
Aromatic Clusters and Hydrogen Storage


1
Department of Education, A. M. School of Educational Sciences, Assam University, Silchar 788011, Assam, India
2
Department of Chemistry, Indian Institute of Technology Kharagpur, Kharagpur 721302, West Bengal, India
*
Author to whom correspondence should be addressed.
Energies 2023, 16(6), 2833; https://doi.org/10.3390/en16062833
Submission received: 10 February 2023
/
Revised: 13 March 2023
/
Accepted: 16 March 2023
/
Published: 18 March 2023
(This article belongs to the Section A5: Hydrogen Energy)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Concurrence of aromaticity and hydrogen trapping potential of some atomic clusters has drawn the attention of scientific community, although in a few cases it has been reported that the partial charges on the constituent atoms of the clusters are probably responsible for H2 trapping via frail van der Waals type of interactions. In this article, an effort is made to review the studies which address the conjunction of aromaticity and hydrogen storage potential of different atomic clusters and the contribution of our research group to this particular topic.
1. Introduction
Continuous increment of machinery for our day-to-day survival as well as the modernization and increment in the number of industries and automobile sector has surged the demand of energy-stock unexpectedly. To bout these huge energy needs, the balance of our environment has been hampered in many ways, including unusual weather, climate change, global warming, etc. [1,2,3,4,5]. A few years back, it was reported that human civilization depends mostly on fossil fuels since most of the needed energy is produced by their combustion; in contrast, such fuels are the primary producer of carbon dioxide and thereby greenhouse gasses [6,7]. Therefore, we need energy resources which would not destroy our environment and fulfil the requirement. Although there are many alternatives to fossil fuels, hydrogen energy is the best option due to its avalanche abundance and pollution-free combustion. Nevertheless, physical storage of H2 is very expensive (needs cryogenic ailment) abreast accident causing [8] and the density of H2 is ~9 × 10−5 g/cm3 at standard ambient temperature and pressure (SATP) [9]. Due to these reasons, many research works worldwide on hydrogen storage technologies as well as on the systems are underway to progress in the finding of potential hydrogen storage material and solve the energy issue. There are many types of hydrogen storage material, but broadly, we can classify them into physical hydrogen storage material and chemical hydrogen storage material. Those materials which involve the trapping of hydrogen via covalent bond formation are termed chemical hydrogen storage materials. Such trapping of hydrogen ensues in the range of 47.8–95.6 kcal/mol [10]. However, such storage of hydrogen possesses a great disadvantage in the liberation as well as further uptake pathways [11]. The physical hydrogen storage can be classified into two types: first is the direct storage of pure H2, but in this case, the necessity of cryogenic ailment and possible hazard restricts the scope; second is the trapping of H2 through weak non-covalent interactions (van der Waals dispersion forces mainly). Such contact occurs by ≤2.4 kcal/mol [12,13].
Hubner et al. reported the variation of interaction of H2 with the electron-deficient and electron-rich aromatic system. They considered a series of aromatic systems for their study, including monosubstituted benzene (C6H5X, where X = CN, CH3, NH2, OH, F, and H), naphthalene and azulene (C10H8), anthracene (C14H10), coronene (C24H12), terephthalic acid (p-C6H4(COOH)2), and dilithium terephthalate (p-C6H4(COOLi)2). They concluded that the binding of molecular H2 occurs feasibly with electron-rich aromatic systems, and, on the other hand, more favourable interaction is noted with larger aromatic systems in comparison to benzene ring [14]. In another report, it was disclosed that the aromaticity of different organic molecular systems directs their binding with alkali metals. Alkali metal-doped organic aromatic systems revealed potential hydrogen binding ability [15]. A few anionic, carbon-based aromatic molecular systems were studied by Bodrenko and co-workers, who reported the stabilization of such systems by alkali and alkaline earth metal cations. Moreover, they disclosed that the nature of the cation monitors the weak bonding interaction between the organometallic compound and molecular hydrogen [16]. So far, in our research group, we have studied many atomic and molecular, neutral as well as ionic clusters in the search of potential hydrogen trapping/encaging systems. These include metal organic framework, neutral and ionic metallic as well as non-metallic clusters, clathrate hydrates, cyanogen cages, etc. Here, in this review, we discuss only those systems which exhibit some sort of aromaticity [13,17,18,19,20,21]. In the following section, theoretical background is discussed briefly.
2. Theoretical Background
Chattaraj and co-workers have been using different density functional theory methodologies to disclose many novel properties of various atomic and molecular systems over the last four decades. Here, we aimed to confer a few of the methodologies which were employed to investigate the clusters that exhibit aromaticity and show potential hydrogen trapping ability concurrently. It is known to almost every scientist that the DFT and conceptual density functional theory (CDFT) can predict the general properties of atoms, molecules, and materials [22,23,24,25,26]. The descriptors derived from DFT to assess the general properties of various atomic, molecular systems and many chemical processes are termed descriptors based on CDFT. Out of these descriptors, the electronegativity (), hardness (), and electrophilicity () help to unveil the global characteristics of a chemical system. On the other hand, to study the reactivity or inertness of a certain atomic center in a molecule the Fukui functions () was developed [27,28,29,30,31,32,33,34,35,36]. We can write the of a system containing N number of electrons as
The chemical potential () can be written as
hardness () as
The equation to calculate electrophilicity () is
One can express the electronegativity and hardness via finite difference method:
χ = (I + A)/2,
η = (I − A).
In the above two equations, I marks the ionization potential and A indicates the electron affinity of a molecular moiety, which can be determined employing the Koopmans’ theorem [37], and .
Here, and mark the energies of the highest occupied molecular orbitals and the lowest unoccupied molecular orbitals.
In the following way, we can calculate the ionization potential and electron affinity of a system via the computation of N and N ± 1 electronic systems’ energy by the ∆SCF technique:
I ≈ E(N − 1) − E(N),
A ≈ E(N) − E(N + 1).
In the above two equations, E(N − 1) is the single point energy of the optimized system containing (N − 1) electrons, whereas the E(N) and E(N + 1) denote the same parameter of the optimized system but of N and (N + 1) electrons, respectively.
Due to the addition or removal of an electron to/from a molecular system (at a constant external potential), a change in electron density occurs, and the same can be determined in terms of the Fukui function [36].
Calculation of local philicity () can help in estimating the selectivity of a particular atomic location. For the kth atomic site, the is
To characterize the nucleophilic attack, the magnitude of would be +, whereas for electrophilic and radical attacks, the values are − and 0, respectively.
The following is the statement of maximum hardness principle (MHP) [39,40,41]: “There seems to be a rule of nature that molecules arrange themselves so as to be as hard as possible”. and the following is the statement of minimum electrophilicity principle (MEP) [42,43,44,45]: “Electrophilicity will be a minimum (maximum) when both chemical potential and hardness are maxima (minima)”. It was concluded that the stability of a molecular system and extremal values of electrophilicity () are correlated. During chemical reactions, molecular vibrations and rotations, many intermediate and transition state species emerge and the evaluation of reveals electrophilicity extremum [42,43,44,45] at certain points in these processes which obey the following condition:
In this equation, indicates reaction coordinate for chemical changes as well as all possible symmetric and asymmetric movements of the component atoms of considered molecular system/s. So far, many research groups have reported the corroboration of MEP and MHP. For the assessment of stability of various molecular systems, such electronic structure principles are utilized worldwide.
When the relative stabilities of cyclohexene, 1.3-cyclohexadiene, hypothetical 1,3,5-cyclohexatriene, and benzene were studied thermodynamically, it became clear that the benzene is much more stable in comparison to the analogous diene and cyclohexene. Such unusual stability can be explained only by the unique property of planar, conjugated, (4n + 2)π electrons containing monocyclic molecular structures, i.e., by aromaticity. Aromaticity has a long history of research of approximately two centuries. The first scratch in this long history was made by Faraday in the year 1825, when he isolated benzene from illuminating gasses. Faraday developed the idea of electron delocalization in benzene molecule [46]. Later, a significant contribution was made by Kekule in the year 1865 with the dazzling concept of alternating single and double bonds in the benzene molecule [47,48,49,50]. Over the last few decades, numerous remarkably stable aromatic systems have been modelled and synthesized worldwide. The following are criteria of a molecule to be aromatic: possession of numbers of conjugated π electrons (, etc.), and cyclic and planar structure. It is worthy to mention here that the electron count was first proposed by Hückel, as one of the mandatory criteria of aromaticity. There are different types of theoretical approaches to ascertain the electron delocalization and aromaticity of a molecular system. Out of the many, Giambiagi et al. first proposed the three-center bond index, and the same was independently proposed by Sannigrahi and Kar [51,52]. Matito and co-workers discussed other electron sharing indexes such as electron sharing index (ESI) of Fluton, Mayer’s bond orders, and delocalization indexes (DI) [53,54,55,56,57,58,59,59,60,61]. The following aromaticity indices were used so far by various research group worldwide to ascertain electron delocalization: para-delocalization index (PDI), aromatic fluctuation index (FLU), MO multicentre bond index (MCI), six-center bond index by Ponec et al. The research group of Ponec et al. further developed the idea of quantitative characterization of homo-aromaticity, nonhomo-aromaticity, and antihomo-aromaticity by the use of MCI [62,63,64,65,66,67,68,69,70]. Chattaraj et al. studied all metal aromatic and antiaromatic systems and disclosed the efficiency of multicenter indices in the study of bonding, aromaticity, and reactivity. Moreover, they used MHP and minimum polarizability principle (MPP) to ascertain the relative stability of aromatic and antiaromatic molecular systems [71,72]. There are many methodologies to study the aromaticity, but mostly a few are used, harmonic oscillator model of aromaticity (HOMA) [73,74,75], nucleus independent chemical shift (NICS) [76], and electron localization function (ELF), etc. In our research group, we mostly used NICS, and particularly for the systems discussed here only NICS was used.
Schleyer et al. first proposed the concept of NICS (in ppm) in the assessment of aromaticity [76]. NICS is computed using a magnetic shielding tensor for a dummy magnetic dipole assumed at the centre of an under-study system. When NICS is computed at the centre of a molecular system, the same is indicated by NICS(0); and when the NICS measurement is conducted at 1 Å above the centre of the ring, it is indicated as NICS(1).
When the magnitude of NICS is negative, the system is aromatic, and when it is positive, the system is antiaromatic. Interestingly, few cage type molecules also reveal (−)Ve NICS values computed at the centre. Such systems, if they possess 2(n + 1)2 valence electrons, exhibit spherical aromaticity [77]. Various organic as well as inorganic cage-like symmetric molecular systems containing π-network validate this rule. Besides this, it is also noted that there are many cage-like molecular systems where one of the faces is open; we may call such a type of system an open-cage system. Stability and reactivity of such a type of open-cage system can be explained by the “Open-Shell Spherical Aromaticity”. Sola et al. introduced this concept for open-cage type systems possessing (2N2 + 2N + 1) numbers of π-electrons [78].
3. Computational Details
The initial coordinates of the guess structures were obtained from the molecular modelling software GaussView 3.0 and GaussView 5.0.8 [79]. Moreover, different molecular orbitals and other output results were obtained and further exploited using this software only. The systems which are identical with the work reported earlier were reoptimized at a new theoretical level using the old geometries, and further computations were performed. Optimizations of different modelled geometries and computations for thermochemistry as well as the magnetic shielding tensors were performed by Gaussian 03 and mostly by Gaussian 09 [80,81]. Various initial guess structures were optimized at different level of theories: B3LYP, M052X, DFT-D-B3LYP, M06, MP2, in combination with the basis sets 6-31G(d), 6-31G(d,p), 6-311+G(d,p), 6-311+G(d), cc-PVDZ as per the chosen molecular system to obtain the stationary points. The obtained stationary point coordinates were further taken to compute the harmonic vibrational frequencies at the used level of theory (optimization) to check the nature of the stationary points. Real values of the harmonic vibrational frequencies confirmed that the optimized systems are at the minima. Few molecular systems were assembled to generate periodic systems and the Vienna ab initio Simulation Package (VASP) [82,83,84,85] was used for the computation. Such periodic systems were modelled using XCrySDen graphical software [86]. As per the component elements of the periodic system, a kinetic energy cut-off of 550 eV was used (PAW potential) [87,88]. To compute the exchange–correlation energy density functional the Generalized Gradient Approximation (GGA) of Perdew–Burke–Ernzerhof was used [89]. A Monkhorst–Pack set of k-points [90] were used for the computations. For a few particular molecular systems, to envisage the effect of electric field on the hydrogen trapping, electric field was applied along the x direction of the modelled system [91,92]. The following equation was used to calculate the change in energy due to the application of the electric field:
In this equation, the term marks the computed total energy of the H2 trapped molecular motif in the electric field applied condition, whereas the term specifies computed energy without the electric field.
4. Atomic Clusters
Mg and Ca Clusters
McNelles and Naumkin studied hydrogen molecule encapsulated (dopped) Mgn clusters (n = 8 to 10) and reported the weak stability of such systems. In the next-to-next year, Chattaraj and co-workers reported further studies on similar systems and their Ca analogues [17]. In this study, they included investigation on the hydrogen molecule trapping ability of such magnesium and calcium clusters with the focus on hydrogen storage and aromaticity. Local minima forms of the modelled molecular hydrogen encaged Mgn and Can clathrate (n = 8–10) are collected in Figure 1. They noticed the interesting fact that the Mgn and Can clusters are unstable, whereas their H2 encapsulated forms are stable. This clarifies that the encapsulation of molecular hydrogen brought stability in them. However, in few cases, the H-H bond of the hydrogen molecule breaks and encaging occurs in the atomic form (Figure 1). Measurement of NICS(0) and NICS(1) at the top and bottom rings (Mg4 and Ca4) of the Mgn and Can forms revealed diatropic ring current. Mulliken population analysis charges on the hydrogen encaged metal centres disclosed positive values with the exception of a few sites of H2Mg9, H2Mg10 and H2Ca10, where negative charges were noted. On the other hand, partial positive charges on the metal centres of Mgn, Can (n = 8–10) were noted at both the upper and lower frames. From this positive partial charge, one may conclude their susceptibility to nucleophilic attack. Interested readers are directed to see Reference [17] for the calculated local reactivity descriptors.
5. Ionic Clusters
5.1. N4Li2 and N6Ca2 Clusters
The moiety N6 had the attention of the scientific community not only for possible use as high energy density material, but also for the structural pattern [93,94,95,96,97]. In 2011, Chattaraj and co-workers reported the aromaticity of N64− and N42− planar cyclic rings. Moreover, they studied these anionic systems with suitable co-ion in the search of a good hydrogen storage material and modelled the N4Li2 and N6Ca2 clusters [18]. A detailed investigation was conducted on the aromaticity of these systems. NICS(0) calculations revealed that both the anionic systems, N64− and N42−, possess aromatic criteria to compare with benzene and cyclobutadiene, respectively. They performed a NICS scan plot for N64−, N42−, benzene, and cyclobutadiene, and detected similar patterns for N64− and benzene. On the other hand, a different nature of plots was obtained for N42− and cyclobutadiene rings. There are ten π-electrons in the N64− cyclic planar system, which were noted to be aromatic. In contrast, surprisingly, the N42− monocyclic system revealed simultaneous appearance of π-aromaticity as well as σ-antiaromaticity, which was then called conflicting aromaticity. They concluded such σ-antiaromaticity due to the obtained positive value of NICS(0) whereas the π-aromaticity was projected from the (−)Ve magnitude of NICS(1). It is possible that the cation–π interaction between Ca2+ ions and N64− rings as well as between Li+ ions and N42− rings stabilized the N6Ca2 and N4Li2 systems, respectively. Such composite systems can interact with up to twelve hydrogen molecules. Local minima forms of hydrogen molecule-bound N6Ca2 and N4Li2 clusters are collected in Figure 2a. The researchers noted that due to the interaction of Ca2+ with the N64− ion, the aromaticity of the latter increases. On the other hand, the N42− monocyclic anion continues to show conflicting aromaticity even after the ionic contact with two Li+. It was mentioned that the binding of hydrogen molecules occurred due to the partial positive charges on the metal center. They noted favorable adsorption energy (ΔEads) for the interaction of hydrogen molecules. The following are adsorption energy data: 1.2 kcal/mol for the interaction with eight hydrogen molecules, with 4 H2 on each Li centers for N4Li2, and 1.3 kcal/mol for the interaction with six hydrogen molecules where each Ca binds 6 H2 in N6Ca2.
5.2. Li3+ and Na3+ Ions
In many molecular systems, aromaticity plays an important role for their stability and reactivity. Keeping in mind the role aromaticity can play, many molecular systems were modelled and designed. In the same line of thought, Chattaraj and co-workers studied the Li3+ and Na3+ ionic moieties and their interactions with molecular hydrogen/s [17]. The local minima forms obtained for the hydrogen molecule-bound equivalents of Li3+ and Na3+ systems are provided in Figure 3. Out of the many trial positions of interaction with H2 molecules, for the Li3+ cluster, trapping at the vertices became mostly feasible. The trigonal plane of the Li3+ cluster can also interact with hydrogen. However, for Na3+, interaction with molecular hydrogen occurs via the vertices of the formed triangle. In the trapping of hydrogen by the ionic clusters, an interesting point was noted. When the interaction between metal ion cluster and hydrogen takes place via the vertices, the hydrogen remains in its molecular form; on the other hand, when the ionic moieties interact via the centre of the triangular ring, atomic hydrogen binding occurs.
For the atomic hydrogen-bound form, the interaction energy is 32.2 kcal/mol (H2Li3+ cluster). Binding energy values fall in the range of 3.6–3.7 for more than one H2-bound cluster. When the interaction of the Li3+ cluster occurs with molecular H2, the Eb values remain in the range of 2.2–2.5 kcal/mol. Nevertheless, for the Na3+ ion, the binding energies are quite low, 0.2–0.7 kcal/mol. Computed results of NICS(0) [= (−)Ve] indicate that the alkali metal cluster cations and their H2 trapped analogues are aromatic. It was noted that the NICS(0) values of Li3+ and H2Li3+ moieties are −8.75 ppm and −14.57 ppm, respectively. The total number of hydrogen molecules adsorbed by the Li3+ moiety is eight via the vertices and molecular plane. Via only vertices, six H2 molecules can be trapped by the same ion. The other cluster ion, Na3+, traps a maximum of ten hydrogen molecules via the vertices of the formed triangular ring. As the number of interacting hydrogen molecules increases, the electrophilicity value decreases. Such trend of electrophilicity denotes increment in the stability of H2-bound ionic forms with cluster size.
5.3. M5Li7+ (M = C, Si, Ge) Clusters
Li-doped star-like clusters obtained the interest of the scientific community due to their stability and aromaticity. Merino et al., Heitjans et al., and Ghosh et al. studied such Li-doped star-like systems [98,99,100,101]. By now, it is known to all that the Li center with a sufficient partial positive charge can trap H2 molecules. With this idea, Chattaraj and co-workers had chosen star-like C5Li7+ and Si5Li7+ ions for detailed investigation. Moreover, they also studied the system Ge5Li7+ as it is the higher congener of the taken system [19]. Both the carbon- and the Si-containing systems’ global minima forms were determined by that time [19]. The simplicity of these structures and their symmetric skeleton (D5h) beside the π-aromaticity were mentioned. In aromaticity, it would be good for the reader to know that the system C5Li7+ is σ-nonaromatic and that Si5Li7+ shows σ-aromaticity. Chattaraj et al. successfully modelled the carbon- and silicon-containing D5h structures, but the Ge analogue was determined to be near D5h. Pauling scale electronegativities of Li, C, Si and Ge are 0.98, 2.55, 1.90, and 2.01, respectively. Due to such differences in electronegativity values, the bonding between C and Li, Si and Li, as well as between Ge and Li is obviously polar in nature. Computation of natural population analysis (NPA) charges revealed that in the C5Li7+ ion, the equatorial Li comprised +0.78e and the axial Li comprised +0.71e charges. Similar positive charges were noted on the Li centres of Si5Li7+ and Ge5Li7+ moieties. It was determined that each of the Li atoms in C5Li7+ can interact and bind three H2 molecules where the binding energy was noted to be −2.8 kcal/mol per H2. So, altogether, the carbon-centered ion can hold up to 21 H2 molecules (Figure 4).
Assessment of hardness and electrophilicity values revealed that as the number of hydrogen molecules increases at the D5h symmetric ions, their stability increases. Such trends of hardness and electrophilicity values validate the MHP and MEP, respectively. It was noted that the Si-centered ion can also trap up to 21 H2 molecules where each of the Li interacts with three H2. Energy of such interaction was determined to be −2.2 kcal/mol per H2 for the axial Li and −3.3 kcal/mol per H2 for the equatorial Li. On the other hand, for the Ge-centered ion, axial Li can trap up to two H2 molecules (interaction energy = −2.5 kcal/mol per H2) and the equatorial Li can hold three hydrogen molecules (interaction energy = −3.4 kcal/mol per H2). Moreover, the lengthening of hydrogen–hydrogen bond in the H2 molecule was noted due to the interaction with the Li centres and simultaneous decrease in the partial charge of the Li centre, and, accordingly, electron density transfer from the σ-bond of molecular hydrogen to the respective Li centres was mentioned. Si5Li7+, Ge5Li7+ and their hydrogen molecule-trapped analogues are provided in Figure 5. When electrophilicities were plotted for the Si- and Ge-centered moieties, it was noted that the obtained trends are in line with the C5Li7+ systems. However, the plot of hardness reflected a different trend, and to justify that, one can utilize scaled hardness per atom or per electron. Calculation of gravimetric hydrogen storage capacity was done and noted 18.3 and 9.3 wt% for the Si5Li7+ and Ge5Li7+ systems, respectively. Moreover, application of electric field (0.001 to 0.005 au) in the direction of the trapped H2 increases the binding energy of adsorption for all the studied systems.
6. Cage-like Clusters
6.1. B12N12 Cage
Inspired by the idea of endohedral and exohedral interaction of H2/H with BN buckyball [102,103,104], Giri et al. modelled the B12N12 moiety and studied the stability, aromaticity, and hydrogen trapping potential [20]. In the endohedral encapsulation of H2 by BN buckyball, the former remains in its molecular form. However, in the case of exohedral encapsulation, the H-H bond ruptures and atomic hydrogen adsorption occurs via each of the B and N centres [103]. In studying B12N12 moiety, Giri et al. explored the various global reactivity descriptors; moreover, the electronic structure principles (MHP, MEP) were also employed and were determined to be significant to gain further insight. B3LYP/6-311+G(d) level of theory was used to perform the geometry optimizations. Optimized geometries were further studied using MP2 level of theory and the same basis set. It was noted that H2 interacts with the B12N12 moiety exohedrally. As N is more electronegative than B, the nitrogen centres possess partial negative charges in the B12N12 cage. It is likely that these partial charges facilitate the interaction with hydrogen. It was reported that the BN cage can hold 12 hydrogen molecules exohedrally (Figure 6).
It was stated that nH2@B12N12 forms are more stable in comparison to the B12N12 cage. Capture of hydrogen molecules by the B12N12 moiety is favourable because of the negative interaction energy per H2 molecule. In addition, the measurement of aromaticity of the H2-bound BN moiety helps to understand the fate of the host as a hydrogen storage material. For all the nH2@B12N12 systems, electronegativity, hardness, and electrophilicity were computed and it was determined that the values of electronegativity decrease with the increasing value of n (number of H2 molecules). The calculated hardness data indicate increment in the magnitude with the increasing values of n, whereas a decreasing trend in the electrophilicity values was noted. NICS was calculated at the centre of B12N12 and nH2@B12N12 clusters. Aromaticity of the modelled moieties was known by the negative NICS values. It was mentioned that the three, six, and nine hydrogen molecule adsorbed B12N12 systems are less stable in comparison to their neighbouring analogues, as noted from the gain in energy values. The gain in energy values were calculated to compare the stabilities of the successive hydrogen-loaded systems. The reported NICS(0) profile indicates higher stability of four, eight, and ten hydrogen molecule adsorbed systems, beyond the steady trend. This unusual higher stability was not justified, neither from the gain in energies, nor from the global reactivity descriptors.
Interaction of one H2 molecule with the BN system was explored from the study of intrinsic reaction coordinate profile (Figure 7). The well-connected reactant, transition state, and product further support the fact that H2 interacts with the N centre of the BN cage. From the pictures of frontier molecular orbitals (FMOs), delocalization of electrons in the nH2@B12N12 systems was noted.
6.2. C12N12 Cage
Chattaraj and co-workers investigated polycyanogen clathrate systems, particularly the C12N12 form and its two other isomers in the search of hydrogen storage molecular frameworks [21]. Such systems are called high energy density materials (HEDM). Three possible isomers of C12N12 were mentioned. These isomers are designated as C12N12-A, C12N12-B, and C12N12-C. The three -A, -B, and -C isomers bear the point group D6d, C3, and C2v, respectively (Figure 8). In the remaining portion of this topic, these three forms are marked as A, B, and C, respectively.
The three isomers have completely different types of morphologies. Isomer A bears two numbers of planar six-membered C6 fragments, each at the end of a cask-like shape. The walls of such casks are formed by five numbers of C2N3 moieties. Constitution of the B form is different; there are two numbers of chairs such as C3N3 hexagonal segments, three numbers of C2N3 parts, and three numbers of C3N2 fragments. The last form, C, is like an open-cage system composed of eight numbers of five membered C3N2 parts. Among these isomers, the C form is the most stable. C is lower in energy by 55.41 kcal/mol compared to the B form. Further, the B form is more stable in comparison to the A isomer by 66.11 kcal/mol. Aromaticity of these three cyanogen forms was revealed by the NICS(0) of −2.601 (A), −5.876 (B), and −4.639 ppm (C). However, it is worthy to mention here that the forms A and B do not obey the rule of spherical aromaticity {2(N + 1)2π rule} [77]. On the other hand, the open-cage isomer C comprises 24 numbers of π electrons and it is clear that the same does not conform to the (2N2 + 2N + 1)π electron rule of open-shell spherical aromaticity [78]. It was noted that all of these forms can interact with more than one molecular H2 exohedrally. However, it is important to mention that a suitable site of trapping molecular hydrogen was obtained via rigorous search. It was reported that out of several possibilities, among the three justifiable sites (1. atop nitrogen atom, 2. atop carbon atom, and 3. atop the midpoint of carbon–nitrogen bond), only those atop the N atom local minima forms of hydrogen-bound analogues were obtained. The binding energy of trapping molecular hydrogen was calculated from the computed energies of different systems () using the following equation (n indicates the number of H2):
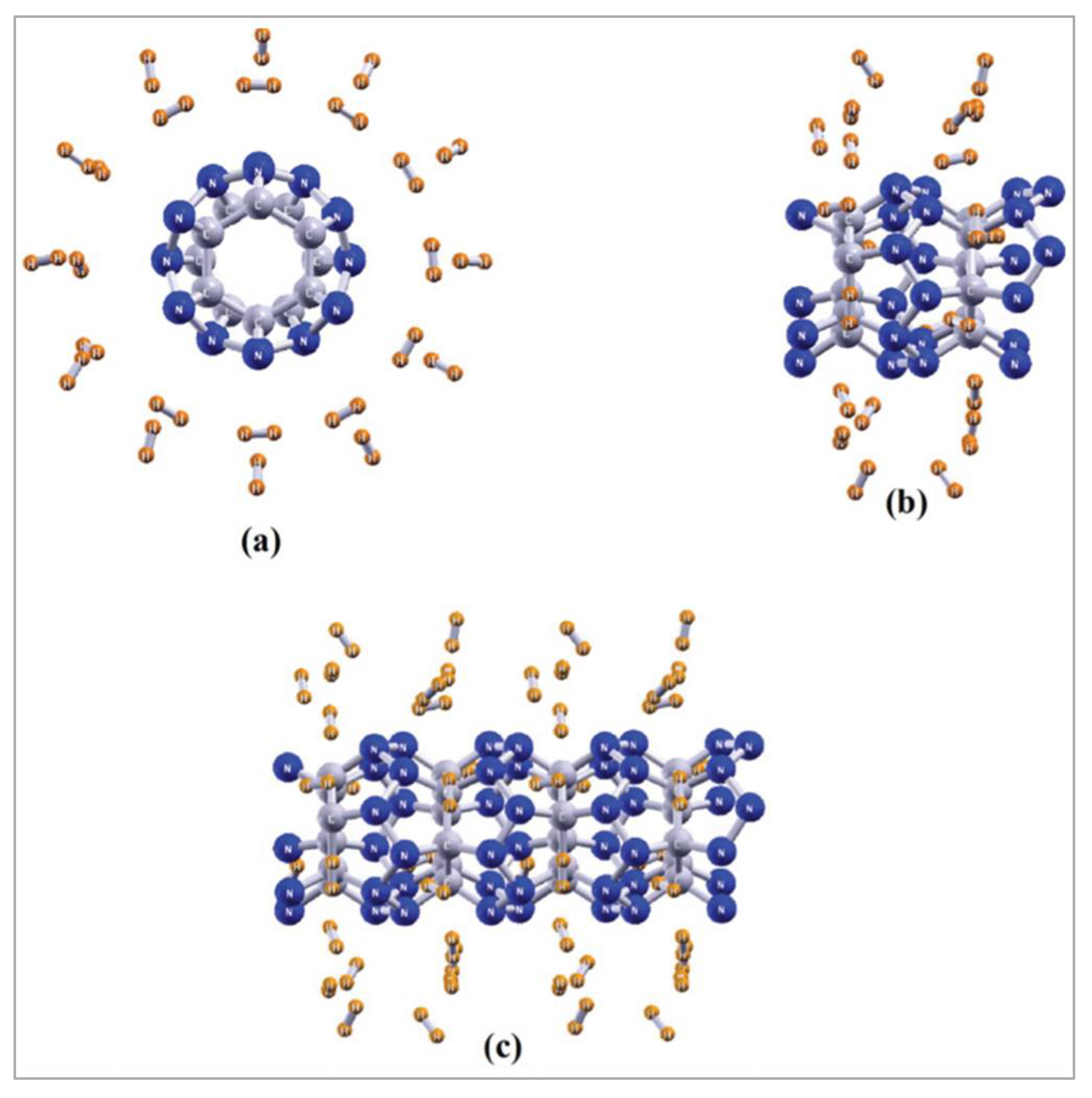
Among all the possible H2-bound forms, it was noted that only the 11 and 12 hydrogen molecule-bound C isomer analogues were not local minima. The feasible region of hydrogen trapping (by the A isomer) was reported from the computed values of free energy at different temperatures and pressures. As the number of trapped hydrogens is same for all the isomers, the gravimetric H2 storage was determined to be 7.2 wt%. The calculated values of hardness mark a nonlinear steady increment for the cases of different numbers of H2-trapped A and C. However, the variation of hardness values for the nH2-loaded B was noted to be random, albeit a correlation was noted between the hardness and binding energy values for the same isomer. Calculated NICS(0) values at the centre of clathrate as well as in their H2-trapped forms show aromaticity. One interesting fact was mentioned: the variations of NICS(0) numbers, binding energy, and hardness for the hydrogen molecule-trapped A forms are in line with each other. Moreover, it was mentioned that application of electric field of 5 × 10−3 a.u. on the molecular hydrogen-bound A form enhances the energy of binding by 0.46 kcal per mol per hydrogen molecule. Desorption process becomes facilitated by the removal of applied electric field. From the most symmetric form, A, C12N24 nanotube was modelled. The skeleton of the C12N24 system comprises a zigzag N12 section which is enclosed by two C6 hexagons (Figure 9). Such systems were modelled by the PBE method. The bond lengths {1.538 (C–C), 1.475 (C–N), and 1.475 (N–N) Å} in the modelled system showed that the type of hybridization could be sp3. In this nanotube unit, the minimum energy position of H2 molecule trapping was also computed, and the most suitable location was determined to be that where the hydrogen molecule interacts with two nitrogen atoms. For the adsorption of 12 and 24 hydrogen molecules, the energy of interaction was computed to be 1.73 and 1.44 kcal/mol per H2, respectively, per unit cell. Calculation of gravimetric H2 storage capacity for such type of binding was noted to be 4.76 and 9.10 wt%, respectively.
7. Conclusions
Density functional theory and conceptual density functional theory revealed that the alkaline earth metal clathrates, Mgn and Can (n = 8–10), may serve the purpose of hydrogen storage. NICS(0,1) of the hydrogen molecule-bound Mgn and Can cages disclosed the aromatic character of such systems. In the ionic clusters, N42−, N62−, Li3+, Na3+, and M5Li7+ (M = C, Si, Ge) show aromaticity. The N64− ring is aromatic with 10π-electrons. However, the N42− ring depicts conflicting aromaticity. Increment in aromaticity occurs for the complexation of N64− ion by Ca2+. In contrast, the N42− unit continues to portray conflicting aromaticity even after binding with two Li+ ions. Li3+ and Na3+ ions show negative NICS(0) value. The D5h symmetric C5Li7+ and Si5Li7+ star-like ions are aromatic in nature. In brief, the carbon-centered star-like ion is π-aromatic and σ-nonaromatic, whereas the Si-centered analogue is π-aromatic as well as σ-aromatic. Hydrogen-trapping potential of all these ions is revealed. Calculated gravimetric hydrogen storage capacity of C5Li7+, Si5Li7+, and Ge5Li7+ ions are 28.0, 18.3, and 9.3 wt%, respectively. In most of the reported ionic systems with the trapping of H2 molecules, the change in aromaticity, hardness, electrophilicity, and interaction energy indicates favourable binding. Good gravimetric storage capacity of hydrogen is also revealed in a few cases. Among the studied neutral cage-like systems, both the B12N12 and C12N12 show aromaticity. Each of these cages can trap up to twelve hydrogen molecules. Among the three possible isomers of C12N12, the most symmetric one (D6d) can store H2 with 7.2 wt%. Moreover, the cluster-assembled material C12N24 can trap hydrogen with 9.1 wt% capacity. For the systems Li3+ and Na3+ and their hydrogen-bound analogues, the local reactivity descriptors reflect reactivity.
Author Contributions
P.K.C. conceptualized the idea of this review article. S.M. wrote the article and P.K.C. reviewed it. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the University Grants Commission, New Delhi for UGC-BSR Research Start-Up-Grant (No. F.30-458/2019(BSR)) and the DST, New Delhi.
Data Availability Statement
All the scientific data reported in this manuscript are available from the author upon reasonable request.
Acknowledgments
S.M. thanks University Grants Commission, New Delhi for UGC-BSR Research Start-Up-Grant (No. F.30-458/2019(BSR)) and his co-workers whose work is presented in this review. P.K.C. would like to thank DST, New Delhi.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Rivard, E.; Trudeau, M.; Zaghib, K. Hydrogen storage for mobility: A review. Materials 2019, 12, 1973. [Google Scholar] [CrossRef] [Green Version]
- Stocker, T.F.; Qin, D.; Plattner, G.-K.; Tignor, M.; Allen, S.K.; Boschung, J.; Nauels, A.; Xia, Y.; Bex, V.; Midgley, P.M. IPCC 2013: Summary for Policymakers. In Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change; Cambridge University Press: Cambridge, UK; New York, NY, USA, 2013. [Google Scholar]
- Walsh, B.S.; Parratt, S.R.; Hoffmann, A.A.; Atkinson, D.; Snook, R.R.; Bretman, A.; Price, T.A.R. The impact of climate change on fertility. Trends Ecol. Evol. 2019, 34, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Raymundo, R.; Asseng, S.; Robertson, R.; Petsakos, A.; Hoogenboom, G.; Quiroz, R.; Hareau, G.; Wolf, J. Climate change impact on global potato production. Eur. J. Agron. 2018, 100, 87–98. [Google Scholar] [CrossRef]
- Mazdiyasni, O.; Kouchak, A.A. Substantial increase in concurrent droughts and heatwaves in the United States. Proc. Natl. Acad. Sci. USA 2015, 112, 11484–11489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- United States Environmental Protection Agency. Inventory of U.S. Greenhouse Gas Emissions and Sinks: 1990–2015. 2017. Available online: https://www.epa.gov/sites/default/files/2017-02/documents/2017_complete_report.pdf (accessed on 8 February 2023).
- BP Statistical Review of World Energy. 2018. Available online: https://www.bp.com/content/dam/bp/business-sites/en/global/corporate/pdfs/energy-economics/statistical-review/bp-stats-review-2018-full-report.pdf (accessed on 8 February 2023).
- Zuttel, A. Hydrogen storage methods. Naturwissenschaften 2004, 91, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Schlapbach, L.; Züttel, A. Hydrogen-storage materials for mobile applications. Nature 2001, 414, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Lubitz, W.; Tumas, W. Hydrogen: An overview. Chem. Rev. 2007, 107, 3900–3903. [Google Scholar] [CrossRef] [PubMed]
- Adeleke, O.A.; Latiff, A.A.A.; Saphira, M.R.; Daud, Z.; Ismail, N.; Ahsan, A.; Aziz, N.A.A.; Al-Gheethi, A.; Kumar, V.; Fadilat, A.; et al. Principles and mechanism of adsorption for the effective treatment of palm oil mill effluent for water reuse. In Nanotechnology in Water and Wastewater Treatment; Elsevier: Amsterdam, The Netherlands, 2019; pp. 1–33. [Google Scholar]
- Tarasov, B.P.; Lototskii, M.V.; Yartys, V.A. Problem of hydrogen storage and prospective uses of hydrides for hydrogen accumulation. Russ. J. Gen. Chem. 2007, 77, 694–711. [Google Scholar] [CrossRef]
- Mondal, S.; Das, P.; Giri, S. Hydrogen Trapping Potential of a Few Novel Molecular Clusters and Ions. In Atomic Clusters with Unusual Structure, Bonding and Reactivity: Theoretical Approaches, Computational Assessment and Applications; Chattaraj, P.K., Pan, S., Merino, G., Eds.; Elsevier Science: Amsterdam, The Netherlands, 2022. [Google Scholar]
- Hubner, O.; Glolss, A.; Fichtner, M.; Klopper, W. On the interaction of dihydrogen with aromatic systems. J. Phys. Chem. A 2004, 108, 3019–3023. [Google Scholar] [CrossRef]
- Srinivasu, K.; Chandrakumar, K.R.S.; Ghosh, S.K. Computational investigation of hydrogen adsorption by alkali metal doped organic molecules: Role of aromaticity. Chem. Phys. Chem. 2009, 10, 427–435. [Google Scholar] [CrossRef]
- Bodrenko, I.V.; Avdeenkov, A.V.; Bessarabov, D.G.; Bibikov, A.V.; Nikolaev, A.V.; Taran, M.D.; Tkalya, E.V. Hydrogen storage in aromatic carbon ring based molecular materials decorated with alkali or alkali-earth metals. J. Phys. Chem. C 2012, 116, 25286–25292. [Google Scholar] [CrossRef]
- Giri, S.; Chakraborty, A.; Chattaraj, P.K. Potential use of some metal clusters as hydrogen storage materials—A conceptual DFT approach. J. Mol. Model. 2011, 17, 777–784. [Google Scholar] [CrossRef] [PubMed]
- Duley, S.; Giri, S.; Sathymurthy, N.; Islas, R.; Merino, G.; Chattaraj, P.K. Aromaticity and hydrogen storage capability of planar N64− and N42− rings. Chem. Phys. Lett. 2011, 506, 315–320. [Google Scholar] [CrossRef]
- Pan, S.; Merino, G.; Chattaraj, P.K. The hydrogen trapping potential of some Li-doped star-like clusters and super-alkali systems. Phys. Chem. Chem. Phys. 2012, 14, 10345–10350. [Google Scholar] [CrossRef] [PubMed]
- Giri, S.; Chakraborty, A.; Chattaraj, P.K. Stability and aromaticity of nH2@B12N12 (n = 1–12) clusters. Nano Rev. 2011, 2, 5767. [Google Scholar] [CrossRef] [PubMed]
- Mondal, S.; Srinivasu, K.; Ghosh, S.K.; Chattaraj, P.K. Isomers of C12N12 as potential hydrogen storage materials and the effect of the electric field therein. RSC Adv. 2013, 3, 6991–7000. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density Functional Theory of Atoms and Molecules; Oxford University Press: New York, NY, USA, 1989. [Google Scholar]
- Geerlings, P.; De Proft, F.; Langenaeker, W. Conceptual density functional theory. Chem. Rev. 2003, 103, 1793–1874. [Google Scholar] [CrossRef]
- Chattaraj, P.K. Chemical Reactivity Theory: A Density Functional View; Taylor and Francis/CRC Press: Boca Raton, FL, USA, 2009. [Google Scholar]
- Cohen, A.J.; Mori-Sanchez, P.; Yang, W. Challenges for density functional theory. Chem. Rev. 2012, 112, 289–320. [Google Scholar] [CrossRef]
- Chakraborty, D.; Chattaraj, P.K. Conceptual density functional theory based electronic structure principles. Chem. Sci. 2021, 12, 6264–6279. [Google Scholar] [CrossRef] [PubMed]
- Mondal, S.; Chattaraj, P.K. Stability and structural dynamics of Be32−cluster. Chem. Phys. Lett. 2014, 593, 128–131. [Google Scholar] [CrossRef]
- Chattaraj, P.K. Electronegativity and hardness: A density functional treatment. J. Indian Chem. Soc. 1992, 69, 173–183. [Google Scholar]
- Chattaraj, P.K.; Roy, D.R. Update 1 of: Electrophilicity index. Chem. Rev. 2007, 107, PR46–PR74. [Google Scholar] [CrossRef]
- Chattaraj, P.K.; Sarkar, U.; Roy, D.R. Electrophilicity index. Chem. Rev. 2006, 106, 2065–2091. [Google Scholar] [CrossRef]
- Mulliken, R.S. Electronic population analysis on LCAO–MO molecular wave functions I. J. Chem. Phys. 1955, 23, 1833. [Google Scholar] [CrossRef] [Green Version]
- Parr, R.G.; Donnelly, R.A.; Levy, M.; Palke, W.E. Electronegativity: The density functional viewpoint. J. Chem. Phys. 1978, 68, 3801. [Google Scholar] [CrossRef]
- Parr, R.G.; Szentpaly, L.V.; Liu, S. Electrophilicity index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Parr, R.G.; Pearson, R.G. Absolute hardness: Companion parameter to absolute electronegativity. J. Am. Chem. Soc. 1983, 105, 7512–7516. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density functional approach to the frontier-electron theory of chemical reactivity. J. Am. Chem. Soc. 1984, 106, 4049–4050. [Google Scholar] [CrossRef]
- Pearson, R.G. Chemical Hardness: Applications from Molecules to Solids; Wiley-VCH: Weinheim, Germany, 1997. [Google Scholar]
- Koopmans, T.A. Über die Zuordnung von Wellenfunktionen und Eigenwerten zu den Einzelnen Elektronen Eines Atoms. Physica 1933, 1, 104–113. [Google Scholar] [CrossRef]
- Yang, W.; Mortier, W.J. The use of global and local molecular parameters for the analysis of the gas-phase basicity of amines. J. Am. Chem. Soc. 1986, 108, 5708–5711. [Google Scholar] [CrossRef]
- Pearson, R.G. Recent advances in the concept of hard and soft acids and bases. J. Chem. Educ. 1999, 64, 561–567. [Google Scholar] [CrossRef]
- Pearson, R.G. The principle of maximum hardness. Acc. Chem. Res. 1993, 26, 250–255. [Google Scholar] [CrossRef]
- Parr, R.G.; Chattaraj, P.K. Principle of maximum hardness. J. Am. Chem. Soc. 1991, 113, 1854–1855. [Google Scholar] [CrossRef]
- Chamorro, E.; Chattaraj, P.K.; Fuentealba, P. Variation of the electrophilicity index along the reaction path. J. Phys. Chem. A 2003, 107, 7068–7072. [Google Scholar] [CrossRef]
- Parthasarathi, R.; Elango, M.; Subramanian, V.; Chattaraj, P.K. Variation of electrophilicity during molecular vibrations and internal rotations. Theor. Chem. Acc. 2005, 113, 257–265. [Google Scholar] [CrossRef]
- Chattaraj, P.K.; Gutierrez-Oliva, S.; Jaque, P.; Toro-Labbé, A. Towards understanding the molecular internal rotations and vibrations and chemical reactions through the profiles of reactivity and selectivity indices: An ab initio SCF and DFT study. Mol. Phys. 2003, 101, 2841–2853. [Google Scholar] [CrossRef]
- Garza, J.; Vargas, R.; Cedillo, A.; Galván, M.; Chattaraj, P.K. Comparison between the frozen core and finite differences approximations for the generalized spin-dependent global and local reactivity descriptors in small molecules. Theor. Chem. Acc. 2006, 115, 257–266. [Google Scholar] [CrossRef]
- Faraday, M.X.X. On new compounds of carbon and hydrogen, and on certain other products obtained during the decomposition of oil by heat. Phil. Trans. R. Soc. 1825, 115, 440–466. [Google Scholar] [CrossRef] [Green Version]
- Kekulè, A. Sur la constitution des substances aromatiques. Bull. Soc. Chim. 1865, 3, 98–100. [Google Scholar]
- Kekulè, A. Untersuchungen uber aromatische verbindungen. Ann. Chem. Pharm. 1866, 137, 129–197. [Google Scholar]
- Kekulè, A. Lehrbuch der Organische Chemie Band 2; Verlag Ferdinand Enke: Erlangen, Germany, 1866; pp. 493–741. [Google Scholar]
- Kekulè, A. Ueber einige condensationsproducte des aldehyds. Liebigs Ann. Chem. 1872, 162, 77–124. [Google Scholar] [CrossRef] [Green Version]
- Giambiagi, M.; De Giambiagi, M.S.; Mundim, K.C. Definition of a multicenter bond index. Struct. Chem. 1990, 1, 423–427. [Google Scholar] [CrossRef]
- Sannigrahi, A.B.; Kar, T. Three-center bond index. Chem. Phys. Lett. 1990, 173, 569–572. [Google Scholar] [CrossRef]
- Matito, E.; Solà, M.; Salvador, P.; Duran, M. Electron sharing indexes at the correlated level. Application to aromaticity calculations. Faraday Discuss. 2007, 135, 325–345. [Google Scholar] [CrossRef] [Green Version]
- Fulton, R.L.; Mixon, S.T. Comparison of covalent bond indexes and sharing indexes. J. Phys. Chem. 1993, 97, 7530–7534. [Google Scholar] [CrossRef]
- Fulton, R.L. Sharing of electrons in molecules. J. Phys. Chem. 1993, 97, 7516–7529. [Google Scholar] [CrossRef]
- Mayer, I. Charge, Bond Order, and Valence in the ab initio SCF Theory. Chem. Phys. Lett. 1983, 97, 270–274. [Google Scholar] [CrossRef]
- Mayer, I. On bond orders and valences in the Ab initio quantum chemical theory. Int. J. Quantum Chem. 1986, 29, 73–84. [Google Scholar] [CrossRef]
- Mayer, I. Bond orders and valences from ab initio wave functions. Int. J. Quantum Chem. 1986, 29, 477–483. [Google Scholar] [CrossRef]
- Mayer, I. Bond order and valence: Relations to Mulliken’s population analysis. Int. J. Quantum Chem. 1984, 26, 151–154, Addendum Int. J. Quantum Chem. 1985, 28, 419–419. [Google Scholar] [CrossRef]
- Fradera, X.; Austen, M.A.; Bader, R.F.W. The Lewis Model and Beyond. J. Phys. Chem. A 1999, 103, 304–314. [Google Scholar] [CrossRef]
- Bader, R.F.W.; Stephens, M.E. Spatial localization of the electronic pair and number distributions in molecules. J. Am. Chem. Soc. 1975, 97, 7391–7399. [Google Scholar] [CrossRef]
- Poater, J.; Fradera, X.; Duran, M.; Sola, M. An Insight into the Local Aromaticities of Polycyclic Aromatic Hydrocarbons and Fullerenes. Chem. Eur. J. 2003, 9, 1113–1122. [Google Scholar] [CrossRef]
- Matito, E.; Duran, M.; Solà, M. The aromatic fluctuation index (FLU): A new aromaticity index based on electron delocalization. J. Chem. Phys. 2005, 122, 014109, Erratum in J. Chem. Phys. 2006, 125, 059901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giambiagi, M.S.; Giambiagi, M.; Fortes, M.S. Multicenter bonds, bond valence and bond charge apportionment. J. Mol. Struct. Theochem 1997, 391, 141–150. [Google Scholar] [CrossRef]
- Giambiagi, M.; De Giambiagi, M.S.; Dos Santos Silva, C.D.; De Figuereido, A.P. Multicenter bond indices as a measure of aromaticity. Phys. Chem. Chem. Phys. 2000, 2, 3381–3392. [Google Scholar] [CrossRef]
- Bultinck, P.; Ponec, R.; Van Damme, S. Multicenter bond indices as a new measure of aromaticity in polycyclic aromatic hydrocarbons. J. Phys. Org. Chem. 2005, 18, 706–718. [Google Scholar] [CrossRef]
- Bultinck, P.; Fias, S.; Ponec, R. Local Aromaticity in Polycyclic Aromatic Hydrocarbons: Electron Delocalization versus Magnetic Indices. Chem.-Eur. J. 2006, 12, 8813–8818. [Google Scholar] [CrossRef]
- Bultinck, P.; Ponec, R.; Carbo-Dorca, R. Aromaticity in linear polyacenes: Generalized population analysis and molecular quantum similarity approach. J. Comput. Chem. 2007, 28, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Bultinck, P.; Rafat, M.; Ponec, R.; Gheluwe, B.V.; Carbo-Dorca, R.; Popelier, P.J. Electron Delocalization and Aromaticity in Linear Polyacenes: Atoms in Molecules Multicenter Delocalization Index. J. Phys. Chem. A 2006, 110, 7642–7648. [Google Scholar] [CrossRef]
- Ponec, R.; Bultinck, P.; Saliner, A.G. Multicenter Bond Indices as a New Means for the Quantitative Characterization of Homoaromaticity. J. Phys. Chem. A 2005, 109, 6606–6609. [Google Scholar] [CrossRef] [PubMed]
- Chattaraj, P.K.; Roy, D.R.; Elango, M.; Subramanian, V. Chemical reactivity descriptor based aromaticity indices applied to Al42− and Al44− systems. J. Mol. Struct. Theochem 2006, 759, 109–110. [Google Scholar] [CrossRef]
- Roy, D.R.; Bultinck, P.; Subramanian, V.; Chattaraj, P.K. Bonding, reactivity and aromaticity in the light of the multicenter indices. J. Mol. Struct. Theochem 2008, 854, 35–39. [Google Scholar] [CrossRef]
- Kruszewski, J.; Krygowski, T.M. Definition of aromaticity basing on the harmonic oscillator model. Tetrahedron Lett. 1972, 36, 3839–3842. [Google Scholar] [CrossRef]
- Krygowski, T.M. Crystallographic studies of inter- and intramolecular interactions reflected in aromatic character of π-electron systems. J. Chem. Inf. Comput. Sci. 1993, 33, 70–78. [Google Scholar] [CrossRef]
- Krygowski, T.M.; Cyranski, M. Structural aspects of aromaticity. Chem. Rev. 2001, 101, 1385–1420. [Google Scholar] [CrossRef] [PubMed]
- Schleyer, P.v.R.; Maerker, C.; Dransfeld, A.; Jiao, H.; Hommes, N.J.R.v.E. Nucleus-independent chemical shifts: A simple and efficient aromaticity probe. J. Am. Chem. Soc. 1996, 118, 6317–6318. [Google Scholar] [CrossRef]
- Hirsch, A.; Chen, Z.; Jiao, H. Spherical aromaticity in Ih symmetrical fullerenes: The 2(N + 1)2 rule. Angew. Chem. Int. Ed. 2000, 39, 3915–3917. [Google Scholar] [CrossRef]
- Poater, J.; Solà, M. Open-shell spherical aromaticity: The 2N2 + 2N + 1 (with S = N + ½) rule. Chem. Commun. 2011, 47, 11647–11649. [Google Scholar] [CrossRef]
- Dennington, R.; Keith, T.A.; Millam, J.M. GaussView, Version 3.0, and 5.0.8; Semichem, Inc.: Shawnee, KS, USA, 2009. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.; Vreven, T., Jr.; Kudin, K.N.; Burant, J.C.N.; et al. Gaussian 03, Revision B.03; Gaussian, Inc.: Pittsburgh, PA, USA, 2003. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Schlegel, H.B.; Scalmani, G.; Barone, V.; Mennucci, B.; et al. Gaussian 09, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal–amorphous-semiconductor transition in germanium. Phys. Rev. B 1994, 49, 14251. [Google Scholar] [CrossRef] [PubMed]
- Kokalj, A. Computer graphics and graphical user interfaces as tools in simulations of matter at the atomic scale. Comp. Mater. Sci. 2003, 28, 155–168. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188. [Google Scholar] [CrossRef]
- Zhou, J.; Wang, Q.; Sun, Q.; Jena, P.; Chen, X.S. Electric field enhanced hydrogen storage on polarizable materials substrates. Proc. Natl. Acad. Sci. USA 2010, 107, 2801–2806. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Hwang, J.-Y.; Shi, S. Hydrogen storage in mesoporous metal oxides with catalyst and external electric field. J. Phys. Chem. C 2010, 114, 7178–7184. [Google Scholar] [CrossRef]
- Vogler, A.; Wright, R.E.; Kunkely, H. Photochemical reductive cis-elimination in cis-diazidobis(triphenylphosphane)platinum(II) evidence of the formation of bis(triphenylphosphane)platinum(0) and hexaazabenzene. Angew. Chem. Ind. Ed. Engl. 1980, 19, 717–718. [Google Scholar] [CrossRef]
- Chung, G.; Schmidt, M.W.; Gordon, M.S. An ab Initio study of potential energy surfaces for N8 isomers. J. Phys. Chem. A 2000, 104, 5647–5650. [Google Scholar] [CrossRef] [Green Version]
- Engelke, R. Ab initio correlated calculations of six nitrogen (N6) isomers. J. Phys. Chem. 1992, 96, 10789–10792. [Google Scholar] [CrossRef]
- Ha, T.-K.; Nguyen, M.T. The identity of the six nitrogen atoms (N6) species. Chem. Phys. Lett. 1992, 195, 179–183. [Google Scholar] [CrossRef]
- Strout, D.L. Acyclic N10 fails as a high energy density material. J. Phys. Chem. A 2002, 106, 816–818. [Google Scholar] [CrossRef]
- Perez-Peralta, N.; Contreras, M.; Tiznado, W.; Stewart, J.K.; Donald, J.; Merino, G. Stabilizing carbon-lithium stars. Phys. Chem. Chem. Phys. 2011, 13, 12975–12980. [Google Scholar] [CrossRef] [PubMed]
- Tiznado, W.; Perez-Peralta, N.; Islas, R.; Toro-Labbe, A.; Ugalde, J.M.; Merino, G. Designing 3-D Molecular Stars. J. Am. Chem. Soc. 2009, 131, 9426–9431. [Google Scholar] [CrossRef]
- Kuhn, A.; Sreeraj, P.; Pöttgen, R.; Wiemhöfer, H.-D.; Wilkening, M.; Heitjans, P. Li NMR Spectroscopy on Crystalline Li12Si7: Experimental Evidence for the Aromaticity of the Planar Cyclopentadienyl-Analogous Si56− Rings. Angew. Chem. Int. Ed. 2011, 50, 12099–12102. [Google Scholar] [CrossRef]
- Jena, N.K.; Srinivasu, K.; Ghosh, S.K. Computational investigation of hydrogen adsorption in silicon-lithium binary clusters. J. Chem. Sci. 2012, 124, 255–260. [Google Scholar] [CrossRef]
- Sun, Q.; Wang, Q.; Jena, P. Storage of molecular hydrogen in B-N cage: Energetics and thermal stability. Nano Lett. 2005, 5, 1273–1277. [Google Scholar] [CrossRef] [PubMed]
- Wen, S.-H.; Deng, W.-Q.; Han, K.-L. Endohedral BN metallofullerene M@B36N36 complex as promising hydrogen storage materials. J. Phys. Chem. C 2008, 112, 12195–12200. [Google Scholar] [CrossRef]
- Cui, X.-Y.; Yang, B.-S.; Wu, H.-S. Ab initio investigation of hydrogenation of (BN)16: A comparison with that of (BN)12. J. Mol. Struct. (Theochem) 2010, 941, 144–149. [Google Scholar] [CrossRef]
Figure 1.
Optimized geometries of H2Mn (where M = Mg, Ca; n = 8, 9, 10) clusters. This figure is reproduced from Reference [17].
Figure 1.
Optimized geometries of H2Mn (where M = Mg, Ca; n = 8, 9, 10) clusters. This figure is reproduced from Reference [17].
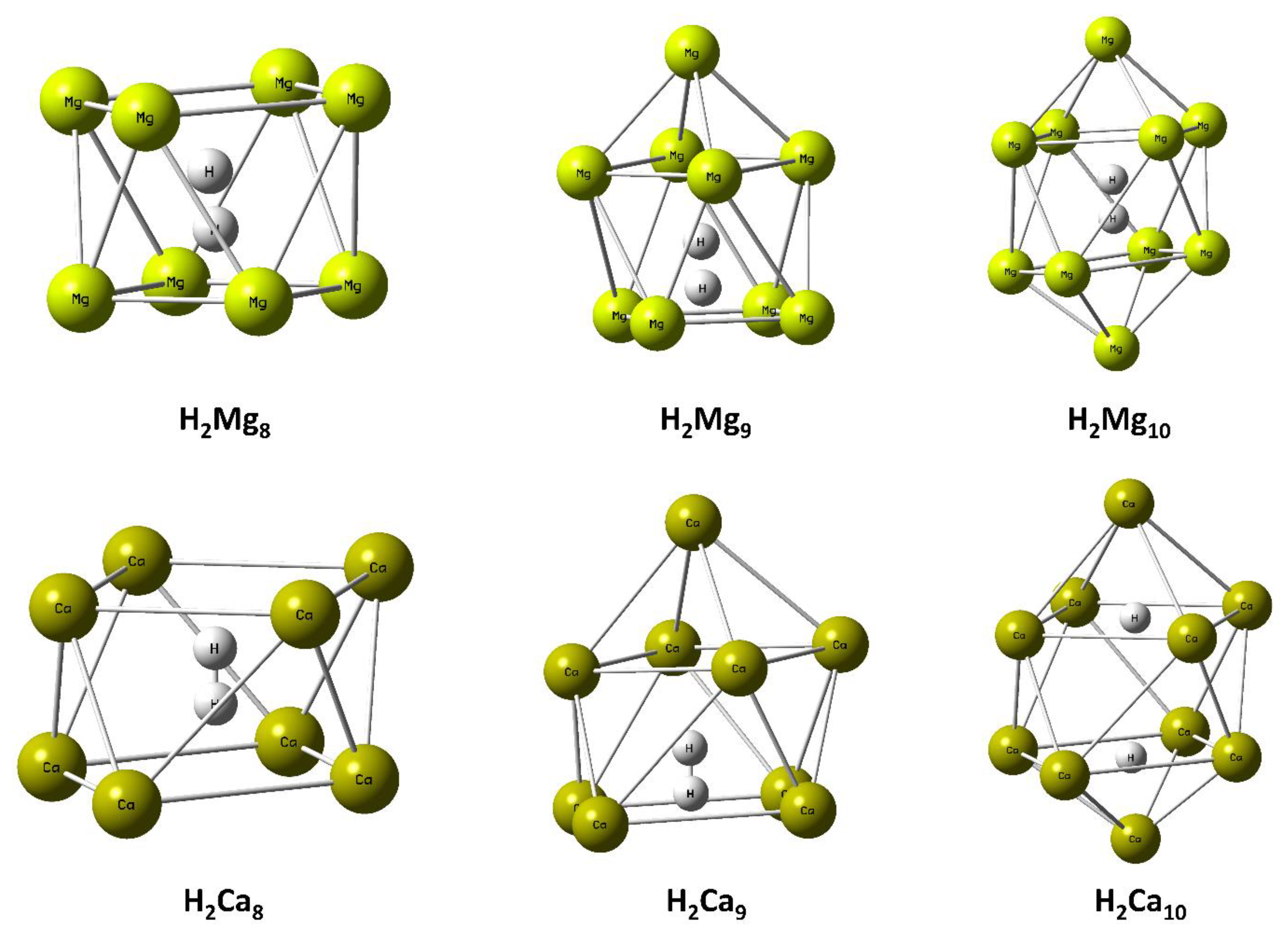
Figure 2.
(a) Local minima forms of N6Ca2 and N4Li2 and their corresponding H2 trapped equivalents. Color code: blue for N, pink for Li, green for Ca, and white for H atoms. (b) NICS-scan plots for N64−, N42−, benzene (Bz), and cyclobutadiene (Cb). This figure is reproduced from Reference [18].
Figure 2.
(a) Local minima forms of N6Ca2 and N4Li2 and their corresponding H2 trapped equivalents. Color code: blue for N, pink for Li, green for Ca, and white for H atoms. (b) NICS-scan plots for N64−, N42−, benzene (Bz), and cyclobutadiene (Cb). This figure is reproduced from Reference [18].
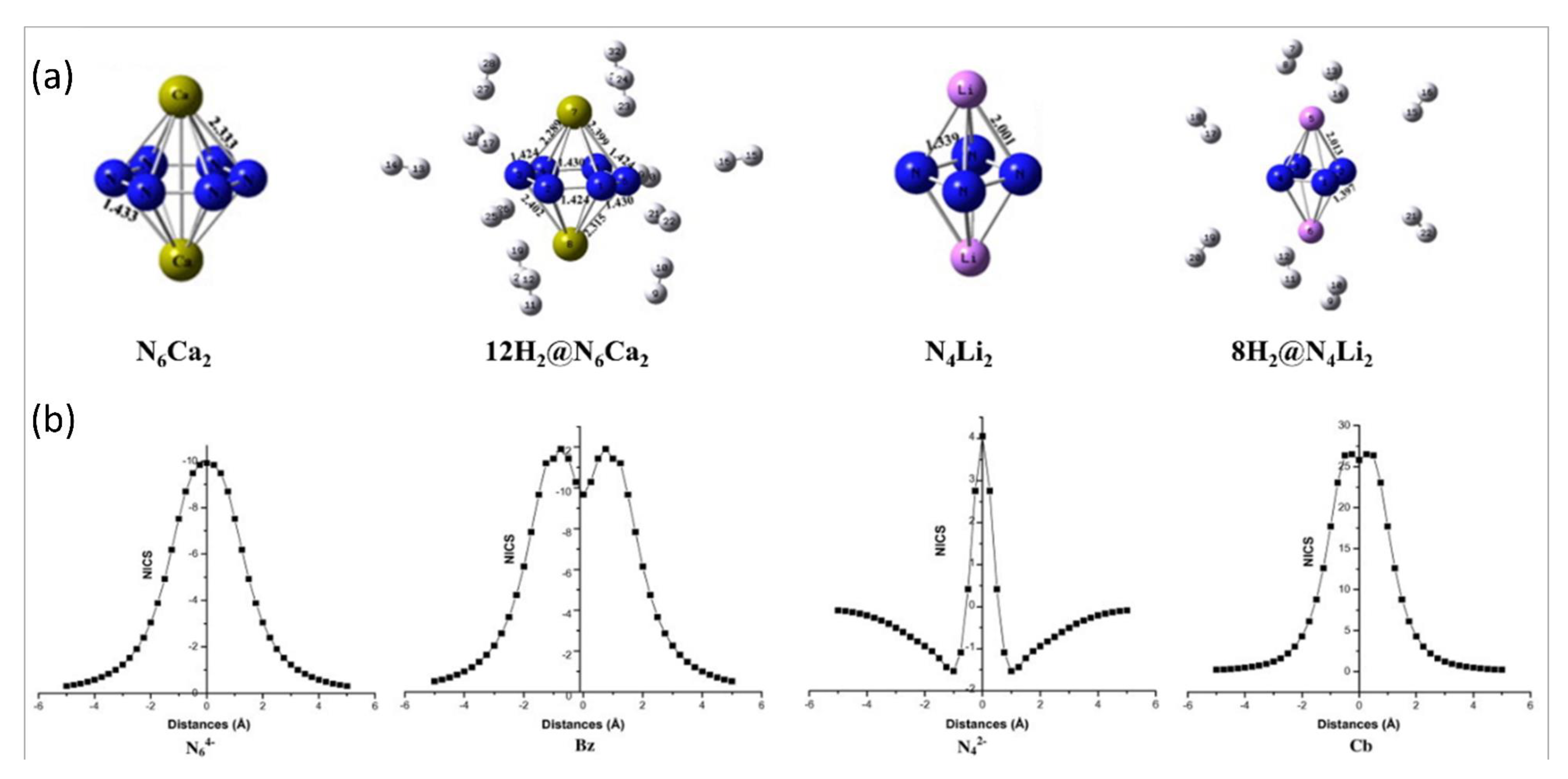
Figure 3.
(a) Forms of H2-bound Li3+ and (b) H2-bound Na3+. This figure is reproduced from Reference [17].
Figure 3.
(a) Forms of H2-bound Li3+ and (b) H2-bound Na3+. This figure is reproduced from Reference [17].
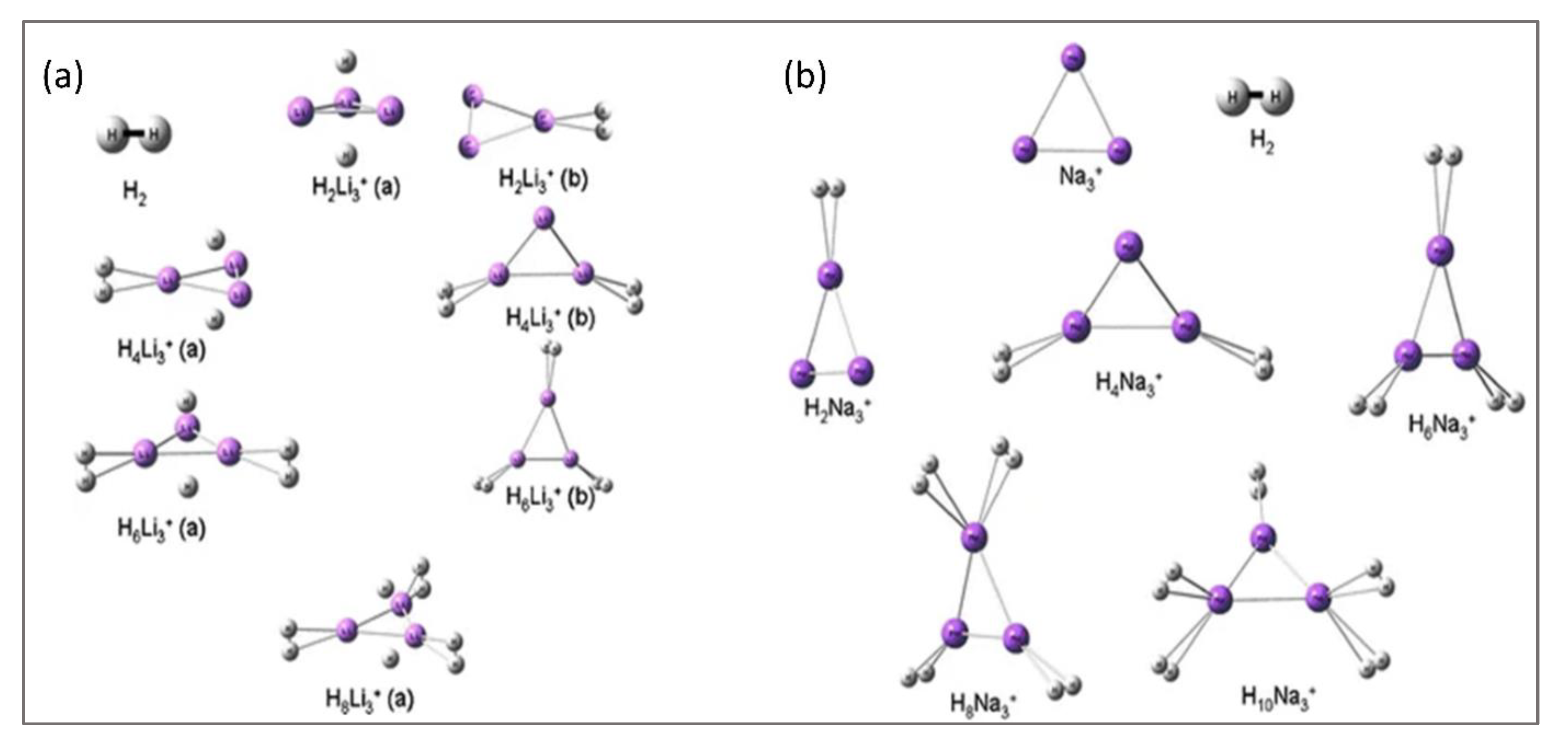
Figure 4.
C5Li7+ and 21 H2@C5Li7+ at the M06/6-311+G(d,p) level. This figure is reproduced from Reference [19].
Figure 4.
C5Li7+ and 21 H2@C5Li7+ at the M06/6-311+G(d,p) level. This figure is reproduced from Reference [19].

Figure 5.
Local minima geometries of M5Li7+ (M = Si, Ge) and their H2 trapped analogues at the M06/6-311+G(d,p) level. This figure is reproduced from the reference [19].
Figure 5.
Local minima geometries of M5Li7+ (M = Si, Ge) and their H2 trapped analogues at the M06/6-311+G(d,p) level. This figure is reproduced from the reference [19].
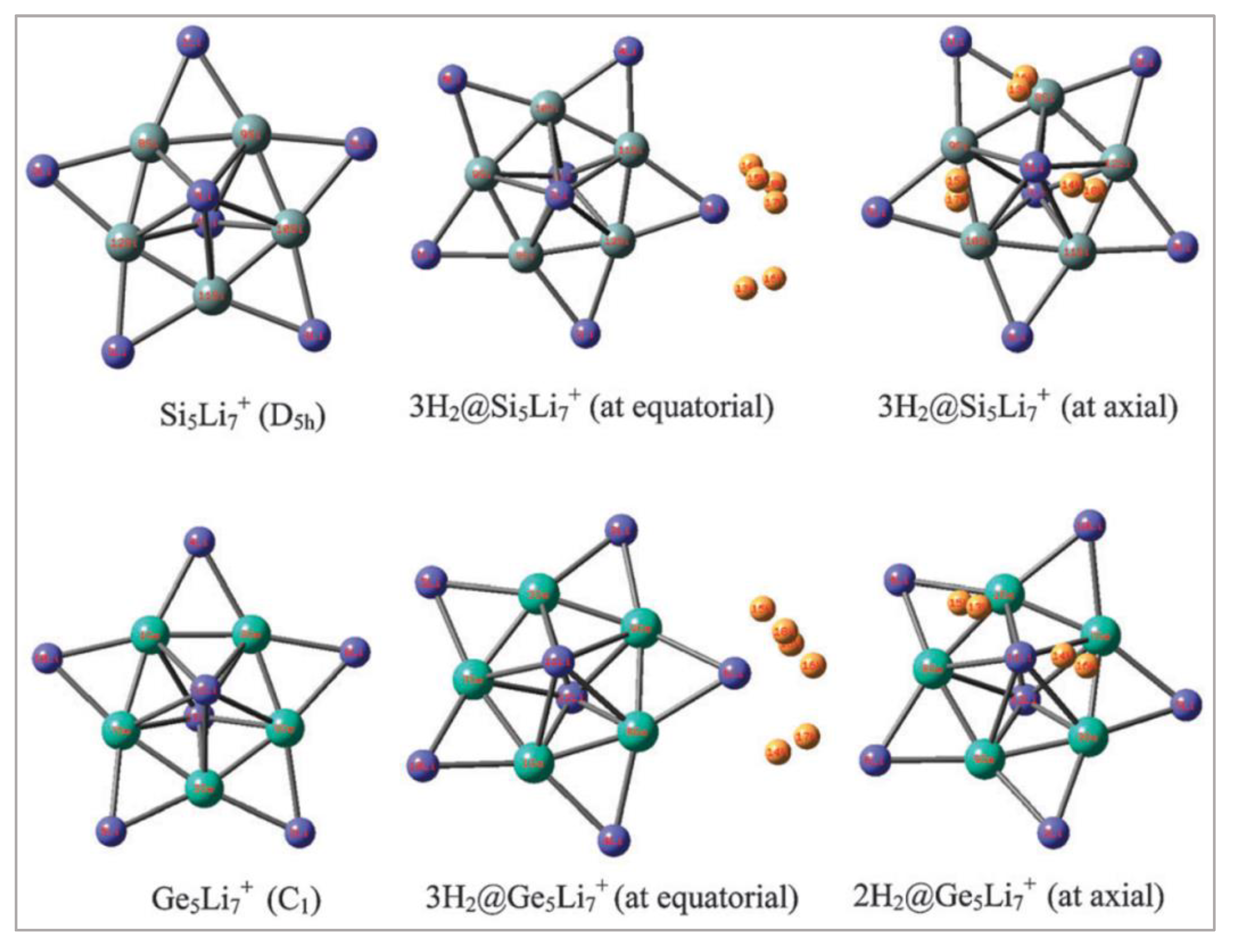
Figure 6.
Energy minima forms (B3LYP/6-311+G(d)) of B12N12 and selected hydrogen molecule-bound analogues. This figure is reproduced from Reference [20].
Figure 6.
Energy minima forms (B3LYP/6-311+G(d)) of B12N12 and selected hydrogen molecule-bound analogues. This figure is reproduced from Reference [20].
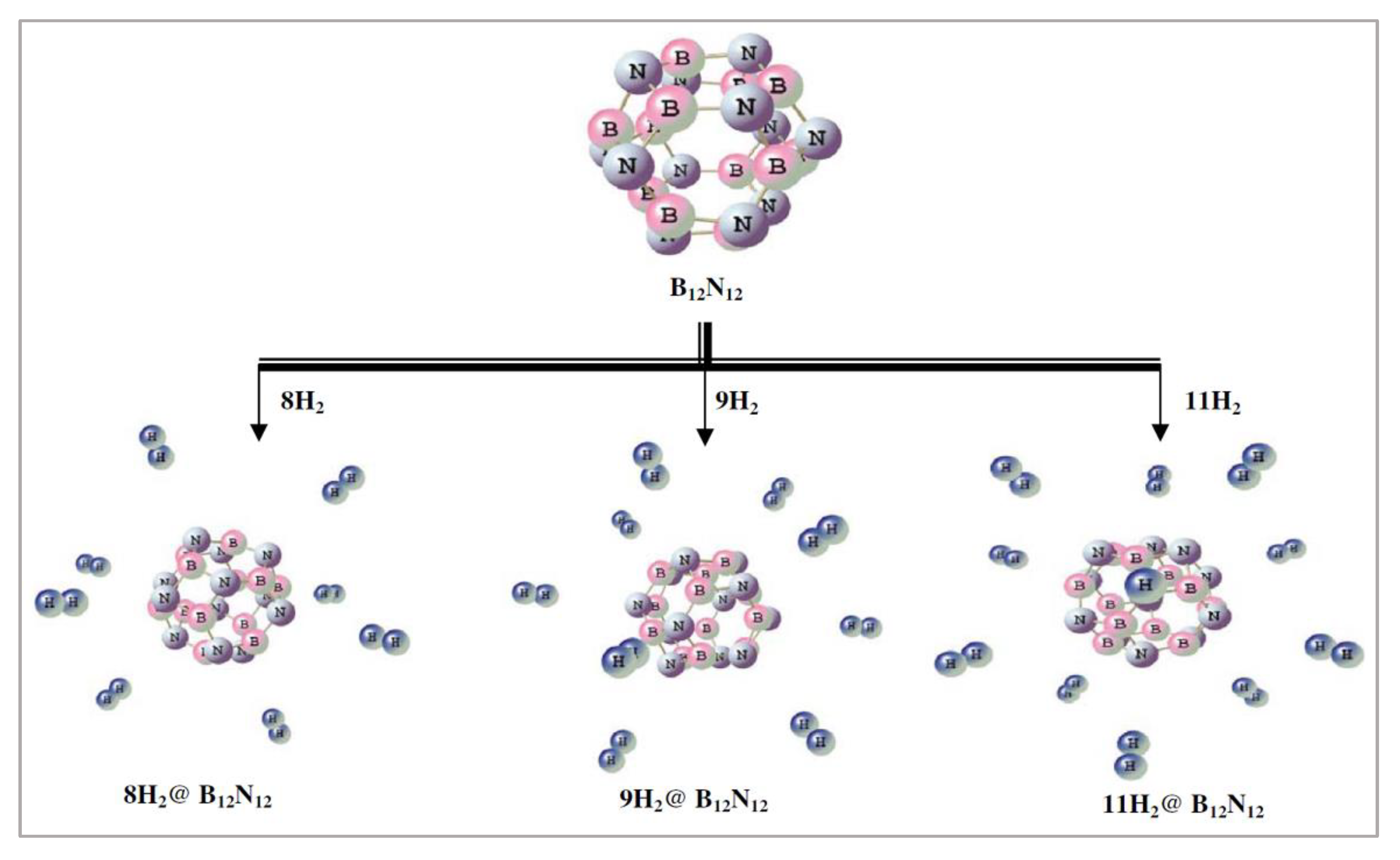
Figure 7.
Relative energy profile for molecular hydrogen absorption on a single BN molecule. This figure is reproduced from Reference [20].
Figure 7.
Relative energy profile for molecular hydrogen absorption on a single BN molecule. This figure is reproduced from Reference [20].

Figure 8.
Local minima form of C12N12-A, C12N12-B and C12N12-C. In (a) panel, the top view is provided, whereas in panel (b), the side view is shown. This figure is reproduced from Reference [21].
Figure 8.
Local minima form of C12N12-A, C12N12-B and C12N12-C. In (a) panel, the top view is provided, whereas in panel (b), the side view is shown. This figure is reproduced from Reference [21].
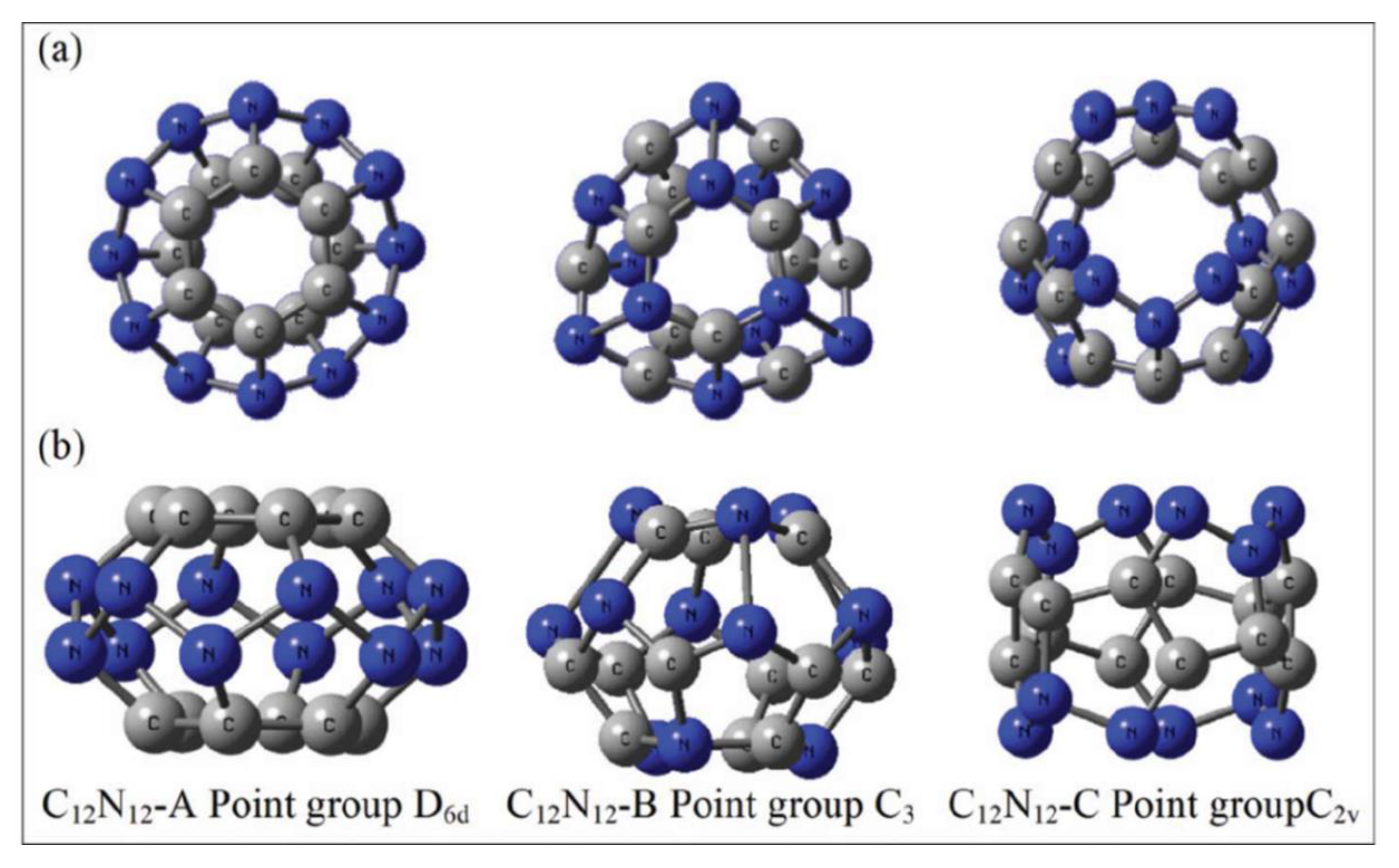
Figure 9.
The segment (a) indicates the top view and the (b) marks the side view. The (c) represents a super cell form of the C12N24(H2)24 unit cell. This figure is reproduced from Reference [21].
Figure 9.
The segment (a) indicates the top view and the (b) marks the side view. The (c) represents a super cell form of the C12N24(H2)24 unit cell. This figure is reproduced from Reference [21].
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Mondal, S.; Chattaraj, P.K. Aromatic Clusters and Hydrogen Storage. Energies 2023, 16, 2833. https://doi.org/10.3390/en16062833
AMA Style
Mondal S, Chattaraj PK. Aromatic Clusters and Hydrogen Storage. Energies. 2023; 16(6):2833. https://doi.org/10.3390/en16062833
Chicago/Turabian StyleMondal, Sukanta, and Pratim Kumar Chattaraj. 2023. "Aromatic Clusters and Hydrogen Storage" Energies 16, no. 6: 2833. https://doi.org/10.3390/en16062833
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.