
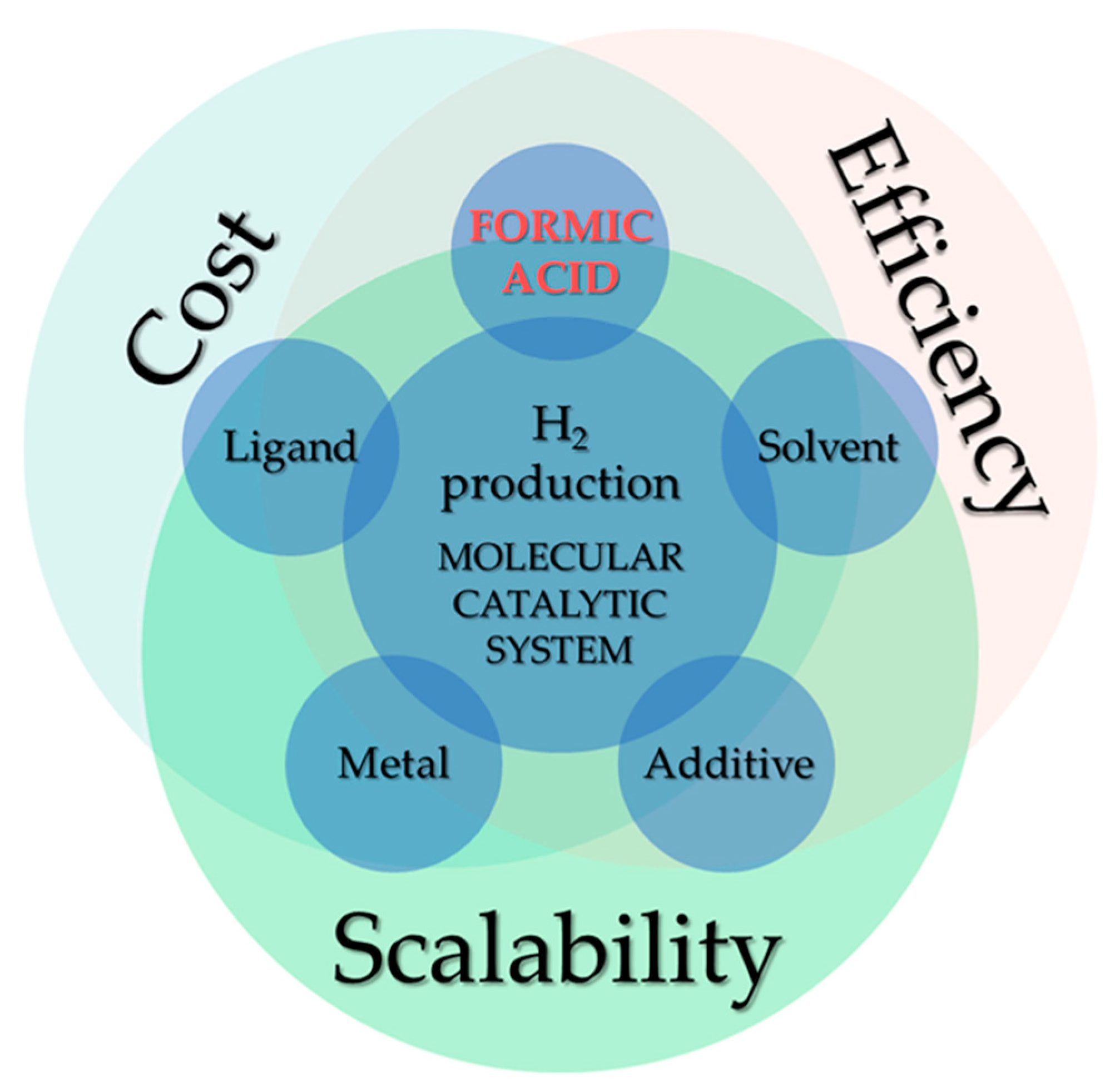
Cost Efficiency Analysis of H2 Production from Formic Acid by Molecular Catalysts
 , , , and
, , , and 
Abstract
:
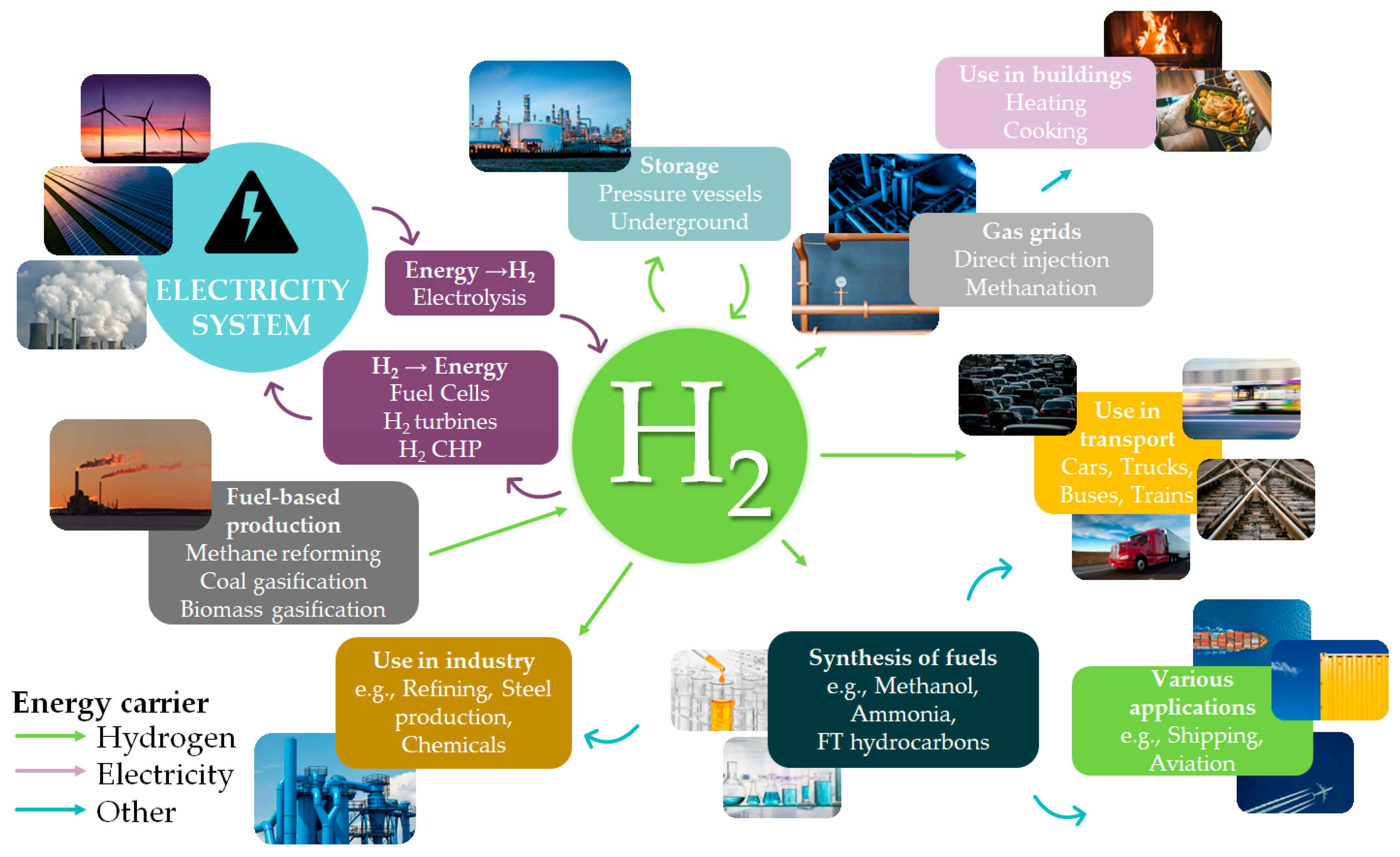
1. Introduction
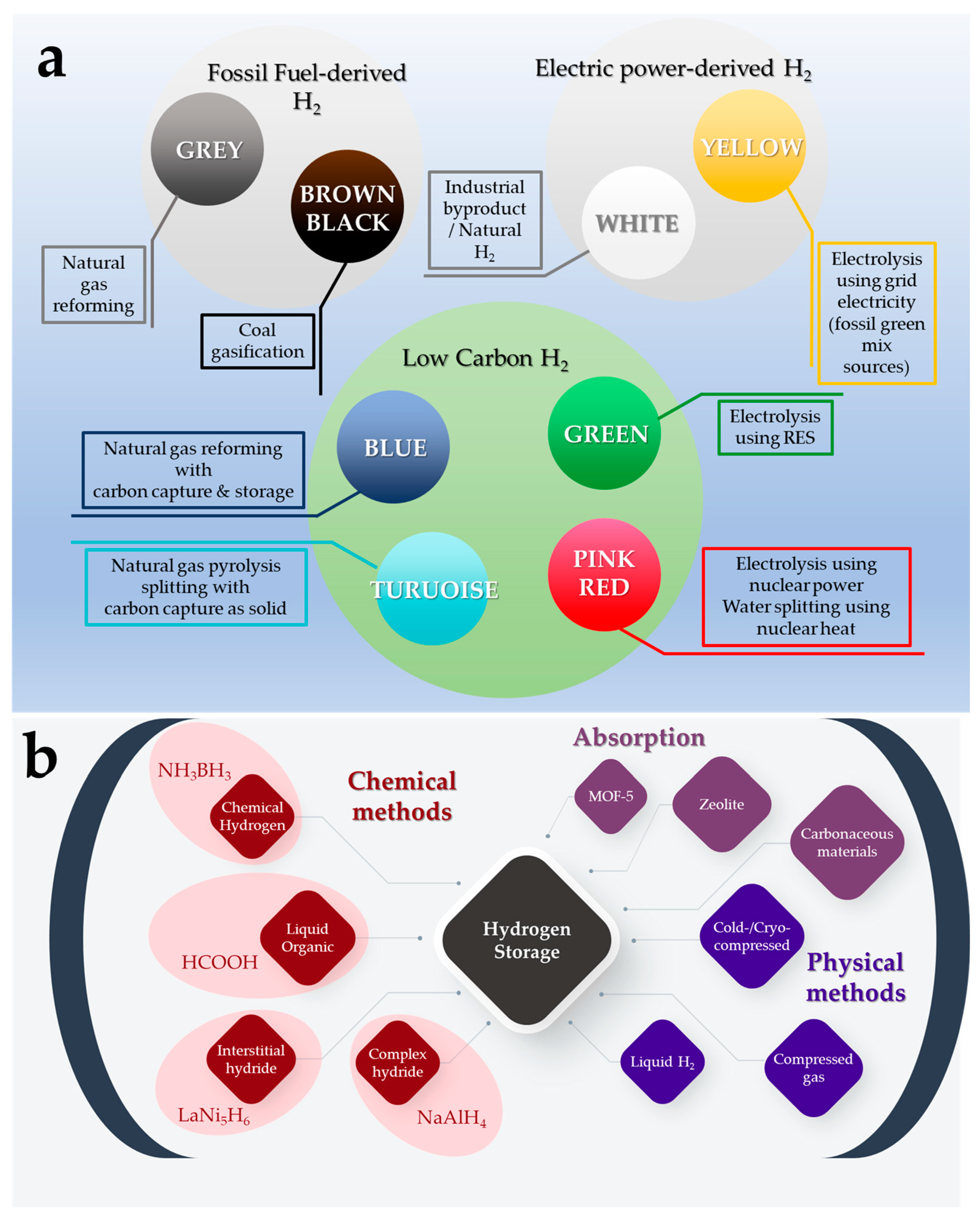
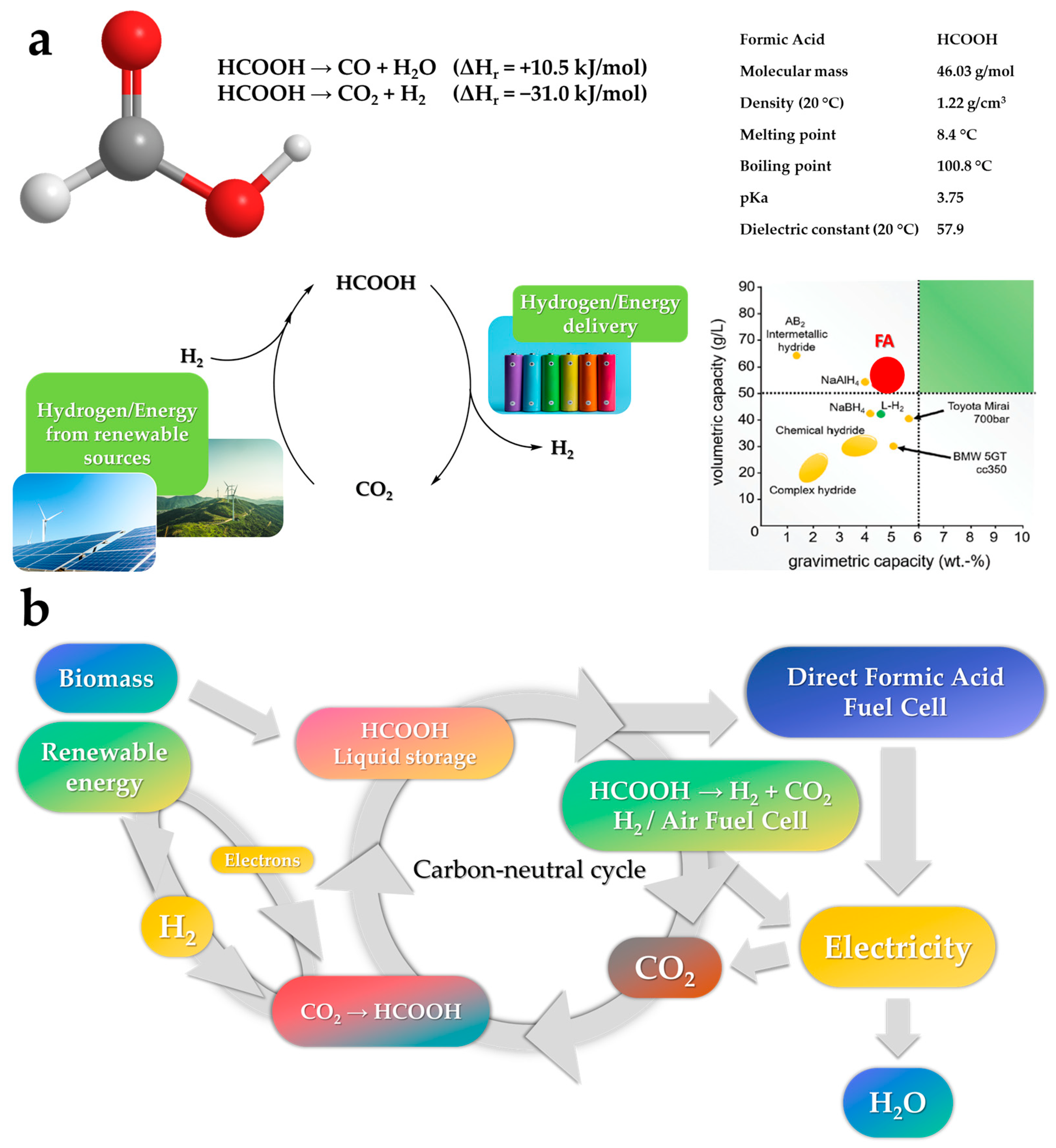
2. Formic Acid: An Efficient Liquid Carrier for H2 Storage
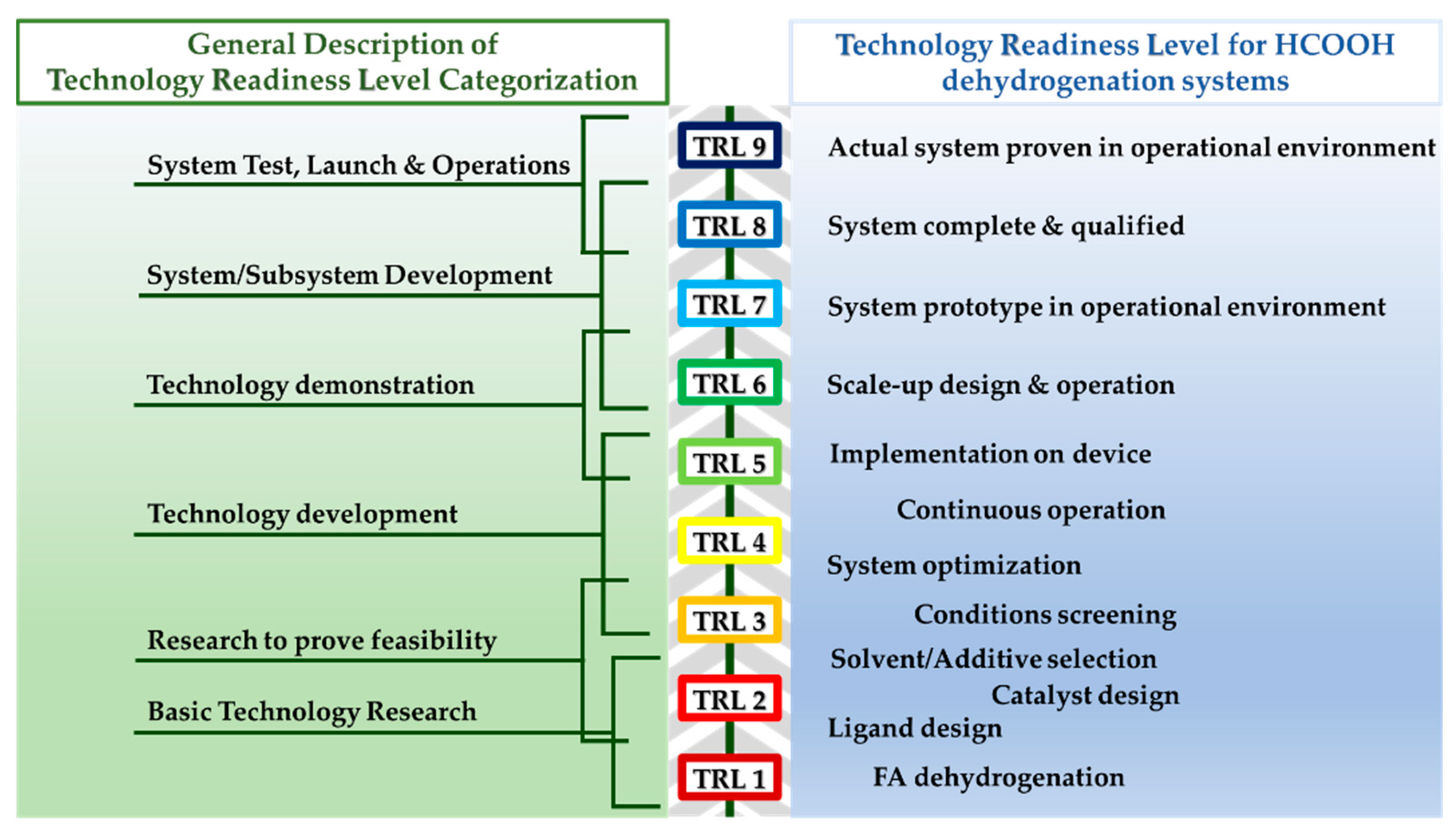
3. Description of Our Cost Analysis Methodology: Lab-Scale and Industrial-Scale Price
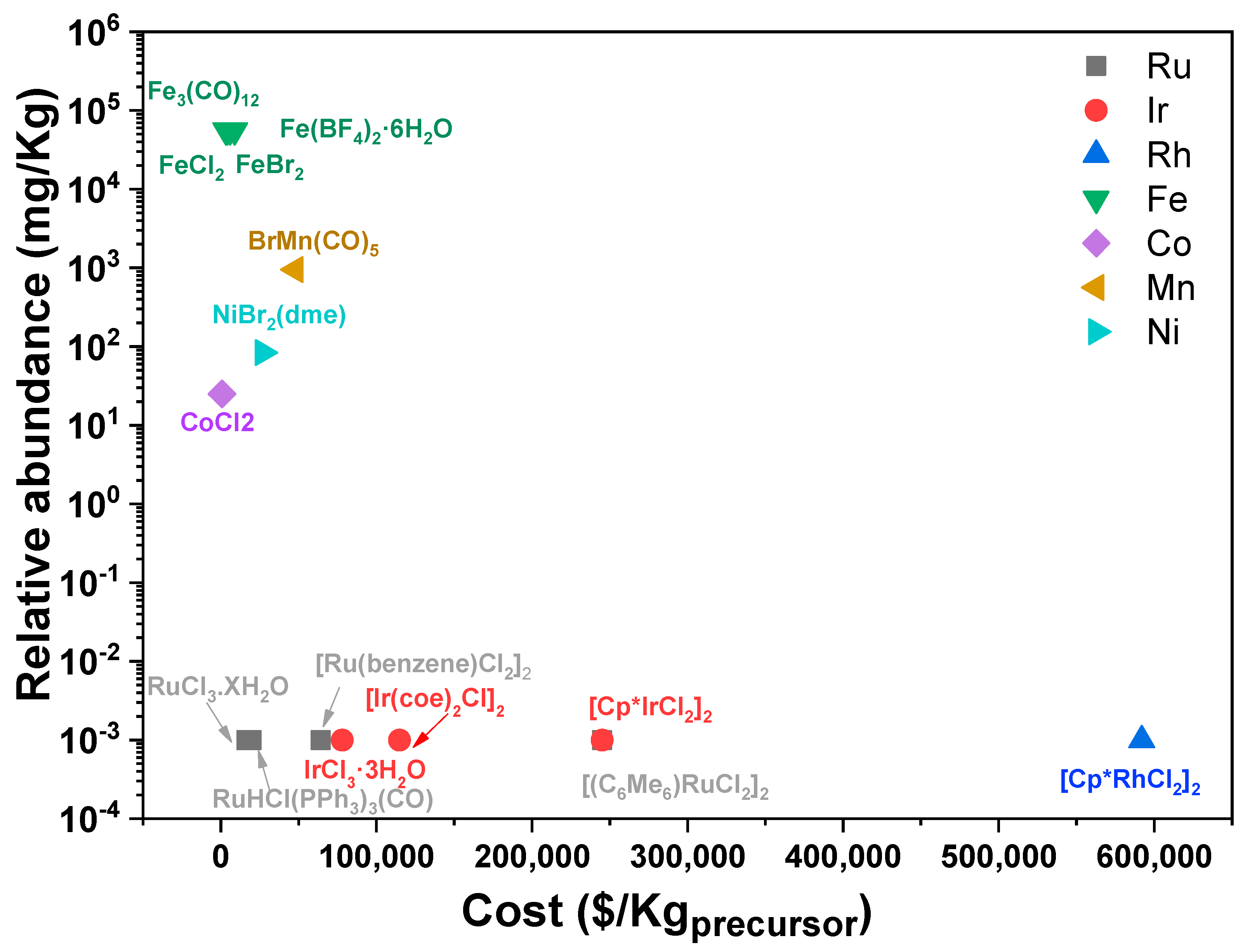
3.1. Catalytic Metals in FA Dehydrogenation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Metal Precursor | Conditions | TONs | Cost ($/kgmetal precursor) | kgH2/kgmetal precursor | 1 $/kgH2 (Metal Precursor) | Ref. |
|---|---|---|---|---|---|---|---|
| #11 | RuHCl(PPh3)3(CO) | 1.42 μmol of catalyst, 35 mL DMF and NH3 (33.5 mmol base), T = 90 °C | 706,500 | 19,920 | 1485 | 1.34 | [51] |
| #12 | [(C6Me6)RuCl2]2 | 1 μmol catalyst (100 mL, 1 mmol) 2 M aqueous FA solution (10 mL), T = 60 °C | 264 | 245,034 | 0.79 | 31,004 | [55] |
| #9 | [RuCl2(benzene)]2 | 9.55 μmol of catalyst (19.1 μmol [Ru]), 115 μmol dppe (Ru/dppe = 1:6), 20 mL DMOA T = 90 °C | 1,000,000 | 64,200 | 4001 | 1.60 | [77] |
| #13 | RuCl3.XH2O | 5.95 μmol of Ru, Ru/PPh3 = 1/3, 5.0 mL,5HCO2H/2NEt3, DMF = 1 mL, T = 40 °C | 700 (after 3 h) | 16,500 | 6.75 | 244 | [77] |
| #14 | [Ir(coe)2Cl]2 | 1 μmol of catalyst, tri-ethylammonium formate = 5 mmol, tBuOH = 1 mL, THF = 0.1 mL, T = 80 °C | 2000 | 114,828 | 4.5 | 2570 | [78] |
| #5 | [Cp*IrCl2]2 | 1 μmol of catalyst, FA = 5 M, 10.0 mL, T = 90 °C | 2,400,000 | 245,108 | 34,761 | 4.07 | [44] |
| #15 | IrCl3·3H2O | 9 mM catalyst, FA = 1250 mM in acetic acid, | >11,000 | 78,000 | 62.43 | 125 | [79] |
| #16 | [Cp*RhCl2]2 | 1 μmol of catalyst (100 mL, 1 mmol) 2 M aqueous FA solution (10 mL), T = 60 °C | 3366 | 592,000 | 11 | 5432 | [55] |
| #3 | FeBr2 | 10 μmol of catalyst, 2 mL 1,4 dioxane, 3 g FA, 10.86 mmol NEt3, T = 40 °C | >100,000 | 5900 | 928 | 0.64 | [68] |
| #4 | FeCl2 | FA (110 μL, 2.91 mmol), LiBF4 = 0.291 mmol, 10 mol%, 5 mL dioxane, T = 80 °C. | 983,642 | 3279 | 4089 | 0.08 | [71] |
| #17 | [Fe(BF4)2]∙6H2O | Fe(BF4)2·6H2O = 74 μmol, PP3 = 296 μmol, 2 mL FA, 50 mL PC, T = 80° C | 92,417 | 4176 | 68.94 | 0.76 | [70] |
| #18 | Fe3(CO)12 | 20 μmol Fe3(CO)12/PPh3/tpy (Fe/PPh3/tpy 1:1:1), 5FA·2NEt3 = 5 mL, 1 mL DMF, T = 60 °C, 300 W xenon lamp (385 nm cutoff) | 126 | 8903 | 0.50 | 1778 | [69] |
| #19 | CoCl2 | 10 μmol of catalyst, FA = 50 mmol, HCOOK = 50 mmol, H2O = 18.1 mL, NaBEt3H = 1.5 mL, T = 80 °C | 2260 | 792 | 35 | 2.27 | [76] |
| #20 | BrMn(CO)5 | 2 mM (10 μmol) of catalyst, 870 mM FA, 5 mL 1,4 dioxane, T = 65 °C | 190 | 45,000 | 1.38 | 3254 | [72] |
| #21 | NiBr2(dme) | 5.3 μmol [Ni], FA/nOctNMe2 = 11:10, 5 mL, PC = 5.0 mL, T = 80 °C | 626 | 27,000 | 4.06 | 665 | [74] |
3.2. Ligands of Catalysts in FA Dehydrogenation
| Entry | Ligand | Conditions | TONs | Cost ($/kgligand) | kgH2/ kgligand | 1 $/kgH2 (Ligand) | Ref. |
|---|---|---|---|---|---|---|---|
| #1 | tBu-PNP (2,6-Bis(di-tert-butylphosphinomethyl)pyridine | 1.0 μmol of catalyst FA = 0.3 mL/h, NEt3 = 1.50 mL, 0.7 mL DMSO, T = 90 °C | 1,100,000 | 158,400 | 5565 | 2.85 | [101] |
| #2 | 4,5-bis(bromomethyl)acridine | 40 µmol (23 mg) catalyst, FA = 5 mL, (0.13 mol), T = 95 °C | 1,701,150 | 599,900 | 9326 | 6.43 | [102] |
| #22 | 1,1,1-tris-(diphenylphosphinomethyl)ethane (triphos) | 12.9 μmol catalyst, 12.9 mmol FA FA:OctNMe2 = 11:10 = (2.73 mL), T = 80 °C | 10,000 (after 6 h) | 39,200 | 32 | 122 | [103] |
| #13 | PPh3 | 5.95 μmol of Ru, Ru/PPh3 = 1/3, 5 HCO2H/2NEt3 = 5 mL, DMF = 1 mL, T = 40 °C | 700 | 107 | 1.78 | 6.00 | [77] |
| #9 | dppe | 9.55 μmol [RuCl2(benzene)]2 (19.1 μmol [Ru]), 115 μmol dppe (Ru/dppe = 1:6), FA added continuously, 20 mL DMOA, T = 60 °C | 1,000,000 | 2760 | 834 | 0.33 | [49] |
| #23 | TPPTS | [Ru(H2O)6](tos)2 = 1.5 mmol, TPPTS = 3 mmol, FA added continuously, 12 mL FA/HCOONa (4 M, 9:1), T = 100 °C | 40,000 | 41,800 | 5281 | 59.36 | [50] |
| #7 | 2,3-Piperazinedione dioxime | Aqueous catalyst stock solution = 0.2 mL, 1.0 µmol final concentration, FA = 10.0 M, 220 mL, T = 70 °C | 5,200,000 | 72,115 | 12,610 | 0.76 | [56] |
| #6 | 4,7-Dihydroxy-1,10-phenanthroline | 1 μmol of catalyst, FA = 40 vol%,10 mol L−1, 22 MPa, T = 80 °C | 5,000,000 | 150,000 | 47,154 | 0.32 | [65] |
| #24 | 4′,4-Dihydroxy-2,2′-bipyridine | 1 μmol catalyst (100 mL, 1 mmol), 2 M aqueous FA solution = 10 mL, T = 60 °C | 18,667 | 99,000 | 199 | 49.9 | [55] |
| #25 | mtppms | 9.8 μmol catalyst, FA = 133 mmol/catalytic cycle HCOONa = 50 mmol, Vtotal = 20.0 mL, T = 100 °C | >67,650 | 41,800 | 371 | 11.28 | [104] |
| #18 | PPh3 | 20 μmol Fe3(CO)12/PPh3/tpy (Fe/PPh3/tpy = 1:1:1), 5FA·2NEt3 = 5 mL, 1 mL DMF, 300 W xenon (385 nm cutoff), T = 60 °C | 126 | 107 | 0.96 | 11.13 | [69] |
| #17 | PP3 | Fe(BF4)2·6H2O = 74 μmol, PP3 = 296 μmol, 2 mL FA, 50 mL PC, T = 80 °C | 92,417 | 131,413 | 68.94 | 191 | [70] |
| #4 | Bis[(2-diisopropylphosphino]ethyl)amine | 0.001 mol% [Fe], FA = 110 μL, (2.91 mmol), LiBF4 = 0.291 mmol, 10 mol%, 5 mL 1,4 dioxane, T = 80 °C | 983,642 | 33,000 | 4089 | 0.81 | [71] |
| #26 | 2-(4,5-dihydro-1H-imidazol-2-yl)pyridine | 0.005 mmol catalyst, FA = 37 mmol, HCOOK = 40 mmol, H2O = 9 mL, triglyme = 4 mL, T = 92.5 °C | 564 (after 3 h) | 112,800 | 7.67 | 7.82 | [73] |
| #26a | 2,2-Bipyridine | 0.005 mmol catalyst, FA = 37 mmol, HCOOK = 40 mmol, H2O = 9 mL, triglyme = 4 mL, T = 92.5 °C | 50 (after 3 h) | 600 | 0.64 | 443 | [73] |
| #26b | 1H,1′H-2,2′-Biimidazole | 0.005 mmol of catalyst, FA = 37 mmol, HCOOK = 40 mmol, H2O = 9 mL, triglyme = 4 mL, T = 92.5 °C | 220 (after 3 h) | 2840 | 3.28 | 3.26 | [73] |
3.3. Additives/Cocatalysts in FA Dehydrogenation
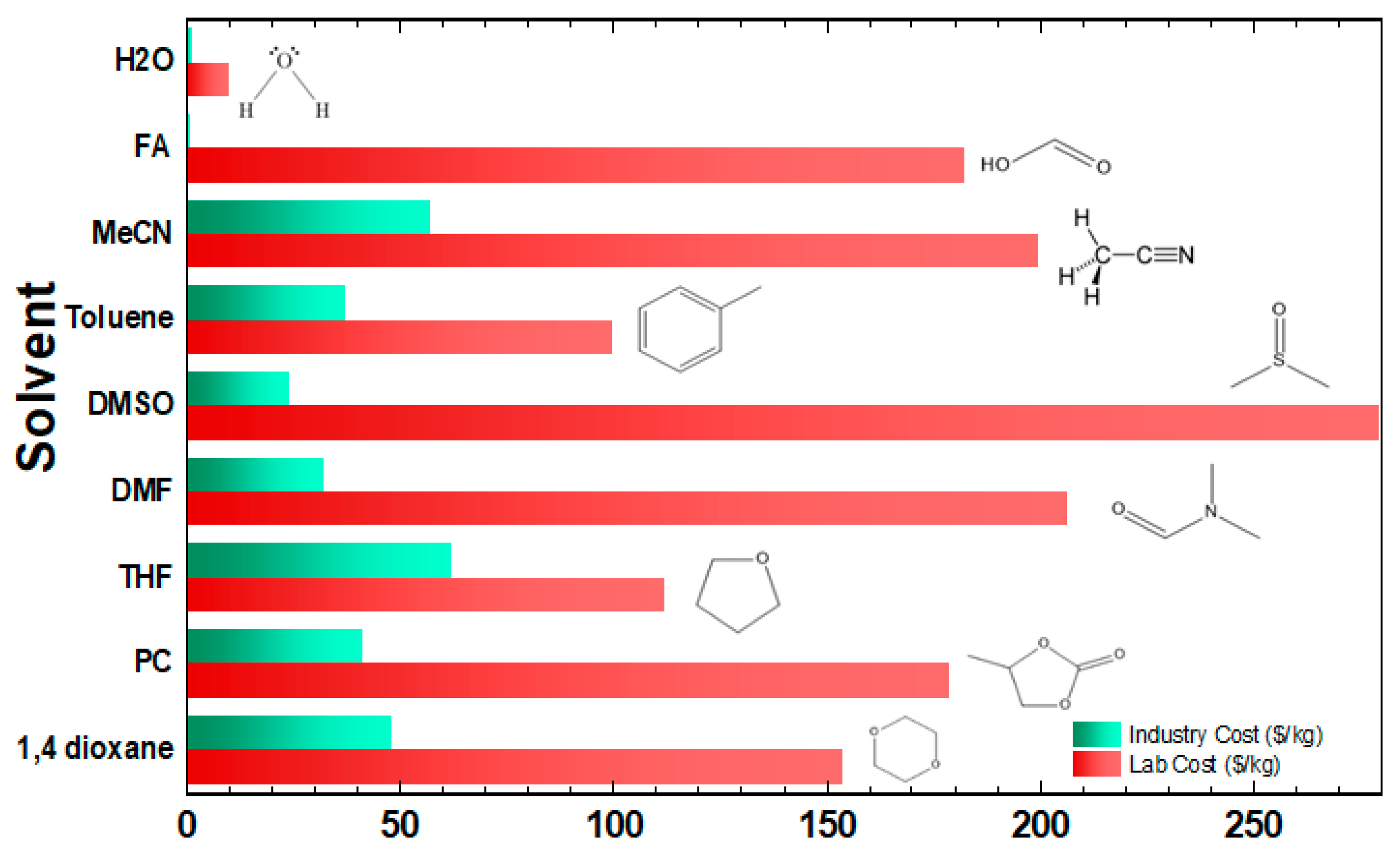
3.4. Solvent Media in FA Dehydrogenation
3.5. Energy Cost in FA Dehydrogenation
4. Selected Catalytic Systems for Overall-Cost Evaluation
5. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- World Energy Balances. Available online: https://www.iea.org/data-and-statistics/data-product/world-energy-balances (accessed on 1 January 2023).
- The Evidence Is Clear: The Time for Action Is Now. We Can Halve Emissions by 2030. Available online: https://www.ipcc.ch/2022/04/04/ipcc-ar6-wgiii-pressrelease/ (accessed on 30 December 2022).
- Quarton, C.J.; Tlili, O.; Welder, L.; Mansilla, C.; Blanco, H.; Heinrichs, H.; Leaver, J.; Samsatli, N.J.; Lucchese, P.; Robinius, M.; et al. The curious case of the conflicting roles of hydrogen in global energy scenarios. Sustain. Energy Fuels 2020, 4, 80–95. [Google Scholar] [CrossRef]
- Pivovar, B.; Rustagi, N.; Satyapal, S. Hydrogen at Scale (H2 @Scale): Key to a Clean, Economic, and Sustainable Energy System. Electrochem. Soc. Interface 2018, 27, 47–52. [Google Scholar] [CrossRef]
- Hanley, E.S.; Deane, J.P.; Gallachóir, B.P.Ó. The role of hydrogen in low carbon energy futures—A review of existing perspectives. Renew. Sustain. Energy Rev. 2018, 82, 3027–3045. [Google Scholar] [CrossRef]
- Yue, M.; Lambert, H.; Pahon, E.; Roche, R.; Jemei, S.; Hissel, D. Hydrogen energy systems: A critical review of technologies, applications, trends and challenges. Renew. Sustain. Energy Rev. 2021, 146, 111180. [Google Scholar] [CrossRef]
- Abe, J.O.; Popoola, A.P.I.; Ajenifuja, E.; Popoola, O.M. Hydrogen energy, economy and storage: Review and recommendation. Int. J. Hydrogen Energy 2019, 44, 15072–15086. [Google Scholar] [CrossRef]
- Anandarajah, G.; McDowall, W.; Ekins, P. Decarbonising road transport with hydrogen and electricity: Long term global technology learning scenarios. Int. J. Hydrogen Energy 2013, 38, 3419–3432. [Google Scholar] [CrossRef]
- Ruffini, E.; Wei, M. Future costs of fuel cell electric vehicles in California using a learning rate approach. Energy 2018, 150, 329–341. [Google Scholar] [CrossRef]
- Hydrogen Refuelling Stations Worldwide. Available online: https://www.h2stations.org/ (accessed on 21 December 2022).
- Hua, T.; Ahluwalia, R.; Eudy, L.; Singer, G.; Jermer, B.; Asselin-Miller, N.; Wessel, S.; Patterson, T.; Marcinkoski, J. Status of hydrogen fuel cell electric buses worldwide. J. Power Source 2014, 269, 975–993. [Google Scholar] [CrossRef]
- Alstom Coradia iLint Distance Run. Available online: https://www.alstom.com/alstom-coradia-ilint-distance-run (accessed on 15 November 2022).
- Zhang, F.; Zhao, P.; Niu, M.; Maddy, J. The survey of key technologies in hydrogen energy storage. Int. J. Hydrogen Energy 2016, 41, 14535–14552. [Google Scholar] [CrossRef]
- Schmidt, O.; Gambhir, A.; Staffell, I.; Hawkes, A.; Nelson, J.; Few, S. Future cost and performance of water electrolysis: An expert elicitation study. Int. J. Hydrogen Energy 2017, 42, 30470–30492. [Google Scholar] [CrossRef]
- Quarton, C.J.; Samsatli, S. The value of hydrogen and carbon capture, storage and utilisation in decarbonising energy: Insights from integrated value chain optimisation. Appl. Energy 2020, 257, 113936. [Google Scholar] [CrossRef]
- Lang, C.; Jia, Y.; Yao, X. Recent advances in liquid-phase chemical hydrogen storage. Energy Storage Mater. 2020, 26, 290–312. [Google Scholar] [CrossRef]
- Hwang, H.T.; Varma, A. Hydrogen storage for fuel cell vehicles. Curr. Opin. Chem. Eng. 2014, 5, 42–48. [Google Scholar] [CrossRef]
- Alazemi, J.; Andrews, J. Automotive hydrogen fuelling stations: An international review. Renew. Sustain. Energy Rev. 2015, 48, 483–499. [Google Scholar] [CrossRef]
- Suh, M.P.; Park, H.J.; Prasad, T.K.; Lim, D.-W. Hydrogen Storage in Metal–Organic Frameworks. Chem. Rev. 2012, 112, 782–835. [Google Scholar] [CrossRef]
- Sordakis, K.; Tang, C.; Vogt, L.K.; Junge, H.; Dyson, P.J.; Beller, M.; Laurenczy, G. Homogeneous Catalysis for Sustainable Hydrogen Storage in Formic Acid and Alcohols. Chem. Rev. 2018, 118, 372–433. [Google Scholar] [CrossRef]
- Viswanathan, B. Hydrogen Storage. In Energy Sources; Elsevier: Amsterdam, The Netherlands, 2017; pp. 185–212. [Google Scholar] [CrossRef]
- Xu, R.; Lu, W.; Toan, S.; Zhou, Z.; Russell, C.K.; Sun, Z.; Sun, Z. Thermocatalytic formic acid dehydrogenation: Recent advances and emerging trends. J. Mater. Chem. A 2021, 9, 24241–24260. [Google Scholar] [CrossRef]
- Dutta, I.; Chatterjee, S.; Cheng, H.; Parsapur, R.K.; Liu, Z.; Li, Z.; Ye, E.; Kawanami, H.; Low, J.S.C.; Lai, Z.; et al. Formic Acid to Power towards Low-Carbon Economy. Adv. Energy Mater. 2022, 12, 2103799. [Google Scholar] [CrossRef]
- Hydrogen and Fuel Cell Technologies Office DOE Technical Targets for Onboard Hydrogen Storage for Light-Duty Vehicles. Available online: https://www.energy.gov/eere/fuelcells/doe-technical-targets-onboard-hydrogen-storage-light-duty-vehicles (accessed on 25 November 2022).
- Álvarez, A.; Bansode, A.; Urakawa, A.; Bavykina, A.V.; Wezendonk, T.A.; Makkee, M.; Gascon, J.; Kapteijn, F. Challenges in the Greener Production of Formates/Formic Acid, Methanol, and DME by Heterogeneously Catalyzed CO2 Hydrogenation Processes. Chem. Rev. 2017, 117, 9804–9838. [Google Scholar] [CrossRef]
- Staffell, I.; Scamman, D.; Velazquez Abad, A.; Balcombe, P.; Dodds, P.E.; Ekins, P.; Shah, N.; Ward, K.R. The role of hydrogen and fuel cells in the global energy system. Energy Environ. Sci. 2019, 12, 463–491. [Google Scholar] [CrossRef]
- Toyota MIRAI, MIRAI Means ‘The Future’. Available online: https://h2.live/en/fuelcell-cars/toyota-mirai/ (accessed on 23 October 2022).
- Formic Acid Market—Growth, Trends, COVID-19 Impact, and Forecasts (2023–2028). Available online: https://www.mordorintelligence.com/industry-reports/formic-acid-market (accessed on 28 November 2022).
- Formic Acid Market by Product Types (Grades of 85%, 94%, 99%, and Others), Applications (Agriculture, Leather & Textile, Rubber, Chemical & Pharmaceuticals, and Others), and Regions (Asia Pacific, North America, Latin America, Europe, and Middle East & Africa)—Global Industry Analysis, Growth, Share, Size, Trends, and Forecast, 2021–2028. Available online: https://dataintelo.com/report/formic-acid-market/ (accessed on 29 November 2022).
- van Putten, R.; Wissink, T.; Swinkels, T.; Pidko, E.A. Fuelling the hydrogen economy: Scale-up of an integrated formic acid-to-power system. Int. J. Hydrogen Energy 2019, 44, 28533–28541. [Google Scholar] [CrossRef]
- Singh, A.K.; Singh, S.; Kumar, A. Hydrogen energy future with formic acid: A renewable chemical hydrogen storage system. Catal. Sci. Technol. 2016, 6, 12–40. [Google Scholar] [CrossRef]
- Czaun, M.; Goeppert, A.; Kothandaraman, J.; May, R.B.; Haiges, R.; Prakash, G.K.S.; Olah, G.A. Formic Acid as a Hydrogen Storage Medium: Ruthenium-Catalyzed Generation of Hydrogen from Formic Acid in Emulsions. ACS Catal. 2013, 4, 311–320. [Google Scholar] [CrossRef]
- Yu, X.; Pickup, P.G. Recent advances in direct formic acid fuel cells (DFAFC). J. Power Source 2008, 182, 124–132. [Google Scholar] [CrossRef]
- Rejal, S.Z.; Masdar, M.S.; Kamarudin, S.K. A parametric study of the direct formic acid fuel cell (DFAFC) performance and fuel crossover. Int. J. Hydrogen Energy 2014, 39, 10267–10274. [Google Scholar] [CrossRef]
- Han, D.; Tsipoaka, M.; Shanmugam, S. A modified cathode catalyst layer with optimum electrode exposure for high current density and durable proton exchange membrane fuel cell operation. J. Power Sources 2021, 496, 229816. [Google Scholar] [CrossRef]
- Chang, J.; Feng, L.; Liu, C.; Xing, W.; Hu, X. An Effective Pd-Ni2P/C Anode Catalyst for Direct Formic Acid Fuel Cells. Angew. Chem. Int. Ed. 2014, 53, 122–126. [Google Scholar] [CrossRef]
- Eppinger, J.; Huang, K.-W. Formic Acid as a Hydrogen Energy Carrier. ACS Energy Lett. 2017, 2, 188–195. [Google Scholar] [CrossRef]
- Guan, C.; Pan, Y.; Zhang, T.; Ajitha, M.J.; Huang, K.-W. An Update on Formic Acid Dehydrogenation by Homogeneous Catalysis. Chem. Asian J. 2020, 15, 937–946. [Google Scholar] [CrossRef]
- U.S. Department of Energy. Technology Readiness Assessment Guide. Available online: https://www.directives.doe.gov/directives-documents/400-series/0413.3-EGuide-04/@@images/file (accessed on 21 October 2022).
- What Are Technology Readiness Levels (TRL)? Available online: https://www.twi-global.com/technical-knowledge/faqs/technology-readiness-levels (accessed on 29 December 2022).
- Gabor, L.; Céline, F.; Paul, D. Hydrogen Production from Formic Acid. EP 1 918 247 A1. 7 May 2008. Available online: https://patentimages.storage.googleapis.com/02/4b/0e/db9b2f842c2658/EP1918247A1.pdf (accessed on 20 October 2022).
- Huang, K.-W.; Zheng, J. Electricity Generation Devices Using Formic Acid. WO 2017/103820 A1. 22 June 2017. Available online: https://repository.kaust.edu.sa/bitstream/handle/10754/625325/WO2017103820A1.pdf?sequence=1&isAllowed=y (accessed on 20 October 2022).
- The world’s First Formic Acid-Based Fuel Cell. Available online: https://actu.epfl.ch/news/the-world-s-first-formic-acid-based-fuel-cell/ (accessed on 12 November 2022).
- Wang, Z.; Lu, S.-M.; Li, J.; Wang, J.; Li, C. Unprecedentedly High Formic Acid Dehydrogenation Activity on an Iridium Complex with anN,N′-Diimine Ligand in Water. Chem. Eur. J. 2015, 21, 12592–12595. [Google Scholar] [CrossRef]
- Belkova, N.V.; Filippov, O.A.; Osipova, E.S.; Safronov, S.V.; Epstein, L.M.; Shubina, E.S. Influence of phosphine (pincer) ligands on the transition metal hydrides reactivity. Coord. Chem. Rev. 2021, 438, 213799. [Google Scholar] [CrossRef]
- Iglesias, M.; Oro, L.A. Mechanistic Considerations on Homogeneously Catalyzed Formic Acid Dehydrogenation. Eur. J. Inorg. Chem. 2018, 2018, 2125–2138. [Google Scholar] [CrossRef]
- Mellmann, D.; Sponholz, P.; Junge, H.; Beller, M. Formic acid as a hydrogen storage material—Development of homogeneous catalysts for selective hydrogen release. Chem. Soc. Rev. 2016, 45, 3954–3988. [Google Scholar] [CrossRef]
- Gao, Y.; Kuncheria, J.; Puddephatt, R.J.; Yap, G.P.A. An efficient binuclear catalyst for decomposition of formic acid. Chem. Commun. 1998, 2365–2366. [Google Scholar] [CrossRef]
- Sponholz, P.; Mellmann, D.; Junge, H.; Beller, M. Towards a Practical Setup for Hydrogen Production from Formic Acid. Chemsuschem 2013, 6, 1172–1176. [Google Scholar] [CrossRef]
- Fellay, C.; Yan, N.; Dyson, P.J.; Laurenczy, G. Selective Formic Acid Decomposition for High-Pressure Hydrogen Generation: A Mechanistic Study. Chem. Eur. J. 2009, 15, 3752–3760. [Google Scholar] [CrossRef]
- Filonenko, G.; van Putten, R.; Schulpen, E.N.; Hensen, E.J.M.; Pidko, E. Highly Efficient Reversible Hydrogenation of Carbon Dioxide to Formates Using a Ruthenium PNP-Pincer Catalyst. Chemcatchem 2014, 6, 1526–1530. [Google Scholar] [CrossRef]
- Prichatz, C.; Trincado, M.; Tan, L.; Casas, F.; Kammer, A.; Junge, H.; Beller, M.; Grützmacher, H. Highly Efficient Base-Free Dehydrogenation of Formic Acid at Low Temperature. Chemsuschem 2018, 11, 3092–3095. [Google Scholar] [CrossRef]
- Solakidou, M.; Theodorakopoulos, M.; Deligiannakis, Y.; Louloudi, M. Double-ligand Fe, Ru catalysts: A novel route for enhanced H2 production from Formic Acid. Int. J. Hydrogen Energy 2020, 45, 17367–17377. [Google Scholar] [CrossRef]
- Theodorakopoulos, M.; Solakidou, M.; Deligiannakis, Y.; Louloudi, M. A Use-Store-Reuse (USR) Concept in Catalytic HCOOH Dehydrogenation: Case-Study of a Ru-Based Catalytic System for Long-Term USR under Ambient O2. Energies 2021, 14, 481. [Google Scholar] [CrossRef]
- Himeda, Y. Highly efficient hydrogen evolution by decomposition of formic acid using an iridium catalyst with 4,4′-dihydroxy-2,2′-bipyridine. Green Chem. 2009, 11, 2018–2022. [Google Scholar] [CrossRef]
- Lu, S.-M.; Wang, Z.; Wang, J.; Li, J.; Li, C. Hydrogen generation from formic acid decomposition on a highly efficient iridium catalyst bearing a diaminoglyoxime ligand. Green Chem. 2018, 20, 1835–1840. [Google Scholar] [CrossRef]
- Wang, W.-H.; Ertem, M.Z.; Xu, S.; Onishi, N.; Manaka, Y.; Suna, Y.; Kambayashi, H.; Muckerman, J.T.; Fujita, E.; Himeda, Y. Highly Robust Hydrogen Generation by Bioinspired Ir Complexes for Dehydrogenation of Formic Acid in Water: Experimental and Theoretical Mechanistic Investigations at Different pH. ACS Catal. 2015, 5, 5496–5504. [Google Scholar] [CrossRef]
- Iguchi, M.; Himeda, Y.; Manaka, Y.; Matsuoka, K.; Kawanami, H. Simple Continuous High-Pressure Hydrogen Production and Separation System from Formic Acid under Mild Temperatures. Chemcatchem 2016, 8, 886–890. [Google Scholar] [CrossRef]
- Wang, S.; Huang, H.; Roisnel, T.; Bruneau, C.; Fischmeister, C. Base-Free Dehydrogenation of Aqueous and Neat Formic Acid with Iridium(III) Cp*(dipyridylamine) Catalysts. Chemsuschem 2019, 12, 179–184. [Google Scholar] [CrossRef]
- Mo, X.-F.; Liu, C.; Chen, Z.-W.; Ma, F.; He, P.; Yi, X.-Y. Metal–Ligand Cooperation in Cp*Ir-Pyridylpyrrole Complexes: Rational Design and Catalytic Activity in Formic Acid Dehydrogenation and CO2 Hydrogenation under Ambient Conditions. Inorg. Chem. 2021, 60, 16584–16592. [Google Scholar] [CrossRef]
- Cheng, S.; Lang, Z.; Du, J.; Du, Z.; Li, Y.; Tan, H.; Li, Y. Engineering of iridium complexes for the efficient hydrogen evolution of formic acid without additives. J. Catal. 2022, 413, 119–126. [Google Scholar] [CrossRef]
- Jongbloed, L.S.; de Bruin, B.; Reek, J.N.H.; Lutz, M.; van der Vlugt, J.I. Reversible cyclometalation at RhI as a motif for metal–ligand bifunctional bond activation and base-free formic acid dehydrogenation. Catal. Sci. Technol. 2016, 6, 1320–1327. [Google Scholar] [CrossRef]
- Fink, C.; Laurenczy, G. A Precious Catalyst: Rhodium-Catalyzed Formic Acid Dehydrogenation in Water. Eur. J. Inorg. Chem. 2019, 2019, 2381–2387. [Google Scholar] [CrossRef]
- Hermosilla, P.; Urriolabeitia, A.; Iglesias, M.; Polo, V.; Casado, M. Efficient solventless dehydrogenation of formic acid by a CNC-based rhodium catalyst. Inorg. Chem. Front. 2022, 9, 4538–4547. [Google Scholar] [CrossRef]
- Iguchi, M.; Himeda, Y.; Manaka, Y.; Kawanami, H. Development of an Iridium-Based Catalyst for High-Pressure Evolution of Hydrogen from Formic Acid. Chemsuschem 2016, 9, 2749–2753. [Google Scholar] [CrossRef]
- Zweig, J.E.; Kim, D.E.; Newhouse, T.R. Methods Utilizing First-Row Transition Metals in Natural Product Total Synthesis. Chem. Rev. 2017, 117, 11680–11752. [Google Scholar] [CrossRef]
- Tamang, S.R.; Findlater, M. Emergence and Applications of Base Metals (Fe, Co, and Ni) in Hydroboration and Hydrosilylation. Molecules 2019, 24, 3194. [Google Scholar] [CrossRef]
- Zell, T.; Butschke, B.; Ben-David, Y.; Milstein, D. Efficient Hydrogen Liberation from Formic Acid Catalyzed by a Well-Defined Iron Pincer Complex under Mild Conditions. Chem. Eur. J. 2013, 19, 8068–8072. [Google Scholar] [CrossRef]
- Boddien, A.; Loges, B.; Gärtner, F.; Torborg, C.; Fumino, K.; Junge, H.; Ludwig, R.; Beller, M. Iron-Catalyzed Hydrogen Production from Formic Acid. J. Am. Chem. Soc. 2010, 132, 8924–8934. [Google Scholar] [CrossRef]
- Boddien, A.; Mellmann, D.; Gärtner, F.; Jackstell, R.; Junge, H.; Dyson, P.J.; Laurenczy, G.; Ludwig, R.; Beller, M. Efficient Dehydrogenation of Formic Acid Using an Iron Catalyst. Science 2011, 333, 1733–1736. [Google Scholar] [CrossRef]
- Bielinski, E.A.; Lagaditis, P.O.; Zhang, Y.; Mercado, B.Q.; Würtele, C.; Bernskoetter, W.H.; Hazari, N.; Schneider, S. Lewis Acid-Assisted Formic Acid Dehydrogenation Using a Pincer-Supported Iron Catalyst. J. Am. Chem. Soc. 2014, 136, 10234–10237. [Google Scholar] [CrossRef]
- Tondreau, A.M.; Boncella, J.M. 1,2-Addition of Formic or Oxalic Acid to –N{CH2CH2(PiPr2)}2-Supported Mn(I) Dicarbonyl Complexes and the Manganese-Mediated Decomposition of Formic Acid. Organometallics 2016, 35, 2049–2052. [Google Scholar] [CrossRef]
- Léval, A.; Agapova, A.; Steinlechner, C.; Alberico, E.; Junge, H.; Beller, M. Hydrogen production from formic acid catalyzed by a phosphine free manganese complex: Investigation and mechanistic insights. Green Chem. 2020, 22, 913–920. [Google Scholar] [CrossRef] [Green Version]
- Enthaler, S.; Brück, A.; Kammer, A.; Junge, H.; Irran, E.; Gülak, S. Exploring the Reactivity of Nickel Pincer Complexes in the Decomposition of Formic Acid to CO2/H2 and the Hydrogenation of NaHCO3 to HCOONa. Chemcatchem 2015, 7, 65–69. [Google Scholar] [CrossRef]
- Neary, M.C.; Parkin, G. Nickel-catalyzed release of H2 from formic acid and a new method for the synthesis of zerovalent Ni(PMe3)4. Dalton Trans. 2016, 45, 14645–14650. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Wei, Z.; Spannenberg, A.; Jiao, H.; Junge, K.; Junge, H.; Beller, M. Cobalt-Catalyzed Aqueous Dehydrogenation of Formic Acid. Chem. Eur. J. 2019, 25, 8459–8464. [Google Scholar] [CrossRef] [PubMed]
- Loges, B.; Boddien, A.; Junge, H.; Beller, M. Controlled Generation of Hydrogen from Formic Acid Amine Adducts at Room Temperature and Application in H2/O2Fuel Cells. Angew. Chem. Int. Ed. 2008, 47, 3962–3965. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, R.; Yamashita, M.; Chung, L.W.; Morokuma, K.; Nozaki, K. Mechanistic Studies on the Reversible Hydrogenation of Carbon Dioxide Catalyzed by an Ir-PNP Complex. Organometallics 2011, 30, 6742–6750. [Google Scholar] [CrossRef]
- Coffey, R.S. The decomposition of formic acid catalysed by soluble metal complexes. Chem. Commun. 1967, 923b–924. [Google Scholar] [CrossRef]
- Nakazawa, H. Chapter 7. Catalysis by Organometallic Complexes. In Coordination Chemistry Fundamentals; Nakazawa, H., Koe, J., Eds.; Royal Society of Chemistry: Cambridge, UK, 2021; pp. 85–96. [Google Scholar] [CrossRef]
- Miranda, P.E. (Ed.) Hydrogen Storage and Transport by Organic Hydrides and Application of Ammonia. In Science and Engineering of Hydrogen-Based Energy Technologies; Elsevier: Amsterdam, The Netherlands, 2019; pp. 229–236. [Google Scholar] [CrossRef]
- Wang, W.-H.; Himeda, Y.; Muckerman, J.T.; Manbeck, G.F.; Fujita, E. CO2 Hydrogenation to Formate and Methanol as an Alternative to Photo- and Electrochemical CO2 Reduction. Chem. Rev. 2015, 115, 12936–12973. [Google Scholar] [CrossRef]
- Piccirilli, L.; Pinheiro, D.L.J.; Nielsen, M. Recent Progress with Pincer Transition Metal Catalysts for Sustainability. Catalysts 2020, 10, 773. [Google Scholar] [CrossRef]
- Boddien, A.; Loges, B.; Junge, H.; Beller, M. Hydrogen Generation at Ambient Conditions: Application in Fuel Cells. Chemsuschem 2008, 1, 751–758. [Google Scholar] [CrossRef]
- Gan, W.; Snelders, D.J.M.; Dyson, P.J.; Laurenczy, G. Ruthenium(II)-Catalyzed Hydrogen Generation from Formic Acid using Cationic, Ammoniomethyl-Substituted Triarylphosphine Ligands. Chemcatchem 2013, 5, 1126–1132. [Google Scholar] [CrossRef]
- Fellay, C.; Dyson, P.; Laurenczy, G. A Viable Hydrogen-Storage System Based On Selective Formic Acid Decomposition with a Ruthenium Catalyst. Angew. Chem. Int. Ed. 2008, 47, 3966–3968. [Google Scholar] [CrossRef]
- Guerriero, A.; Bricout, H.; Sordakis, K.; Peruzzini, M.; Monflier, E.; Hapiot, F.; Laurenczy, G.; Gonsalvi, L. Hydrogen Production by Selective Dehydrogenation of HCOOH Catalyzed by Ru-Biaryl Sulfonated Phosphines in Aqueous Solution. ACS Catal. 2014, 4, 3002–3012. [Google Scholar] [CrossRef]
- Fukuzumi, S.; Kobayashi, T.; Suenobu, T. Efficient Catalytic Decomposition of Formic Acid for the Selective Generation of H2and H/D Exchange with a Water-Soluble Rhodium Complex in Aqueous Solution. Chemsuschem 2008, 1, 827–834. [Google Scholar] [CrossRef]
- Guo, J.; Yin, C.K.; Zhong, D.L.; Wang, Y.L.; Qi, T.; Liu, G.H.; Shen, L.T.; Zhou, Q.S.; Peng, Z.H.; Yao, H.; et al. Formic Acid as a Potential On-Board Hydrogen Storage Method: Development of Homogeneous Noble Metal Catalysts for Dehydrogenation Reactions. Chemsuschem 2021, 14, 2655–2681. [Google Scholar] [CrossRef]
- Morales-Morales, D. (Ed.) Pincer Compounds: Chemistry and Applications; Elsevier: Amsterdam, The Netherlands, 2018. [Google Scholar]
- Liu, C.-C.; Liu, Q.-L.; Wu, Z.-Y.; Chen, Y.-C.; Xie, H.-J.; Lei, Q.-F.; Fang, W.-J. Mechanistic insights into small molecule activation induced by ligand cooperativity in PCcarbeneP nickel pincer complexes: A quantum chemistry study. J. Mol. Model. 2015, 21, 242. [Google Scholar] [CrossRef]
- Adams, G.M.; Weller, A.S. POP-type ligands: Variable coordination and hemilabile behaviour. Coord. Chem. Rev. 2018, 355, 150–172. [Google Scholar] [CrossRef]
- Gradiski, M.V.; Tsui, B.T.H.; Lough, A.J.; Morris, R.H. PNN′ & P2NN′ ligands via reductive amination with phosphine aldehydes: Synthesis and base-metal coordination chemistry. Dalton Trans. 2019, 48, 2150–2159. [Google Scholar] [CrossRef]
- Li, H.; Zheng, B.; Huang, K.-W. A new class of PN3-pincer ligands for metal–ligand cooperative catalysis. Coord. Chem. Rev. 2015, 293–294, 116–138. [Google Scholar] [CrossRef] [Green Version]
- Mellone, I.; Gorgas, N.; Bertini, F.; Peruzzini, M.; Kirchner, K.; Gonsalvi, L. Selective Formic Acid Dehydrogenation Catalyzed by Fe-PNP Pincer Complexes Based on the 2,6-Diaminopyridine Scaffold. Organometallics 2016, 35, 3344–3349. [Google Scholar] [CrossRef]
- Hsu, M.S.S.-F.; Rommel, D.-C.S.; Eversfield, M.S.P.; Muller, K.; Klemm, E.; Thiel, W.R.; Plietker, B. A Rechargeable Hydrogen Battery Based on Ru Catalysis. Angew. Chem. Int. Ed. 2014, 53, 7074–7078. [Google Scholar] [CrossRef]
- Anderson, N.H.; Boncella, J.; Tondreau, A.M. Manganese-Mediated Formic Acid Dehydrogenation. Chem. Eur. J. 2019, 25, 10557–10560. [Google Scholar] [CrossRef]
- Stathi, P.; Solakidou, M.; Louloudi, M.; Deligiannakis, Y. From Homogeneous to Heterogenized Molecular Catalysts for H2 Production by Formic Acid Dehydrogenation: Mechanistic Aspects, Role of Additives, and Co-Catalysts. Energies 2020, 13, 733. [Google Scholar] [CrossRef]
- Mellone, I.; Bertini, F.; Peruzzini, M.; Gonsalvi, L. An active, stable and recyclable Ru(ii) tetraphosphine-based catalytic system for hydrogen production by selective formic acid dehydrogenation. Catal. Sci. Technol. 2016, 6, 6504–6512. [Google Scholar] [CrossRef]
- Bertini, F.; Mellone, I.; Ienco, A.; Peruzzini, M.; Gonsalvi, L. Iron(II) Complexes of the Linear rac-Tetraphos-1 Ligand as Efficient Homogeneous Catalysts for Sodium Bicarbonate Hydrogenation and Formic Acid Dehydrogenation. ACS Catal. 2015, 5, 1254–1265. [Google Scholar] [CrossRef]
- Pan, Y.; Pan, C.-L.; Zhang, Y.; Li, H.; Min, S.; Guo, X.; Zheng, B.; Chen, H.; Anders, A.; Lai, Z.; et al. Selective Hydrogen Generation from Formic Acid with Well-Defined Complexes of Ruthenium and Phosphorus–Nitrogen PN 3 -Pincer Ligand. Chem. Asian J. 2016, 11, 1357–1360. [Google Scholar] [CrossRef] [PubMed]
- Kar, S.; Rauch, M.; Leitus, G.; Ben-David, Y.; Milstein, D. Highly efficient additive-free dehydrogenation of neat formic acid. Nat. Catal. 2021, 4, 193–201. [Google Scholar] [CrossRef]
- Mellone, I.; Peruzzini, M.; Rosi, L.; Mellmann, D.; Junge, H.; Beller, M.; Gonsalvi, L. Formic acid dehydrogenation catalysed by ruthenium complexes bearing the tripodal ligands triphos and NP3. Dalton Trans. 2013, 42, 2495–2501. [Google Scholar] [CrossRef] [PubMed]
- Papp, G.; Ölveti, G.; Horváth, H.; Kathó, Á.; Joó, F. Highly efficient dehydrogenation of formic acid in aqueous solution catalysed by an easily available water-soluble iridium(iii) dihydride. Dalton Trans. 2016, 45, 14516–14519. [Google Scholar] [CrossRef]
- DuBois, M.R.; DuBois, D.L. The role of pendant bases in molecular catalysts for H2 oxidation and production. Comptes Rendus Chim. 2008, 11, 805–817. [Google Scholar] [CrossRef]
- Li, X.; Ma, X.; Shi, F.; Deng, Y. Hydrogen Generation from Formic Acid Decomposition with a Ruthenium Catalyst Promoted by Functionalized Ionic Liquids. Chemsuschem 2010, 3, 71–74. [Google Scholar] [CrossRef]
- Kothandaraman, J.; Czaun, M.; Goeppert, A.; Haiges, R.; Jones, J.-P.; May, R.B.; Prakash, G.K.S.; Olah, G.A. Amine-Free Reversible Hydrogen Storage in Formate Salts Catalyzed by Ruthenium Pincer Complex without pH Control or Solvent Change. Chemsuschem 2015, 8, 1442–1451. [Google Scholar] [CrossRef]
- Boddien, A.; Loges, B.; Junge, H.; Gärtner, F.; Noyes, J.R.; Beller, M. Continuous Hydrogen Generation from Formic Acid: Highly Active and Stable Ruthenium Catalysts. Adv. Synth. Catal. 2009, 351, 2517–2520. [Google Scholar] [CrossRef]
- Majewski, A.; Morris, D.J.; Kendall, K.; Wills, M. A Continuous-Flow Method for the Generation of Hydrogen from Formic Acid. Chemsuschem 2010, 3, 431–434. [Google Scholar] [CrossRef]
- Solakidou, M.; Deligiannakis, Y.; Louloudi, M. Heterogeneous amino-functionalized particles boost hydrogen production from Formic Acid by a ruthenium complex. Int. J. Hydrogen Energy 2018, 43, 21386–21397. [Google Scholar] [CrossRef]
- Fukuzumi, S.; Kobayashi, T.; Suenobu, T. Unusually Large Tunneling Effect on Highly Efficient Generation of Hydrogen and Hydrogen Isotopes in pH-Selective Decomposition of Formic Acid Catalyzed by a Heterodinuclear Iridium−Ruthenium Complex in Water. J. Am. Chem. Soc. 2010, 132, 1496–1497. [Google Scholar] [CrossRef]
- Grubel, K.; Jeong, H.; Yoon, C.W.; Autrey, T. Challenges and opportunities for using formate to store, transport, and use hydrogen. J. Energy Chem. 2020, 41, 216–224. [Google Scholar] [CrossRef]
- Su, J.; Yang, L.; Lu, M.; Lin, H. Highly Efficient Hydrogen Storage System Based on Ammonium Bicarbonate/Formate Redox Equilibrium over Palladium Nanocatalysts. Chemsuschem 2015, 8, 813–816. [Google Scholar] [CrossRef]
- Iguchi, M.; Guan, C.; Huang, K.-W.; Kawanami, H. Solvent effects on high-pressure hydrogen gas generation by dehydrogenation of formic acid using ruthenium complexes. Int. J. Hydrogen Energy 2019, 44, 28507–28513. [Google Scholar] [CrossRef]
- Iturmendi, A.; Rubio-Pérez, L.; Pérez-Torrente, J.J.; Iglesias, M.; Oro, L.A. Impact of Protic Ligands in the Ir-Catalyzed Dehydrogenation of Formic Acid in Water. Organometallics 2018, 37, 3611–3618. [Google Scholar] [CrossRef]
- Luque-Gómez, A.; García-Abellán, S.; Munarriz, J.; Polo, V.; Passarelli, V.; Iglesias, M. Impact of Green Cosolvents on the Catalytic Dehydrogenation of Formic Acid: The Case of Iridium Catalysts Bearing NHC-phosphane Ligands. Inorg. Chem. 2021, 60, 15497–15508. [Google Scholar] [CrossRef]
- Younas, M.; Rezakazemi, M.; Arbab, M.S.; Shah, J.; Rehman, W.U. Green hydrogen storage and delivery: Utilizing highly active homogeneous and heterogeneous catalysts for formic acid dehydrogenation. Int. J. Hydrogen Energy 2022, 47, 11694–11724. [Google Scholar] [CrossRef]
- Curley, J.B.; Bernskoetter, W.H.; Hazari, N. Additive-Free Formic Acid Dehydrogenation Using a Pincer-Supported Iron Catalyst. Chemcatchem 2020, 12, 1934–1938. [Google Scholar] [CrossRef]
- Celaje, J.J.A.; Lu, Z.; Kedzie, E.; Terrile, N.J.; Lo, J.N.; Williams, T.J. A prolific catalyst for dehydrogenation of neat formic acid. Nat. Commun. 2016, 7, 11308. [Google Scholar] [CrossRef] [PubMed]
- Junge, H.; Boddien, A.; Capitta, F.; Loges, B.; Noyes, J.R.; Gladiali, S.; Beller, M. Improved hydrogen generation from formic acid. Tetrahedron Lett. 2009, 50, 1603–1606. [Google Scholar] [CrossRef]
- Pfister, S.; Bayer, P.; Koehler, A.; Hellweg, S. Environmental Impacts of Water Use in Global Crop Production: Hotspots and Trade-Offs with Land Use. Environ. Sci. Technol. 2011, 45, 5761–5768. [Google Scholar] [CrossRef] [PubMed]
- Matsunami, A.; Kayaki, Y.; Ikariya, T. Enhanced Hydrogen Generation from Formic Acid by Half-Sandwich Iridium(III) Complexes with Metal/NH Bifunctionality: A Pronounced Switch from Transfer Hydrogenation. Chem. Eur. J. 2015, 21, 13513–13517. [Google Scholar] [CrossRef]
- Mellmann, D.; Barsch, E.; Bauer, M.; Grabow, K.; Boddien, A.; Kammer, A.; Sponholz, P.; Bentrup, U.; Jackstell, R.; Junge, H.; et al. Base-Free Non-Noble-Metal-Catalyzed Hydrogen Generation from Formic Acid: Scope and Mechanistic Insights. Chem. Eur. J. 2014, 20, 13589–13602. [Google Scholar] [CrossRef]
- Montandon-Clerc, M.; Dalebrook, A.F.; Laurenczy, G. Quantitative aqueous phase formic acid dehydrogenation using iron(II) based catalysts. J. Catal. 2016, 343, 62–67. [Google Scholar] [CrossRef]
- Picquet, M.; Tkatchenko, I.; Tommasi, I.; Wasserscheid, P.; Zimmermann, J. Ionic Liquids, 3. Synthesis and Utilisation of Protic Imidazolium Salts in Homogeneous Catalysis. Adv. Synth. Catal. 2003, 345, 959–962. [Google Scholar] [CrossRef]
- Pârvulescu, V.I.; Hardacre, C. Catalysis in Ionic Liquids. Chem. Rev. 2007, 107, 2615–2665. [Google Scholar] [CrossRef]
- Berger, M.E.M.; Assenbaum, D.; Taccardi, N.; Spiecker, E.; Wasserscheid, P. Simple and recyclable ionic liquid based system for the selective decomposition of formic acid to hydrogen and carbon dioxide. Green Chem. 2011, 13, 1411–1415. [Google Scholar] [CrossRef]
- Yasaka, Y.; Wakai, C.; Matubayasi, N.; Nakahara, M. Slowdown of H/D Exchange Reaction Rate and Water Dynamics in Ionic Liquids: Deactivation of Solitary Water Solvated by Small Anions in 1-Butyl-3-Methyl-Imidazolium Chloride. J. Phys. Chem. A 2007, 111, 541–543. [Google Scholar] [CrossRef]
- Anderson, J.L.; Ding, J.; Welton, T.; Armstrong, D.W. Characterizing Ionic Liquids On the Basis of Multiple Solvation Interactions. J. Am. Chem. Soc. 2002, 124, 14247–14254. [Google Scholar] [CrossRef]
- Yasaka, Y.; Wakai, C.; Matubayasi, N.; Nakahara, M. Controlling the Equilibrium of Formic Acid with Hydrogen and Carbon Dioxide Using Ionic Liquid. J. Phys. Chem. A 2010, 114, 3510–3515. [Google Scholar] [CrossRef]
- Bates, E.D.; Mayton, R.D.; Ntai, I.; Davis, J.H. CO2 Capture by a Task-Specific Ionic Liquid. J. Am. Chem. Soc. 2002, 124, 926–927. [Google Scholar] [CrossRef]
- Zhang, Z.; Xie, Y.; Li, W.; Hu, S.; Song, J.; Jiang, T.; Han, B. Hydrogenation of Carbon Dioxide is Promoted by a Task-Specific Ionic Liquid. Angew. Chem. 2008, 120, 1143–1145. [Google Scholar] [CrossRef]
- Scholten, J.D.; Prechtl, M.H.G.; Dupont, J. Decomposition of Formic Acid Catalyzed by a Phosphine-Free Ruthenium Complex in a Task-Specific Ionic Liquid. Chemcatchem 2010, 2, 1265–1270. [Google Scholar] [CrossRef]
- Geldbach, T.J.; Dyson, P.J. A Versatile Ruthenium Precursor for Biphasic Catalysis and Its Application in Ionic Liquid Biphasic Transfer Hydrogenation: Conventional vs Task-Specific Catalysts. J. Am. Chem. Soc. 2004, 126, 8114–8115. [Google Scholar] [CrossRef]
- Baaqel, H.; Tulus, V.; Chachuat, B.; Guillén-Gosálbez, G.; Hallett, J. Uncovering the True Cost of Ionic Liquids using Monetization. In Computer Aided Chemical Engineering; Elsevier: Amsterdam, The Netherlands, 2020; Volume 48, pp. 1825–1830. [Google Scholar] [CrossRef]
- The Cost of Water and Its Pricing General Principles. Available online: https://wikiwater.fr/the-cost-of-water-and-its-pricing (accessed on 27 November 2022).
- Onishi, N.; Kanega, R.; Kawanami, H.; Himeda, Y. Recent Progress in Homogeneous Catalytic Dehydrogenation of Formic Acid. Molecules 2022, 27, 455. [Google Scholar] [CrossRef]
- Leon, A. Hydrogen Technology: Mobile and Portable Applications; Springer: Berlin/Heidelberg, Germany, 2008. [Google Scholar]
- Lewis, N.S.; Nocera, D.G. Powering the planet: Chemical challenges in solar energy utilization. Proc. Natl. Acad. Sci. USA 2006, 103, 15729–15735. [Google Scholar] [CrossRef]
- Whitesides, G.M.; Crabtree, G.W. Don’t Forget Long-Term Fundamental Research in Energy. Science 2007, 315, 796–798. [Google Scholar] [CrossRef]
- Gao, Y.; Kuncheria, J.K.; Jenkins, H.A.; Puddephatt, R.J.; Yap, G.P.A. The interconversion of formic acid and hydrogen/carbon dioxide using a binuclear ruthenium complex catalyst. J. Chem. Soc. Dalton Trans. 2000, 3212–3217. [Google Scholar] [CrossRef]
- Dicks, A.L.; Rand, D.A.J. Fuel Cell Systems Explained, 3rd ed.; Wiley: Hoboken, NJ, USA, 2018. [Google Scholar] [CrossRef]
- Czaun, M.; Kothandaraman, J.; Goeppert, A.; Yang, B.; Greenberg, S.; May, R.B.; Olah, G.A.; Prakash, G.K.S. Iridium-Catalyzed Continuous Hydrogen Generation from Formic Acid and Its Subsequent Utilization in a Fuel Cell: Toward a Carbon Neutral Chemical Energy Storage. ACS Catal. 2016, 6, 7475–7484. [Google Scholar] [CrossRef]
- Hydrozine, Liquid Organic Hydrogen Carrier. Available online: https://www.dens.one/ (accessed on 21 October 2022).
- Satyapal, S.; U.S. Department of Energy, (DoE). Hydrogen and Fuel Cell Technologies Office, 2021 AMR Plenary Session. Available online: https://www.hydrogen.energy.gov/pdfs/review21/plenary5_satyapal_2021_o.pdf (accessed on 3 January 2023).
- Kayfeci, M.; Keçebaş, A.; Bayat, M. Hydrogen production. In Solar Hydrogen Production; Elsevier: Amsterdam, The Netherlands, 2019; pp. 45–83. [Google Scholar] [CrossRef]
- Ruth, M.; Jadun, P.; Gilroy, N.; Connelly, E.; Boardman, R.; Simon, A.J.; Elgowainy, A.; Zuboy, J. The Technical and Economic Potential of the H2@Scale Hydrogen Concept within the United States. 2020. Available online: https://www.nrel.gov/docs/fy21osti/77610.pdf (accessed on 4 January 2023).
- IEA. The Future of Hydrogen, Seizing Today’s Opportunities, Report Prepared by the IEA for the G20, Japan. June 2019. Available online: https://iea.blob.core.windows.net/assets/9e3a3493-b9a6-4b7d-b499-7ca48e357561/The_Future_of_Hydrogen.pdf (accessed on 6 January 2023).
- Stathi, P.; Deligiannakis, Y.; Avgouropoulos, G.; Louloudi, M. Efficient H2 production from formic acid by a supported iron catalyst on silica. Appl. Catal. A Gen. 2015, 498, 176–184. [Google Scholar] [CrossRef]
- Ahn, Y.; Byun, J.; Kim, D.; Kim, B.-S.; Lee, C.-S.; Han, J. System-level analysis and life cycle assessment of CO2 and fossil-based formic acid strategies. Green Chem. 2019, 21, 3442–3455. [Google Scholar] [CrossRef]





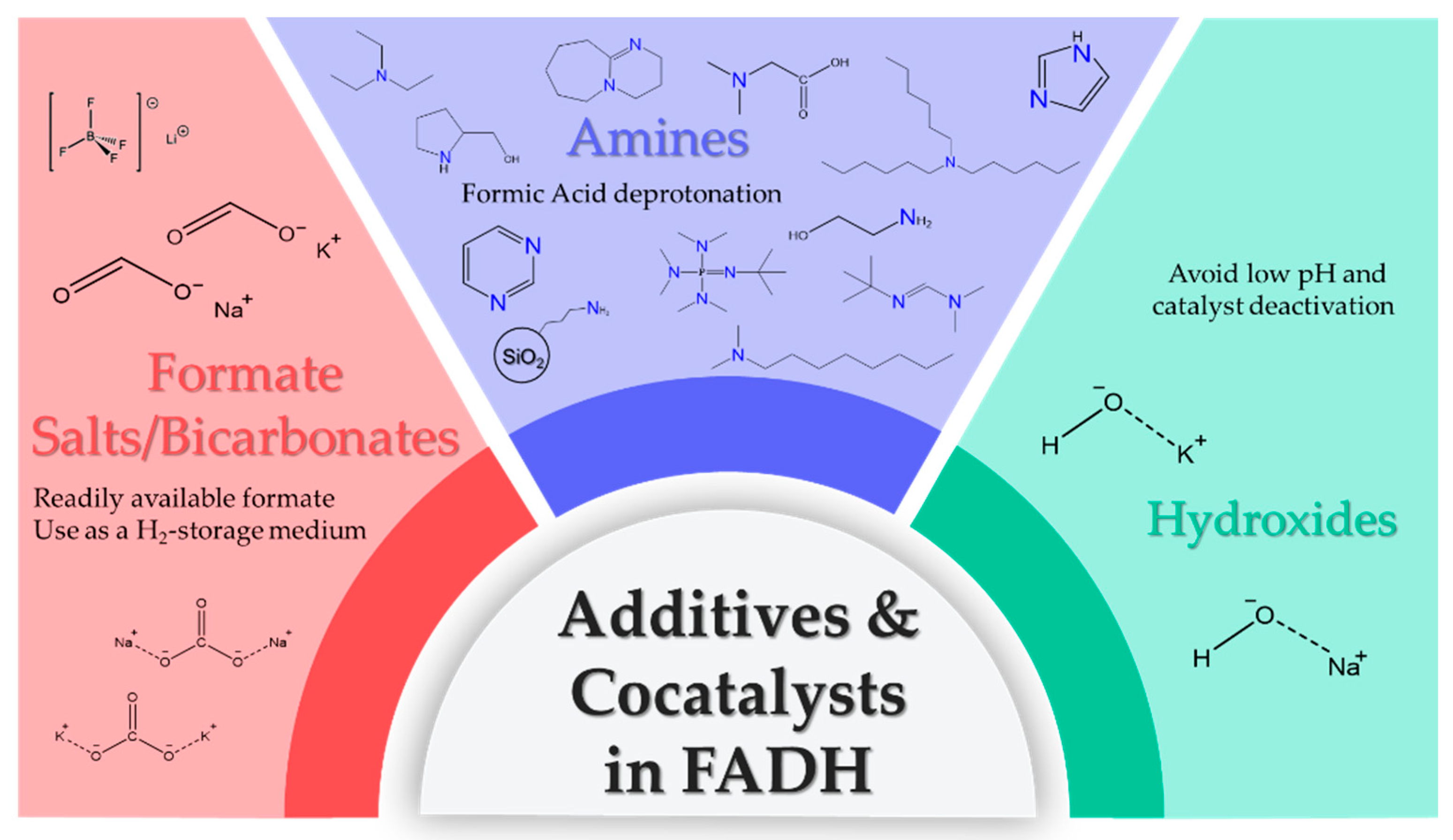


| Entry | Additive | Conditions | TONs | Cost ($/kgadditive) | kgH2 /kgadditive | 2 $/kgH2 (additive) | Ref. |
|---|---|---|---|---|---|---|---|
| #3a | NBu3 | 21.8 μmol catalyst, FA = 1 g, 10.86 mmol NBu3, 2 mL THF, T = 40 °C | 1 50,000 | 106 | 1.08 | 9.78 | [68] |
| #3 | Net3 | 10 μmol catalyst, FA = 3 g, 10.86 mmol NEt3, 2 mL 1,4 dioxane T = 40 °C | 100,000 | 63 | 1.82 | 3.46 | [68] |
| #9 | nOctNMe2 | 9.55 μmol [RuCl2(benzene)]2 (19.1 μmol [Ru]), 115 μmol dppe (Ru/dppe = 1:6), 20 mL nOctNMe2, T = 60 °C | 1,000,000 | 200 | 2.5 | 8 | [49] |
| #9a | Me2NHex | 9.55 μmol [RuCl2(benzene)]2 (19.1 μmol [Ru]), 115 μmol dppe (Ru/dppe = 1:6), 20 mL Me2NHex, T = 60 °C | 1 1,041,355 | 3166 | 2.68 | 116 | [49] |
| #9b | NHex3 | 9.55 μmol [RuCl2(benzene)]2 (19.1 μmol [Ru]), 115 μmol dppe (Ru/dppe = 1:6), 20 mL NHex3, T = 60 °C | 1 378,675 | 154 | 0.95 | 16.3 | [49] |
| #9c | Me2NDec | 9.55 μmol [RuCl2(benzene)]2 (19.1 μmol [Ru]), 115 μmol dppe (Ru/dppe = 1:6), 20 mL Me2NDec | 1 358,744 | 84 | 0.88 | 9.53 | [49] |
| #11 | NHex3 | 1.42 μmol of catalyst, continuous flow of FA, NHex3 = 33.5 mmol, 35 mL DMF, T = 90 °C | 706,500 | 154 | 0.21 | 73.39 | [51] |
| #9d | DBU | 1.42 μmol of catalyst, continuous flow of FA, DBU = 33.5 mmol, 35 mL DMF, T = 90 °C | 310,000 | 46 | 0.16 | 28.2 | [51] |
| #9e | Net3 | 1.42 μmol of catalyst, continuous flow of FA, Net3 = 33.5 mmol, 35 mL DMF, T = 90 °C | 326,000 | 63 | 0.26 | 24.37 | [51] |
| #4 | LiBF4 | 0.001 mol% [Fe], FA = 110 μL, (2.91 mmol), LiBF4 = 0.291 mmol, 10 mol%, 5 mL 1,4 dioxane, T = 80 °C | 983,642 | 3760 | 7215 | 0.05 | [71] |
| #25 | HCOONa | 9.8 μmol catalyst, FA = 133 mmol/catalytic cycle HCOONa = 50 mmol, Vtotal = 20.0 mL, T = 100 °C | >67,650 | 14 | 0.39 | 3.6 | [104] |
| #26c | HCOOK | 0.005 mmol catalyst, FA = 37 mmol, HCOOK = 40 mmol, H2O = 9 mL, triglyme = 4 mL, T = 92.5 °C | 5746 (after 45 h) | 27 | 0.21 | 12.6 | [73] |
| #26d | KOH | 0.37 mmol of catalyst, FA = 5 mmol, KOH = 40 mmol, H2O = 9 mL, triglyme = 4 mL, T = 92.5 °C | 2895 | 120 | 1.42 | 8.4 | [73] |
| Entry | Solvent | Conditions | TONs | Cost ($/kgsolvent) | kgH2/kgsolvent | 2 $/kgH2 (Solvent) | Ref. |
|---|---|---|---|---|---|---|---|
| #1 | DMSO | 1.0 μmol of catalyst FA = 0.3 mL/h, NEt3 = 1.50 mL, 0.7 mL DMSO, T = 90 °C | 1,100,000 | 24 | 0.2 | 12 | [101] |
| #1a | DMF | 1.0 μmol of catalyst FA = 0.3 mL/h, NEt3 = 1.50 mL, 10 mL DMSO, T = 90 °C | 93,000 | 32 | 0.02 | 161 | [101] |
| #1b | Toluene | 1.0 μmol of catalyst FA = 0.3 mL/h, NEt3 = 1.50 mL, 10 mL toluene, T = 90 °C | 1 23,158 | 37 | 0.01 | 343 | [101] |
| #1c | MeCN | 1.0 μmol of catalyst FA = 0.3 mL/h, NEt3 = 1.50 mL, 5 mL MeCN, T = 90 °C | 1 55,579 | 57 | 0.03 | 201 | [101] |
| #1d | THF | 1.0 μmol of catalyst FA = 0.3 mL/h, NEt3 = 1.50 mL, 5 mL THF, T = 90 °C | 1 42,581 | 62 | 0.02 | 323 | [101] |
| #3 | 1,4 dioxane | 10 μmol catalyst, FA = 3 g, 10.86 mmol NEt3, 2 mL 1,4 dioxane, T = 40 °C | 100,000 | 48 | 0.97 | 4.94 | [68] |
| #3b | THF | 10 μmol catalyst, FA = 3 g, 10.86 mmol NEt3, 2 mL THF, T = 40 °C | 1 79,632 | 62 | 0.9 | 6.90 | [68] |
| #3c | PC | 10 μmol catalyst, FA = 3 g, 10.86 mmol NEt3, 2 mL PC, T = 40 °C | 1 47,933 | 41 | 0.4 | 10.3 | [68] |
| #3d | H2O | 10 μmol catalyst, FA = 3 g, 10.86 mmol NEt3, 2 mL H2O, T = 40 °C | 1 12,711 | 11 | 0.13 | 8.3 | [68] |
| Catalytic System | Cost ($/kgH2) Metal | Cost ($/kgH2) Ligand | Cost ($/kgH2) Additive | Cost ($/kgH2) FA | Cost ($/kgH2) Solvent | 1 Total Cost ($/kgH2) | Ref. |
|---|---|---|---|---|---|---|---|
| #1 | 0.86 | 11.9 | 3.11 | 0.04 | 2.85 | 18.85 | [101] |
| #2 | 0.55 | 6.43 | 0.00 | 1.61 | 0.00 | 8.60 | [102] |
| #3 | 0.64 | 31.31 | 3.46 | 1.38 | 4.94 | 41.72 | [68] |
| #4 | 0.08 | 0.81 | 0.05 | 0.0004 | 0.13 | 1.06 | [71] |
| #5 | 4.07 | 1.79 | 0.00 | 0.029 | 0.74 | 6.82 | [44] |
| #6 | 1.95 | 32.98 | 0.00 | 1.38 | 0.32 | 36.62 | [65] |
| #7 | 1.94 | 28.94 | 0.00 | 1.21 | 0.76 | 32.85 | [56] |
| #8 | 3.73 | 3.91 | 0.45 | 1.38 | 0.00 | 9.46 | [119] |
| #9 | 1.6 | 0.33 | 8.00 | 1.50 | 0.00 | 11.44 | [49] |
| #10 | 11.28 | 20.31 | 3.58 | 1.38 | 12.06 | 48.63 | [104] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Solakidou, M.; Gemenetzi, A.; Koutsikou, G.; Theodorakopoulos, M.; Deligiannakis, Y.; Louloudi, M. Cost Efficiency Analysis of H2 Production from Formic Acid by Molecular Catalysts. Energies 2023, 16, 1723. https://doi.org/10.3390/en16041723
Solakidou M, Gemenetzi A, Koutsikou G, Theodorakopoulos M, Deligiannakis Y, Louloudi M. Cost Efficiency Analysis of H2 Production from Formic Acid by Molecular Catalysts. Energies. 2023; 16(4):1723. https://doi.org/10.3390/en16041723
Chicago/Turabian StyleSolakidou, Maria, Aikaterini Gemenetzi, Georgia Koutsikou, Marinos Theodorakopoulos, Yiannis Deligiannakis, and Maria Louloudi. 2023. "Cost Efficiency Analysis of H2 Production from Formic Acid by Molecular Catalysts" Energies 16, no. 4: 1723. https://doi.org/10.3390/en16041723