAC (Co-NG) was prepared by using nitrogen-doped graphene as a support ], which exhibits very low overpotential (30 mV) in hydrogen generation. Besides carbon nitride, some studies focus on the doping of transition metal phosphides (TMPs) with appropriate elements to enhance stability. Wu et al. [137] designed a W-doped CoP nanoarray (W-Cop), which exhibits excellent stability during HER under different pH solutions [139]. Moreover, a different approach involves defect engineering to design stable HER SACs through vacancies and dopants [139].
Additionally, single atomic sites minimize the size of metal particles, and, generally, the concentration is relatively low, i.e., <0.5%, to prevent single atomic agglomeration. Because of this low concentration, there is a long way to go before we jump from the laboratory use to the industrial and actual production. In 2016, Zheng et al. approached this problem by synthesizing a Pd/TiO2 catalyst with a high Pd single atom concentration of 1.5% stabilized due to the formation of ethylene glycol free radical induced by UV treatment, which facilitates the formation of Pd–O bonds and subsequently stabilized the individual Pd atom.
6. Conclusions
Palladium deposited on different solid supports can adopt quite distinct species, which, in turn, can promote different catalytic reaction paths. Within palladium nanoparticles, atomic {Pdn+-X} states, where n = 0, 1, 2, 3, and X = atom or hydride, can be stabilized. These can behave as single-site-catalysts that demonstrate considerable degree of chemical reactivity. In the present review, we put focus on the distinction of these {Pdn+-X} states vs. pure atomic states, i.e., isolated Pd-atoms on a solid surface and their pertinence on catalytic processes related to energy storage or energy conversion, H2 production reaction (HRR), oxygen reduction reaction (ORR), and water-splitting. Successful synthesis of {Pdn+-X} states and their detection methodologies are currently standalone challenges. We consider that future state-of-the-art synthesis methods will enrich this dynamic landscape, while detection methods could shed light on the very interesting, quantum phenomena at the {nanoparticle–particle-to-atom} interface.
Author Contributions
Writing—original draft preparation P.S., M.S., A.Z., L.B. and Y.D.; review, editing. P.S., Y.D. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Hellenic Foundation for Research and Innovation (H.F.R.I.) under the “First Call for H.F.R.I. Research Projects to support Faculty members and Researchers and the procurement of high-cost research equipment grant” (HFRI-FM17-1888).
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Liu, L.; Corma, A. Metal Catalysts for Heterogeneous Catalysis: From Single Atoms to Nanoclusters and Nanoparticles. Chem. Rev. 2018, 118, 4981–5079. [Google Scholar] [CrossRef] [PubMed]
- Stathi, P.; Solakidou, M.; Louloudi, M.; Deligiannakis, Y. From Homogeneous to Heterogenized Molecular Catalysts for H 2 Production by Formic Acid Dehydrogenation: Mechanistic Aspects, Role of Additives, and Co-Catalysts. Energies 2020, 13, 733. [Google Scholar] [CrossRef]
- Schalow, T.; Brandt, B.; Laurin, M.; Schauermann, S.; Guimond, S.; Kuhlenbeck, H.; Libuda, J.; Freund, H.J. Formation of interface and surface oxides on supported Pd nanoparticles. Surf. Sci. 2006, 600, 2528–2542. [Google Scholar] [CrossRef]
- Mondal, S.; Ballav, T.; Biswas, K.; Ghosh, S.; Ganesh, V. Exploiting the Versatility of Palladium Catalysis: A Modern Toolbox for Cascade Reactions. Eur. J. Org. Chem. 2021, 2021, 4566–4602. [Google Scholar] [CrossRef]
- Gates, B.C.; Flytzani-Stephanopoulos, M.; Dixon, D.A.; Katz, A. Catalysis Science & Technology Atomically dispersed supported metal catalysts: Perspectives and suggestions for future research. Catal. Sci. Technol. 2017, 7, 4259. [Google Scholar]
- Wolfe, P.J.; Li, J. Chapter 1 An introduction to palladium catalysis. Tetrahedron Org. Chem. Ser. 2007, 26, 1–35. [Google Scholar]
- Zhang, S.; Bai, B.; Liu, J.; Zhang, J. Atomically dispersed catalytic sites: A new frontier for cocatalyst/photocatalyst composites toward sustainable fuel and chemical production. Catalysts 2021, 11, 1168. [Google Scholar] [CrossRef]
- Tauster, S.J.; Fung, S.C.; Baker, R.T.K.; Horsley, J.A. Strong interactions in supported-metal catalysts. Science 1981, 211, 1121–1125. [Google Scholar] [CrossRef]
- Van Vaerenbergh, B.; Lauwaert, J.; Vermeir, P.; De Clercq, J.; Thybaut, J.W. Synthesis and Support Interaction Effects on the Palladium Nanoparticle Catalyst Characteristics, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2019; Volume 65, ISBN 9780128171011. [Google Scholar]
- Su, J.; Ge, R.; Dong, Y.; Hao, F.; Chen, L. Recent progress in single-atom electrocatalysts: Concept, synthesis, and applications in clean energy conversion. J. Mater. Chem. A 2018, 6, 14025–14042. [Google Scholar] [CrossRef]
- Zhang, Y.; Guo, L.; Tao, L.; Lu, Y.; Wang, S. Defect-Based Single-Atom Electrocatalysts. Small Methods 2019, 3, 1800406. [Google Scholar] [CrossRef]
- Cotton, F.A.; Wilkinson, C.; Murillo, C.A.; Manfred, B. Chemistry of Transition Elements. In Advanced Inorganic Chemistry, 4th ed.; John Wiley & Sons: Hoboken, NJ, USA, 1980; pp. 749–753. [Google Scholar]
- Negishi, E.; Meijere, A. Handbook of Organopalladium Chemistry for Organic Synthesis, 4th ed.; John Wiley & Sons: Hoboken, NJ, USA, 2002; p. 279. [Google Scholar]
- Murata, K.; Kosuge, D.; Ohyama, J.; Mahara, Y.; Yamamoto, Y.; Arai, S.; Satsuma, A. Exploiting Metal-Support Interactions to Tune the Redox Properties of Supported Pd Catalysts for Methane Combustion. ACS Catal. 2020, 10, 1381–1387. [Google Scholar] [CrossRef]
- Trzeciak, A.M.; Augustyniak, A.W. The role of palladium nanoparticles in catalytic C–C cross-coupling reactions. Coord. Chem. Rev. 2019, 384, 1–20. [Google Scholar] [CrossRef]
- Saldan, I.; Semenyuk, Y.; Marchuk, I.; Reshetnyak, O. Chemical synthesis and application of palladium nanoparticles. J. Mater. Sci. 2015, 50, 2337–2354. [Google Scholar] [CrossRef]
- Fujiwara, K.; Müller, U.; Pratsinis, S.E. Pd Subnano-Clusters on TiO2 for Solar-Light Removal of NO. ACS Catal. 2016, 6, 1887–1893. [Google Scholar] [CrossRef]
- Pinto, F.M.; Suzuki, V.Y.; Silva, R.C.; La Porta, F.A. Oxygen Defects and Surface Chemistry of Reducible Oxides. Front. Mater. 2019, 6, 260. [Google Scholar] [CrossRef]
- Puigdollers, A.R.; Schlexer, P.; Tosoni, S.; Pacchioni, G. Increasing oxide reducibility: The role of metal/oxide interfaces in the formation of oxygen vacancies. ACS Catal. 2017, 7, 6493–6513. [Google Scholar] [CrossRef]
- Wendt, S.; Sprunger, P.T.; Lira, E.; Madsen, G.K.H.; Li, Z.; Hansen, J.; Matthiesen, J.; Blekinge-Rasmussen, A.; Lægsgaard, E.; Hammer, B.; et al. The role of interstitial sites in the Ti3d defect state in the band gap of titania. Science 2008, 320, 1755–1759. [Google Scholar] [CrossRef]
- Park, J.B.; Graciani, J.; Evans, J.; Stacchiola, D.; Ma, S.; Liu, P.; Nambu, A.; Sanz, J.F.; Hrbek, J.; Rodriguez, J.A. High catalytic activity of Au/CeOx/TiO2(110) controlled by the nature of the mixed-metal oxide at the nanometer level. Proc. Natl. Acad. Sci. USA 2009, 106, 4975–4980. [Google Scholar] [CrossRef]
- Bernal, S.; Calvino, J.J.; Cauqui, M.A.; Gatica, J.M.; Larese, C.; Pérez Omil, J.A.; Pintado, J.M. Some recent results on metal/support interaction effects in NM/CeO2 (NM: Noble metal) catalysts. Catal. Today 1999, 50, 175–206. [Google Scholar] [CrossRef]
- Akbayrak, S.; Tonbul, Y.; Özkar, S. Nanoceria supported palladium(0) nanoparticles: Superb catalyst in dehydrogenation of formic acid at room temperature. Appl. Catal. B Environ. 2017, 206, 384–392. [Google Scholar] [CrossRef]
- Gniewek, A.; Ziółkowski, J.J.; Trzeciak, A.M.; Zawadzki, M.; Grabowska, H.; Wrzyszcz, J. Palladium nanoparticles supported on alumina-based oxides as heterogeneous catalysts of Suzuki–Miyaura reaction. J. Catal. 2008, 254, 121–130. [Google Scholar] [CrossRef]
- Stathi, P.; Belles, L.; Deligiannakis, Y. Multipotent Atomic Palladium Species Pd1+, Pd2+-O2-, and Pd3+Formed at the Interface of Pd/TiO2 Nanoparticles: Electron Paramagnetic Resonance Study. J. Phys. Chem. C 2022, 126, 14125–14137. [Google Scholar] [CrossRef]
- Liu, K.; Qin, R.; Zhou, L.; Liu, P.; Zhang, Q.; Jing, W.; Ruan, P.; Gu, L.; Fu, G.; Zheng, N. Cu2O-supported atomically dispersed Pd catalysts for semihydrogenation of terminal alkynes: Critical role of oxide supports. CCS Chem. 2019, 1, 207–214. [Google Scholar] [CrossRef]
- Tan, H.; Xu, Y.P.; Rong, S.; Zhao, R.; Cui, H.; Chen, Z.N.; Xu, Z.N.; Zhang, N.N.; Guo, G.C. Enhanced metal-support interaction between Pd and hierarchical Nb2O5via oxygen defect induction to promote CO oxidative coupling to dimethyl oxalate. Nanoscale 2021, 13, 18773–18779. [Google Scholar] [CrossRef] [PubMed]
- Moussa, S.; Siamaki, A.R.; Gupton, B.F.; El-Shall, M.S. Pd-partially reduced graphene oxide catalysts (Pd/PRGO): Laser synthesis of Pd nanoparticles supported on PRGO nanosheets for carbon-carbon cross coupling reactions. ACS Catal. 2012, 2, 145–154. [Google Scholar] [CrossRef]
- Parker, S.F.; Walker, H.C.; Callear, S.K.; Grünewald, E.; Petzold, T.; Wolf, D.; Möbius, K.; Adam, J.; Wieland, S.D.; Jiménez-Ruiz, M.; et al. The effect of particle size, morphology and support on the Van formation of palladium hydride in commercial catalysts. Chem. Sci. 2019, 10, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Devivaraprasad, R.; Nalajala, N.; Bera, B.; Neergat, M. Electrocatalysis of Oxygen Reduction Reaction on Shape-Controlled Pt and Pd Nanoparticles—Importance of Surface Cleanliness and Reconstruction. Front. Chem. 2019, 7, 648. [Google Scholar] [CrossRef] [PubMed]
- Keresszegi, C.; Grunwaldt, J.D.; Mallat, T.; Baiker, A. In situ EXAFS study on the oxidation state of Pd/Al2O3 and Bi-Pd/Al2O3 during the liquid-phase oxidation of 1-phenylethanol. J. Catal. 2004, 222, 268–280. [Google Scholar] [CrossRef]
- Chenakin, S.P.; Melaet, G.; Szukiewicz, R.; Kruse, N. XPS study of the surface chemical state of a Pd/(SiO2+TiO2) catalyst after methane oxidation and SO2 treatment. J. Catal. 2014, 312, 1–11. [Google Scholar] [CrossRef]
- Nilsson, J.; Carlsson, P.A.; Fouladvand, S.; Martin, N.M.; Gustafson, J.; Newton, M.A.; Lundgren, E.; Grönbeck, H.; Skoglundh, M. Chemistry of Supported Palladium Nanoparticles during Methane Oxidation. ACS Catal. 2015, 5, 2481–2489. [Google Scholar] [CrossRef]
- Michalik, J.; Heming, M.; Kevan, L. Trivalent and monovalent palladium cations in PdNa-X zeolite: Electron spin resonance and electron spin echo modulation spectroscopic studies. J. Phys. Chem. 1986, 90, 2132–2136. [Google Scholar] [CrossRef]
- Zina, M.S.; Ghorbel, A. ESR study of MoY and PdMoY reduction. Magn. Reson. Chem. 2004, 42, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Narayana, M.; Michalik, J.; Contarini, S.; Kevan, L. Determination of the chemical state of palladium in PdNa-X zeolite by Electron Spin Resonance and X-ray Photoelectron Spectroscopy. J. Phys. Chem. 1985, 89, 3895–3899. [Google Scholar] [CrossRef]
- Yu, J.S.; Comets, J.M.; Kevan, L. Electron paramagnetic resonance and electron spin echo modulation spectroscopic studies on the location and adsorbate interactions of paramagnetic Pd ion species in PdII-exchanged K-L zeolite. J. Chem. Soc. Faraday Trans. 1993, 89, 4397–4403. [Google Scholar] [CrossRef]
- Kevan, L. Formation and adsorbate interactions of paramagnetic Pd(I) species in Pd(II)-exchanged NaK- and H-clinoptilolite. J. Phys. Chem. B 2000, 104, 3608–3615. [Google Scholar]
- Ghosh, A.K.; Kevan, L. Electron spin resonance and electron spin echo spectroscopic studies of catalytic dimerization of ethylene on palladium-exchanged Na-X and Ca-X zeolites. Proposed reaction mechanism for ethylene dimerization. J. Am. Chem. Soc. 1988, 110, 8044–8050. [Google Scholar] [CrossRef]
- Michalik, J.; Lee, H.; Kevan, L. Ethylene dimerization on Pd-Ca-X zeolite: Gas chromatographic and electron spin resonance studies. J. Phys. Chem. 1985, 89, 4282–4285. [Google Scholar] [CrossRef]
- Yu, J.S.; Kevan, L. Adsorbate Interactions of Paramagnetic Palladium(I) Species in Pd(II)-Exchanged K-L Zeolite. Langmuir 1995, 11, 1617–1625. [Google Scholar] [CrossRef]
- Stokes, L.S.; Murphy, D.M.; Farley, R.D.; Rowlands, C.C.; Bailey, S. EPR investigation of Pd(I) species in palladium-exchanged ZSM-5 and beta zeolites. Phys. Chem. Chem. Phys. 1999, 1, 621–628. [Google Scholar] [CrossRef]
- Schalow, T.; Brandt, B.; Starr, D.E.; Laurin, M.; Schauermann, S.; Shaikhutdinov, S.K.; Libuda, J.; Freund, H.J. Oxygen-induced restructuring of a Pd/Fe3O4 model catalyst. Catal. Lett. 2006, 107, 189–196. [Google Scholar] [CrossRef]
- Su, R.; Dimitratos, N.; Liu, J.; Carter, E.; Althahban, S.; Wang, X.; Shen, Y.; Wendt, S.; Wen, X.; Niemantsverdriet, H.; et al. Mechanistic Insight into the Interaction Between a Titanium Dioxide Photocatalyst and Pd Cocatalyst for Improved Photocatalytic Performance. ACS Catal. 2016, 6, 4239–4247. [Google Scholar] [CrossRef]
- Luna, A.L.; Dragoe, D.; Wang, K.; Beaunier, P.; Kowalska, E.; Ohtani, B.; Bahena Uribe, D.; Valenzuela, M.A.; Remita, H.; Colbeau-Justin, C. Photocatalytic Hydrogen Evolution Using Ni-Pd/TiO2: Correlation of Light Absorption, Charge-Carrier Dynamics, and Quantum Efficiency. J. Phys. Chem. C 2017, 121, 14302–14311. [Google Scholar] [CrossRef]
- Saputro, A.G.; Putra, R.I.D.; Maulana, A.L.; Karami, M.U.; Pradana, M.R.; Agusta, M.K.; Dipojono, H.K.; Kasai, H. Theoretical study of CO2 hydrogenation to methanol on isolated small Pdx clusters. J. Energy Chem. 2019, 35, 79–87. [Google Scholar] [CrossRef]
- Ou, Z.; Ran, J.; Niu, J.; Qin, C.; He, W.; Yang, L. A density functional theory study of CO2 hydrogenation to methanol over Pd/TiO2 catalyst: The role of interfacial site. Int. J. Hydrogen Energy 2020, 45, 6328–6340. [Google Scholar] [CrossRef]
- Guo, Q.; Ghadiri, R.; Weigel, T.; Aumann, A.; Gurevich, E.L.; Esen, C.; Medenbach, O.; Cheng, W.; Chichkov, B.; Ostendorf, A. Comparison of in Situ and ex Situ Methods for Synthesis of Two-Photon Polymerization Polymer Nanocomposites. Polymers 2014, 6, 2037–2050. [Google Scholar] [CrossRef]
- Piermatti, O. Green Synthesis of Pd Nanoparticles for Sustainable and Environmentally Benign Processes. Catalysts 2021, 11, 1258. [Google Scholar] [CrossRef]
- Chen, A.; Ostrom, C. Palladium-Based Nanomaterials: Synthesis and Electrochemical Applications. Chem. Rev. 2015, 115, 11999–12044. [Google Scholar] [CrossRef]
- Rezaee, S.; Ghobadi, N. Synthesis of Ag-Cu-Pd alloy thin films by DC-magnetron sputtering: Case study on microstructures and optical properties. Results Phys. 2018, 9, 1148–1154. [Google Scholar] [CrossRef]
- Barzola-Quiquia, J.; Schulze, S.; Esquinazi, P. Transport Properties and Atomic Structure of Ion-Beam-Deposited W, Pd and Pt Nanostructures. Nanotechnology 2009, 20, 165704–165711. [Google Scholar] [CrossRef]
- Bhuvana, T.; Kulkarni, G.U. Highly Conducting Patterned Pd Nanowires by Direct-Write Electron Beam Lithography. ACS Nano 2008, 2, 457–462. [Google Scholar] [CrossRef]
- Ocola, L.E.; Rue, C.; Maas, D. High-Resolution Direct-Write Patterning Using Focused Ion Beams. MRS Bull. 2014, 39, 336–341. [Google Scholar] [CrossRef]
- Wei, C.; Zhang, Z.; Cheng, D.; Sun, Z.; Zhu, M.; Li, L. An overview of laser-based multiple metallic material additive manufacturing: From macro: From micro-scales. Int. J. Extrem. Manuf. 2021, 3, 012003. [Google Scholar] [CrossRef]
- Fonseca, J.; Lu, J. Single-Atom Catalysts Designed and Prepared by the Atomic Layer Deposition Technique. ACS Catal. 2021, 11, 7018–7059. [Google Scholar] [CrossRef]
- Piernavieja-Hermida, M.; Lu, Z.; White, A.; Low, K.B.; Wu, T.; Elam, J.W.; Wu, Z.; Lei, Y. Towards ALD thin film stabilized single-atom Pd1 catalysts. Nanoscale 2016, 8, 15348–15356. [Google Scholar] [CrossRef] [PubMed]
- Muravev, V.; Spezzati, G.; Su, Y.Q.; Parastaev, A.; Chiang, F.K.; Longo, A.; Escudero, C.; Kosinov, N.; Hensen, E.J.M. Interface dynamics of Pd–CeO2 single-atom catalysts during CO oxidation. Nat. Catal. 2021, 4, 469–478. [Google Scholar] [CrossRef]
- Xie, B.; Xiong, Y.; Chen, R.; Chen, J.; Cai, P. Catalytic Activities of Pd−TiO Film towards the Oxidation of Formic Acid. Catal. Commun. 2005, 6, 699–704. [Google Scholar] [CrossRef]
- Tian, M.; Malig, M.; Chen, S.; Chen, A. Synthesis and Electrochemical Study of TiO2-Supported PdAu Nanoparticles. Electrochem. Commun. 2011, 13, 370–373. [Google Scholar] [CrossRef]
- Zhao, R.; Ding, R.; Yuan, S.; Jiang, W.; Liang, B. Palladium Membrane on TiO2 Nanotube Arrays-Covered Titanium Surface by Combination of Photocatalytic Deposition and Modified Electroless Plating Processes and Its Hydrogen Permeability. Int. J. Hydrogen Energy 2011, 36, 1066–1073. [Google Scholar] [CrossRef]
- Cookson, J. The preparation of palladium nanoparticles. Platin. Met. Rev. 2012, 56, 83–98. [Google Scholar] [CrossRef]
- Andelman, T.; Tan, M.C.; Riman, R.E. Thermochemical engineering of hydrothermal crystallisation processes. Mater. Res. Innov. 2013, 14, 9–15. [Google Scholar] [CrossRef]
- Navlani-García, M.; Martis, M.; Lozano-Castelló, D.; Cazorla-Amorós, D.; Mori, K.; Yamashita, H. Investigation of Pd nanoparticles supported on zeolites for hydrogen production from formic acid dehydrogenation. Catal. Sci. Technol. 2014, 5, 364–371. [Google Scholar] [CrossRef]
- Astruc, D.; Lu, F.; Aranzaes, J.R. Nanoparticles as Recyclable Catalysts: The Frontier between Homogeneous and Heterogeneous Catalysis. Angew. Chem. Int. Ed. 2005, 44, 7852–7872. [Google Scholar] [CrossRef] [PubMed]
- Ott, L.S.; Finke, R.G. Transition-metal nanocluster stabilization for catalysis: A critical review of ranking methods and putative stabilizers. Coord. Chem. Rev. 2007, 251, 1075–1100. [Google Scholar] [CrossRef]
- Adams, B.D.; Wu, G.; Nlgro, S.; Chen, A. Facile synthesis of Pd-Cd nanostructures with high capacity for hydrogen storage. J. Am. Chem. Soc. 2009, 131, 6930–6931. [Google Scholar] [CrossRef] [PubMed]
- Yi, Q.; Huang, W.; Liu, X.; Xu, G.; Zhou, Z.; Chen, A. Electroactivity of Titanium-Supported Nanoporous Pd−Pt Catalysts towards Formic Acid Oxidation. J. Electroanal. Chem. 2008, 619−620, 197–205. [Google Scholar] [CrossRef]
- Guo, P.; Wei, Z.; Ye, W.; Qin, W.; Wang, Q.; Guo, X.; Lu, C.; Zhao, X.S. Preparation and Characterization of Nanostructured Pd with High Electrocatalytic Activity. Colloids Surf. A 2012, 395, 75–81. [Google Scholar] [CrossRef]
- Yi, Q.; Niu, F.; Sun, L. Fabrication of Novel Porous Pd Particles and Their Electroactivity towards Ethanol Oxidation in Alkaline Media. Fuel 2011, 90, 2617–2623. [Google Scholar] [CrossRef]
- Yuan, M.; Liu, A.; Zhao, M.; Dong, W.; Zhao, T.; Wang, J.; Tang, W. Bimetallic PdCu Nanoparticle Decorated Three-Dimensional Graphene Hydrogel for Non-Enzymatic Amperometric Glucose Sensor. Sens. Actuators B 2014, 190, 707–714. [Google Scholar] [CrossRef]
- Kuai, L.; Yu, X.; Wang, S.; Sang, Y.; Geng, B. Au−Pd Alloy and Core−Shell Nanostructures: One-Pot Coreduction Preparation, Formation Mechanism, and Electrochemical Properties. Langmuir 2012, 28, 7168–7173. [Google Scholar] [CrossRef]
- Zhuang, W.; Liu, X.; Chen, L.; Liu, P.; Wen, H.; Zhou, Y.; Wang, J. One-pot hydrothermal synthesis of ultrafine Pd clusters within Beta zeolite for selective oxidation of alcohols. Green Chem. 2020, 22, 4199–4209. [Google Scholar] [CrossRef]
- Zhang, H.; Ke, D.; Cheng, L.; Feng, X.; Hou, X.; Wang, J.; Li, Y.; Han, S. CoPt-Co hybrid supported on amino modified SiO2 nanospheres as a high performance catalyst for hydrogen generation from ammonia borane. Prog. Nat. Sci. Mater. Int. 2019, 29, 1–974. [Google Scholar] [CrossRef]
- Sreedhar, B.; Yada, D.; Reddy, P. Nanocrystalline Titania-Supported Palladium (0) Nanoparticles for Suzuki–Miyaura Cross-Coupling of Aryl and Heteroaryl Halides. Adv. Synth. Catal. 2011, 353, 2823–2836. [Google Scholar] [CrossRef]
- Thanh, N.T.K.; Maclean, N.; Mahiddine, S. Mechanisms of nucleation and growth of nanoparticles in solution. Chem. Rev. 2014, 114, 7610–7630. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Kumamoto, Y.; Hashiguchi, T.; Mamba, T.; Murayama, H.; Yamamoto, E.; Ishida, T.; Honma, T.; Tokunaga, M. Wacker Oxidation of Terminal Alkenes Over ZrO2-Supported Pd Nanoparticles Under Acid- and Cocatalyst-Free Conditions. ChemSusChem 2017, 10, 3482–3489. [Google Scholar] [CrossRef]
- Cui, X.J.; Junge, K.; Dai, X.C.; Kreyenschulte, C.; Pohl, M.M.S.; Wohlrab, F.; Shi, A. Bruckner and M. Beller, Synthesis of single atom based heterogeneous platinum catalysts: High selectivity and activity for hydrosilylation reactions. ACS Cent. Sci. 2017, 3, 580–585. [Google Scholar] [CrossRef]
- Guo, J.; Liu, H.; Li, D.; Wang, J.; Djitcheu, X.; He, D.; Zhang, Q. A minireview on the synthesis of single atom catalysts. RSC Adv. 2022, 12, 9373–9394. [Google Scholar] [CrossRef]
- Kim, H.E.; Lee, I.H.; Cho, J.; Shin, S.; Ham, H.C.; Kim, J.Y.; Lee, H. Palladium Single-Atom Catalysts Supported on C@C3N4 for Electrochemical Reactions. ChemElectroChem 2019, 6, 4757–4764. [Google Scholar] [CrossRef]
- Zhang, C.; Oliaee, S.N.; Hwang, S.Y.; Kong, X.; Peng, Z. A Generic Wet Impregnation Method for Preparing Substrate-Supported Platinum Group Metal and Alloy Nanoparticles with Controlled Particle Morphology. Nano Lett. 2016, 16, 164–169. [Google Scholar] [CrossRef]
- Lai, W.H.; Zhang, L.F.; Hua, W.B.; Indris, S.; Yan, Z.C.; Hu, Z.; Zhang, B.; Liu, Y.; Wang, L.; Liu, M.; et al. General π-Electron-Assisted Strategy for Ir, Pt, Ru, Pd, Fe, Ni Single-Atom Electrocatalysts with Bifunctional Active Sites for Highly Efficient Water Splitting. Angew. Chem. Int. Ed. 2019, 58, 11868–11873. [Google Scholar] [CrossRef]
- Lee, C.-H.; Wang, S.-C.; Yuan, C.-J.; Wen, M.-F.; Chang, K.-S. Comparison of Amperometric Biosensors Fabricated by Palladium Sputtering, Palladium Electrodeposition and Nafion/carbon Nanotube Casting on Screen-Printed Carbon Electrodes. Biosens. Bioelectron. 2007, 22, 877–884. [Google Scholar] [CrossRef]
- Zhao, Y.; Qin, S.-J.; Li, Y.; Deng, F.-X.; Liu, Y.-Q.; Pan, G.-B. Electrodeposition of Dendritic Pd Nanoarchitectures on NGaN(0001): Nucleation and Electrocatalysis for Direct Formic Acid Fuel Cells. Electrochim. Acta 2014, 145, 148–153. [Google Scholar] [CrossRef]
- Deligiannakis, Y.; Tsikourkitoudi, V.; Stathi, P.; Wegner, K.; Papavasiliou, J.; Louloudi, M. PdO/Pd0/TiO2Nanocatalysts Engineered by Flame Spray Pyrolysis: Study of the Synergy of PdO/Pd0on H2Production by HCOOH Dehydrogenation and the Deactivation Mechanism. Energy Fuels 2020, 34, 15026–15038. [Google Scholar] [CrossRef]
- Zhang, L.; Wu, W.; Jiang, Z.; Fang, T. A review on liquid-phase heterogeneous dehydrogenation of formic acid: Recent advances and perspectives. Chem. Pap. 2018, 72, 2121–2135. [Google Scholar] [CrossRef]
- Li, Z.; Xu, Q. Metal-Nanoparticle-Catalyzed Hydrogen Generation from Formic Acid. Acc. Chem. Res. 2017, 50, 1449–1458. [Google Scholar] [CrossRef]
- Iost, K.N.; Borisov, V.A.; Temerev, V.L.; Surovikin, Y.V.; Pavluchenko, P.E.; Trenikhin, M.V.; Arbuzov, A.B.; Shlyapin, D.A.; Tsyrulnikov, P.G.; Vedyagin, A.A. Carbon support hydrogenation in Pd/C catalysts during reductive thermal treatment. Int. J. Hydrogen Energy 2018, 43, 17656–17663. [Google Scholar] [CrossRef]
- Kim, J.D.; Choi, M.Y.; Choi, H.C. Catalyst activity of carbon nanotube supported Pd catalysts for the hydrogenation of nitroarenes. Mater. Chem. Phys. 2016, 173, 404–411. [Google Scholar] [CrossRef]
- Lam, E.; Luong, J.H.T. Carbon materials as catalyst supports and catalysts in the transformation of biomass to fuels and chemicals. ACS Catal. 2014, 4, 3393–3410. [Google Scholar] [CrossRef]
- Hattori, M.; Einaga, H.; Daio, T.; Tsuji, M. Efficient hydrogen production from formic acid using TiO2-supported AgPd@Pd nanocatalysts. J. Mater. Chem. A 2015, 3, 4453–4461. [Google Scholar] [CrossRef]
- Wang, N.; Niu, F.; Wang, S.; Huang, Y. Catalytic activity of flame-synthesized Pd/TiO2 for the methane oxidation following hydrogen pretreatments. Particuology 2018, 41, 58–64. [Google Scholar] [CrossRef]
- Yuranov, I.; Moeckli, P.; Suvorova, E.; Buffat, P.; Kiwi-Minsker, L.; Renken, A. Pd/SiO2 catalysts: Synthesis of Pd nanoparticles with the controlled size in mesoporous silicas. J. Mol. Catal. A Chem. 2003, 192, 239–251. [Google Scholar] [CrossRef]
- Boudjahem, A.G.; Redjel, A.; Mokrane, T. Preparation, characterization and performance of Pd/SiO 2 catalyst for benzene catalytic hydrogenation. J. Ind. Eng. Chem. 2012, 18, 303–308. [Google Scholar] [CrossRef]
- Kumar, M.; Bhati, V.S.; Ranwa, S.; Singh, J.; Kumar, M. Pd/ZnO nanorods based sensor for highly selective detection of extremely low concentration hydrogen. Sci. Rep. 2017, 7, 1–9. [Google Scholar]
- Bahruji, H.; Bowker, M.; Hutchings, G.; Dimitratos, N.; Wells, P.; Gibson, E.; Jones, W.; Brookes, C.; Morgan, D.; Lalev, G. Pd/ZnO catalysts for direct CO2hydrogenation to methanol. J. Catal. 2016, 343, 133–146. [Google Scholar] [CrossRef]
- Bagheri, S.; Muhd Julkapli, N.; Bee Abd Hamid, S. Titanium dioxide as a catalyst support in heterogeneous catalysis. Sci. World J. 2014, 2014, 727496. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Tan, S.; Moon, J.; Yan, Z.; Fung, V.; Li, N.; Yang, S.Z.; Cheng, Y.; Abney, C.W.; Wu, Z.; et al. Harnessing strong metal–support interactions via a reverse route. Nat. Commun. 2020, 11, 3042. [Google Scholar] [CrossRef] [PubMed]
- Baker, R.T.K.; Prestridge, E.B.; Murrell, L.L. Electron microscopy of supported metal particles. III. The role of the metal in an SMSI interaction. J. Catal. 1983, 79, 348–358. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, H.; Wang, L.; Ali, S.; Wang, C.; Wang, L.; Meng, X.; Li, B.; Su, D.S.; Xiao, F.S. Wet-Chemistry Strong Metal-Support Interactions in Titania-Supported Au Catalysts. J. Am. Chem. Soc. 2019, 141, 2975–2983. [Google Scholar] [CrossRef]
- Wang, J.; Tan, H.; Jiang, D.; Zhou, K. Enhancing H 2 evolution by optimizing H adatom combination and desorption over Pd nanocatalyst. Nano Energy 2017, 33, 410–417. [Google Scholar] [CrossRef]
- Li, J.; Chen, W.; Zhao, H.; Zheng, X.; Wu, L.; Pan, H.; Zhu, J.; Chen, Y.; Lu, J. Size-dependent catalytic activity over carbon-supported palladium nanoparticles in dehydrogenation of formic acid. J. Catal. 2017, 352, 371–381. [Google Scholar] [CrossRef]
- Lv, Q.; Meng, Q.; Liu, W.; Sun, N.; Jiang, K.; Ma, L.; Peng, Z.; Cai, W.; Liu, C.; Ge, J.; et al. Pd-PdO Interface as Active Site for HCOOH Selective Dehydrogenation at Ambient Condition. J. Phys. Chem. C 2018, 122, 2081–2088. [Google Scholar] [CrossRef]
- Meher, S.; Rana, R.K. A rational design of a Pd-based catalyst with a metal-metal oxide interface influencing molecular oxygen in the aerobic oxidation of alcohols. Green Chem. 2019, 21, 2494–2503. [Google Scholar] [CrossRef]
- Wang, T.; Tao, X.; Li, X.; Zhang, K.; Liu, S.; Li, B. Synergistic Pd Single Atoms, Clusters, and Oxygen Vacancies on TiO2 for Photocatalytic Hydrogen Evolution Coupled with Selective Organic Oxidation. Small 2021, 17, 1–10. [Google Scholar] [CrossRef]
- Liu, L.; Wu, X.; Wang, L.; Xu, X.; Gan, L.; Si, Z.; Li, J.; Zhang, Q.; Liu, Y.; Zhao, Y.; et al. Atomic palladium on graphitic carbon nitride as a hydrogen evolution catalyst under visible light irradiation. Commun. Chem. 2019, 2, 2–9. [Google Scholar] [CrossRef]
- Cao, S.; Li, H.; Tong, T.; Chen, H.C.; Yu, A.; Yu, J.; Chen, H.M. Single-Atom Engineering of Directional Charge Transfer Channels and Active Sites for Photocatalytic Hydrogen Evolution. Adv. Funct. Mater. 2018, 28, 1802169. [Google Scholar] [CrossRef]
- Liu, G.; Lv, H.; Zeng, Y.; Yuan, M.; Meng, Q.; Wang, Y.; Wang, C. Single-Atom Pd–N3 Sites on Carbon-Deficient g-C3N4 for Photocatalytic H2 Evolution. Trans. Tianjin Univ. 2021, 27, 139–146. [Google Scholar] [CrossRef]
- Li, W.; Chu, X.S.; Wang, F.; Dang, Y.Y.; Liu, X.Y.; Ma, T.H.; Li, J.-Y.; Wang, C.-Y. Pd single-atom decorated CdS nanocatalyst for highly efficient overall water splitting under simulated solar light. Appl. Catal. B Environ. 2022, 304, 121000–121009. [Google Scholar] [CrossRef]
- Zhou, P.; Li, N.; Chao, Y.; Zhang, W.; Lv, F.; Wang, K.; Yang, W.; Gao, P.; Guo, S. Thermolysis of Noble Metal Nanoparticles into Electron-Rich Phosphorus-Coordinated Noble Metal Single Atoms at Low Temperature. Angew. Chem. Int. Ed. 2019, 58, 14184–14188. [Google Scholar] [CrossRef] [PubMed]
- Nørskov, J.K.; Bligaard, T.; Rossmeisl, J.; Christensen, C.H. Towards the computational design of solid catalysts. Nat. Chem. 2009, 1, 37–46. [Google Scholar] [CrossRef]
- Stamenkovic, V.; Simon Mun, B.; J Mayrhofer, K.J.; Ross, P.N.; Markovic, N.M.; Rossmeisl, J.; Greeley, J.; Nørskov, J.K.; Rossmeisl, J.; Greeley, J.; et al. Changing the Activity of Electrocatalysts for Oxygen Reduction by Tuning the Surface Electronic Structure. Angew. Chem. Int. Ed. 2006, 45, 2897–2901. [Google Scholar] [CrossRef] [PubMed]
- Lang, P.; Yuan, N.; Jiang, Q.; Zhang, Y.; Tang, J. Recent Advances and Prospects of Metal-Based Catalysts for Oxygen Reduction Reaction. Energy Technol. 2020, 8, 1900984. [Google Scholar] [CrossRef]
- Hwang, J.; Noh, S.H.; Han, B. Design of active bifunctional electrocatalysts using single atom doped transition metal dichalcogenides. Appl. Surf. Sci. 2019, 471, 545–552. [Google Scholar] [CrossRef]
- Jiao, Y.; Zheng, Y.; Jaroniec, M.; Qiao, S.Z. Design of electrocatalysts for oxygen- and hydrogen-involving energy conversion reactions. Chem. Soc. Rev. 2015, 44, 2060–2086. [Google Scholar] [CrossRef] [PubMed]
- Von Weber, A.; Baxter, E.T.; White, H.S.; Anderson, S.L. Cluster size controls branching between water and hydrogen peroxide production in electrochemical oxygen reduction at Ptn/ITO. J. Phys. Chem. C 2015, 119, 11160–11170. [Google Scholar] [CrossRef]
- Chen, R.; Li, H.; Chu, D.; Wang, G. Unraveling oxygen reduction reaction mechanisms on carbon-supported fe-phthalocyanine and co-phthalocyanine catalysts in alkaline solutions. J. Phys. Chem. C 2009, 113, 20689–20697. [Google Scholar] [CrossRef]
- Peng, Y.; Lu, B.; Chen, S. Carbon-Supported Single Atom Catalysts for Electrochemical Energy Conversion and Storage. Adv. Mater. 2018, 30, 1801995. [Google Scholar] [CrossRef]
- Yang, X.F.; Wang, A.; Qiao, B.; Li, J.; Liu, J.; Zhang, T. Single-atom catalysts: A new frontier in heterogeneous catalysis. Acc. Chem. Res. 2013, 46, 1740–1748. [Google Scholar] [CrossRef]
- Chen, Y.; Huang, Z.; Ma, Z.; Chen, J.; Tang, X. Fabrication, characterization, and stability of supported single-atom catalysts. Catal. Sci. Technol. 2017, 7, 4250–4258. [Google Scholar] [CrossRef]
- Wang, Q.; Huang, X.; Zhao, Z.L.; Wang, M.; Xiang, B.; Li, J.; Feng, Z.; Xu, H.; Gu, M. Ultrahigh-Loading of Ir Single Atoms on NiO Matrix to Dramatically Enhance Oxygen Evolution Reaction. J. Am. Chem. Soc. 2020, 142, 7425–7433. [Google Scholar] [CrossRef]
- Xiang, W.; Zhao, Y.; Jiang, Z.; Li, X.; Zhang, H.; Sun, Y.; Ning, Z.; Du, F.; Gao, P.; Qian, J.; et al. Palladium single atoms supported by interwoven carbon nanotube and manganese oxide nanowire networks for enhanced electrocatalysis. J. Mater. Chem. A 2018, 6, 23366–23377. [Google Scholar] [CrossRef]
- Li, J.; Cheng, Y.; Zhang, J.; Fu, J.; Yan, W.; Xu, Q. Confining Pd Nanoparticles and Atomically Dispersed Pd into Defective MoO3 Nanosheet for Enhancing Electro- A nd Photocatalytic Hydrogen Evolution Performances. ACS Appl. Mater. Interfaces 2019, 11, 27798–27804. [Google Scholar] [CrossRef]
- Liu, Q.; Peng, Y.; Li, Q.; He, T.; Morris, D.; Nichols, F.; Mercado, R.; Zhang, P.; Chen, S. Atomic Dispersion and Surface Enrichment of Palladium in Nitrogen-Doped Porous Carbon Cages Lead to High-Performance Electrocatalytic Reduction of Oxygen. ACS Appl. Mater. Interfaces 2020, 12, 17641–17650. [Google Scholar] [CrossRef] [PubMed]
- Stephens, J.A.; Hwang, G.S. Atomic arrangements in AuPt/Pt(100) and AuPd/Pd(100) surface alloys: A Monte Carlo study using first principles-based cluster expansions. J. Phys. Chem. C 2011, 115, 21205–21210. [Google Scholar] [CrossRef]
- Jirkovský, J.S.; Panas, I.; Ahlberg, E.; Halasa, M.; Romani, S.; Schiffrin, D.J. Single atom hot-spots at Au-Pd nanoalloys for electrocatalytic H2O2 production. J. Am. Chem. Soc. 2011, 133, 19432–19441. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, H.; Liu, S.; Norouzi Banis, M.; Song, Z.; Li, J.; Yang, L.; Markiewicz, M.; Zhao, Y.; Li, R.; et al. Pt/Pd Single-Atom Alloys as Highly Active Electrochemical Catalysts and the Origin of Enhanced Activity. ACS Catal. 2019, 9, 9350–9358. [Google Scholar] [CrossRef]
- Horiuti, J.; Polanyi, M. Grundlinien einer Theorie der Protonübertragung. Acta Physicochim. 1935, 4, 505–532. [Google Scholar]
- Ge, Z.; Fu, B.; Zhao, J.; Li, X.; Ma, B.; Chen, Y. A review of the electrocatalysts on hydrogen evolution reaction with an emphasis on Fe, Co and Ni-based phosphides. J. Mater. Sci. 2020, 55, 14081–14104. [Google Scholar] [CrossRef]
- Tavares, M.C.; Machado, S.A.S.; Mazo, L.H. Study of hydrogen evolution reaction in acid medium on Pt microelectrodes. Electrochim. Acta 2001, 46, 4359–4369. [Google Scholar] [CrossRef]
- Zheng, Y.; Jiao, Y.; Chen, J.; Liu, J.; Liang, J.; Du, A.; Zhang, W.; Zhu, Z.; Smith, S.C.; Jaroniec, M.; et al. Nanoporous graphitic-C3N4@carbon metal-free electrocatalysts for highly efficient oxygen reduction. J. Am. Chem. Soc. 2011, 133, 20116–20119. [Google Scholar] [CrossRef]
- Luo, Z.; Ouyang, Y.; Zhang, H.; Xiao, M.; Ge, J.; Jiang, Z.; Wang, J.; Tang, D.; Cao, X.; Liu, C.; et al. Chemically activating MoS2 via spontaneous atomic palladium interfacial doping towards efficient hydrogen evolution. Nat. Commun. 2018, 9, 2120. [Google Scholar] [CrossRef]
- Gao, X.; Yu, G.; Zheng, L.; Zhang, C.; Li, H.; Wang, T.; An, P.; Liu, M.; Qiu, X.; Chen, W. Strong Electron Coupling from the Sub-Nanometer Pd Clusters Confined in Porous Ceria Nanorods for Highly Efficient Electrochemical Hydrogen Evolution Reaction. ACS Appl. Energy Mater. 2019, 2, 966–973. [Google Scholar] [CrossRef]
- Lau, T.H.M.; Wu, S.; Kato, R.; Wu, T.S.; Kulhavý, J.; Mo, J.; Zheng, J.; Foord, J.S.; Soo, Y.L.; Suenaga, K.; et al. Engineering Monolayer 1T-MoS2 into a Bifunctional Electrocatalyst via Sonochemical Doping of Isolated Transition Metal Atoms. ACS Catal. 2019, 9, 7527–7534. [Google Scholar] [CrossRef]
- Zhang, Z.; Ma, C.; Tu, Y.; Si, R.; Wei, J.; Zhang, S.; Wang, Z.; Li, J.F.; Wang, Y.; Deng, D. Multiscale carbon foam confining single iron atoms for efficient electrocatalytic CO2 reduction to CO. Nano Res. 2019, 12, 2313–2317. [Google Scholar] [CrossRef]
- Fei, H.; Dong, J.; Arellano-Jiménez, M.J.; Ye, G.; Kim, N.D.; Samuel, E.; Peng, Z.; Zhu, Z.; Qin, F.; Bao, J.; et al. Atomic cobalt on nitrogen-doped graphene for hydrogen generation. Nat. Commun. 2015, 6, 8668. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Han, N.; Ning, S.; Chen, T.; Zhu, C.; Pan, C.; Wu, H.; Pennycook, S.J.; Guan, C. Single-Atom Tungsten-Doped CoP Nanoarrays as a High-Efficiency pH-Universal Catalyst for Hydrogen Evolution Reaction. ACS Sustain. Chem. Eng. 2020, 8, 14825–14832. [Google Scholar] [CrossRef]
- Liu, S.; Chen, C.; Zhang, Y.; Zheng, Q.; Zhang, S.; Mu, X.; Chen, C.; Ma, J.; Mu, S. Vacancy-coordinated hydrogen evolution reaction on MoO3-: X anchored atomically dispersed MoRu pairs. J. Mater. Chem. A 2019, 7, 14466–14472. [Google Scholar] [CrossRef]
- Liu, P.; Zhao, Y.; Qin, R.; Mo, S.; Chen, G.; Gu, L.; Chevrier, D.M.; Zhang, P.; Guo, Q.; Zang, D.; et al. Catalysis: Photochemical route for synthesizing atomically dispersed palladium catalysts. Science 2016, 352, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Ou, H.; Wang, D.; Li, Y. How to select effective electrocatalysts: Nano or single atom? Nano Sel. 2021, 2, 492–511. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).