Abstract
Some of the issues concerning energy security and climate change can be addressed by employing nuclear power (NP) to supply the energy required for the conversion of carbon dioxide (CO2) into chemicals, products, and materials. Nuclear energy represents a neutral carbon source that can be generated sustainably, reliably, and consistently. Nuclear power plants (NPPs) could supply energy in the form of heat, electricity, and ionizing radiation to drive CO2 chemical reactions underpinning NP-to-X type of pathways. CO2 conversion processes are either commercially available or emerging technologies at different developmental maturity stages. This work reviews the published literature (articles and patents) that reports R&D results and the understanding and development of chemical reactions and processes, as well as the efforts in integrating NPPs and chemical processes (CPs). As will be made evident, a new industrial era for the manufacturing of decarbonized chemicals, products, and materials will be possible by developing and implementing new (more energy- and carbon-efficient) processes responding to the NP-to-X pathways. This new decarbonizing platform not only contributes to achieving net zero goals but also broadens the NPP product beyond electricity.
1. Introduction
Some of the most significant concerns in the 21st century have been energy security and climate change. Ensuring energy availability to underpin economic growth has been based on an exacerbated reliance on fossil resources, which are readily available and globally accessible. However, the detrimental impacts on the climate of the consequential CO2 emissions have driven R&D activities on the development of mitigation and removal technologies. Therefore, the mitigation of carbon emissions and the use of renewable or carbon (C) neutral energy systems are intrinsically connected to attaining energy security under stable future climate systems [1]. However, renewable energy generation inherently depends on weather conditions causing power fluctuations, intermittency, and/or interruptions, which in turn have brought about the development of energy storage technologies. In this regard, chemical compounds could be employed as energy carriers to facilitate storage and transportation (energy transmission). The identification of compounds exhibiting high energy density and with low production costs started over a decade ago when methane was identified. However, the development of production processes with low energy and carbon intensity is still needed. A new R&D area emerged and was coined “Power-to-Gas” (PtG), with the electrochemical methanation reaction as the process of choice. Thus, the production of methane using renewable energy and a renewable resource, such as CO2, gave rise to a new renewable fuel, i.e., renewable natural gas (RNG). Alternatively, and in terms of their energy density and transportability, other energy carriers became of interest, such as dimethyl ether (DME) and, more particularly, liquids, e.g., methanol and Fischer–Tropsch products. The economic benefits and competitive value-added of chemicals production broadened the range of energy carriers and expanded the topic beyond methane-to-“X” products, i.e., PtX pathways. In some instances, other acronyms include “V” for value in PtV and “L” for liquids in PtL, in which case, attention was focused on fuels and, more specifically, methanol. In summary, the requirements for the selected energy carrier include high energy density, low energy intensity, low carbon intensity, low production and storage costs, and easy transportation. Since power is supplied in the form of electricity, the type of chemical process (CP) receiving the most attention is the electrochemical process.
Opposite to renewable energy, nuclear power (NP) appeals include consistency, reliability, and security, and NP is an energy resource considered C-neutral. These characteristics make nuclear energy an excellent solution to replace part of the energy demand currently being met by fossil sources (e.g., coal and natural gas). Additionally, nuclear power plants (NPP) provide a more versatile energy mix in the form of heat (thermal load), electricity (off-peak current), and radiation (neutrons, fast electrons, and gamma/X-rays). The progress and deployment potentialities of micro- and small modular nuclear reactors enable the application of multiple approaches for the implementation of decarbonization strategies. The various nuclear reactor technologies (e.g., pebble bed reactors, accelerator-driven systems, nuclear battery reactors, advanced nuclear fuel designs, super-critical CO2 reactors, and others operated at higher temperatures, such as fusion reactors, molten salt reactors, liquid metal-cooled fast reactors, fluoride salt-cooled high-temperature reactors, and high-temperature gas reactors) [2] offer a variety of options to harness and adapt to energy integration into CPs. Larger-scale nuclear reactors of Generation IV [3] are specifically interested in providing heat within a temperature range of 550 °C–850 °C [4]. More demanding chemical reactions may find grounds for integration into Very-High-Temperature Reactors (VHTR) for temperatures in the range of 1000 °C or the Ultra-High-Temperature Reactor (UHTR) for temperatures of 1300 °C [5]. The heat provided at these temperature ranges might represent a cost-effective solution and a more competitive and environmentally amicable manner for industrial heat than fossil fuel combustion. The safety features of many of the Generation IV reactors are thought to be the worthiest characteristics to consider for integration into CPs. Regardless of the great R&D advances reached by these attractive reactors, their commercial implementation is further into the future, and the testing of built prototypes has brought up both developmental insights but also challenges to be addressed. Nevertheless, all these advances and potentialities justify R&D efforts in the development of NP-to-X pathways, which will require the development of integrated energy systems (IES) coupling NP to chemical processes. Currently, these efforts are minimal and nonexistent within the CO2 valorization area.
Japan [6] and, more recently, Europe [7] initiated efforts on PtG pathways to develop long-term storage solutions for renewable electricity. The goal was the production of high energy-containing gases, such as CH4 and H2, from carbon sources and renewable electricity. In the Japanese conception, CO2 is recycled [8] via the methanation reaction ((R1), the Sabatier reaction) [9,10] using H2 produced by the electrolysis of seawater [11] and powered by renewable energy [12]. Process development and pilot plant testing took place during the 1980s–1990s [12].
CO2 + 4H2 → CH4 + 2H2O
The European process concept follows the Japanese one, though their R&D efforts broaden the water electrolysis window to a wider range of electrolyzers. In this case, rather than considering a recycling of CO2, they also generalize it to emission valorization. Thus, the PtG concept developed in Japan, as well as in Germany, Switzerland, Denmark, and France, can be represented by the scheme shown in Figure 1. The main drawbacks of PtG are relatively low efficiency and high costs. The process economy was largely influenced by RNG production costs, which were strongly dependent on the annual operational time and the electricity price [13].
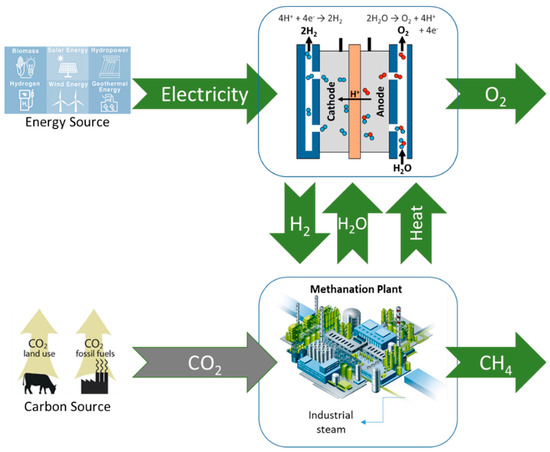
Figure 1.
General PtG concept developed in Japan and Europe.
As mentioned above, this work directly concerns NP–chemical-processing IESs, which integrate the energy offered by an NPP with that demanded by a chemical plant (CP), which forms the basis for NP-to-X pathways. This type of IES has a first consequence in the diversification of the NPP product slate beyond electricity into chemicals, which can reach more distant markets than those accessible by transmission lines. NPP integration into CPs is not new, and a long history of efforts dates back to the 1970s, with two significant projects, namely Nuclear Hydrogen (NH) [14] and ADAM and EVA (A&E) [15]. These two programs are the most relevant examples of devoted efforts on the technology development of NP-CP IES that underpinned the potentialities for the development of NP-to-X pathways. The early studies on NH were high-temperature applications and were based on the thermochemical production of bulk hydrogen. Hydrogen can be produced from the splitting of the water molecule, which is an energy-intense reaction. In fact, at standard conditions, DG° = 237.2 kJ/mol and DH° = 285.8 kJ/mol, and only at temperatures above 4400 °C does DG become negative. This thermodynamic limitation has been overcome by designing multi-reaction cyclic pathways, which enable the process chemistry to drive at temperatures above 550 °C [16]. Examples of the investigated thermochemical water splitting cycles include (i) iodine–sulfur, (ii) iron chlorides, (iii) hybrid sulfur, (iv) Ca–Br, and (v) Cu–Cl. To lower the process temperature, a cycle is created, coupling a high-temperature endothermic reaction with a low-temperature exothermic one [16]. These production pathways were considered by the Nuclear Hydrogen Program and are characterized as being stoichiometric reactions, and the required energy was supplied by an NPP [17,18,19,20,21,22,23,24,25,26]. However, the energy intensity of these hydrogen production technologies is not better than that of the existing commercially operated technologies. Importantly, then, it is important for intensification/integration efforts to avoid leveraging inefficiencies of either the candidate CP or nuclear resources. Therefore, the development of CPs with better energy efficiency is needed.
Another early study was the A&E process based on the chemical heat pipe concept (see Figure 2), which involves producing a chemical energy carrier through an endothermic reaction and transporting it to a remote location where it could be converted back to energy or to other chemical products. It was first tested in Germany using heat from a high-temperature gas-cooled nuclear reactor for the first half cycle (EVA, from the German term “Einzelrohr-Versuchs-Anlage”, meaning single-tube experimental facility) to produce synthesis gas (syngas, a mixture of carbon monoxide and hydrogen), by methane steam reforming (SMR, the endothermic reaction (R2)). The Adam half-cycle of the process was run in a distant location and consisted of the methanation reaction of syngas (an exothermic reaction, (R3)). The methanation heat of reaction could be used for electricity generation or any other process heat [27].
CH4 + H2O → CO + 3H2 ΔH298K = 206 kJ/mol
CO + 3H2 → CH4 + H2O ΔH298K = −206 kJ/mol
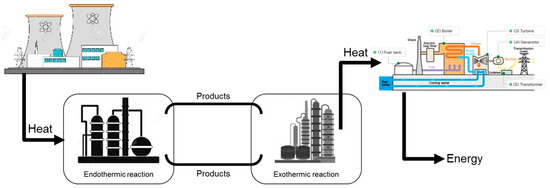
Figure 2.
The chemical heat pipe concept (Based on concepts from [15,27]).
As can be derived from the above-referred description of the NH and A&E projects, great advances have been achieved in NP-CP IES, which can be used to undertake further development of NP-to-X pathways. In fact, current efforts are devoted to green hydrogen production, but unfortunately, nothing has been reported in regard to CO2 valorization. In this regard and for the purpose of this work, CO2 valorization will be seen as its chemical transformation into value-added products. In connection with NE, the reported CO2 valorization work using available nuclear energy is related to driving or inducing reactions by means of ionizing radiation, for which chemistry and chemical reactions were revised recently [28]. In particular, dry reforming, the reaction of methane with CO2 to produce syngas, has been proven to be radiation-induced [29]. Additionally, the opportunities for employing the energy mix (heat, electricity, and radiation) available from NPPs have been identified [30], which further emphasizes the existing potential of implementing NP-to-X pathways in the immediate future.
It is important to point out that CO2 exhibits high stability and/or low reactivity, and its chemical transformation involves significant energy consumption, ergo energy-intense processes. Accordingly, to accomplish its conversion typically requires severe conditions, such as high temperatures and pressures. Therefore, any new process cannot rely on fossil energy resources (e.g., coal and natural gas), in which case a significant quantity of greenhouse gases (GHGs) would be released. Consequently, new processes need to be developed not only to take advantage of neutral-C energy generation but also to attain cost-effectiveness through more energy-efficient processes. In fact, efficiency improvement is one of the recommended approaches for emission reduction purposes. However, emissions from industrial processes (e.g., cement, steel, refining, etc.) and from high-temperature heat (e.g., >1000 °C) have been recognized as being difficult to cost-effectively avoid [31]. Technological solutions have been developed, but these are unsustainable, absurdly expensive, or have not reached commercial scale [32].
Thus, CO2 keeps accumulating in the atmosphere, reaching a concentration of 417 parts per million (ppm), with global emissions of over 37 GtonCO2/y in 2022 [33]. The industrial and energy sectors are responsible for nearly 70% of these emissions [34]. The magnitude of CO2 emissions and the difficulties in their reduction support their categorization as renewable feedstock [35,36,37,38,39]. However, prior to feeding it to any chemical reactor, emissions need to be captured. Although many capture technologies have been commercially available for over 50 years, the installed capacity was about 40–50 Mton/y in 2019. Since then, this capacity has undergone a steady growth of over 57% (57–68%) interannually to reach a level of 361 Mton/y by 31 July 2023 [40]. The CCS projects tracking that the International Energy Agency (IEA) carries out indicated that, most likely, the total amount of CO2 potentially captured by 2030 would be around 435 Mton/y, and the storage capacity would increase to around 615 Mton/y [41]. Therefore, in the best-case scenario and considering that during the last five years, emissions have remained very close to the 37 Gton/y range, CCS alone will contribute to mitigating less than 2% of the global emissions. This number increases favorably (up to 60%) when the 2050 net-zero goal is considered since this goal assigned only 1 Gton/y to be captured and stored by 2050 [41]. The significant role that carbon capture (CC) plays in decarbonization is clear, but it is not enough, as has already been established by the Intergovernmental Panel on Climate Change (IPCC) [42] and the IEA [43]. Consequently, they have recommended the incorporation of additional approaches to the decarbonization strategy, including energy demand reduction, energy efficiency improvements, and commercial implementation of conversion processes [42,43,44]. In fact, using emitted CO2 to produce fuels, chemicals, and materials could complement CCS role while creating revenues, which will incentivize industry by providing economic sustainability [45], particularly those recognized as difficult to decarbonize (e.g., cement, steel, refining, and high-temperature heat) [31].
The integration of chemical CO2-utilization processes to CC technologies might serve as a vehicle to close the C-cycle and underpin a C-based circular economy (CCE). Although closing the C-cycle represents a big challenge on its own, doing it sustainably requires the use of particular methods and tools to implement the circularization strategy [46]. A goal to establish a sustainable CCE (SCCE) needs to keep the circular economy (CE) definition in our mind all the way through: “a CE is one where nothing is wasted and where natural resources are managed sustainably, and biodiversity is protected, valued and restored in ways that enhance our society’s resilience; and our low-carbon growth has long been decoupled from resource use, setting the pace for a safe and sustainable global society” [47].
This work will review the published literature concerning R&D and technology development to valorize CO2 using energy (heat, electricity, or radiation) that could be provided by NPPs. Special attention will be given to compounds whose production might attain sustainability. The factors considered in the search for sustainability are (i) the use or consumption of massive amounts of CO2, (ii) a long lifecycle of the product, (iii) the economy of scale, and/or (iv) a reduction in energy intensity from the currently employed production process technology. The chemical conversion of CO2 into intermediates or building blocks and final products will be reviewed in the following sections.
2. Chemical Pathways to Valorize CO2
Abundant chemical reactions of CO2 have been the subject of research and the core of the development of process technologies. Although CO2 utilization includes rendering a service and/or an application, the present work will only consider its chemical conversion into value-added products, such as fuels, bulk or commodity chemicals, materials, and specialty products. Vast R&D efforts have been devoted to the study and technological development of these reactions [48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95], though the level of progress and maturity differ among them, with only a few becoming commercially available [96,97]. Currently, some of the proposed technological solutions are unsustainable, extremely expensive, or have not reached commercial scale [32].
2.1. Synthesis Gas (Syngas)
Synthesis gas or syngas is a mixture of carbon monoxide (CO) and hydrogen (H2), which, under certain conditions, reacts to produce a variety of products, both oxygenates and hydrocarbons (Figure 3). Some of these compounds are raw materials in the synthesis of other chemicals and materials, giving rise to multiple chemical pathways and a broad product slate.
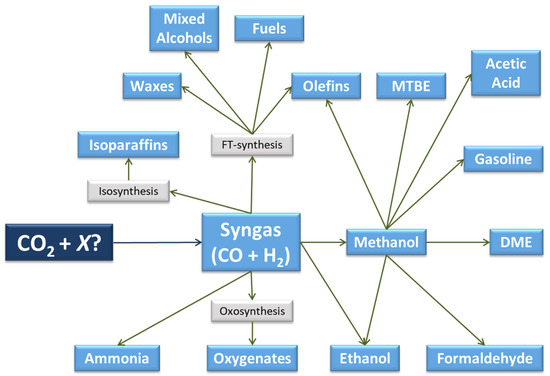
Figure 3.
Pathways and products from CO2 via syngas or methanol (adapted from [30]).
Evidently, syngas is a very versatile and valuable intermediate as well as the preferred source of H2. Commercially available technologies are based on the reforming of hydrocarbons, partial oxidation, or gasification of residues or coal. The selection of technology is based on the final product desired, which depends on the H2/CO ratio (syngas ratio, see Figure 4) or the application given to the syngas. Production is mostly carried out either by SMR (R2) or coal gasification. SMR is the preferred technology when H2 is the desired product, while coal gasification is selected for power generation applications.
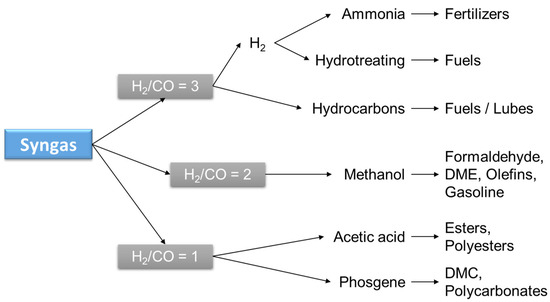
Figure 4.
Preferred syngas ratio for different products.
By 2019, global production of syngas was 261.02 million normal cubic meters per hour (Mnm3/h), with a value of $43.6 bln [98]. Global syngas production consumes about 500 exaJ, constituting 29% from natural gas, 39% from crude oil, and 32% from coal [99].
The development of cost-effective technology for its production from CO2 would open a path for a vast consumption of this compound while satisfying the market of a great variety of products. Several pathways involving different forms of energy have been proposed for the production of syngas from CO2, such as thermocatalytic pathways and chemical loops (driven by heat), electrochemical (driven by electricity) pathways, and radiation-induced (driven by ionizing radiation) pathways, which are discussed in the following sections.
2.1.1. Thermocatalytic Pathway
The thermocatalytic pathway for producing syngas from CO2 is based on the methane dry reforming (MDR) reaction (R4). The favorable environmental impact of this reaction is obvious since it consumes the two top carbon-containing GHGs (CO2 and methane, CH4). However, MDR has severe thermodynamic limitations, starting with a high thermodynamic potential (ΔG°298 = 174.6 kJmol−1) that becomes negative only at very high temperatures, e.g., ΔG°1073 = −44.76 kJ mol−1 [100,101]. Additionally, the endothermicity of the reaction (ΔH° = 247 kJ/mol) also sets a requirement for high temperatures (800–1000 °C) [102]. Under these conditions, not only does the employed catalyst deactivate rapidly, but selectivity is also very low, leading to undesirable products and reducing economic competitiveness.
CO2 + CH4 → 2CO + 2H2 ΔH298K = 247 kJ/mol
The MDR intensification with the partial oxidation reaction, adding small amounts of oxygen or water, contributes to overcoming thermodynamic limitations through the exothermicity of the oxidation reactions (R5). The O2 partial pressure needs to be carefully controlled to avoid total oxidation (R6). A net reduction in energy requirements results in a more energy-efficient MDR process [103] that also contributes to the reduction of carbon formation [104,105,106].
CH4 + ½O2 → CO + 2H2 ΔH298K = −36 kJ/mol
CH4 + 2O2 → CO2 + 2H2O ΔH298K = −802 kJ/mol
Carbon formation is one of the reasons behind catalyst deactivation. The reacting mixture containing CO2, CO, and CH4 is prompted to form carbon through the methane decomposition reaction (R7) and the Boudouard reaction (R8) [107]. While deactivation caused by carbon deposition could be remediated with catalyst regeneration, the high operating temperatures also trigger irreversible deactivation, shortening the life cycle of the catalyst. Irreversible structural transformations, such as sintering and recombination of the active components, take place at high temperatures and accelerate deactivation. More stable catalysts [108,109] have been studied, although the problem still remains.
CH4 → C + 2H2 ΔH298K = 74.85 kJ/mol
2CO → C + CO2 ΔH298K = −172 kJ/mol
Among the undesirable products, water is formed through a side reaction, the reverse water gas shift (RWGS, (R9)), consuming part of the produced H2 and forming additional CO that results in H2/CO ratios < 1 [110,111,112]. However, water is known to suppress coke formation probably through the steam carbon gasification reaction (R10), and from this viewpoint, RWGS could be beneficial. In fact, a really “dry” MDR will be difficult to realize at a large scale with realistic feedstocks and at the high severity the MDR reaction requires [113].
CO2 + H2 → CO + H2O ΔH298K = 41.15 kJ/mol
C + H2O → CO + H2 ΔH298K= 131 kJ/mol
The lack of selectivity and the fast deactivation are the main drivers for the vast efforts devoted to catalysis R&D. New catalyst designs and formulations [104,114,115,116,117,118,119,120,121,122,123,124], new synthetic methods [108,109,125,126,127,128,129], and the study of deactivation [100,114,117,127,128,129,130,131,132,133,134,135,136,137,138,139,140,141,142,143] have been the main R&D subjects. Noble metal catalysts are more stable since, by exhibiting higher activity, they can perform at lower temperatures [144]. However, regardless of their instability, the low cost of base metals places Co and Ni at the top of the preferences list for industrial catalyst development [145], particularly as good prospects for H2 production [146].
These catalyst issues, together with the energy demands of the MDR reaction, contribute to an unfavorable economy. While technological improvements are being addressed but not yet achieved, alternative approaches have considered integration and/or intensification of other processes. These intensified, combined, and/or integrated technologies have been techno-economically assessed, and some examples are cited here. As can be deduced from the (R4) stoichiometry, the H2/CO ratio of MDR syngas is lower than that produced from SMR (~3) and could be increased by the intensification with POX. A lower ratio favors alkenes, oxygenates, hydroformylation and acetic acid [147], and methanol synthesis, including so its derivative products (e.g., gasoline and paraffins, olefins, and aromatics) [148] (see Figure 3). Experimental data of a combined MDR–oxidative coupling of methane (OCM) process from a mini-scale unit showed good potentialities by lowering energy consumption through a 40% autothermal behavior [149]. Lower OPEX and CAPEX were assessed for methanol production when MDR instead of SMR was the source of syngas to the methanol synthesis [134]. Although MDR is preferably considered for syngas production, there might be favorable situations in which to incentivize H2 production [146]; for instance, a membrane reactor can be used to produce ultra-pure H2 [150].
Regarding technology development, two processes were developed in the 1990s: the Sulfur PAssivated ReforminG (SPARG™) process [113,151,152] and the CALCOR™ process [153,154,155]. And more recently, two others are under development: the R&D activities from the Gas Technology Institute (GTI) [156] and the DRYREF™ process [157,158]. In the SPARG™ process (originally designed by Haldor-Topsoe), sulfur is used to selectively poison active sites, leading to disassembling metal clusters, favoring reforming and inhibiting coke formation [152]. The poisoning effect of sulfur on Ni SMR catalysts has been reported [159,160], as well as in MDR [132]. Although the retarding effect on CH4 decomposition was shown [132], absolute proof of a dual effect on MDR and coke formation has not been published. The inhibiting carbon formation feature was demonstrated in an SMR-type pilot unit [161]. This process is operated within the temperature range of 875–945 °C to produce syngas at a ratio between 0.55 and 3.2 (preferably 1.8–3) by using a H2O/CH4 ratio of 0.1–0.26 in the pre-reformer unit and a CO2/CH4 ratio of 0.07–2.5 in the MDR unit. In this process, the severity of the operation determines the carbon-formation tendency. Since carbon deposition increases with decreasing CO2/CH4 and H2O/CH4 ratios [162], high severity is defined by an operation far beyond the carbon limit curve (the carbon limit curves are plots indicative of the deposition of carbon normalized to the amount of converted carbon sources at the thermodynamic equilibrium, as a function of process conditions) [163]. Sterling Chemical Inc. built, started (1987), and continuously operated a commercial demonstration unit for four years in Texas, USA [152]. The coke suppression of the SPARG™ process was better than the use of water for the same purposes. It was estimated that 133,000 Nm3/h of water would have been required to attain a similar effect [113]. However, as mentioned above, the total absence of water was not recommended, and most SPARG plants were designed to feed significant amounts of water to decrease severity and mitigate carbon formation as dictated by the limit for carbon limit curve. More details on the SPARG process and the industrial experience on the different sites where it was tested and demonstrated can be found in [113].
Caloric GmbH developed the multistage CALCOR™ process based on a Ni catalyst to produce CO-enriched syngas (H2/CO < 1, ~0.42) [154,155]. Coke deposition is minimized by varying feedstock compositions and catalysts (reactivity and geometry) through the stages of the process. Since CO2 in the feed exceeds the stoichiometric requirements, the gas product is mainly CO, with a minor proportion of CO2, H2, water, and traces of CH4. This unreacted CO2 is then separated and recycled back into the reformer. Hydrogen is also recovered separately to finally collect a stream that might contain up to 99.95% CO [164], depending on the downstream application (e.g., acetic acid and phosgene production [165]).
The GTI’s R&D activities have been focused on the study, design, and development of a nanocatalyst that would enable smooth and stable operations in the absence of steam [156]. The process would convert captured CO2 in a compact reactor, utilizing a nano-engineered catalyst on hollow fiber tubes [114,115,116,117,125]. A long-run (400-h) laboratory-scale continuous test showed stable catalyst performance [118]. The process is thought to be integrated into a dimethyl ether (DME) synthesis process. Based on energy density, pipeline transportability, and use as a drop-in fuel, DME was considered. However, further research was announced to be needed to prove the integrated concept at the same scale [156].
The development of the DRYREF™ process has been carried out by a large industrial academia alliance that includes Lynde, BASF, hte, Karlsruhe Institute of Technology, the Technical University of Munich, the University of Leipzig, and DECHEMA Forschungsinstitut [158] through a €1 billion project funded by the German Ministry for Economic Affairs and Energy [166]. This technology can be seen as an attempt to improve the energy efficiency of the SMR process by using an innovative Ni-based oxide-supported catalyst (SYNSPIRE™ G1-110) that can operate at higher pressures (20–40 bar) and by including steam (at a steam-to-carbon (S/C) ratio of 0.9–1.8) to minimize coke formation. The presence of steam, besides promoting the RWGS reaction (R9), also brings the possibility of the occurrence of the SMR (R2), resulting in a more variable syngas H2/CO ratio depending on the S/C ratio [167].
A simulation of a relevant MDR process (DRYREF type) integrated into the Fischer–Tropsch synthesis indicated that the production of 93 ton/h of a synthetic fuel composed of 72% diesel, 26% gasoline, and 2% LPG looks environmentally attractive by consuming 330 ton/h of CO2 even though the economy did not show great promises [168]. Linde advanced the development to the pilot stage at Pullach, Germany, in 2015–2017 and demonstrated its suitability for integration into downstream processes such as aldehydes, methanol, acetic, and formic acid synthesis [169]. The pilot test at an S/C of 0.9 not only demonstrated energy savings but also an extended catalyst lifecycle due to a performance exempted from carbon deposition. With these results, Linde Engineering estimated savings of $12 M in its first five years of operation in the costs for building a new plant with a capacity of 50,000 scf/h. Compared to SMR, Linde’s DRYREF represented overall reductions in OPEX and CAPEX of, respectively, 5% and 3% [157]. Although Linde announced the technology would be commercially ready by 2018 after the Linde Pilot Reformer commissioning [169], no further information has been provided.
Although processes have reached a commercial scale, no investment has been made in their implementation. One of the reasons is the materials needed to handle the required high temperatures. Therefore, technological improvements in both catalysts and processes that either can withstand reaction temperatures or perform at less severe conditions without detrimental effects on competitiveness would be essential for a commercial application. Nuclear power might have an opportunity, particularly to lower reaction temperatures, as it has been recently demonstrated that radiation could induce the MDR reaction at conditions close to ambient (room temperature and absolute pressures of 100 psi) (more details are in Section 2.1.4) [29].
2.1.2. Chemical Looping Pathway
Chemical looping (CL) is a type of thermochemical cycle to transport oxygen within a reacting system. The original conception of the thermochemical cycles was to combine heat sources with chemical reactions that disaggregate molecules. Chemical compounds are continuously recycled in these processes, creating closed loops by integrating two hemi-cycles [170]. Buelens et al. [171] have described the particular case involving CO2 in CL through three types of processes, namely, thermal redox, chemical redox, and carbonation. As illustrated in Figure 5, in the first half cycle (hemicycle 1, (R11)), an oxidant (redox CLs) or CO2 (carbonation CL) reacts with a metal (Me) or a metal oxide (MeO), respectively, in the so-called “Air Reactor”. Meanwhile, in hemicycle 2 (R12), a reductant (chemical) or heat (thermal and carbonation) is used to restore the initial compounds in the Fuel Reactor.
| Hemicycle 1 (Reduction step) | Hemicycle 2 (Oxidation step) | |
| (R11) | (R12) | |
| Thermal Redox | Me + Oxidant → MeO + Reduced-oxidant | MeO + Heat → Me + ½O2 |
| Chemical Redox | Me + Oxidant → MeO + Reduced-oxidant | MeO + Reductant → Me + Oxidant |
| Carbonation | MeO + (CO2)sol → MeCO3 | MeCO3 + Heat → MeO + Pure CO2 |
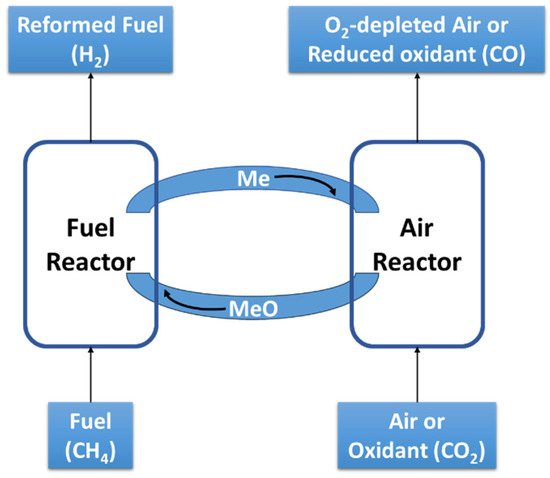
Figure 5.
CL process general scheme, illustrating CL MDR.
The carbonation CL is applied as CC means, with a direct recovery of concentrated CO2. Meanwhile, the redox-based CLs can be applied to CO, H2, or syngas production. Carbonation CL reduces the energy and costs of a CC application, and CO production has been mentioned as being economically sustainable [171]. The high efficiency and reduction in costs and energy demands for the power and industrial sectors, as well as the lack of commercial experience, have created barriers to CL growth [172].
Syngas production through CL may require the combination of multiple reactions, including methane oxidation (R13), CO2 reduction (R14), and steam/water reduction (R15)—reactions that are described below and seen in the net reaction (R16). Hemicycle 2 is the reoxidation of the metal (R17) to restore the starting oxide. A CL-MDR process requires oxygen carriers with high oxygen storage capacity (e.g., perovskite-type materials and oxides and mixed oxides of Fe, Co, Ni, Cu, Ce, etc.) [173,174]. It has also been postulated that methane POX (R18) can occur rather than methane total oxidation (R13), in which case a H2-rich syngas could be recovered (H2/CO ~ 2) from hemicycle 1. In this case, the CO2 reduction reaction (R14) can be used as hemicycle 2 [175], from which pure CO can be separated, and a high syngas ratio of hemicycle 1 is desired.
4MeO + CH4 → 4Me + CO2 + 2H2O Methane oxidation
2Me + 2CO2 → 2MeO + 2CO CO2 reduction
2Me + 2H2O → 2MeO + 2H2 Steam reduction
CH4 + CO2 → 2CO + 2H2 Net reaction (R13 + R14 + R15)
2Me + O2 → 4MeO Metal oxidation with air
MeO + CH4 → Me + CO + 2H2 Methane POX
An investigation of mixed oxides of Fe, Co, or Ni with ceria or zirconia showed the superiority of the Ni-CeO2 system in terms of both activity and stability (over 60 cycles). Co ranked second with slightly lower activity and stability (over 12 cycles), but Fe also showed poor performance and stability (over 12 cycles). The analysis of the spent materials indicated that in the case of Ni and Co, these metals act as catalysts and ceria as the oxygen-transporting material since the former metals, once reduced during hemicycle 1, do not re-oxidize again when going through hemicycle 2 [176].
One of the benefits of CL MDR derives from taking advantage of the redox properties of a Me/MeO pair rather than limiting MDR to thermodynamics and CO2 reactivity. A thermodynamic assessment indicated that for a MeO/CH4 ratio < 1 and Fe2O3 as MeO, the POX reaction dominates (R18). However, MeO/CH4 ratio > 1 would be undesirable since methane combustion would overcome CO and H2 production, and CO2 and H2O would become the major products [177].
Another advantage of CL MDR comes from the stepwise operation. During hemicycle 1 ((R16) or (R18)), some decomposition might take place, and coke will deposit on the MeO/Me, but it will be burnt off during hemicycle 2 by the presence of the oxidant. Since H2 is produced in hemicycle 1, it will never be in contact with CO2, and the RWHS reaction (R9) could be avoided, with concomitant H2 savings and increased syngas ratio [178,179]. Trimetallic mixed oxides (Ni-Fe-Al) exhibited better stability than binary systems, as proven over 20 CL MDR cycles and in conventional MDR [180]. A perovskite-type system (La0.9Sr0.1FeO3 supported on yttria stabilized-zirconia) also showed stable performance over 7 h operation of 20 min CL MDR cycles [181].
No report on the environmental impact of standalone CL units could be found. Most of the reported assessments regard applications such as a combustion system (chemical looping combustion, CLC) [182,183,184,185], IESs [186,187,188], decarbonization of power plants (coal or natural gas) [182,183,184,185,189,190], and on reforming [191,192]. All these assessments report environmental benefits ranging from very small (e.g., thermal natural gas power plants) to becoming negative carbon-emitting facilities (e.g., coal power plant–CL CC–Sequestration). Additionally, economic and environmental sustainability was evaluated through a simulation of an IES consisting of biomass gasification and syngas CL to poly-generate power, DME, and ammonia. The results showed that the system thermal and exergy efficiencies were 60.15% and 46.70%, respectively, with a payback time of 8 years and net present value of nearly $1.6 M for a 20-year life cycle [188]. Regarding CL reforming, both hydrogen [191] and syngas [192] production have been evaluated. Compared to conventional reforming technologies (SMR, POX or autothermal, ATR), CL reforming offers higher energy efficiencies, lower carbon emissions, and lower operating costs [192]. The carbon footprint, acidification, freshwater eutrophication, ozone depletion, photochemical ozone formation, and depletion of minerals and metals have been evaluated for a CL MDR process as a measure of its environmental performance. The leakage of biogas from the anaerobic digestion plants was the most determinant factor for the environmental viability of green hydrogen production by CL MDR. The perovskite-based material employed in this system environmentally outperformed when benchmarked against a conventional DRM [191].
Regardless of the progress reached by CL in general and CL MDR specifically, further R&D is needed in heat integration, new reactor designs, process integration, oxygen carriers with increased capacity by improved formulations and/or structures, further testing for longer runs at larger scales (pilot and demonstration), and comprehensive TEA and LCA. Regarding heat integration, a couple of works on radiation-induced reductive decomposition of carbonates and oxides could be considered to couple NP to thermochemical cycles as a means to improve energy efficiency [193,194].
2.1.3. Electrochemical Pathway
The electrification strategy for decarbonization has been pushing high pressure into the development of electrochemical processes for CO2 conversion. CO2 electrolysis is based on its reductive decomposition to produce CO and O2, taking advantage of catalysts and electric power to overcome the energy barrier for CO2 activation and reduce the energy required for its reduction reaction [195]. CO2 electrochemical reduction has been an R&D subject for decades, and the thousands of published articles have been the basis for the developed electrolyzers. Commercial electrolyzers are available for bench-top scale for both liquid- and gas-phase CO2 reduction R&D, e.g., [196]. On the industrial scale, some companies have announced commercial availability for electrolyzers producing higher reduction products, e.g., [197,198]. Regardless of these announcements, technology development is still in progress and trying to overcome identified challenges such as high selectivity electrocatalysts at high current density (high conversion), reliability, durability, productivity, and energy efficiency [199]. Although the combination of a water electrolyzer with another electrolyzer for CO2 can be used for syngas production and/or green hydrogen production by electrolysis is receiving great attention, the revision of this work will fall away from the scope of the present work (various colors are used to categorize the magnitude of the net carbon emissions during a production process: a brown process technology uses heat and energy generated from the combustion of fossil fuel; in a blue process technology, CC, CCS, and/or CCU technologies are incorporated to mitigate emissions; in a green process technology, the heat and energy are generated from renewable, low-C or neutral-C sources; and in some instances, gray is used as an intermediate emitting scale between brown and blue—other, less-used colors include pink, turquoise, white, and yellow);. It is the simultaneous or co-electrolysis of CO2 and water that is relevant here, and currently, within the net zero proposed strategies, co-electrolysis represents the most studied electrochemical alternative.
The electrochemical CO2 reduction processes, in general, can be separated into two ranges of operating temperatures: low-temperature electrolysis (LTE) (T < 100 °C) and high-temperature electrolysis (HTE) (T ≤ 600 °C) processes [200]. The development of suitable ion-conductive materials is defined by the selected temperature range.
In HTE, the co-electrolysis of steam and CO2 produces syngas [201]. Initially, many studies were focused on this operation mode [201,202,203,204,205,206,207,208,209,210,211], introducing improvements by considering co-production (e.g., chlorine [212]) or integration with other plants (e.g., biomass gasification [205]). In order to decrease the syngas ratio, part of the electrochemically produced H2 can be used in RWGS reaction to simultaneously increase C-utilization, for instance [213]. CO2 HTE is the option of choice when a combination with thermocatalytic processes is desired, e.g., CO2 total reduction [214]. The reduction of the equilibrium potential and the improved reaction kinetics are significant advantages of HTE operation compared with LTE. However, in cases of unavailability of external heat or when fossil-sourced heat is undesired, LTE might become the only option.
LTE follows two approaches based on the CO2 phase employed as feedstock. The first concerns the gas phase, and the other concerns the liquid phase. The gas-phase operation requires high CO2 concentrations to avoid electrode flooding (deactivation) and ensure good conversion [215,216]. Reactor architectures to deliver gas-phase CO2 to the cathode rather than dissolved in a liquid electrolyte are recommended to improve current density [217]. Similarly, electrodes that facilitate the mass transport of CO2 gas to catalytic sites pushed the development of gas diffusion electrodes (GDEs), which also control water flux and inhibit side reactions [218]. GDEs enable operationsat current densities over 200 mA/cm2 to produce CO and formate [218].
The high over-potential of CO2 LTE probably represents the greatest challenge faced in advancing it toward scaling. The high activation energy (slow kinetics) and other sources of energy losses and inefficiencies are responsible for that overpotential. The design and development of new electrocatalysts represents only one of the approaches that address the issues and contribute to overcoming the challenge. Extensive catalysis R&D has focused on understanding the catalytic phenomenon and the development of new catalysts [219,220,221]. Catalysts based on metals, metal alloys, metal oxides/chalcogenides, carbon nanotubes, and enzymes have been proposed [222].
Both HTE and gas-phase LTE will require CO2 to be captured, recovered, and purified from emitting sources prior to feeding to the electrochemical cell. Under the current state of technology, the low conversion and poor selectivity leave unconverted reactants mixed into the products, creating needs for separation, recovery, purification, and/or recycling, which affect the economy of the technology [200,223,224]. Process intensification under the LTE mode of operation may be the improvement approach to choose. The reported studies include the electrolysis of bicarbonate (mostly KHCO3) solutions [225,226] and the direct co-electrolysis of captured CO2 solutions. Bicarbonate solutions were tested to evaluate the change of the anodic reaction in decreasing the overpotential. An operating voltage of 2.3 V was achieved by running the hydrogen oxidation reaction (HOR) at the anode instead of the oxygen evolution reaction (OER) [227]. The electrocatalytic selectivity of Zn-Al layered double hydroxides for bicarbonate reduction at a current density of 15 mA/cm2 and −1.4 V vs. RHE was 77% for CO and 94% for syngas [228]. The lifecycle and economic assessment of the electrochemical conversion of bicarbonate solutions for formate and CO production was reported, and in both cases, this capture–conversion integration rendered economic and environmental benefits, leading to an eco-efficient categorization of this C-utilization approach [229]. In general, electrochemical testing using bicarbonate solutions has gained acceptance as a rapid method for assessing the impact of different variables on the performance during the direct conversion of captured CO2, facilitating the evaluation and understanding of catalytic parameters [230,231,232,233] of feedstock composition [226,234,235,236,237], and of operating conditions [225,238]. In a demonstration of direct co-electrolysis of captured CO2, the intensification of solvent recovery and conversion within the electrochemical unit was considered [239], achieving conversions better than 70% under a current density of 200 mA/cm2 [240]. In this case, a switch polarity solvent (1-cyclohexylpiperidine) was used as capture media. The performance for the co-electrolysis of monoethanolamine-captured CO2 was not as good as the previously discussed, with a lower current density of 50 mA/cm2 and faradaic efficiency (FE) of 72% [241]. (The faradaic efficiency (FE) of the CO2 electrochemical reduction is the ratio of the measured amount of produced main reduction product (e.g., CO) and its theoretical amount evaluated using Faraday’s Law. Thus, it can be calculated as FE(%) = Fniχiṅ/I, where ni is the number of the electrons involved in the considered CO2 reduction, F is the Faraday constant, χi is the volume fraction of the considered product, I is the electrical current, and ṅ is the molar CO2 gas flow rate. The FE was lower (45%) when 2-amino-2-methyl-1-propanol (AMP) was used as a capture solvent and electrolyte for CO2 reduction to CO [242]. Syngas was selectively produced from CO2 captured in 1.0 M alcohol amine solutions, using electrocatalysts of Au or Cu supported on MgAl-layered double hydroxides. The hydrogen evolution reaction (HER) was enhanced toward negative potential under employed conditions [226]. The effect of different amine-based deep eutectic solvents used as electrolytes for CO2 reduction on the cell performance was evaluated. A synergistic effect among nano-size agglomerate dispersion on Ag-surface, bicarbonate formation, exchange current density, and Cl− ions present in the deep eutectic solvent facilitated CO2 reduction to CO [243]. Recovery of the capture solvent was the focus of the study as a way to leverage intensification [244].
The electrochemical pathways and electrocatalytic reduction of CO2 to produce syngas have been the subject of patents and patent applications. Reported concepts and schemes include multistep, integrated, and intensified processes [245,246,247,248,249,250,251,252,253,254,255,256,257,258,259,260,261,262,263,264,265,266,267,268].
The lifecycle of a power-to-syngas integrated system (renewable energy-based) was assessed for an HTE operation mode. This study considered CO2 from direct air capture (DAC) as feedstock. In comparison with an SMR-based system, the HTE showed lower global warming potential (GWP) and fossil fuel depletion, as well as negative net GWP in cradle-to-gate LCA when HTE was exclusively operated on renewable power. However, continuous HTE operation would require additional fossil electricity, under which circumstances achievement of negative net GWP is limited, and other environmental issues (human toxicity, acidification, particulate matter, or metal depletion) became worse than for SMR [269].
2.1.4. Radiation-Induced Pathways
The effects of ionizing radiation on molecules include excitation, ionization, neutralization, activation, and conversion via chemical reactions. At this point, it is worth defining some radiation-related parameters. The absorbed dose is the amount of radiation absorbed by an atom or molecule, and it is measured either using the gray (Gy, SI unit, J/kg) or the rad (CGS unit, 100 erg/g, 0.01 Gy). The magnitude of the radiation effect on a chemical transformation is parametrically defined by the G-value. The G-value is the number of molecules, atoms, or free radicals formed or converted per 100 eV of energy absorbed.
The radiolysis of CO2 has been studied for over a century, and the main reaction that occurs is reductive decomposition (R19), with CO as the main product [270]. The maximum decomposition observed was 0.1% on solid CO2, with a G value of 9 to 10. This low yield (G value) has been explained as due to a greater reverse reaction, known as backreaction, i.e., recombination of products. Nonetheless, a reaction network has been proposed to explain the low yields, as well [271,272].
CO2 → CO + O
Since ionizing radiation has additional effects than just chemical conversion, the presence of other molecules can result in the production of other species besides CO. Some of the various chemistries that ionizing radiation can induce on CO2 have been described and discussed previously [28]. Regarding syngas production and based on the (R4)–(R6) and (R9) reactions described above, it seems possible to combine the CO2 radiolytic reactions with those of CH4, O2, and/or water to induce the formation of the desired product. The radiation-induced reactivity and reactions of this reacting system have not been studied systematically. Instead, the reaction with water and with methane have been studied in more detail.
The individual radiolysis of CO2 and H2O was found to be affected by the phase state of the reactant, the dose rate, and the ionizing radiation type. The effect of irradiation conditions was stronger for CO2 than for water [273]. Both atomic hydrogen [274,275] and H2 [276,277,278,279,280] have been observed to be formed from water radiolysis. It can be expected that the ion radicals or even the nascent hydrogen formed under irradiation will be highly reactive and react with other molecules present. Conflicting G(CO) results have been reported for the reaction with water; in one instance, water increased G(CO) [281], while in another, a decrease was observed [282]. These studies did not report H2 formation. High reported CO yields were explained as due to the formation of weakly bound H2O···CO2 complexes that decomposed into CO. This study also postulated the presence of hydrogen atoms [283]. The reported radiolysis studies of CO2 and H2O mixtures indicated the formation of CO and H2, though these products were supposed to be formed from the radiolysis of the individual molecules [284,285,286]. These radiation-induced reactions (probably due to the potentiality of the formed products) have given rise to patents [287,288,289].
In the reaction with methane (or with heavier hydrocarbons [290,291]), yields and product distribution varied. Reported results include the deposition of reactive carbon [292], the formation of a formaldehyde polymer wax [293], and a mixture of hydrogen, ethane, higher alkanes, ethene, acetylene, and polymeric (CnH2n) liquid [293,294]. More recently, the radiation-induced MDR reaction (R4) was proven at temperatures close to ambient, demonstrating further that a small amount of methane can stop the backreactions from occurring, with a consequent CO yield increase. Furthermore, radiation-induced catalytic promotion was also proven to take place at the same operating conditions [29].
2.2. Methanol
Conventionally, methanol synthesis is carried out from syngas as a ratio of 2 (R20) [295], using commercially available and currently operative, mature catalytic (e.g., [296,297,298,299,300]) process (e.g., [71,301]) technologies. However, the fact that methanol synthesis is an energy- and carbon-intense process has prompted the search for a sustainable alternative [302].
CO + 2H2 ⇌ CH3OH ΔH298K = −91 kJ/mol
The uses of methanol (e.g., fuel, chemical, solvent, etc.) [303] as well as its applications (reactant, building block, fuel cells, etc.) [304] are increasing and pressing its industrial demand into a continuous growth cycle. An important methanol attribute is to be a liquid at ambient conditions that makes it pipeline transportable, handled, and distributed using existing infrastructure. It can be used as a fuel or blendstock in the gasoline range. Otherwise, it can be converted into dimethyl ether (DME) that can be used as a fuel or blendstock in the diesel range or replace liquefied natural gas (LNG) and liquified petroleum gas (LPG) [154]. For instance, methanol is the main component in feedstock for the synthesis of other intermediates and building blocks (e.g., olefins, ethanol, acetic acid, etc., see Figure 3). Technologies for olefin production from methanol (MTO process) are already mature technology [305,306,307]. In summary, methanol can be used as vehicular fuel, it can generate electricity in fuel cells, and it is an important raw material in the chemical and petrochemical industry [153]. Hence, methanol is an important energy carrier for a clean and sustainable energy future, which, within the NPtX scenario, appears as a convenient solution for efficient energy storage on a large scale and has an emission reduction potential if produced from CO2. Methanol and these derivative products already have a well-established (large) market, and if produced from CO2, both the intermediate product and its derivatives will represent a vast consumption of this GHG compound. Commercially operating technologies are currently used for the production of methanol derivatives; hence, no further discussion is provided here on these mature technologies. Instead, the work regarding methanol synthesis from CO2 follows.
2.2.1. Thermocatalytic Pathways
The thermo-catalytic CO2 hydrogenation can produce methanol in a single step characterized by reaction (R21) or in two steps involving the production of CO by RWGS (R9), as the first step, and feeding additional H2, in a second step, after intermediate H2O removal. This second step proceeds in a conventional methanol synthesis reactor.
CO2 + 3H2 → CH3OH + H2O ΔH298K = −49.4 kJ/mol
At this point, it is worth noting that CO2 hydrogenation gives rise to the so-called “hydrogen conundrum” (the term conundrum is used here to refer to a problem that is difficult to solve because its solution might take it back to the causes of the problem) since, traditionally, H2 is produced by SMR, which, as mentioned above, is a CO2-emitting process. Hydrogenation reactions are generally exothermic, and in the case of the methanol-producing (R21) reaction, the methanation reaction (shown as reaction (R1)) can also take place under similar conditions. Therefore, promoting either reaction requires the development of highly selective catalysts, which in the case of CO2 hydrogenation has not received enough effort, and most of the published results concern the use of conventional methanol synthesis catalysts.
Carbon Recycling International (CRI) licenses technology based on the Olah single-step process (R21), the Emissions-to-Liquids (EtL) technology [308]. An EtL demonstration plant was commissioned in 2005 and has been operative since then in Svartsengi, Iceland [309]. Renewable (geothermal) power and green hydrogen produced from water electrolysis are employed to produce methanol using CO2 captured from the geo-power plant. In February 2024, CRI started up a larger EtL plant in Anyang City, Henan Province, China, to produce 110 kton of methanol to be used as a renewable fuel in the plant fleet [310]. The plant is expected to consume 160 ktonCO2/y, captured from an adjacent coal power plant.
Lurgi also offers another single-step option [311]. This process has undergone subsequent improvements, including the addition of stages, reactors, heat exchangers, separators, etc. [312,313,314,315,316], after its initial development.
Regarding two-step processes, a technology option is commercially available: the CAMERE technology [317,318]. In the first step, the RWGS reactor is coated with a water perm-selective membrane to improve water removal. In this way, CO2 reduction increases, improving CO yield [319,320]. As mentioned above, the second step considers a hydrogen addition to proceed according to typical methanol synthesis conditions (R20) [321].
The use of conventional methanol synthesis catalysts (CO hydrogenation catalysts) in all these processes creates drawbacks derived from poor selectivity and low conversion that jeopardize the process economy. Some of the side reactions involved from the lack of selectivity include the Boudouard (R8), the methanation (R1), and WGS/RWGS (R9) reactions, which have been mentioned already. Worth emphasizing is the Sabatier methanation reaction (R1) [322], with ∆H = −165 kJ/mol, which, although being an energy-intense reaction, has received great attention due to its role in reviving the PtG program [323]. Other side reactions lead to the formation of alcohols, ketones, esters, and oligomers, such as the following:
CO2 + 2H2 → CH4 + O2 3CO2 + 3H2O → C3H6O + 4O2
nCO2 + 3nH2 → (CH2)n + 2nH2O 2CO2 + 2H2O → 2CH3OH + 3O2
CO2 + 2H2O → CH4 + 2O2 2CO2 + 2H2O → 2HCOOH + O2
Additionally, water formation not only limits the CO yield but also represents hydrogen and exergy losses [324]. Therefore, more active and selective catalysts (not for CO but rather for CO2 hydrogenation) are needed together with more energy-efficient processes. Some of the efforts devoted to heterogeneous catalysts can be found in [325,326,327,328,329,330,331,332], photocatalysts [333], biocatalysts [334,335], and processes [58,222,336,337,338,339,340,341,342,343,344,345,346,347]. Additional improvements have been reached by integration to make power plants more energy-efficient [348] and by CO2 recycling [349].
2.2.2. Electrochemical Pathways
The hydrogen conundrum has prompted the incorporation of green hydrogen in CO2 hydrogenation applications [350]. The production of green hydrogen falls outside the scope of the present work, and although these pathways are sometimes regarded as electrochemical pathways, in this work, we will consider only those in which CO2 is electrochemically converted.
The electrocatalytic processes have been gaining track and include CO2 reduction (e.g., [351]), co-electrolysis (to syngas followed by methanol synthesis), and hydrogenation processes [352,353,354,355,356]. The electrochemical CO2 reduction to form methanol has been mainly studied for gas-phase systems [357,358,359], with substantial efforts devoted to electrocatalytic studies. Improved gas dispersion was attained when using electrocatalyst support conjugated microporous polymers (e.g., TPE-CMP) mixed with carbon nanotubes (CNT) to ensure a high electronic conductivity. Pt nanoparticles were doped onto the support material as an active phase [360]. CO2 adsorption may be strongly enhanced due to the pore structure of the p-conjugated polymer. Good performance measured as liquid product yield was thought to be due to the high local concentration of CO2 on the polymer surface, enabling access to the active metal nanoparticles [360].
Improvements have been attempted by the incorporation of biocatalysts [334,335] and the development of bio-electrochemical processes [222]. One of the former examples of bio-electrocatalyst consisted of immobilized enzymes for the reduction of CO2 and reported a very low FE (<10%) for methanol formation [334,361]. The low solubility of CO2 in aqueous media is a barrier to high throughput operation of bio-electrocatalytic technologies.
Electrocatalytic hydrogenation to higher-reduced products has been claimed in the patent literature [250,251,253,254,256,260,352,353,354,355,356,362] based on studies carried out at bench-top scale. Results can be characterized by low yields, poor selectivity, high over-potential, low FE, and current densities. Based on the revised literature (articles and patents), the electrochemical route needs improvements on electrocatalysts [360,363,364] and on cell potential, current density, and durability of materials and components [359].
2.2.3. Economic and Environmental Assessments
The methanol economy [71,365] centers this molecule as the core to drive the production of a variety of products (fuels, chemicals, and materials). It has been around for more than 15 years, and although all products are commercially produced, such an economy has not emerged. Implementing this economy based on the existing technologies will bear drastic environmental consequences due to their energy intensity and carbon footprint. Instead, using CO2 for its production represents a compelling alternative to massively utilize the carbon emissions [71]. Nevertheless, when considering CO2 as a feedstock, only a few of the needed technologies are commercially available; some need improvements [366], and many are under development [357,367]; however, many for derivative products and materials are mature [71]. The technological maturity of both thermocatalytic [357,368,369,370,371,372,373,374,375] and electrocatalytic pathways [247,248,357,376,377,378] vary but still offer a myriad of opportunities for a decarbonized and decarbonizing technology platform to start implementing the methanol economy.
The economic and environmental assessment of the CO2-based processes are rarely reported. Improved energetic efficiency was assessed for methanol as an energy carrier considering the overall energy conversion-storage system, including methanol as the storage medium. The study, based on thermodynamic data for methanol synthesis, showed the exergy of the heat released in the synthesis reactions of 16.2–20.0%, depending on the applied conversion technology [324].
The capture costs and the energy (electricity) costs contributed together to make the electrochemical production of methanol from CO2 extremely expensive in Germany [379]. This study demonstrated how capture costs (and thus feedstock), energy source, and location affect the methanol production economy. Nevertheless, a production cost comparison among different technologies and feedstocks was reported without including capture cost, feedstock production costs, and geographical location impacts [380].
Exothermic reactions like methanol synthesis offer an opportunity for energy integration between the capture and the synthesis units. A supply chain optimization in Germany assessed economic profits of 999.62–1568.17 €/tonCO2 captured and utilized. Utilization was exemplified with the production of methanol, DME, formic acid, acetic acid, urea, and polypropylene carbonate [381]. However, in some instances, these energy savings are not enough to compensate the economy, and it was shown that a 33% energy saving in solvent regeneration still resulted economically unfeasible when the energy cost was 70 €/kWh since the production cost was 1.5 times the evaluated revenues [344]. Energy savings were also mentioned for the integration of a biomass-fired oxy-combustor and a solid oxide electrochemical electrolyzer, but economic and/or environmental impacts were not reported [382].
Seven different integrating configurations/scenarios were considered in the environmental performance of methanol (and urea) production using CO2 emitted from a blast furnace of a steel mill. The global warming impact (GWI) reductions evaluated across the whole functional unit (FU) varied from 0.70 to 4.62 MtCO2-eq/FU within the seven scenarios [383].
2.3. Urea
In nature, mammals synthesize urea (CO(NH2)2) when metabolizing proteins. Industrially, it is produced using the Bosch–Meiser process [384,385], which was developed in 1922. This technology consists of a two-step process characterized by reactions (R22) and (R23). The first step is the reaction between liquid ammonia (NH3) and gaseous CO2 (R22) to form carbamate that decomposes through reaction (R23) in the second step, yielding urea and water. While the carbamate formation reaction (R22) is an exothermic reaction, its decomposition (R23) to yield urea is endothermic. Currently, the industrial production of urea consumes the largest CO2 volume by any CO2 conversion process (about 130 MtonCO2/y, globally) [386].
2NH3 + CO2 ⇌ H2N-COONH4 ΔH298K = −117 kJ/mol
H2N-COONH4 ⇌ (NH2)2CO + H2O ΔH298K = 15.5 kJ/mol
The urea market is determined mainly by its use as a source of nitrogen in fertilizers and, in a minor proportion, by its use as a co-reactant or intermediate in the chemical industry. In fact, around 90% of the global urea production (~184 ton/y in 2022) is consumed by the fertilizer sector [385]. Excessive consumption of urea for fertilizer applications would have an environmental impact directly in the agricultural sector and indirectly through its production in the industrial sector. In the chemical industry, urea is applied as a building block for producing two materials: urea–formaldehyde and urea–melamine–formaldehyde resins [52,387,388,389,390,391].
The large CO2 consumption during urea synthesis cannot compensate for the volume emitted from the ammonia production processes. The energy and carbon intensity of the ammonia-producing technology is partially due to the high severity employed and the large hydrogen consumption. Hydrogen production is also an energy- and carbon-intense process. The ammonia synthesis process is one of the top energy-intense technologies. Ammonia synthesis, together with benzene, methanol, and steam cracking, is one of the four top processes responsible for the high CO2 emissions of the chemical industry [392]. Not all urea plants include an ammonia plant, and not all ammonia plants include a hydrogen plant; therefore, only a few cases can recycle part of the ammonia plant-emitted CO2 to feed the urea synthesis process. Green ammonia production will represent a big step toward the sustainability of the urea synthesis technology. The search for more energy-efficient and less severe processes for ammonia synthesis has received significant R&D efforts for many decades. More recently, the electrochemical synthesis of ammonia appeared as one of the green alternatives that could emerge in the near future (e.g., [393,394,395,396,397,398]). Cleaner alternatives have been announced by the major technology licensor for converting some ammonia plants from brown to blue and building new commercial-scale plants of green ammonia (e.g., [399,400]). Then, it seems that the efforts on green ammonia and hydrogen production will shorten the wait for sustainable urea production.
These opportunities toward large-scale sustainable production of urea (and so of its derivatives) appear with barriers and limitations, some of which have been identified in sustainability assessments and studies. For instance, the unfeasibility of completing the decarbonization of the ammonia industry by 2050 was revealed in a prospective LCA study [401]. Regardless of the potentialities of electrolysis-based green ammonia, replacing fossil-based ammonia will require expansion of renewable energy, reduction in the urea demand, infrastructure for CO2 transport and storage, and securing supply of raw materials. Additionally, considering the longevity of existing plants, the growth of ammonia as a fuel will favor the economy [401]. Other points of attention for the urea plant that require improvements include the following: the material preparation stage, synthesis stage, and waste treatment stage, while for the hydrogen production unit, steam generation, replacement of fossil fuels burning, and energy efficiency measures [402]. Optimization to maximize efficiency and minimize costs is needed for the amine CC unit to avoid exergy destruction since an exergoeconomic analysis showed this unit as responsible for the environmental impact of the plant [403]. The environmental impact of urea production has been assessed as a way to reflect its potential in GHG emission mitigation. As an example, steel mills might reduce more than 65% of their emissions by integrating them into a urea plant run on wind energy [383].
The environmental impact of urea agriculture application could be the most relevant. The environmental impact of urea production and use collectively includes not only GHG emissions but also eutrophication and acidification [404]. Synthetic fertilizers (including urea) were found to be environmentally better than organic-derived ones when 18 environmental indicators were evaluated in terms of kg of nutrients rather than per kg of product [405]. The incorporation of renewable resources and sustainable management are needed to attain sustainable agriculture, as revealed by an integrated LCA and Emergy. (Emergy is a thermodynamics-based concept that provides a holistic environmental accounting system. An emergy assessment identifies and measures all energy and matter inputs into a given system to evaluate the environmental cost of a given resource, expressed in a common unit, the solar emergy joule (sej). The unit emergy value, UEV, is the emergy per unit product.) The authors of [406] evaluated the environmental costs and impacts of resources (including urea) in agricultural production [407]. The environmental impact of urea production has also been evaluated for other fertilizer/agricultural applications. One of these was the production of a bio-based binder derived from hemp, cultivated using urea-based fertilizer. In this case, urea production topped the contributions to carbon emissions (21%) due to the energy consumption of the urea plant (30.1 GJ/tonurea, [402]) [408]. Nonetheless, some benefits have also been reported, this time for the blending of controlled-release urea that increased agronomic product yield and decreased associated emissions [409]. At a global scale, sustainability improvement measures might include the use of renewable feedstocks and raw materials, eco-efficient upstream processes, and renewable energy sources [410]; here, one may add C-neutral sources and a note of attention for NP.
Versatile resins can be synthesized by the co-polymerization of urea with formaldehyde. The complex chemistry involved in the polymerization reaction includes addition and condensation reactions. The urea molecule undergoes the addition of hydroxymethyl oligomers formed by condensation and cross-linking reactions of the hydroxymethyl moiety of formaldehyde [411,412]. Both the kinetics and the properties of the produced resin are affected by temperature, pH, and the formaldehyde-to-urea (F/U) molar ratio [413], which determine the extent of branching and type and proportion of chemical bonds [414]. Urea-formaldehyde (UF) resins are mostly used as wood adhesives (85% of the ~11 Mton/y global adhesive market [415]) in the manufacture of plywood, particleboard, fiberboard, and oriented strand board [416,417]. The post-war housing efforts created a market opportunity for plywood and particleboard [418]. However, soon after this popular application escalated, the worst environmental impact caused by formaldehyde releases over time was discovered, leading to indoor pollution that threatened the health of exposed humans [419,420,421,422,423,424,425]. Mitigation of formaldehyde emissions and improvements to product properties were achieved through changes in the polymerization conditions and in the manufacturing process [415,419,426,427,428,429].
The possibilities for green/renewable urea production leave the challenge to sustainable formaldehyde production before any attempt for sustainability can be made for UF resin production. Pathways from CO2 [222,352,430,431] and from biomass [432,433,434] have been investigated for UF resin production, but development is in its infancy. The comparison of the environmental impact of UF resins with bio-based adhesives is controversial. LCA studies showed lower environmental impact and functional advantages on composite adhesives [435,436], but another study concerned with bio-cementitious materials found a greater environmental (eutrophication) impact [437].
Abating formaldehyde emissions was also attempted by reformulating the resin to improve stability through the incorporation of melamine, a cyanamide trimer (C3H6N6) [438,439,440,441,442]. Due to its toxicity, melamine use and commercialization are highly regulated [443]. Though a wide range of applications is found in tableware and building materials [444], additional applications are given to the products manufactured with melamine copolymerized UF resins [445,446,447,448,449,450] in reinforced wood panels, construction, shopfitting industry furniture, interior decoration, etc. These low-cost melamine-UF resins advantageously offer high strength, high dimensional stability, isotropicity, and enhanced processability. Compared to UF resins, melamine-UF resins exhibit lower photochemical oxidation, ecotoxicity, and human toxicity [451], and the environmental impact of its production is mainly due to energy consumption and the factor derived from the UF resin production [452,453]. Therefore, sustainable melamine-UF resins will pass first through the sustainable production of UF resins and then sustainable melamine production, involving (green) urea (e.g., [388,391,454]) and (green) NH3 (e.g., [387,389,390]), will be pursued.
2.4. Polymers
The synthesis of oxygenated building blocks can be achieved by carboxylation reactions, which add a carboxylic group (-COO formed, for instance, from CO2) in other organic molecules. Various organic compounds can be obtained, e.g., acids [455], esters, lactones, carbamates [456,457,458], and carbonates [459]. Among the chemical reactions of CO2, carboxylation is the least energy-intense, and, in return, compounds with high energy value, e.g., strained cyclic molecules, or unsaturated compounds can be produced. Similarly, in hydrocarboxylation, CO2 reacts with olefins and/or alcohols in the presence of H2 to yield carboxylic acids or esters [460].
Polymeric materials such as polyacids, polyurethanes, and polycarbonates can be derived, respectively, from carboxylic acids, esters, and organic carbonates produced by carboxylation [461,462]. These high-molecular-weight materials fixate CO2 in their matrix as long as their lifecycle or service cycle lasts. The equivalent materials, currently produced from fossil feedstocks, could be replaced by these decarbonized options that are additionally biodegradable, renewable materials with low toxicity. In conclusion, carboxylation reactions can use massive volumes of CO2 to produce commodity and special materials.
2.4.1. Thermocatalytic Pathways
Carboxylation of alcohols produces dialkyl carbonates, according to reaction (R24), which occurs under base-catalyzed conditions at high pressures. Tin oxide, zirconia [463], and alkoxides of Sn (IV) and Ti (IV) [464] are examples of studied catalysts.
CO2 + 2ROH → RO-COO-R + H2O
Dimethyl carbonate (DMC) constitutes the most relevant example of an organic carbonate monomer based on its possibilities for sustainable manufacturing via the carboxylation of methanol (R25). Since green methanol can be synthesized sustainably, as discussed above, and CO2 is considered a renewable feedstock then, DMC can become sustainable in the short term.
CO2 + 2CH3OH → CH3OCO2CH3 + H2O DH°298K = −27.90 kJ/mol | DG°298K = 26.21 kJ/mol
The exothermicity of the (R25) reaction could advantageously be used for energy integration and to develop a more energy-efficient process. However, thermodynamic limitations (DG°298K) preclude the reaction from occurring spontaneously at ambient temperatures and pressures [465], calling for process intensification. Reactive distillation increased conversion from 10% to 99.5% and also reduced energy consumption [466] (reactive distillation is an intensified process that combines distillation with chemical conversion in a single unit, the distillation tower; a catalytic system is placed conveniently within the tower to separate one of the products from the reacting mixture and favor conversion or shift the equilibrium to the right). Product recovery is another process difficulty since DMC and methanol form an azeotrope. Extractive distillation processes, commonly used for azeotropic separations, consume large volumes of solvents and energy. Economic benefits (70% savings in total annual costs) and positive environmental impact were found when product recovery was carried out with ionic liquids, though some evaluated solvents thermally decomposed under employed conditions [467]. DMC electrochemical synthesis, in the presence of 1-butyl-3-methylimidazolium bromide and potassium methoxide, showed better performance (within the limitations of product recovery) and environmental impact than the currently commercially used technology (ENI process) [468]. Commercial DMC production is based on the carbonylation of methanol reaction (R26), which has been replacing the original technology based on the reaction between phosgene and methanol, forming methyl chloroformate as intermediate (R27) that then decomposes into DMC and hydrochloric acid (R28).
CO + ½O2 + 2CH3OH → CH3OCO2CH3 + H2O
COCl2 + CH3OH → CH3OCOCl + HCl
CH3OCOCl + CH3OH → CH3OCO2CH3 + HCl
The catalytic active phase for the DMC synthesis is an amphoteric pair of sites, on which the acid sites promote methylation from methanol while the basic sites activate CO2. Examples of this type of catalysts include CeO2 [469], ZrO2 [470], hydrotalcite [471], Y2O3 [465], and [Nb(OMe)5]2 [472].
The DMC market is largely determined by its use as a methylating agent [473,474]. However, this use is threatened by its consequent CO2 formation, which increases atmospheric releases, though new developments in methylating processes to mitigate emissions have started [475]. Regardless of these threats, DMC demand has been continuously growing, underpinned by its advantageous properties and attributes that include non-toxicity, reactivity, and derivatives (e.g., aromatic polycarbonates, solvents, and fuels) [476]. The search for a sustainable production pathway is incentivized by this growing market and by the interest in DMC’s advantageous properties. One example is urea methanolysis [477], which produces methyl carbamate, from which DMC results through a reaction with methanol [478]. This route showed slight environmental advantages but more technical limitations. The sustainability of studied/developed pathways (six different pathways, Path 1 through 6, described below) has been compared by assessing the environmental impact based on the stoichiometry of involved reactions [479]. As can be noticed, CO2 can be consumed directly in Paths 4 (for urea synthesis), 5, and 6.
| Path 1 | (R27) + (R28) | Phosgene, coproducing HCl | |
| Path 2 | (R29) | 2NO + 2CH3OH → 2CH3ONO + H2 | via methyl nitrite, coproducing CO and NO |
| (R30) | 2CH3ONO + CO → CH3OCO2CH3 + 2NO | ||
| Path 3 | (R26) | Methanol carbonylation | |
| Path 4 | (R31) | CO(NH2)2 + CH3OH → CH3OCONH2 + NH3 | Urea pathway, coproducing NH3 |
| (R32) | CH3OCONH2 + CH3OH → CH3OCO2CH3 + NH3 | ||
| Path 5 | (R33) | (CH2)2O + CO2 → (CH2)2OCO2 | Ethylene oxide (EO) pathway, coproducing ethylene glycol (EG) |
| (R34) | (CH2)2OCO2 + 2CH3OH → CH3OCO2CH3 + (CH2)2(OH)2 | ||
| Path 6 | (R24) | Methanol carboxylation |
The indicators used in the assessment of the environmental impact were the potential for ozone depletion; for photochemical oxidation; and for acidification; GWP; and human, terrestrial, and aquatic toxicity. A total score was evaluated for each path, considering the environmental indicators and the economic and toxicity analyses. Based on these total scores, the paths were ranked 3 → 6 → 2 → 5 → 4 → 1. The economics of Path 5 was found to be the best, while the environmental assessment favored Paths 3 and 6. However, Paths 1, 2, and 3 do not represent decarbonizing options and should not be considered for further development if industrial decarbonization is the purpose. In fact, Path 1 is already being phased out. Therefore, methanol carboxylation (Path 6) and the EO Path 5 pathways should be kept on the developmental plate, although the authors ruled out Path 6 on unexplained industrial unfeasibility grounds [479]. Optimized data were obtained from Path 4 and Path 5 simulations and used for evaluating sustainability indexes such as material index, energy index, and ecoefficiency. This new evaluation confirmed the superiority of Path 5 over Path 4 [480]. By-product recycling was considered for improving Path 5 and resulted in environmental (10% impact reduction) and economic benefits of a 20% increase in DMC yield, a five-fold increase in the net present value, and a reduction of the payback period from 8.3 years to 3.6 years [481].
DMC can be one of the starting reactants in the production of polyurethanes (PUs) if a sustainable pathway is desired. Its reaction with amines produces carbamates [482], which can be converted into isocyanates [483]. The isocyanate NCO groups react with polyol OH groups to form PUs. Alternatively, DMC could react with aliphatic diols to form oligo-carbonate diols and also polycarbonates [484,485]. The nearly 26 Mton/y PUs’ global demand is defined by their uses in manufacture (e.g., footwear, sponges, pillows, mattresses, seats, cast objects, bezels, structural parts, etc.) and in construction (e.g., thermal insulation boards, structure and sandwich panels, foams, etc.). Nevertheless, construction applications are limited by their flammability. New applications are also emerging, such as the chemical uses of oxadiazinetriones [486]. The positive environmental impact of PUs lies inarises from their chemical inertia and non-carcinogenicity. However, their decomposition products are hazardous and toxic, and some can even be lethal. Therefore, the assessment of the Pus’ environmental impact needs to involve both production methods and applications and to consider human exposure to toxic and hazardous raw materials and/or compounds [487]. The evaluation of these health impacts should be as comprehensive as possible, including all materials, products, and compounds and their life cycle stages (e.g., production stage, storage stage, service life, disposal stage, etc.). Comprehensive evaluations have not been reported; instead, specific stages for particular applications can be found, and some examples are as follows: PU-containing tiles [488] (e.g., gypsum ceiling tiles [489]), PUs’ production stage (e.g., as building material [490], insulating panels [491], waterborne PU dispersions [492]), service life stage (e.g., insulation applications) [493]. All these analyses concerned petrochemically produced PUs, and the takeaway message is that environmental benefits can be obtained from PU applications, but a need for a sustainable production pathway exists. One approach is forming urethane groups from the reaction of cyclic carbonates with bifunctional primary or secondary amines. This approach has been considered for the synthesis of thermosetting plastic networks [494], which can be used as binder to replace Portland cement in concrete for building applications [495]. Another approach that considers biomass and CO2 as starting materials has been investigated to formulate new composite materials. Some of these investigated pathways have been economically and environmentally assessed [496,497,498,499,500,501]. Glycerol carbonate emerges as a potential candidate for renewable starting material, particularly considering the increasing glycerol surplus arising from biodiesel production [502]. In fact, glycerol appears economically attractive to underpin a production platform for renewable chemicals and products [503,504]. Glycerol carbonate can be obtained from the reaction of glycerol with CO2 [505] using tin complexes as homogenous catalysts [506,507] or with urea in the presence of metal-impregnated zeolites [508], zinc salts [509], and lanthanum oxide [510]. Glycerol carbonate can be used in the manufacture of PUs, polyesters, and polyamides as well as solvent, emulsifier, and chemical intermediate [511]. The economic and environmental impacts of biomass-involving pathways and polymer products have been reported [496,497,512,513,514,515,516,517,518,519].
Specialty chemicals and polycarbonates can be produced from cyclic carbonates, economically and environmentally. The thermocatalytic production of polycarbonates from the reaction of CO2 with epoxides has been studied using organic bases, alkalis, metal halides, metal complexes, ionic liquids, zeolites, and metal oxides [481]. The industrial scalability of the potential processes was challenged by catalyst stability and/or recovery [520]. Reported studies on the catalysts did not address these scalability-associated issues; instead, kinetics and selectivity were focused on [458,521]. R&D activities leading to the development of catalysts and processes for CO2 chemical utilization started at Bayer during the first decade of the 2000s [522]. An overview of the considered chemical technologies was reported in [459]. A variety of polymers could be produced from the developed and scaled-up processes, e.g., polycarbonates, polyethercarbonates, polyacrylates, and PUs [82]. The reaction of cyclic acid anhydrides and epoxides with CO2 produced terpolymers and polyalkylenecarbonates, respectively [462].
2.4.2. Radiation-Induced Pathways
Additionally, to thermocatalytic pathways and methods described above for the production of polymers, ionizing radiation has been considered in the search for more energy-efficient processes. The radiation-induced carboxylation reactions have been employed to produce salicylic acid from phenol [523,524], malonic acid from acetic acid [524,525,526,527], glycolic acid [523], and amino acids from amines [528]. The radiation-induced carboxylation of methanol produces formic, glycolic, and oxalic acids, as well as ethylene glycol and formaldehyde [529]. Meanwhile, radiation-induced carboxylation of formic acid produced preferentially oxalic acid [530]. The conversion of CO2 has been found to produce transient species such as COO−/COOH and organic (hydrocarbonaceous) radicals, leading to the production of carboxylic acids, esters, or organic carbonates [528].
Although polymerization reactions are known to be initiated by free radicals and these key reactive species can be created by γ-radiation, only a few works have been reported for radiation-induced polymerization, e.g., vinyl monomers [531], styrene and methyl styrene [532], succinic acid [533], methylmethacrylate [531], and carboxylic acids [534]. On the other hand, CO2 can be grafted as carboxylated functional groups on linear low-density polyethylene by γ-irradiation under supercritical conditions. Thus, radiation-induced carboxylation can be used to functionalize polymers [535]. Other organic compounds have been grafted on polymers, e.g., maleic anhydride [536], that can be synthesized using CO2 as a (co-)reactant.
Assuming that (i) carboxylation reactions can be induced by ionizing radiation, (ii) the products of the carboxylation reactions can polymerize, and (iii) polymerization reactions can be induced by radiation, it could be expected that there might be reaction conditions under which carboxylation and polymerization can be carried out in a single chemical process step under the effects of ionizing radiation. The monomer production and its consumption through polymerization in one pot will overcome the low carboxylation yield that is thermodynamically limited by shifting the equilibrium to the right. Even though the production of value-added polymers is one of the economically rewarding massive chemical utilization of CO2 routes and all the involved reactions have been proven to occur, the development of CPs to carry these reactions out is still in its infancy. Moreover, the recent discovery that polymers containing CO2 moiety are responsive to a variety of stimuli/effects has highly increased the value of these polymers.
2.5. Carbonates and Bicarbonates
Carbonation is a naturally occurring reaction between CO2 and alkaline or alkaline earth elements or their oxides, leading to the formation of mineral materials, which is also known as mineralization. Carbonation moves carbon from atmospheric gases to Earth’s solid constituents as part of the C-cycle. This reaction on mineral deposits in geological formations has been used industrially with sequestration purposes to store CO2 [56,59,87,537,538,539,540,541,542,543]. The experience gained from sequestration identified kinetic limitations and high energy consumption as the top challenges to address for a wider range of applications to be developed.
Inexpensive silicate minerals (e.g., serpentine, olivine, and wollastonite) are considered raw materials for the production of carbonates and bicarbonates using CO2 in an acid solution. Metal cations (Me) lixiviate from the mineral matrix and react with the dissolved CO2, resulting in the consumption of protons (R35) and solid precipitation (R36).
2H+ + H2O + MeSiO3 → Me2+ + H4SiO4
H2CO3 + Me2+ → MeCO3 + 2H+
The carbonation heat of reaction (ΔH298K of (R36)) ranges from −60 to −100 kJ/mol. This exothermicity does not compensate the kinetic limitations [544]. Cations leaching is the reason for the slow kinetics, moving R&D to investigate the effect of mineral type and composition, metal element, pH, and temperature on considered minerals and approaches: reacting phases (gas–solid vs. aqueous), resource (raw material and CO2) selection, and raw material pre-treatment and/or activation [540]. For instance, ammonium salts can be used to pretreat the raw material to facilitate Mg extraction [545], and serpentinite and metaperidotite minerals are selected based on their high Mg content [546]. Increasing the temperature to improve the kinetics is not a solution for the aqueous-phase option since dissolved CO2 will decrease. Thus, increasing temperature has to be accompanied by an increase in pressure as well to avoid the CO2 move from the liquid to the gas phase, e.g., pressurized fluidized bed suggested by Abo Akademi [547]. However, solving the kinetic problems (by increasing severity) seems to aggravate the energy intensity problem.
The energy intensity problem was originally due to waste and mineral treatments (mechanical separation, crushing, milling, and grinding), material flow management, CO2 compression, and/or process conditions [548]. Energy intensity has been addressed through efficiency improvements, process integration, and intensification. The integration of the heat released by (R36) could be used to balance the energy demanded by metal extraction/digestion, i.e., lixiviation (R35), in a sort of autothermal operation [549]. The economic benefits of cheap mineral resources bring other inefficiencies to the process. The metal content of these minerals and the carbonation stoichiometry imply that a large amount of mineral is required to remove or bind CO2 (2–3 tonrock/tonCO2).
The development of the direct aqueous mineralization method started in the 1990s at Los Alamos National Laboratory [550] and continued at the National Energy Technology Laboratory (NETL), leading to [551,552,553] to produce Ca and/or Mg carbonates. This process follows the two steps described by (R35) and (R36) reactions [554]. The high energy consumed by the process was supplied by a coal-fired power plant, which made the process impracticable due to the large CO2 emissions [553]. The economic drawback was identified as associated with the product’s low price and elevated production costs [555].
This perception of economic disadvantages was later confirmed by sequestration experiences and reported analyses/assessments [556]. For instance, carbonation costs using olivine ranged from $50 to $100/tonCO2, which increased power generation by 30–50% when applied as a CCS method for power plants [78]. The process economy could be improved by enhancing energy efficiency [557], improving materials management [558], and by intensification [559]. Cambridge Carbon Capture claims to have improved both economic [560] and environmental [561] impacts by incorporating an alkaline pretreatment step [562,563], using a cheaper mineral [564] and CO2 source [565], co-producing electricity [566], and valorizing all products and by-products [567]. This company emphasized the most value-adding aspects: the optimization of production costs and the valorization of all products [560]. The former aspect considers the use of cheaper materials [564], mostly industrial wastes (e.g., cement bypass and kiln dust, waste cement, construction and demolition mineral waste, red mud, fly ash [568], steel/blast furnace/electric arc furnace slags [569], mine tailings, municipal waste incinerator ash, and water treatment sludge [570]). Meanwhile, for the latter, the relevant products include calcium and magnesium oxides, carbonate mineral powders, silica, and high-purity metals [567]. Products’ value and valorization have been considered in the development of methods to produce specialty nanoparticle materials, e.g., calcium, vanadium, silver, copper, nickel, etc., by integration into different industrial processes and carbonating industrial wastes [571,572,573], e.g., steel mills [574], combination with RWGS [561], cement [575], and incineration plants [576]. Besides the economic benefits of nanomaterials production, the environmental assessment also had a positive result.
Electrochemical carbonation pathways have been employed to produce bicarbonates (e.g., ammonium [577]) and co-produce electricity [578] or hydrogen [579]. This type of pathway has been considered for recycling scrap metal sources, such as wasted aluminum foil [578,579].
Carbonation of industrial wastes might bear logistic limitations since, in some instances, wastes might be generated in a different location from where CO2 is emitted. However, the metal (steel) and cement industries, two of the largest CO2 emitters, are excellent candidates for implementing carbonation within their decarbonization strategies. In these cases, the direct use of flue gases [545,580] constitutes an intensification option by eliminating the capture step [560] and will contribute to economic improvements by reducing the required Capex.
Carbonation technological developments have been covered in patents, e.g., [563,581,582,583,584]. Assuming that economic and technical hurdles are overcome and carbonation processes are implemented, the market of the products is limited by their current uses and applications. Among the multiple uses and applications of carbonates and bicarbonates (health, cosmetics, pulp and paper, chemicals, hygiene, glass, photography, textiles, construction, etc. [45,585,586]), most relevant to this work is the nuclear sector, for which molten salts are used as cooling fluid in certain reactors [587,588,589,590,591,592]. Regardless of these many uses and applications, implementation of a potentially sustainable and decarbonizing production option will change market dynamics and will affect product prices and, in consequence, process economy. Therefore, new applications and uses need to be developed to contribute to demand growth and to keep the price from at least not dropping.
3. Integration to Nuclear Power
Although the idea of integrating an NPP into a CP is not new, it has not been accomplished, and the remaining challenges need to be addressed to further develop and realize NP-CP IES that could underpin the implementation of NP-to-X pathways. These challenges, for the purpose of this work, concern energy integration, which, in the case of heat and electricity integration, can be considered a mature technical area. However, the particularities of the heat and electrical current (intensity and voltage, for instance) that the NPP might offer and the CP might demand will be the subject of analysis, design, and implementation for the corresponding integration project.
Nevertheless, relevant experience was gained in Germany, Japan, Russia, and the USA on the heat integration of chemical heat pipes (Figure 2) during the development and demonstration of A&E and NH projects [593]. In German EVA-I tests, the primary loop of the high-temperature gas (helium) reactor (HTGR) was directly integrated into an SMR reactor. Although these tests provided valuable data on heat transfer, mechanical design, and behavior of reformer materials in helium with regard to H2 and tritium permeation and corrosion [594], they also raised questions on the validity of a single-tube reactor. Thus, a multi-tubular (18 and 30 tubes) EVA-II reactor was then built and tested under normal (nuclear) operation conditions to assess thermal, process, and mechanical design; material properties and selection; as well as transient behavior under fluctuating conditions (load variations, start-up, and shut down) [595], over 5200 h [596]. Heat transfer was studied for two different heating arrangements: a baffle structure (30 tubes), an annular structure (18 tubes), and an external electric heater. In the case of He, which was used as heating medium, it entered the reformer tubes at 950 °C and exited the reactor at 650 °C [597]. It was found that heat transfer occurred at >90% by convection and less than 10% by radiation and that it could be improved by increasing He temperature or pressure, increasing pressure drop, or incorporating fins on the tubes [598]. The selection of materials for building the different process components was based on tests run for over 10,000 h [599,600,601]. Similarly, the ADAM I and II units were built and tested at 650 °C for 2344 h and 6000 h, respectively [602]. The integrated A&E plant transported energy of 5.72 MW(th) through the pipeline, receiving an input of 10.78 MW(e) total power, mostly provided by the helium flow (heat). The ADAM plant was operated with an energy efficiency of 72% (5.42 MW of heat released and 2.34 MW used heat). This experience demonstrated the possibilities of transporting 300 kW of nuclear power [603,604,605] and nuclear heat over long distances [15,593,597,606,607] for 10,150 h. The drawbacks of the technology included contamination by solid fission products and increased corrosion of the core and the fuel elements [608].
In the A&E German development, an intermediate heat exchanger (IHX) was deemed unnecessary within the initial assumptions [595]. After thorough analyses of the found drawbacks, this initial assumption was ruled as wrong. Instead, the Japanese decided to include an IHX in their HTTR project, which also involved commercially available SMR. The use of the IHX reduced hydrogen and tritium permeation rates to negligible values, avoiding product contamination. In this case, the Japanese could concentrate the R&D efforts on hydrogen (and tritium) permeation [609,610] as well as acquiring heat transfer data while still addressing materials design, selection, and testing [611,612]. The observation of He leaks [613,614] and the possibilities of explosion and fires [615,616] elevated safety concerns [617] and were favorably used to develop guidelines to design safer and more reliable reformers [618,619,620,621]. Scaling (1/30 of industrial scale) and integration to demonstrate performance were carried out, using electrical heating to model nuclear heat and to produce 110 m3/h of H2 [622]. Testing of this unit was used to develop start-up and shut-down protocols and to collect data on materials’ behavior, performance (under transient and steady state), and operating conditions and to define SMR design premises [622], including control instrumentation [623]. An SMR to produce 4200 Nm3/h of hydrogen, using a conventional Ni-catalyst and consuming about 10 MW of thermal energy was designed [619], and a dynamic simulation code to predict heat and temperature fluctuations of the system was developed [623]. Although drastic drawbacks were never reported, the shifting of hydrogen production R&D to the study of the sulfur–iodine thermochemical cycle indicates that some would have been found [624,625].
Heat integration has been proposed to be carried out either using spent nuclear fuel (SNF) [626] or employing open cycles [627] rather than the closed-cycle type used by A&E or any other heating pipe. In summary, heat management within energy integration between NP and CPs might not be as simple as expected from the maturity of the technology but instead would create new challenges to be addressed in the immediate future.
Energy integration of electrochemical reactions to NPPs would bring benefits of a combined option of heat and electrical power to the electrochemical plant. The scale-up attempts of emerging CO2 electrolysis technologies were recently compared [361,628], pointing out some of the scalability issues of concepts that have been demonstrated at a laboratory scale. High over-potential, low FE, and low current densities are the main challenges to overcome to avoid additional costs prior to any attempt to scale up [629]. Current density can be improved in HTE CO2 reduction, leading to performance improvements as well in solid oxide electrochemical cells [200]. In HTE operations, part of the energy requirements is provided by thermal energy, and lower electric power is consumed by the electrolysis process. In fact, HTE energy consumption, in CO2 co-electrolysis, can be 30–35% lower than that of LTE, and the estimated H2 production cost was as low as $0.660/gge (gallon of gasoline equivalent) [630]. The high temperature employed creates opportunities to manufacture other products beyond CO as well as to process intensification by introducing a combination of electro- and heterogeneous catalysts and/or a combination of physical and chemical processes. The experience gained through the integrating efforts of LTE or HTE for H2 production to NPPs [23,631,632,633,634] should serve as a knowledge platform when other electrochemical units need to be integrated, and the barriers of economic feasibility have to be overcome. In the H2 case, cogeneration of electricity by reversible operation of an LTE or HTE as fuel cell (FC) has been suggested as an economic improvement of the integrated system [635]. HTE operations are most benefitted from integration to NPPs that would provide both heat and electricity. However, for HTE, material stability represents higher risks as operating temperature increases [636,637,638,639]. NPP’s integration will require endurance of long-term stability of the electrolysis system under industrial operation conditions [200]. Finally, combination of electrochemical process with radiation-induced chemistry could further contribute to decreasing the activation potential for electrochemical reduction.
Meanwhile, the use of radiation as energy source through integration of the chemical process to an NPP is new and not a straightforward step. The use of ionizing radiation to drive CPs gives rise to new and complex challenges. The first question to ask is whether such integration is worth an investment. Schmeda-Lopez et al. tried to answer this question through an economic evaluation of a process concept that combines a propane radiolysis process (to produce propylene) with a molten salt nuclear power reactor. The ionizing radiation employed was γ-ray from the radioactive gaseous fission products that is either dissipated or wasted within the NPP. The economy of the NP-integrated process resulted in unfavorable returns on investment for small NPPs, but it was more than acceptable for larger NPPs [640].
Livingstone proposed to supply the γ-radiation from rod assemblies of SNFs to reduce CO2 [641]. The theoretical assumptions for designing the chemical reactor were (i) direct contact between the spent fuel and the reactor walls and (ii) the critical role of the fast Compton electrons in inducing the reaction [641]. This proposal takes advantage of employing the radiation emitted by SNF during its cooling process in the storage pond. The activity of a conventional 60Co source (~1014 Bq), typically used in commercial applications, could be four orders of magnitude lower than that of SNFs (~1019 Bq). Additional advantageous characteristics of the SNF γ-radiation are the almost neutron-free nature of the radiation, its mean energy of 578.4 keV, and an energy spectrum extending from a few hundred keV to over 2 MeV. Comparatively, the energy (γ-radiation) from SNFs will induceto completion chemical reactions in seconds or minutes that might take hours to complete by exposure to conventional radiation sources.
In terms of availability, the NE sector follows and takes strict measures for the safe management, storage, and disposal of SNFs. As can be deduced from the previous paragraph, once the energy generation lifecycle is complete (15–18 months), the SNF rod assemblies are removed from the reactor core with a significant remaining residual radioactivity since only about 2% of the fissionable fuel is consumed. The residual radioactivity (~98%) would take decades to be released in the form of heat from the SNF rods, which are regarded as “hot”. These hot SNF assemblies need to be “cool” enough to safely be handled and transported. During this cooling process, substantial amounts of energy are released from the SNF rods, and this energy is safely dissipated and unused. Thus, this cooling is carried out in water pools that every NPP provides to initially store the SNF assemblies. While the pool temperature increases, its water content decreases by evaporation losses, so energy (to cool down the pool) and water are consumed. Water needs to be replenished since, by regulation, storage has to be at least under20 feet of water [642]. Since the SNF storage in the cooling pools lasts for 10–15 years, the storage capacity of existing cooling pools has begun to be exhausted [643]. In the United States (US), for instance, 60,000 metric tons of heavy metal (MTHM) of SNF were stored at nationwide sites by 2008 [643], for a yearly accumulation of 2000 MTHM [644]. Thus, if the 104 nuclear reactors in the US remain operative by 2050, then the inventory would increase to about 130,000 MTHM [645]. From the initial storage at the NPP location, the SNF should be moved to a permanent storage facility, which commonly is a geological formation where it should remain for at least 10,000 years. However, these permanent geological repositories are difficult to license and implement, leaving the SNF at their generating NPP for longer times than the cooling requires. Nonetheless, at this point, it is worth mentioning the NE sector’s efforts of SNF reprocessing for recycling purposes [645,646]. Reprocessing facilities are operative in France, the United Kingdom, and Russia. France reprocesses SNFs domestically and has reprocessed SNFs from Japan, Germany, Switzerland, Belgium, Italy, Spain, the Netherlands, and Italy. SNFs from Japan, Germany, Switzerland, Belgium, Italy, the Netherlands, Sweden, and Canada have been reprocessed in the United Kingdom, while Russia has reprocessed SNFs from Ukraine. The recycling in the NE sector is currently limited by economics, making storage the disposal alternative [647].
It is clear that the ionizing radiation emitted by SNF in cooling ponds, otherwise wasted, could offer a reliable energy source to drive CPs. Additionally, if the considered chemical reaction is endothermic, the heat of reaction could contribute to cooling the pond and will not only save on energy consumption but also prevent water losses, an important move toward eco-efficiency and sustainability.
Radiation-induced conversion pathways for CO2 could be more energy-efficient processes, benefiting from the added synergy, which lowers the overall energy requirements for CO2 conversion by utilizing the limited value of SNF ionizing radiation at NPP facilities. However, this integration concept brings about new challenges, such as the need to increase the radiolytic conversion efficiency of CO2 and to find means for controlling the propagation over the termination reactions. The radiolysis of CO2 has found very little to no application due to the lack of fundamental understanding of these two main scientific challenges. It is known that the low radical yield is mainly due to recombination reactions (backreactions but also to the inherent low reactivity of CO2. Backreactions are termination reactions that compete with the propagation reactions that cascade into more radical formation. Knowledge on the mechanisms to improve radical yields, either by the use of additives or identification of favorable conditions for radical formation, is still needed. Radicals are highly reactive but rather than recombining reactions (backreactions or termination reactions), what is needed are conditions to favor the propagation reaction to increase radical yield.
Additionally, integration also demands attention regarding certain aspects of the radiation-induced CP. On the one hand, the rational design of chemical pathways and on the other, radiation-tolerant materials and components are technical areas calling for R&D. Controlling reaction pathways, besides the issues pointed out above, also includes taking apart, tuning, or stopping undesired pathways while accelerating specific ones. This might require (radiation-derived) heat management and/or equipment and reactor design [648,649]. The effects of radiation-induced reacting mixture and reaction products on the corrosion of reactor materials [650] also require further investigation.
The CO2 valorization through NP-to-X pathways represents decarbonizing approaches that connect the NE industry to a decarbonized chemical sector and enable a smooth, consistent energy transition of a hard-to-decarbonize industry. This work has examined a variety of possible conversion pathways for CO2, as well as the importance of framing conversion within the carbon cycle and using it as the cycle-closing mechanism to enable a circular economy, which has been reported as a way to procure sustainability [96,651]. Along the text, scientific questions, issues, gaps, and challenges that need to be addressed through R&D are mentioned. The challenges to be addressed, as well as the issues that need to be solved, have also been discussed. The unique energy mix provided by NPPs creates a platform of opportunities for the development of processes and methodologies that support the NP-to-X pathways. CO2 valorization provides the economic incentives for decarbonization.
The enormous gap between current emissions and utilization calls for a suite of chemical process technologies since there is not a single answer or a single solution. Then, there cannot be a single processing solution for the 37 Gton of emitted CO2, and instead, a suite of process technologies meeting certain requirements is needed. Thus, these processes are required to minimize hydrogen consumption and produce value-added products using low-carbon-emitting energy sources, ultimately resulting in cost-effective technologies. In order to avoid leveraging inefficiencies, the new processes being developed should be more energy-efficient than those currently operative. Process intensification needs to be present in these NP-to-X pathways through the combination, coupling, and/or integration of processes that take advantage of potential synergies among the available forms of energy (heat, electrons, and radiation).
Since energy is the key integrating factor in the NP-CP IES to enable NP-to-X pathways, no risk should be taken to jeopardize energy security, affordability, and accessibility, which are the basis for human development. Finally, in the valorization of CO2 through NP-to-X pathways, sustainability has to be the core driver since the lack of sustainable pathways in the past is the reason for having to face the current situation.
4. Future Potentialities
CO2 valorization, as part of a decarbonizing strategy, relies on the development of process technologies with the highest energy and carbon efficiency. In terms of energy, and due to the intrinsic energy intensity involved in CO2 chemical reactions, not only is high efficiency required, but also the energy has to be provided by a C-neutral source. Then, regarding C-efficiency, it can be satisfied partially by the use of C-neutral sources and additionally by reactions consuming large CO2 amounts and rendering products that retain carbon for long periods of time. Furthermore, the development of these CP targets a sustainable future on the horizon by addressing the identified economic and environmental challenges. For instance, hydrogenation reactions should employ green H2 and additionally should either minimize its consumption or develop an intensified process that generates hydrogen in the reacting medium.
Concerning CO2 as feedstock, a first consideration is the holistic examination of the magnitude of the emissions (Gton/y scale) and of the (high) emitters (global 104 scale), the CO2 concentration in the different effluents, emitters’ throughput, contaminants present, and geographical location. It becomes evident that a universal technology of the one-size-fits-all solution type does not exist and probably cannot be developed. Moreover, those feedstock attributes give rise to composition, concentration, and logistic mismatches between the emitter and the potential conversion plant. The mismatches in composition and concentration are defined by a potential discrepancy between the feedstock specification to be utilized and the concentration of undesired compounds and CO2 in the emitted stream. A logistic mismatch is found when the emitter is located at an uneconomical distance from the using location. Finally, the dynamics and scale of the emissions throughput at a given emitter location may differ from that required to satisfy the market size of the proposed product(s).
This work has examined some chemical pathways that fall within the above-mentioned preferred attributes, though improvements in energy and C-efficiency are required to further develop into sustainable process technologies. Examples of awarded patents and published patent applications have been included in the discussion. The chemical reactions discussed were categorized according to the type of energy employed to satisfy the high energy requirements of CO2 conversion reactions, i.e., thermal/heat, electricity, and ionizing radiation. These three types of energy can become available from NPPs and create a new (decarbonizing) platform for the production of (decarbonized) chemicals, products, and materials derived from CO2. However, the integration of CPs to NPPs will bring new challenges. Although heat or electricity integration is a mature technical area, the particularities of each considered plant, as has been pointed out above, need to be addressed and sorted out. The former involves further development and implementation of chemical heat pipes, and the application of gained experiences and knowledge on new processes. Similarly, electrochemical process integration is not just a plug-and-play step, and some of the particularities of each process have already become an R&D subject. The low deployment of electrochemical processes is a result of their lack of economic sustainability, mainly due to a continuously increasing electricity cost. Thus, the challenges found within the electrochemical pathways concern those aspects associated with improving energy efficiency, such as overcoming reactions overpotential by developing more active electrocatalysts, improving faradaic efficiency by developing more selective electrocatalysts and intensifying processes by reducing number of process steps, for instance.
Radiation-induced chemistries go beyond the study of radiolytic reactions into tailor-designed pathways for the production of value-added products. New concepts for either new synthesis methods, new deconstruction methods, and/or new ways of approaching the development of radiation-induced CPs can be created by addressing identified scientific challenges. The use of catalysts and/or electrocatalysts calls for dynamic experimentation under continuous flow at high dose rates. Still, the definition of the fundamental principles behind the competition between propagation and termination reaction needs to be investigated, for which new approaches will need to be defined. Therefore, the broadest challenges probably come from the development and integration of radiation-induced CPs. Regarding the chemical process itself, there are aspects in which fundamental knowledge needs to be created. A comprehensive understanding of the interactions between ionizing radiation with the reacting molecules, with the surface of (radiation-induced-activated?) catalysts or electrocatalysts, and/or with adsorbed species on catalysts/electrocatalysts is key for controlling the propagation reactions (formation of ion radicals) and the termination reactions (backreactions or disappearance of ion radicals). The application of the knowledge derived from the understanding of the chemical transformations of ion radicals/free radicals could be directed to design reaction pathways for the production of specific compounds. The catalysis knowledge is not only restricted on the activating (increase in catalytic activity) effect of ionizing radiation but also catalyst development needs to address effects on stability and on integrity, as well as the safe use and management of catalysts that have been exposed to ionizing radiation. The identification of knowledge gaps and the approaches and attempts to fill these have been reviewed not long ago [28]. The development of protocols for the safe use of ionizing radiation expands into the full development of radiation-induced CPs. The application of ionizing radiation sourced from SNFs during the cooling process has been proposed but needs further efforts and attention before considering scaling to a commercial application. Similar to the suggested R&D work with catalysts, products and materials that have been exposed to radiation will require handling under strict safety protocols. Radiation-induced processes might open new opportunities for other wastes besides CO2 since the combination of feedstocks is also possible, as it would be the use of CO2 for radiolytically functionalizing plastic wastes [28] as an upcycling mechanism.
Overall, the NP-to-X pathways are of particular interest in the development of new CPs for dealing with emitted CO2 and also for handling renewable, non-recyclable wastes, for which radiation, heat, and/or electricity can drive such reactions. NPPs’ energy mix would also give opportunities for finding synergies between the energy forms by combining heat and electricity (e.g., high-temperature electrolysis), radiation and heat (e.g., radiation-induced thermocatalytic reactions), radiation and electricity (e.g., radiation-induced electrocatalytic reactions), etc.
Finally, the synergistic use of the three different types of energy (e.g., radiation and (electro)catalysis, radiation and thermal-catalysis, or radiation and high-temperature electrocatalysis) collectively gives rise to new and overwhelming R&D challenges. Process design and development will lead to the definition of whether simultaneous or consecutive application of the radiation type is applied. In some instances, simultaneity might result obvious when lifetime or reactivity of ion radicals implies so. However, in cases where the stability and integrity of components, catalysts/electrocatalysts, products, and materials are suspected or jeopardized, then overcoming the existing challenges in the development of consecutive processes becomes the option.
5. Concluding Remarks
Nuclear power represents one of the means to reliably provide three different forms of C-free energy, namely, heat, electricity, and ionizing radiation, at a large scale from a single point. The utilization of part of this energy to drive chemical reactions to produce a variety of CO2-derived products not only broadens the market of NPPs beyond electricity generation but also significantly benefits the environment, giving use to noxious emissions while creating value from it. The new markets will include industrial and commercial sectors such as manufacturing, chemical, construction, and buildings. An additional benefit of the diversification of nuclear energy interests is its revitalization, supported by new partners, markets, customers, and end-users.
However, the integration of NP with CP is not a straightforward step, and scientific and technical problems are found along the way. Greater benefits would be obtained from energy-intense chemical transformations. Currently, high temperatures and heat are provided by the combustion of fossil fuels, leading to significant CO2 emissions. Therefore, heat integration into an NPP will bring economic and environmental benefits. These benefits could be better if, in addition to heat, the desired chemical transformation can be induced by ionizing radiation and/or carried out electrochemically.
Although low yields are observed from radiation-induced CO2 reactions, the presence of other compounds (e.g., methane or water) and catalysts in the reacting medium improve yields. Catalyst and electrocatalyst research can be designed to guide radical reactions, scale the potential of electrochemical processes to final conversion into desirable products, and control the propagation and termination reactions to attain the desired product distribution. Ideally, full synergy among heat, electricity, and ionizing radiation would make the best out of the NP-to-X pathways.
The development of decarbonizing processes responding to the NP-to-X pathways gives rise to a new industrial era for the manufacture of decarbonized chemicals, products, and materials with higher efficiency that will lower the contribution of industry to the emission of polluting compounds into the atmosphere. The progress made in the development of nuclear reactors dealing with higher-temperature heat or delivering smaller and/or modular amounts of energy can be advantageously used to implement distributed platforms for these new manufacturing schemes while contributing to the achievement of net zero goals.
Funding
This manuscript was prepared within activities performed at the Idaho National Laboratory—Battelle Energy Alliance, LLC—under DOE Idaho Operations Office contract No. DE-AC07-05ID14517.
Acknowledgments
The author acknowledges that the Idaho National Laboratory—Battelle Energy Alliance, LLC—has granted permission to publish this work.
Conflicts of Interest
The author declares no conflicts of interest.
Abbreviations and Acronyms
| Adam & Eva | A&E | integrated energy systems | IES |
| 2-amino-2-methyl-1-propanol | AMP | intermediate heat exchanger | IHX |
| Carbon Recycling International | CRI | hydrogen evolution reaction | HER |
| Chemical looping | CL | life cycle assessment | LCA |
| chemical process | CP | metal | Me |
| dimethyl carbonate | DMC | metal oxide | MeO |
| dimethyl ether | DME | methane dry reforming | MDR |
| Emergy Accounting | EMA | metric tons of heavy metal | MTHM |
| Emissions-to-Liquids | EtL | nuclear energy | NE |
| Ethylene glycol | EG | nuclear hydrogen | NH |
| Ethylene oxide | EO | nuclear power plants | NPP |
| faradaic efficiency | FE | oxidative coupling of methane | OCM |
| formaldehyde-to-urea | F/U | power to gas | PtG |
| fuel cell | FC | power to liquids | PtL |
| functional unit | FU | power to value | PtV |
| gas diffusion electrodes | GDE | power to any energy-carrier | PtX |
| Gas Technology Institute | GTI | renewable natural gas | RNG |
| global warming impact | GWI | spent nuclear fuel | SNF |
References
- Chen, X.; McElroy, M.B.; Wu, Q.; Shu, Y.; Xue, Y. Transition towards higher penetration of renewables: An overview of interlinked technical, environmental and socio-economic challenges. J. Mod. Power Syst. Clean Energy 2019, 7, 1–8. [Google Scholar] [CrossRef]
- Milko, J.; Allen, T.; Fitzpatrick, R. Keeping up with the Advanced Nuclear Industry; Third Way: Washington, DC, USA, 2018; 3p, Available online: https://www.thirdway.org/graphic/keeping-up-with-the-advanced-nuclear-industry (accessed on 8 December 2018).
- Fütterer, M.A.; Fu, L.; Sink, C.; de Groot, S.; Pouchon, M.; Kim, Y.W.; Carré, F.; Tachibana, Y. Status of the very high temperature reactor system. Prog. Nucl. Energy 2014, 77, 266–281. [Google Scholar] [CrossRef]
- US Department of Energy; Nuclear Energy Research Advisory Committe; Generation IV International Forum. A Technology Roadmap for Generation IV Nuclear Energy Systems; GIF-002-00 Report; USA Department of Energy: Washington, DC, USA, 2002; 97p.
- Fumizawa, M.; Kosuge, Y.; Horiuchi, H. Thermal evaluation for a ultra high temperature reactor with pebble type fuels. In Proceedings of the 17th International Conference on Nuclear Engineering, ICONE17, Brussels, Belgium, 12–16 July 2009; Volume 1, pp. 865–872. [Google Scholar] [CrossRef]
- Hashimoto, K.; Yamasaki, M.; Fujimura, K.; Matsui, T.; Izumiya, K.; Komori, M.; El-Moneim, A.A.; Akiyama, E.; Habazaki, H.; Kumagai, N.; et al. Global CO2 recycling—Novel materials and prospect for prevention of global warming and abundant energy supply. Mater. Sci. Eng. A 1999, 267, 200–206. [Google Scholar] [CrossRef]
- Buchholz, O.S.; Van Der Ham, A.G.J.; Veneman, R.; Brilman, D.W.F.; Kersten, S.R.A. Power-to-Gas: Storing surplus electrical energy a design study. In Proceedings of the 12th International Conference on Greenhouse Gas Control Technologies, GHGT 2014; Elsevier Ltd.: Amsterdam, The Netherlands, 2014; Volume 63, pp. 7993–8009. [Google Scholar] [CrossRef]
- Hashimoto, K.; Akiyama, E.; Habazaki, H.; Kawashima, A.; Shimamura, K.; Komori, M.; Kumagai, N. Global CO2 Recycling. Zair.-Kankyo 1996, 45, 614–620. [Google Scholar] [CrossRef][Green Version]
- Habazaki, H.; Yamasaki, M.; Kawashima, A.; Hashimoto, K. Methanation of carbon dioxide on Ni/(Zr-Sm)O(x) catalysts. Appl. Organomet. Chem. 2000, 14, 803–808. [Google Scholar] [CrossRef]
- Hashimoto, K.; Kato, Z.; Kumagai, N.; Izumiya, K. Key materials and systems for the use of renewable energy in the form of methane. In Proceedings of the 28th International Conference on Ocean, Offshore and Arctic Engineering, OMAE2009, Honolulu, HI, USA, 31 May–5 June 2009; Volume 4, pp. 1003–1011. [Google Scholar] [CrossRef]
- Kato, Z.; Izumiya, K.; Kumagai, N.; Hashimoto, K. Energy-saving seawater electrolysis for hydrogen production. J. Solid State Electrochem. 2009, 13, 219–224. [Google Scholar] [CrossRef]
- Hashimoto, K.; Kumagai, N.; Izumiya, K.; Takano, H.; Kato, Z. The production of renewable energy in the form of methane using electrolytic hydrogen generation. Energy Sustain. Soc. 2014, 4, 17. [Google Scholar] [CrossRef]
- Götz, M.; Lefebvre, J.; Mörs, F.; McDaniel Koch, A.; Graf, F.; Bajohr, S.; Reimert, R.; Kolb, T. Renewable Power-to-Gas: A technological and economic review. Renew. Energy 2016, 85, 1371–1390. [Google Scholar] [CrossRef]
- Gomberg, H.J.; Gordus, A.A. Hydrogen for synthetic fuels via nuclear energy. J. Fusion Energy 1982, 2, 319–339. [Google Scholar] [CrossRef][Green Version]
- Harth, R.E.; Boltendahl, U. The chemical heat pipe EVA and ADAM. Interdiscip. Sci. Rev. 1981, 6, 221–228. [Google Scholar] [CrossRef]
- Orhan, M.F.; Dincer, I.; Rosen, M.A.; Kanoglu, M. Integrated hydrogen production options based on renewable and nuclear energy sources. Renew. Sustain. Energy Rev. 2012, 16, 6059–6082. [Google Scholar] [CrossRef]
- El-Emam, R.S.; Khamis, I. Advances in nuclear hydrogen production: Results from an IAEA international collaborative research project. Int. J. Hydrogen Energy 2019, 44, 19080–19088. [Google Scholar] [CrossRef]
- Sink, C.J., Jr.; Taylor, A. An overview of the Nuclear hydrogen initiative. In Proceedings of the Topical Conference on Hydrogen 2006, Held at the 2006 AIChE Spring National Meeting, Orlando, FL, USA, 23–27 April 2006; pp. 40–45. [Google Scholar]
- Sakaba, N.; Ohashi, H.; Sato, H.; Hara, T.; Kato, R.; Kunitomi, K. Hydrogen production by high-temperature gas-cooled reactor; conceptual design of advanced process heat exchangers of the HTTR-is hydrogen production system. Trans. At. Energy Soc. Jpn. 2008, 7, 242–256. [Google Scholar] [CrossRef]
- Naterer, G.F.; Dincer, I.; Zamfirescu, C. Hydrogen Production from Nuclear Energy; Springer: London, UK, 2013; Volume 9781447149385, pp. 1–492. [Google Scholar] [CrossRef]
- Elder, R.; Allen, R. Nuclear heat for hydrogen production: Coupling a very high/high temperature reactor to a hydrogen production plant. Prog. Nucl. Energy 2009, 51, 500–525. [Google Scholar] [CrossRef]
- Revankar, S.T. Nuclear hydrogen production. In Storage and HYBRIDIZATION of Nuclear Energy: Techno-Economic Integration of Renewable and Nuclear Energy; Elsevier: Amsterdam, The Netherlands, 2018; pp. 49–117. [Google Scholar] [CrossRef]
- Yan, X.L.; Konishi, S.; Hori, M.; Hino, R. Nuclear hydrogen production: An overview. In Nuclear Hydrogen Production Handbook; CRC Press: Boca Raton, FL, USA, 2016; pp. 47–82. [Google Scholar]
- Petrescu, F.I.T.; Petrescu, R.V.V. Nuclear hydrogen structure and dimensions. Int. J. Hydrogen Energy 2019, 44, 10833–10837. [Google Scholar] [CrossRef]
- O’Brien, J.E. Review of the potential of nuclear hydrogen for addressing energy security and climate change. Nucl. Technol. 2012, 178, 55–65. [Google Scholar] [CrossRef]
- Khan, S.U.D. Using next generation nuclear power reactors for development of a techno-economic model for hydrogen production. Int. J. Energy Res. 2019, 43, 6827–6839. [Google Scholar] [CrossRef]
- Höhlein, B.; Menzer, R.; Range, J. High temperature methanation in the long-distance nuclear energy transport system. Appl. Catal. 1981, 1, 125–139. [Google Scholar] [CrossRef]
- Ramirez-Corredores, M.M.; Gadikota, G.; Huang, E.E.; Gaffney, A.M. Radiation-induced chemistry of carbon dioxide: A pathway to close the carbon loop for a circular economy. Front. Energy Res. 2020, 8, 17. [Google Scholar] [CrossRef]
- Ramirez-Corredores, M.M.; Rollins, H.W.; Morco, R.P.; Zarzana, C.A.; Diaz, L.A. Radiation-induced dry reforming: A negative emission process. J. Clean. Prod. 2023, 429, 139539. [Google Scholar] [CrossRef]
- Ramirez-Corredores, M.M.; Diaz, L.A.; Gaffney, A.M.; Zarzana, C.A. Identification of opportunities for integrating chemical processes for carbon (dioxide) utilization to nuclear power plants. Renew. Sustain. Energy Rev. 2021, 150, 111450. [Google Scholar] [CrossRef]
- Luderer, G.; Vrontisi, Z.; Bertram, C.; Edelenbosch, O.Y.; Pietzcker, R.C.; Rogelj, J.; De Boer, H.S.; Drouet, L.; Emmerling, J.; Fricko, O.; et al. Residual fossil CO2 emissions in 1.5–2 °C pathways. Nat. Clim. Change 2018, 8, 626–633. [Google Scholar] [CrossRef]
- Rissman, J.; Bataille, C.; Masanet, E.; Aden, N.; Morrow, W.R.; Zhou, N.; Elliott, N.; Dell, R.; Heeren, N.; Huckestein, B.; et al. Technologies and policies to decarbonize global industry: Review and assessment of mitigation drivers through 2070. Appl. Energy 2020, 266, 114848. [Google Scholar] [CrossRef]
- Global Carbon Project. Global Carbon Atlas: CO2 Emissions. Available online: http://www.globalcarbonatlas.org/en/CO2-emissions (accessed on 15 February 2024).
- Crippa, M.; Guizzardi, D.; Schaaf, E.; Monforti-Ferrario, F.; Quadrelli, R.; Risquez Martin, A.; Rossi, S.; Vignati, E.; Muntean, M.; Brandao De Melo, J.; et al. GHG Emissions of All World Countries—2023; Publications Office of the European Union, European Commission Joint Research Centre: Luxembourg, 2023; 268p. [Google Scholar] [CrossRef]
- Aresta, M.; Dibenedetto, A. Utilisation of CO2 as a chemical feedstock: Opportunities and challenges. Dalton Trans. 2007, 28, 2975–2992. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K.; Kumagai, N.; Izumiya, K.; Kato, Z. Carbon dioxide, the feedstock for using renewable energy. In Proceedings of the European Materials Research Society (E-MRS) 2010 Fall Meeting, Warsaw, Poland, 13–17 September 2010. [Google Scholar]
- Dibenedetto, A.; Angelini, A.; Stufano, P. Use of carbon dioxide as feedstock for chemicals and fuels: Homogeneous and heterogeneous catalysis. J. Chem. Technol. Biotechnol. 2014, 89, 334–353. [Google Scholar] [CrossRef]
- Grignard, B.; Gennen, S.; Jérôme, C.; Kleij, A.W.; Detrembleur, C. Advances in the use of CO2 as a renewable feedstock for the synthesis of polymers. Chem. Soc. Rev. 2019, 48, 4466–4514. [Google Scholar] [CrossRef]
- Müller, L.J.; Kätelhön, A.; Bringezu, S.; McCoy, S.; Suh, S.; Edwards, R.; Sick, V.; Kaiser, S.; Cuéllar-Franca, R.; El Khamlichi, A.; et al. The carbon footprint of the carbon feedstock CO2. Energy Environ. Sci. 2020, 13, 2979–2992. [Google Scholar] [CrossRef]
- Global CCS Institute. The Global Status of CCS 2023; Global CCS Institute: Melbourne, Australia, 2023; 98p, Available online: https://status23.globalccsinstitute.com/ (accessed on 15 January 2024).
- Budinis, S.; Fajardy, M.; Greenfield, C. Carbon Capture, Utilisation and Storage. Available online: https://www.iea.org/energy-system/carbon-capture-utilisation-and-storage (accessed on 15 June 2024).
- Allen, M.; Babiker, M.; Chen, Y.; de Coninck, H.; Connors, S.; van Diemen, R.; Dube, O.P.; Ebi, K.L.; Engelbrecht, F.; Ferrat, M.; et al. Global Warming of 1.5 °C; Special Report; Intergovernmental Panel on Climate Change (IPCC); Cambridge University Press: Cambridge, UK, 2018; 540p, Available online: https://www.ipcc.ch/sr15/ (accessed on 20 September 2021).
- Fernández, A.; Spencer, T.; McGlade, C.; Remme, U.; Brent, W.; D’Ambrosio, D. Net Zero Roadmap: A Global Pathway to Keep the 1.5 °C Goal in Reach; International Energy Agency: online, 2023; 226p, Available online: https://www.iea.org/reports/net-zero-roadmap-a-global-pathway-to-keep-the-15-0c-goal-in-reach (accessed on 2 November 2023).
- Riahi, K.; Schaeffer, R.; Arango, J.; Calvin, K.; Guivarch, C.; Hasegawa, Y.; Jiang, K.; Kriegler, E.; Matthews, R.; Peters, G.P.; et al. Mitigation pathways compatible with Long-term goals. In Climate Change 2022: Mitigation of Climate Change; Shukla, P.R., Skea, J., Slade, R., Eds.; Intergovernmental Panel on Climate Change (IPCC): Cambridge, UK; New York, NY, USA, 2023; pp. 295–408. Available online: https://www.ipcc.ch/report/ar6/wg3/downloads/ (accessed on 9 April 2024).
- Ramirez-Corredores, M.M.; Goldwasser, M.R.; Falabella de Sousa Aguiar, E. Decarbonization. In Decarbonization as a Route towards Sustainable Circularity; Springer International Publishing: Cham, Switzerland, 2023; pp. 15–101. [Google Scholar] [CrossRef]
- Ramirez-Corredores, M.M.; Goldwasser, M.R.; Falabella de Sousa Aguiar, E. Sustainable Circularity. In Decarbonization as a Route towards Sustainable Circularity; Springer International Publishing: Cham, Switzerland, 2023; pp. 103–125. [Google Scholar] [CrossRef]
- General Union Environment Action Programme to 2020 ‘Living Well, within the Limits of Our Planet’, in 1386/2013/EU. The 7th Environment Action Programme: European Union. 2013. 30p. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX:32013D1386 (accessed on 24 June 2022).
- National Research Council. Carbon Management: Implications for R&D in the Chemical Sciences and Technology; The National Academies Press: Washington, DC, USA, 2001; 236p. [CrossRef]
- Mahajan, D.; Song, C.; Scaroni, A.W. Catalytic Reduction of CO2 into Liquid Fuels: Simulating Reactions under Geologic Formation Conditions. In CO2 Conversion and Utilization; American Chemical Society: Washington, DC, USA, 2002; Volume 809, pp. 166–180. [Google Scholar] [CrossRef]
- Song, C.; Gaffney, A.F.; Fujimoto, K. CO2 Conversion and Utilization; ACS symposium series; Wiley: Washington, DC, USA, 2002; Volume 809, 427p. [Google Scholar]
- Aresta, M. Carbon Dioxide as Chemical Feedstock; Wiley-VCH: Washington, DC, USA, 2010; 394p. [Google Scholar] [CrossRef]
- Fink, J.K. Reactive Polymers Fundamentals and Applications: A Concise Guide to Industrial Polymers, 2nd ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2013; pp. 1–535. [Google Scholar] [CrossRef]
- Centi, G.; Perathoner, S. Green Carbon Dioxide: Advances in CO2 Utilization; Wiley Blackwell: Hoboken, NJ, USA, 2014; Volume 9781118590881, pp. 1–303. [Google Scholar]
- Muradov, N. Liberating energy from carbon: Introduction to decarbonization. In Lecture Notes in Energy; Wiley Blackwell: Hoboken, NJ, USA, 2014; Volume 22, 450p. [Google Scholar]
- Nakagawa, Y.; Honda, M.; Tomishige, K. Direct synthesis of organic carbonates from CO2 and alcohols using heterogeneous oxide catalysts. In Green Carbon Dioxide: Advances in CO2 Utilization; Wiley Blackwell: Hoboken, NJ, USA, 2014; Volume 9781118590881, pp. 119–148. [Google Scholar] [CrossRef]
- Fennell, P.; Anthony, B. Calcium and Chemical Looping Technology for Power Generation and Carbon Dioxide (CO2) Capture; Woodhead Publishing: Cambridge, UK, 2015; 446p. [Google Scholar]
- Aresta, M.; Dibenedetto, A.; Quaranta, E. Reaction Mechanisms in Carbon Dioxide Conversion; Springer: Berlin, Germany, 2016; 423p. [Google Scholar] [CrossRef]
- Kiss, A.A.; Pragt, J.J.; Vos, H.J.; Bargeman, G.; de Groot, M.T. Enhanced process for methanol production by CO2 Hydrogenation. In Computer Aided Chemical Engineering; Kravanja, Z., Bogataj, M., Eds.; Elsevier: Amsterdam, The Netherlands, 2016; Volume 38, pp. 985–990. [Google Scholar] [CrossRef]
- Chiang, P.-C.; Pan, S.-Y. Carbon Dioxide Mineralization and Utilization; Springer: Berlin/Heidelberg, Germany, 2017; 456p. [Google Scholar]
- Karamé, I.; Shaya, J.; Srour, H. Carbon Dioxide Chemistry, Capture and Oil Recovery; InTech Open: Online publication, 2018; 253p. [Google Scholar]
- Aresta, M.; Karimi, I.; Kawi, S. An Economy Based on Carbon Dioxide and Water, Potential of Large Scale Carbon Dioxide Utilization; Springer: Cham, Switzerland, 2019; 436p. [Google Scholar] [CrossRef]
- Klitkou, A.; Fevolden, M.; Capasso, M. From Waste to Value: Valorisation Pathways for Organic Waste Streams in Circular Bioeconomies; Taylor and Francis: Abingdon, UK, 2019; pp. 1–306. [Google Scholar] [CrossRef]
- North, M.; Styring, P. Carbon Dioxide Utilization; De Gruyter: Berlin, Germany, 2019; Volume 1 Fundamentals. [Google Scholar] [CrossRef]
- National Academies of Sciences-Engineering-Medicine. Gaseous Carbon Waste Streams Utilization: Status and Research Needs; The National Academies Press: Washington, DC, USA, 2019; 256p. [CrossRef]
- National Academies of Sciences-Engineering-Medicine. Gaseous carbon waste resources. In Gaseous Carbon Waste Streams Utilization: Status and Research Needs; The National Academies Press: Washington, DC, USA, 2019; pp. 27–38. [Google Scholar] [CrossRef]
- Rafiee, A.; Khalilpour, K.R.; Milani, D. CO2 Conversion and utilization pathways. In Polygeneration with Polystorage for Chemical and Energy Hubs; Khalilpour, K.R., Ed.; Academic Press: Cambridge, MA, USA, 2019; pp. 213–245. [Google Scholar] [CrossRef]
- National Academies of Sciences-Engineering-Medicine. Accelerating Decarbonization of the U.S. Energy System; The National Academies Press: Washington, DC, USA, 2021; 268p. [CrossRef]
- Ramirez-Corredores, M.M.; Goldwasser, M.R.; Falabella de Sousa Aguiar, E. Decarbonization as a Route Towards Sustainable Circularity, 1st ed.; Springer: Cham, Switzerland, 2023; Volume XXIX, 153p. [Google Scholar] [CrossRef]
- Aresta, M. Carbon dioxide reduction and uses as a chemical feedstock. In Activation of Small Molecules: Organometallic and Bioinorganic Perspectives; John Wiley and Sons: Hoboken, NJ, USA, 2006; pp. 1–41. [Google Scholar] [CrossRef]
- Aresta, M.; Dibenedetto, A. Artificial carbon sinks: Utilization of carbon dioxide for the synthesis of chemicals and technological applications. In Greenhouse Gas Sinks; CABI Publishing: Oxfordshire, UK, 2007; pp. 98–114. [Google Scholar]
- Olah, G.A.; Goeppert, A.; Prakash, G.K.S. Beyond Oil and Gas: The Methanol Economy, 2nd ed.; Wiley-VCH: Washington, DC, USA, 2009; 334p. [Google Scholar] [CrossRef]
- Aresta, M. Carbon dioxide: Utilization options to reduce its accumulation in the atmosphere. In Carbon Dioxide as Chemical Feedstock; Wiley-VCH: Washington, DC, USA, 2010; pp. 1–13. [Google Scholar] [CrossRef]
- Aresta, M.; Dibenedetto, A. Industrial utilization of carbon dioxide (CO2). In Developments and Innovation in Carbon Dioxide (CO2) Capture and Storage Technology; Elsevier Ltd.: Amsterdam, The Netherlands, 2010; Volume 2, pp. 377–410. [Google Scholar] [CrossRef]
- Suib, S.L. New and Future Developments in Catalysis. Activation of Carbon Dioxide; Elsevier B.V.: Amsterdam, The Netherlands, 2013; Volume 7, 644p. [Google Scholar] [CrossRef]
- Shi, F.; Morreale, B. Novel Materials for Carbon Dioxide Mitigation Technology; Elsevier: Amsterdam, The Netherlands, 2015; pp. 207–229. [Google Scholar]
- Styring, P.; Quadrelli, E.A.; Armstrong, K. Carbon Dioxide UtiIisation: Closing the Carbon Cycle; Elsevier: Amsterdam, The Netherlands, 2015; 336p. [Google Scholar]
- Aresta, M. The carbon dioxide problem. In An Economy Based on Carbon Dioxide and Water, Potential of Large Scale Carbon Dioxide Utilization; Aresta, M., Karimi, I., Kawi, S., Eds.; Springer: Cham, Switzerland, 2019; pp. v–xi. [Google Scholar]
- Mazzotti, M.; Abanades, J.C.; Allam, R.; Lackner, K.S.; Meunier, F.; Rubin, E.; Sanchez, J.C.; Yogo, K.; Zevenhoven, R. Mineral carbonation and industrial uses of carbon dioxide. In Special Report on Carbon Dioxide Capture and Storage; Metz, B., Davidson, O., de Coninck, H., Loos, M., Meyer, L., Eds.; Intergovernmental Panel on Climate Change (IPCC); Cambridge University Press: Cambridge, UK, 2005; pp. 319–338. [Google Scholar]
- Aresta, M.; Dibenedetto, A. Carbon dioxide: A valuable source of carbon for chemicals, fuels and materials. In Catalytic Process Development for Renewable Materials; Wiley-VCH: Washington, DC, USA, 2013; pp. 355–385. [Google Scholar] [CrossRef]
- Centi, G.; Perathoner, S. Catalytic transformation of CO2 to fuels and chemicals, with reference to biorefineries. In The Role of Catalysis for the Sustainable Production of Bio-Fuels and Bio-Chemicals; Triantafyllidis, K., Lappas, A., Stöcker, M., Eds.; Elsevier: Amsterdam, The Netherlands, 2013; pp. 529–555. [Google Scholar] [CrossRef]
- Hall, P.J.; Wilson, I.A.G.; Rennie, A. CO2-derived fuels for energy storage. In Carbon Dioxide UtiIisation: Closing the Carbon Cycle; Styring, P., Quadrelli, E.A., Armstrong, K., Eds.; Elsevier: Amsterdam, The Netherlands, 2015; pp. 33–44. [Google Scholar]
- Langanke, J.; Wolf, A.; Peters, M. Polymers from CO2—An industrial perspective. In Carbon Dioxide Utilisation; Styring, P., Quadrelli, E.A., Armstrong, K., Eds.; Elsevier: Amsterdam, The Netherlands, 2015; pp. 59–71. [Google Scholar] [CrossRef]
- Aresta, M.; Nocito, F. Large scale utilization of carbon dioxide: From its reaction with energy rich chemicals to (Co)-processing with water to afford energy rich products. Opportunities and barriers. In An Economy Based on Carbon Dioxide and Water, Potential of Large Scale Carbon Dioxide Utilization; Aresta, M., Karimi, I., Kawi, S., Eds.; Springer: Cham, Switzerland, 2019; pp. 2–34. [Google Scholar]
- Leonzio, G. State-of-the-art overview of CO2 conversions. In Carbon Dioxide Utilization to Sustainable Energy and Fuels; Inamuddin, Boddula, R., Ahamed, M.I., Khan, A., Eds.; Springer Nature: Berlin/Heidelberg, Germany, 2022; pp. 335–353. [Google Scholar] [CrossRef]
- Ushikoshi, K.; Moria, K.; Watanabe, T.; Takeuchi, M.; Saito, M. A 50 kg/day class test plant for methanol synthesis from CO2 and H2. In Studies in Surface Science and Catalysis; Inui, T., Anpo, M., Izui, K., Yanagida, S., Yamaguchi, T., Eds.; Elsevier: Amsterdam, The Netherlands, 1998; Volume 114, pp. 357–362. [Google Scholar] [CrossRef]
- Song, C. CO2 Conversion and utilization: An overview. In CO2 Conversion and Utilization; Song, C., Gaffney, A.F., Fujimoto, K., Eds.; ACS Symposium Series: Washington, DC, USA, 2002; Volume 809, pp. 2–30. [Google Scholar]
- Huijgen, W.J.J.; Comans, R.N.J. Carbon Dioxide Sequestration by Mineral Carbonation: Literature Review; ECN-C-03-016; Energy Research Centre of The Netherlands: Petten, The Netherlands, 2003; 53p. [Google Scholar]
- Gattrell, M.; Gupta, N.; Co, A. A review of the aqueous electrochemical reduction of CO2 to hydrocarbons at copper. J. Electroanal. Chem. 2006, 594, 1–19. [Google Scholar] [CrossRef]
- Ma, J.; Sun, N.; Zhang, X.; Zhao, N.; Xiao, F.; Wei, W.; Sun, Y. A short review of catalysis for CO2 conversion. Catal. Today 2009, 148, 221–231. [Google Scholar] [CrossRef]
- Budzianowski, W.M. Mass-recirculating systems in CO2 capture technologies: A review. Recent Pat. Eng. 2010, 4, 15–43. [Google Scholar] [CrossRef]
- Mikkelsen, M.; Jørgensen, M.; Krebs, F.C. The teraton challenge. A review of fixation and transformation of carbon dioxide. Energy Environ. Sci. 2010, 3, 43–81. [Google Scholar] [CrossRef]
- Fan, M.S.; Abdullah, A.Z.; Bhatia, S. Catalytic technology for carbon dioxide reforming of methane to synthesis gas. ChemCatChem 2009, 1, 192–208. [Google Scholar] [CrossRef]
- Seemann, M.; Thunman, H. Methane synthesis. In Substitute Natural Gas from Waste; Materazzi, M., Foscolo, P.U., Eds.; Academic Press: Cambridge, MA, USA, 2019; pp. 221–243. [Google Scholar] [CrossRef]
- Chauvy, R.; De Weireld, G. CO2 Utilization Technologies in Europe: A Short Review. Energy Technol. 2020, 8, 2000627. [Google Scholar] [CrossRef]
- Siddique, M.H.; Maqbool, F.; Shahzad, T.; Waseem, M.; Rasul, I.; Hayat, S.; Afzal, M.; Faisal, M.; Muzammil, S. Recent advances in carbon dioxide utilization as renewable energy. In Green Sustainable Process for Chemical and Environmental Engineering and Science: Carbon Dioxide Capture and Utilization; Elsevier: Amsterdam, The Netherlands, 2023; pp. 197–210. [Google Scholar] [CrossRef]
- Ramirez-Corredores, M.M. A carbon dioxide refinery: The core of a sustainable carbon-based circular economy. Highlights Sustinability 2024, 3, 205–239. [Google Scholar] [CrossRef]
- Dziejarski, B.; Krzyżyńska, R.; Andersson, K. Current status of carbon capture, utilization, and storage technologies in the global economy: A survey of technical assessment. Fuel 2023, 342, 127776. [Google Scholar] [CrossRef]
- Makaryan, I.A.; Sedov, I.V. Progress of Commercial Technologies for Producing Syngas and Hydrogen from Hydrocarbon Gases. Russ. J. Appl. Chem. 2023, 96, 619–642. [Google Scholar] [CrossRef]
- BP. Statistical Review of World Energy; BP: London, UK, 2020; 68p, Available online: https://www.bp.com/en/global/corporate/energy-economics/statistical-review-of-world-energy.html (accessed on 15 November 2021).
- Ginsburg, J.M.; Piña, J.; El Solh, T.; De Lasa, H.I. Coke formation over a nickel catalyst under methane dry reforming conditions: Thermodynamic and kinetic models. Ind. Eng. Chem. Res. 2005, 44, 4846–4854. [Google Scholar] [CrossRef]
- Nikoo, M.K.; Amin, N.A.S. Thermodynamic analysis of carbon dioxide reforming of methane in view of solid carbon formation. Fuel Process. Technol. 2011, 92, 678–691. [Google Scholar] [CrossRef]
- Burkart, M.D.; Hazari, N.; Tway, C.L.; Zeitler, E.L. Opportunities and Challenges for Catalysis in Carbon Dioxide Utilization. ACS Catal. 2019, 9, 7937–7956. [Google Scholar] [CrossRef]
- Jensen, C.; Duyar, M.S. Thermodynamic Analysis of Dry Reforming of Methane for Valorization of Landfill Gas and Natural Gas. Energy Technol. 2021, 9, 2100106. [Google Scholar] [CrossRef]
- Li, Y.; Wang, Y.; Zhang, Z.; Hong, X.; Liu, Y. Oxidative reformings of methane to syngas with steam and CO2 catalyzed by metallic Ni based monolithic catalysts. Catal. Commun. 2008, 9, 1040–1044. [Google Scholar] [CrossRef]
- Gao, J.; Hou, Z.; Liu, X.; Zeng, Y.; Luo, M.; Zheng, X. Methane autothermal reforming with CO2 and O2 to synthesis gas at the boundary between Ni and ZrO2. Int. J. Hydrogen Energy 2009, 34, 3734–3742. [Google Scholar] [CrossRef]
- Múnera, J.F.; Carrara, C.; Cornaglia, L.M.; Lombardo, E.A. Combined oxidation and reforming of methane to produce pure H2 in a membrane reactor. Chem. Eng. J. 2010, 161, 204–211. [Google Scholar] [CrossRef]
- Hunt, J.; Ferrari, A.; Lita, A.; Crosswhite, M.; Ashley, B.; Stiegman, A.E. Microwave-Specific Enhancement of the Carbon–Carbon Dioxide (Boudouard) Reaction. J. Phys. Chem. C 2013, 117, 26871–26880. [Google Scholar] [CrossRef]
- Bell, A.T. The impact of nanoscience on heterogeneous catalysis. Science 2003, 299, 1688–1691. [Google Scholar] [CrossRef]
- Rivas, I.; Alvarez, J.; Pietri, E.; Pérez-Zurita, M.J.; Goldwasser, M.R. Perovskite-type oxides in methane dry reforming: Effect of their incorporation into a mesoporous SBA-15 silica-host. Catal. Today 2010, 149, 388–393. [Google Scholar] [CrossRef]
- Kim, D.K.; Stöwe, K.; Müller, F.; Maier, W.F. Mechanistic study of the unusual catalytic properties of a new NiCe mixed oxide for the CO2 reforming of methane. J. Catal. 2007, 247, 101–111. [Google Scholar] [CrossRef]
- Wang, Y.-H.; Liu, H.-M.; Xu, B.-Q. Durable Ni/MgO catalysts for CO2 reforming of methane: Activity and metal–support interaction. J. Mol. Catal. A Chem. 2009, 299, 44–52. [Google Scholar] [CrossRef]
- Liu, D.; Cheo, W.N.E.; Lim, Y.W.Y.; Borgna, A.; Lau, R.; Yang, Y. A comparative study on catalyst deactivation of nickel and cobalt incorporated MCM-41 catalysts modified by platinum in methane reforming with carbon dioxide. Catal. Today 2010, 154, 229–236. [Google Scholar] [CrossRef]
- Mortensen, P.M.; Dybkjær, I. Industrial scale experience on steam reforming of CO2-rich gas. Appl. Catal. A Gen. 2015, 495, 141–151. [Google Scholar] [CrossRef]
- Shang, Z.; Li, S.; Li, L.; Liu, G.; Liang, X. Highly active and stable alumina supported nickel nanoparticle catalysts for dry reforming of methane. Appl. Catal. B Environ. 2017, 201, 302–309. [Google Scholar] [CrossRef]
- Shang, Z.; Li, S.; Wang, Q.; Gu, X.; Liang, X. Nano-engineered nickel catalysts supported on 4-channel A-Al2O3 hollow fibers for dry reforming of methane. AIChE J. 2018, 64, 2625–2631. [Google Scholar] [CrossRef]
- Jin, B.; Shang, Z.; Li, S.; Jiang, Y.B.; Gu, X.; Liang, X. Reforming of methane with carbon dioxide over cerium oxide promoted nickel nanoparticles deposited on 4-channel hollow fibers by atomic layer deposition. Catal. Sci. Technol. 2020, 10, 3212–3222. [Google Scholar] [CrossRef]
- Jin, B.; Li, S.; Liang, X. Enhanced activity and stability of MgO-promoted Ni/Al2O3 catalyst for dry reforming of methane: Role of MgO. Fuel 2021, 284, 119082. [Google Scholar] [CrossRef]
- Jin, B.; Li, S.; Liang, X. High-performance catalytic four-channel hollow fibers with highly dispersed nickel nanoparticles prepared by atomic layer deposition for dry reforming of methane. Ind. Eng. Chem. Res. 2022, 61, 10377–10386. [Google Scholar] [CrossRef]
- Shi, C.; Zhang, A.; Li, X.; Zhang, S.; Zhu, A.; Ma, Y.; Au, C. Ni-modified Mo2C catalysts for methane dry reforming. Appl. Catal. A Gen. 2012, 431–432, 164–170. [Google Scholar] [CrossRef]
- Nagaraja, B.M.; Bulushev, D.A.; Beloshapkin, S.; Chansai, S.; Ross, J.R.H. Potassium-doped Ni-MgO-ZrO2 catalysts for dry reforming of methane to synthesis gas. Top Catal. 2013, 56, 1686–1694. [Google Scholar] [CrossRef]
- Elsayed, N.H.; Roberts, N.R.M.; Joseph, B.; Kuhn, J.N. Low temperature dry reforming of methane over Pt-Ni-Mg/ceria-zirconia catalysts. Appl. Catal. B Environ. 2015, 179, 213–219. [Google Scholar] [CrossRef]
- Rezaei, R.; Moradi, G.; Sharifnia, S. Dry Reforming of Methane over Ni-Cu/Al2O3 Catalyst Coatings in a Microchannel Reactor: Modeling and Optimization Using Design of Experiments. Energy Fuels 2019, 33, 6689–6706. [Google Scholar] [CrossRef]
- Dang, C.; Luo, J.; Yang, W.; Li, H.; Cai, W. Low-temperature catalytic dry reforming of methane over Pd promoted Ni–CaO–ca12al14o33 Multifunctional Catalyst. Ind. Eng. Chem. Res. 2021, 60, 18361–18372. [Google Scholar] [CrossRef]
- Yadav, P.K.; Patrikar, K.; Mondal, A.; Sharma, S. Ni/Co in and on CeO2: A comparative study on the dry reforming reaction. Sustain. Energy Fuels 2023, 7, 3853–3870. [Google Scholar] [CrossRef]
- Jin, B.; Li, S.; Liu, Y.; Liang, X. Engineering metal-oxide interface by depositing ZrO2 overcoating on Ni/Al2O3 for dry reforming of methane. Chem. Eng. J. 2022, 436, 135195. [Google Scholar] [CrossRef]
- Rosen, B.A.; Gileadi, E.; Eliaz, N. Electrodeposited Re-promoted Ni foams as a catalyst for the dry reforming of methane. Catal. Commun. 2016, 76, 23–28. [Google Scholar] [CrossRef]
- Zubenko, D.; Singh, S.; Rosen, B.A. Exsolution of Re-alloy catalysts with enhanced stability for methane dry reforming. Appl. Catal. B Environ. 2017, 209, 711–719. [Google Scholar] [CrossRef]
- Meng, J.; Pan, W.; Gu, T.; Bu, C.; Zhang, J.; Wang, X.; Liu, C.; Xie, H.; Piao, G. One-pot synthesis of a highly active and stable ni-embedded hydroxyapatite catalyst for syngas production via dry reforming of methane. Energy Fuels 2021, 35, 19568–19580. [Google Scholar] [CrossRef]
- Huang, L.; Li, D.; Tian, D.; Jiang, L.; Li, Z.; Wang, H.; Li, K. Optimization of Ni-based catalysts for dry reforming of methane via alloy design: A review. Energy Fuels 2022, 36, 5102–5151. [Google Scholar] [CrossRef]
- Praserthdam, S.; Somdee, S.; Rittiruam, M.; Balbuena, P.B. Computational study of the evolution of ni-based catalysts during the dry reforming of methane. Energy Fuels 2020, 34, 4855–4864. [Google Scholar] [CrossRef]
- Wang, C.; Sun, N.; Zhao, N.; Wei, W.; Sun, Y.; Sun, C.; Liu, H.; Snape, C.E. Coking and deactivation of a mesoporous Ni-CaO-ZrO2 catalyst in dry reforming of methane: A study under different feeding compositions. Fuel 2015, 143, 527–535. [Google Scholar] [CrossRef]
- Osaki, T.; Horiuchi, T.; Suzuki, K.; Mori, T. Suppression of carbon deposition in CO2-reforming of methane on metal sulfide catalysts. Catal. Lett. 1995, 35, 39–43. [Google Scholar] [CrossRef]
- Arora, S.; Prasad, R. An overview on dry reforming of methane: Strategies to reduce carbonaceous deactivation of catalysts. RSC Adv. 2016, 6, 108668–108688. [Google Scholar] [CrossRef]
- Mondal, K.; Sasmal, S.; Badgandi, S.; Chowdhury, D.R.; Nair, V. Dry reforming of methane to syngas: A potential alternative process for value added chemicals—A techno-economic perspective. Environ. Sci. Pollut. Res. 2016, 23, 22267–22273. [Google Scholar] [CrossRef] [PubMed]
- Gallego, J.; Batiot-Dupeyrat, C.; Barrault, J.; Mondragón, F. Severe deactivation of a LaNiO3 perovskite-type catalyst precursor with H2S during methane dry reforming. Energy Fuels 2009, 23, 4883–4886. [Google Scholar] [CrossRef]
- Sokolov, S.; Kondratenko, E.V.; Pohl, M.M.; Rodemerck, U. Effect of calcination conditions on time on-stream performance of Ni/La2O3-ZrO2 in low-temperature dry reforming of methane. Int. J. Hydrogen Energy 2013, 38, 16121–16132. [Google Scholar] [CrossRef]
- Wang, C.; Sun, N.; Zhao, N.; Wei, W.; Zhang, J.; Zhao, T.; Sun, Y.; Sun, C.; Liu, H.; Snape, C.E. The properties of individual carbon residuals and their influence on the deactivation of Ni-CaO-ZrO2 catalysts in CH4 dry reforming. ChemCatChem 2014, 6, 640–648. [Google Scholar] [CrossRef]
- Pawar, V.; Ray, D.; Subrahmanyam, C.; Janardhanan, V.M. Study of short-term catalyst deactivation due to carbon deposition during biogas dry reforming on supported Ni catalyst. Energy Fuels 2015, 29, 8047–8052. [Google Scholar] [CrossRef]
- Zhang, R.-j.; Xia, G.-f.; Li, M.-f.; Wu, Y.; Nie, H.; Li, D.-d. Effect of support on the performance of Ni-based catalyst in methane dry reforming. J. Fuel Chem. Technol. 2015, 43, 1359–1365. [Google Scholar] [CrossRef]
- Lou, Y.; Steib, M.; Zhang, Q.; Tiefenbacher, K.; Horváth, A.; Jentys, A.; Liu, Y.; Lercher, J.A. Design of stable Ni/ZrO2 catalysts for dry reforming of methane. J. Catal. 2017, 356, 147–156. [Google Scholar] [CrossRef]
- Li, R.; Xu, W.; Deng, J.; Zhou, J. Coke-resistant Ni-Co/ZrO2-CaO-based microwave catalyst for highly effective dry reforming of methane by microwave catalysis. Ind. Eng. Chem. Res. 2021, 60, 17458–17468. [Google Scholar] [CrossRef]
- Wang, Y.B.; He, L.; Zhou, B.C.; Tang, F.; Fan, J.; Wang, D.Q.; Lu, A.H.; Li, W.C. Hydroxyapatite nanorods rich in [Ca-O-P] sites stabilized Ni species for methane dry reforming. Ind. Eng. Chem. Res. 2021, 60, 15064–15073. [Google Scholar] [CrossRef]
- Cherbański, R.; Kotkowski, T.; Molga, E. Thermogravimetric analysis of coking during dry reforming of methane. Int. J. Hydrogen Energy 2023, 48, 7346–7360. [Google Scholar] [CrossRef]
- Pakhare, D.; Spivey, J. A review of dry (CO2) reforming of methane over noble metal catalysts. Chem. Soc. Rev. 2014, 43, 7813–7837. [Google Scholar] [CrossRef]
- Aziz, M.A.A.; Setiabudi, H.D.; Teh, L.P.; Annuar, N.H.R.; Jalil, A.A. A review of heterogeneous catalysts for syngas production via dry reforming. J. Taiwan Inst. Chem. Eng. 2019, 101, 139–158. [Google Scholar] [CrossRef]
- de Medeiros, F.G.M.; Lopes, F.W.B.; Rego de Vasconcelos, B. Prospects and technical challenges in hydrogen production through dry reforming of methane. Catalysts 2022, 12, 363. [Google Scholar] [CrossRef]
- Du, C.; Lu, P.; Tsubaki, N. Efficient and new production methods of chemicals and liquid fuels by carbon monoxide hydrogenation. ACS Omega 2020, 5, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Bjørgen, M.; Joensen, F.; Spangsberg Holm, M.; Olsbye, U.; Lillerud, K.P.; Svelle, S. Methanol to gasoline over zeolite H-ZSM-5: Improved catalyst performance by treatment with NaOH. Appl. Catal. A Gen. 2008, 345, 43–50. [Google Scholar] [CrossRef]
- Godini, H.R.; Xiao, S.; Jašo, S.; Stünkel, S.; Salerno, D.; Son, N.X.; Song, S.; Wozny, G. Techno-economic analysis of integrating the methane oxidative coupling and methane reforming processes. Fuel Process. Technol. 2013, 106, 684–694. [Google Scholar] [CrossRef]
- Kim, S.; Ryi, S.K.; Lim, H. Techno-economic analysis (TEA) for CO2 reforming of methane in a membrane reactor for simultaneous CO2 utilization and ultra-pure H2 production. Int. J. Hydrogen Energy 2018, 43, 5881–5893. [Google Scholar] [CrossRef]
- Sharifian, R.; van der Wal, H.C.; Wagterveld, R.M.; Vermaas, D.A. Fouling management in oceanic carbon capture via in-situ electrochemical bipolar membrane electrodialysis. Chem. Eng. J. 2023, 458, 141407. [Google Scholar] [CrossRef]
- Udengaard, N.R. Sulfur passivated reforming process lowers syngas H2/CO ratio. Oil Gas J. 1992, 90, 62–67. [Google Scholar]
- Teuner, S.C.; Neumann, P.; Von Linde, F. CO through CO2 reforming—The Calcor standard and Calcor economy processes. Oil Gas Eur. Mag. 2001, 27, 44–46. [Google Scholar]
- Kurz, G.; Teuner, S. Calcor process for CO production. Erdoel Und Kohle Erdgas Petrochem. 1990, 43, 171–172. [Google Scholar]
- Teuner, S.C.; Neumann, P.; Von Linde, F. CO through CO2 reforming: The Calcor standard and Calcor economy processes. Erdoel Erdgas Kohle 2001, 117, 580–582. [Google Scholar]
- GTI. Dry Reforming of Methane to Produce Syngas and Reduce CO2 Emissions. Available online: https://www.gti.energy/dry-reforming-of-methane-to-produce-syngas-and-reduce-CO2-emissions/ (accessed on 3 June 2024).
- Bartesch, T.; Wawrzinek, K.; Behrens, A.; Peschel, A. DRYREF & SYNSPIRE innovation for HyCO applications. In Proceedings of the Global Syngas Technologies Conference, Virtual, 20–21, 27–28 October 2020; Available online: https://globalsyngas.org/wp-content/conference-presentations/2020/2020-w1-d2-m2-DRYREF-Global-Syngas-Conference.pdf (accessed on 4 July 2024).
- Linde Engineering. Smaller Carbon Footprint. Higher Process Efficiency. Available online: https://www.engineering.linde.com/dryref (accessed on 16 March 2024).
- Rostrup-Nielsen, J.R. Sulfur-passivated nickel catalysts for carbon-free steam reforming of methane. J. Catal. 1984, 85, 31–43. [Google Scholar] [CrossRef]
- Rostrup-Nielsen, J.R. Promotion by poisoning. In Studies in Surface Science and Catalysis; Elsevier: Amsterdam, The Netherlands, 1991; Volume 68, pp. 85–101. [Google Scholar] [CrossRef]
- Dibben, H.C.; Olesen, P.; Rostrup-Nielsen, J.R.; Tøttrup, P.B.; Volengaard, N.R. Make low H2/CO syngas using sulfur passivated reforming. Hydrocarb. Process 1986, 65, 71–74. [Google Scholar]
- Mustafaev, I.I.; Gurbanov, M.A.; Hajiev, H.M. Gasification of different types of carbon in steam and carbon dioxide in the presence of γ-radiation. Carbon 1988, 26, 125–128. [Google Scholar] [CrossRef]
- Steib, M. Dry Reforming of Methane—Understanding Metal Support Interactions and Evaluation of Regeneration Protocols. Ph.D. Thesis, Technische Universität München, Munich, Germany, 2017. [Google Scholar]
- York, A.P.E.; Xiao, T.C.; Green, M.L.H.; Claridge, J.B. Methane oxyforming for synthesis gas production. Catal. Rev.-Sci. Eng. 2007, 49, 511–560. [Google Scholar] [CrossRef]
- Shah, Y.T.; Gardner, T.H. Dry reforming of hydrocarbon feedstocks. Catal. Rev.-Sci. Eng. 2014, 56, 476–536. [Google Scholar] [CrossRef]
- Green Car Congress. Linde Pilot Testing Dry Reforming Process to Generate Syngas from CO2 and Methane for Production of Fuels and Chemicals. Available online: https://www.greencarcongress.com/2015/10/20151016-lpr.html (accessed on 8 March 2022).
- Schwab, E.; Milanov, A.; Schunk, S.A.; Behrens, A.; Schödel, N. Dry reforming and reverse water gas shift: Alternatives for syngas production? Chem.-Ing.-Tech. 2015, 87, 347–353. [Google Scholar] [CrossRef]
- Er-Rbib, H.; Bouallou, C.; Werkoff, F. Production of synthetic gasoline and diesel fuel from dry reforming of methane. In Proceedings of the 19th World Hydrogen Energy Conference, WHEC 2012, Toronto, ON, Canada, 3–7 June 2019; pp. 156–165. [Google Scholar] [CrossRef]
- Engineering India. Linde Technologies That Do More with Less. Available online: https://www.linde-engineering.in/en/about_linde_engineering/success-stories/technologies-more-with-less.html? (accessed on 5 June 2024).
- Boretti, A.; Nayfeh, J.; Al-Maaitah, A. Hydrogen production by solar thermochemical water-splitting cycle via a beam down concentrator. Front. Energy Res. 2021, 9, 5. [Google Scholar] [CrossRef]
- Buelens, L.C.; Poelman, H.; Marin, G.B.; Galvita, V.V. 110th anniversary: Carbon dioxide and chemical looping: Current research trends. Ind. Eng. Chem. Res. 2019, 58, 16235–16257. [Google Scholar] [CrossRef]
- Fang, H.; Haibin, L.; Zengli, Z. Advancements in development of chemical-looping combustion: A review. Int. J. Chem. Eng. 2009, 2009, 710515. [Google Scholar] [CrossRef]
- Moghtaderi, B. Review of the recent chemical looping process developments for novel energy and fuel applications. Proc. Energy Fuels 2012, 26, 15–40. [Google Scholar] [CrossRef]
- Barros do Nascimento, R.A.; Pimenta de Macedo, H.; Melo, D.M.A.; Santiago, R.C.; Rodrigues de Araújo, T.; Medeiros, R.L.B.A.; Adánez, J. Structure and reactivity of brazilian iron ores as low-cost oxygen carriers for chemical looping combustion. Ind. Eng. Chem. Res. 2022, 61, 2469–2482. [Google Scholar] [CrossRef]
- Zhu, M.; Song, Y.; Chen, S.; Li, M.; Zhang, L.; Xiang, W. Chemical looping dry reforming of methane with hydrogen generation on Fe2O3/Al2O3 oxygen carrier. Chem. Eng. J. 2019, 368, 812–823. [Google Scholar] [CrossRef]
- Guerrero-Caballero, J.; Kane, T.; Haidar, N.; Jalowiecki-Duhamel, L.; Löfberg, A. Ni, Co, Fe supported on Ceria and Zr doped Ceria as oxygen carriers for chemical looping dry reforming of methane. Catal. Today 2019, 333, 251–258. [Google Scholar] [CrossRef]
- Chein, R.Y.; Hsu, W.H. Thermodynamic analysis of syngas production via chemical looping dry reforming of methane. Energy 2019, 180, 535–547. [Google Scholar] [CrossRef]
- Löfberg, A.; Kane, T.; Guerrero-Caballero, J.; Jalowiecki-Duhamel, L. Chemical looping dry reforming of methane: Toward shale-gas and biogas valorization. Chem. Eng. Process. Process Intensif. 2017, 122, 523–529. [Google Scholar] [CrossRef]
- Li, D.; Xu, R.; Gu, Z.; Zhu, X.; Qing, S.; Li, K. Chemical-looping conversion of methane: A review. Energy Technol. 2020, 8, 1900925. [Google Scholar] [CrossRef]
- Kim, Y.; Lim, H.S.; Lee, M.; Lee, J.W. Ni-Fe-Al mixed oxide for combined dry reforming and decomposition of methane with CO2 utilization. Catal. Today 2021, 368, 86–95. [Google Scholar] [CrossRef]
- Sastre, D.; Galván, C.Á.; Pizarro, P.; Coronado, J.M. Enhanced performance of CH4 dry reforming over La0.9Sr0.1FeO3/YSZ under chemical looping conditions. Fuel 2022, 309, 122122. [Google Scholar] [CrossRef]
- Petrakopoulou, F.; Tsatsaronis, G.; Boyano, A.; Morosuk, T. Exergoeconomic and exergoenvironmental evaluation of power plants including CO2 capture. Chem. Eng. Res. Des. 2011, 89, 1461–1469. [Google Scholar] [CrossRef]
- Petrakopoulou, F.; Tsatsaronis, G. Can carbon dioxide capture and storage from power plants reduce the environmental impact of electricity generation? Energy Fuels 2014, 28, 5327–5338. [Google Scholar] [CrossRef]
- Petrescu, L.; Cormos, C.C. Environmental assessment of IGCC power plants with pre-combustion CO2 capture by chemical & calcium looping methods. J. Clean. Prod. 2017, 158, 233–244. [Google Scholar] [CrossRef]
- Bareschino, P.; Mancusi, E.; Urciuolo, M.; Paulillo, A.; Chirone, R.; Pepe, F. Life cycle assessment and feasibility analysis of a combined chemical looping combustion and power-to-methane system for CO2 capture and utilization. Renew. Sustain. Energy Rev. 2020, 130, 109962. [Google Scholar] [CrossRef]
- Chirone, R.; Paulillo, A.; Coppola, A.; Scala, F. Carbon capture and utilization via calcium looping, sorption enhanced methanation and green hydrogen: A techno-economic analysis and life cycle assessment study. Fuel 2022, 328, 125255. [Google Scholar] [CrossRef]
- Peres, C.B.; Resende, P.M.R.; Nunes, L.J.R.; Morais, L.C.D. Advances in carbon capture and use (CCU) technologies: A comprehensive review and CO2 mitigation potential analysis. Clean. Technol. 2022, 4, 1193–1207. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, Y.; Zhang, Y.; Wu, Q.; Xin, L.; Zhou, Y.; Yin, K.; Wang, Y.; Li, X.; Cui, P. A novel power, DME, and ammonia polygeneration system using Aspen plus based on the integration of biomass gasification and syngas chemical looping. Energy Convers. Manag. 2024, 299, 117808. [Google Scholar] [CrossRef]
- Xiang, D.; Li, P.; Yuan, X.; Cao, H.; Liu, L.; Liu, Y. Energy consumption and greenhouse gas emissions of shale gas chemical looping reforming process integrated with coal gasification for methanol production. Appl. Therm. Eng. 2021, 193, 116990. [Google Scholar] [CrossRef]
- Baker, R.; Alizadeh Sahraei, O.; Dal-Cin, M.M.; Bensebaa, F. A technology development matrix for carbon capture: Technology status and R&D gap assessment. Front. Energy Res. 2022, 10, 908658. [Google Scholar] [CrossRef]
- Martínez-Ramón, N.; Romay, M.; Iribarren, D.; Dufour, J. Life-cycle assessment of hydrogen produced through chemical looping dry reforming of biogas. Int. J. Hydrogen Energy 2024, 78, 373–381. [Google Scholar] [CrossRef]
- Virla, L.D.; de Oliveira, C.F.; Camacho, F.G.; Kaur, M.; Mahinpey, N. Chemical looping reforming (CLR) for syngas production. In Advances in Synthesis Gas. Methods, Technologies and Applications; Syngas Production and Preparation; Elsevier: Amsterdam, The Netherlands, 2022; Volume 1, pp. 149–177. [Google Scholar] [CrossRef]
- Hosokawa, Y.; Kajiya, S.; Ohshima, A.; Ishida, N.; Washio, M.; Usuki, A. CO2 conversion by high-dose rate electron beam irradiation: One-step, metal-free and simultaneous production of H2, CO, CH4, c2h6 and organic acids from an acid-decomposed CaCO3/additive EtOH mixture. Green Chem. 2019, 21, 3091–3098. [Google Scholar] [CrossRef]
- Meyer, W.; La-Belle, D.W.; Rassoul, S.A. The effects of Co-60 gamma radiation on the rmal decomposition of carbonate compounds. Int. J. Appl. Radiat. Isot. 1973, 24, 315–318. [Google Scholar] [CrossRef]
- Álvarez, A.; Borges, M.; Corral-Pérez, J.J.; Olcina, J.G.; Hu, L.; Cornu, D.; Huang, R.; Stoian, D.; Urakawa, A. CO2 Activation over catalytic surfaces. ChemPhysChem 2017, 18, 3135–3141. [Google Scholar] [CrossRef]
- Dioxide Materials. CO2 Electrolyzers. Available online: https://dioxidematerials.com/technology/CO2-electrolysis/ (accessed on 25 January 2024).
- OCO Chem. Carbon FluX Electrolyzer™. Available online: https://ocochem.com/product/ (accessed on 25 January 2024).
- Twelve. Opus™ System. Available online: https://www.twelve.co/post/how-the-opus-system-by-twelve-turns-CO2-into-products (accessed on 25 January 2024).
- Chen, C.; Khosrowabadi Kotyk, J.F.; Sheehan, S.W. Progress toward commercial application of electrochemical carbon dioxide reduction. Chem 2018, 4, 2571–2586. [Google Scholar] [CrossRef]
- Küngas, R. Review—Electrochemical CO2 reduction for CO production: Comparison of low- and high-temperature electrolysis technologies. J. Electrochem. Soc. 2020, 167, 044508. [Google Scholar] [CrossRef]
- O’Brien, J.E.; McKellar, M.G.; Harvego, E.A.; Stoots, C.M. High-temperature electrolysis for large-scale hydrogen and syngas production from nuclear energy—Summary of system simulation and economic analyses. Int. J. Hydrogen Energy 2010, 35, 4808–4819. [Google Scholar] [CrossRef]
- Hartvigsen, J.; Elangovan, S.; O’Brien, J.; Stoots, C.; Herring, J. Operation of high temperature steam electrolyzer module. ECS Trans. 2007, 7, 357–363. [Google Scholar] [CrossRef]
- Delacourt, C.; Ridgway, P.L.; Kerr, J.B.; Newman, J. Design of an electrochemical cell making syngas (CO+ H2) from CO2 and H2O reduction at room temperature. J. Electrochem. Soc. 2008, 155, B42–B49. [Google Scholar] [CrossRef]
- McKellar, M.G.; O’Brien, J.E.; Stoots, C.M.; Hawkes, G.L. Process model for the production of syngas via high temperature co-electrolysis. In Proceedings of the ASME International Mechanical Engineering Congress and Exposition, IMECE 2007, Seattle, WA, USA, 11–15 November 2007; pp. 691–699. [Google Scholar] [CrossRef]
- McKellar, M.G.; Hawkes, G.L.; O’Brien, J.E. The production of syngas via high temperature electrolysis and biomass gasification. In Proceedings of the ASME International Mechanical Engineering Congress and Exposition, IMECE 2008, Boston, MA, USA, 31 October–6 November 2008; pp. 229–235. [Google Scholar] [CrossRef]
- O’Brien, J.E.; McKellar, M.G.; Stoots, C.M.; Herring, J.S.; Hawkes, G.L. Parametric study of large-scale production of syngas via high-temperature co-electrolysis. Int. J. Hydrogen Energy 2009, 34, 4216–4226. [Google Scholar] [CrossRef]
- Stoots, C.; O’Brien, J.; Hartvigsen, J. Results of recent high temperature coelectrolysis studies at the Idaho National Laboratory. Int. J. Hydrogen Energy 2009, 34, 4208–4215. [Google Scholar] [CrossRef]
- Stoots, C.M.; O’Brien, J.E.; Herring, J.S.; Hartvigsen, J.J. Syngas production via high-temperature coelectrolysis of steam and carbon dioxide. J. Fuel Cell Sci. Technol. 2009, 6, 0110141–01101412. [Google Scholar] [CrossRef]
- Fu, Q.; Mabilat, C.; Zahid, M.; Brisse, A.; Gautier, L. Syngas production via high-temperature steam/CO2 co-electrolysis: An economic assessment. Energy Environ. Sci. 2010, 3, 1382–1397. [Google Scholar] [CrossRef]
- Stoots, C.M.; O’Brien, J.E.; Condie, K.G.; Hartvigsen, J.J. High-temperature electrolysis for large-scale hydrogen production from nuclear energy—Experimental investigations. Int. J. Hydrogen Energy 2010, 35, 4861–4870. [Google Scholar] [CrossRef]
- Redissi, Y.; Bouallou, C. Valorization of carbon dioxide by co-electrolysis of CO2/H2O at high temperature for syngas production. In Proceedings of the 11th International Conference on Greenhouse Gas Control Technologies, GHGT 2012, Kyoto, Japan, 18–22 November 2012; pp. 6667–6678. [Google Scholar] [CrossRef]
- Lister, T.E.; Dufek, E.J. Chlor-syngas: Coupling of electrochemical technologies for production of commodity chemicals. Energy Fuels 2013, 27, 4244–4249. [Google Scholar] [CrossRef]
- Foit, S.R.; Vinke, I.C.; de Haart, L.G.J.; Eichel, R.-A. Power-to-Syngas: An Enabling Technology for the Transition of the Energy System? Angew. Chem. Int. Ed. 2017, 56, 5402–5411. [Google Scholar] [CrossRef]
- Hernández, S.; Amin Farkhondehfal, M.; Sastre, F.; Makkee, M.; Saracco, G.; Russo, N. Syngas production from electrochemical reduction of CO2: Current status and prospective implementation. Green Chem. 2017, 19, 2326–2346. [Google Scholar] [CrossRef]
- Ripatti, D.S.; Veltman, T.R.; Kanan, M.W. Carbon monoxide gas diffusion electrolysis that produces concentrated C2 products with high single-pass conversion. Joule 2019, 3, 240–256. [Google Scholar] [CrossRef]
- Leonard, M.E.; Clarke, L.E.; Forner-Cuenca, A.; Brown, S.M.; Brushett, F.R. Investigating electrode flooding in a flowing electrolyte, gas-fed carbon dioxide electrolyzer. ChemSusChem 2020, 13, 400–411. [Google Scholar] [CrossRef]
- Weekes, D.M.; Salvatore, D.A.; Reyes, A.; Huang, A.; Berlinguette, C.P. Electrolytic CO2 reduction in a flow cell. Acc. Chem. Res. 2018, 51, 910–918. [Google Scholar] [CrossRef]
- Higgins, D.; Hahn, C.; Xiang, C.; Jaramillo, T.F.; Weber, A.Z. Gas-diffusion electrodes for carbon dioxide reduction: A new paradigm. ACS Energy Lett. 2019, 4, 317–324. [Google Scholar] [CrossRef]
- Durrani, J. Can Catalysis Save Us from Our CO2 Problem? Available online: https://www.chemistryworld.com/news/can-catalysis-save-us-from-our-CO2-problem/3010555.article (accessed on 24 June 2022).
- Zhang, Y.; Guo, S.-X.; Zhang, X.; Bond, A.M.; Zhang, J. Mechanistic understanding of the electrocatalytic CO2 reduction reaction—New developments based on advanced instrumental techniques. Nano Today 2020, 31, 100835. [Google Scholar] [CrossRef]
- Lim, R.J.; Xie, M.; Sk, M.A.; Lee, J.-M.; Fisher, A.; Wang, X.; Lim, K.H. A review on the electrochemical reduction of CO2 in fuel cells, metal electrodes and molecular catalysts. Catal. Today 2014, 233, 169–180. [Google Scholar] [CrossRef]
- Zhang, W.; Hu, Y.; Ma, L.; Zhu, G.; Wang, Y.; Xue, X.; Chen, R.; Yang, S.; Jin, Z. Progress and Perspective of Electrocatalytic CO2 Reduction for Renewable Carbonaceous Fuels and Chemicals. Adv. Sci. 2018, 5, 1700275. [Google Scholar] [CrossRef] [PubMed]
- Küngas, R.; Blennow, P.; Heiredal-Clausen, T.; Holt, T.; Rass-Hansen, J.; Primdahl, S. Systematic lifetime testing of stacks in CO2 electrolysis. ECS Trans. 2017, 78, 2895–2905. [Google Scholar] [CrossRef]
- Artz, J.; Müller, T.E.; Thenert, K.; Kleinekorte, J.; Meys, R.; Sternberg, A.; Bardow, A.; Leitner, W. Sustainable conversion of carbon dioxide: An integrated review of catalysis and life cycle assessment. Chem. Rev. 2018, 118, 434–504. [Google Scholar] [CrossRef]
- Li, T.; Lees, E.W.; Goldman, M.; Salvatore, D.A.; Weekes, D.M.; Berlinguette, C.P. Electrolytic conversion of bicarbonate into co in a flow cell. Joule 2019, 3, 1487–1497. [Google Scholar] [CrossRef]
- Li, L.; Yang, J.; Li, L.; Huang, Y.; Zhao, J. Electrolytic reduction of CO2 in KHCO3 and alkanolamine solutions with layered double hydroxides intercalated with gold or copper. Electrochim. Acta 2022, 402, 139523. [Google Scholar] [CrossRef]
- Zhang, Z.; Lees, E.W.; Ren, S.; Mowbray, B.A.W.; Huang, A.; Berlinguette, C.P. Conversion of Reactive Carbon Solutions into CO at Low Voltage and High Carbon Efficiency. ACS Cent. Sci. 2022, 8, 749–755. [Google Scholar] [CrossRef]
- Yamaguchi, N.; Nakazato, R.; Matsumoto, K.; Kakesu, M.; Rosero-Navarro, N.C.; Miura, A.; Tadanaga, K. Electrocatalytic property of Zn-Al layered double hydroxides for CO2 electrochemical reduction. J. Asian Ceram. Soc. 2023, 11, 406–411. [Google Scholar] [CrossRef]
- Yue, P.; Kang, Z.; Fu, Q.; Li, J.; Zhang, L.; Zhu, X.; Liao, Q. Life cycle and economic analysis of chemicals production via electrolytic (bi)carbonate and gaseous CO2 conversion. Appl. Energy 2021, 304, 117768. [Google Scholar] [CrossRef]
- Zhu, W.; Michalsky, R.; Metin, Ö.; Lv, H.; Guo, S.; Wright, C.J.; Sun, X.; Peterson, A.A.; Sun, S. Monodisperse au nanoparticles for selective electrocatalytic reduction of CO2 to CO. J. Am. Chem. Soc. 2013, 135, 16833–16836. [Google Scholar] [CrossRef] [PubMed]
- Mistry, H.; Reske, R.; Zeng, Z.; Zhao, Z.-J.; Greeley, J.; Strasser, P.; Cuenya, B.R. Exceptional size-dependent activity enhancement in the electroreduction of CO2 over Au nanoparticles. J. Am. Chem. Soc. 2014, 136, 16473–16476. [Google Scholar] [CrossRef] [PubMed]
- Ham, Y.S.; Choe, S.; Kim, M.J.; Lim, T.; Kim, S.K.; Kim, J.J. Electrodeposited Ag catalysts for the electrochemical reduction of CO2 to CO. Appl. Catal. B Environ. 2017, 208, 35–43. [Google Scholar] [CrossRef]
- Lees, E.W.; Goldman, M.; Fink, A.G.; Dvorak, D.J.; Salvatore, D.A.; Zhang, Z.; Loo, N.W.X.; Berlinguette, C.P. Electrodes Designed for Converting Bicarbonate into CO. ACS Energy Lett. 2020, 5, 2165–2173. [Google Scholar] [CrossRef]
- Verma, S.; Lu, X.; Ma, S.; Masel, R.I.; Kenis, P.J.A. The effect of electrolyte composition on the electroreduction of CO2 to CO on Ag based gas diffusion electrodes. Phys. Chem. Chem. Phys. 2016, 18, 7075–7084. [Google Scholar] [CrossRef]
- Larrea, C.; Torres, D.; Avilés-Moreno, J.R.; Ocón, P. Multi-parameter study of CO2 electrochemical reduction from concentrated bicarbonate feed. J. CO2 Util. 2022, 57, 101878. [Google Scholar] [CrossRef]
- Pimlott, D.J.; Jewlal, A.; Kim, Y.; Berlinguette, C.P. Oxygen-resistant CO2 reduction enabled by electrolysis of liquid feedstocks. J. Am. Chem. Soc. 2023, 145, 25933–25937. [Google Scholar] [CrossRef]
- Pimlott, D.J.D.; Jewlal, A.; Mowbray, B.A.W.; Berlinguette, C.P. Impurity-Resistant CO2 Reduction Using Reactive Carbon Solutions. ACS Energy Lett. 2023, 8, 1779–1784. [Google Scholar] [CrossRef]
- Kim, Y.; Lees, E.W.; Berlinguette, C.P. Permeability matters when reducing CO2 in an electrochemical flow cell. ACS Energy Lett. 2022, 7, 2382–2387. [Google Scholar] [CrossRef]
- Diaz, L.A.; Gao, N.; Adhikari, B.; Lister, T.E.; Dufek, E.J.; Wilson, A.D. Electrochemical production of syngas from CO2 captured in switchable polarity solvents. Green Chem. 2018, 20, 620–626. [Google Scholar] [CrossRef]
- Gao, N.; Quiroz Arita, C.; Diaz, L.A.; Lister, T.E. Intensified co-electrolysis process for syngas production from captured CO2. J. CO2 Util. 2020, 43, 101365. [Google Scholar] [CrossRef]
- Lee, G.; Li, Y.C.; Kim, J.Y.; Peng, T.; Nam, D.H.; Sedighian Rasouli, A.; Li, F.; Luo, M.; Ip, A.H.; Joo, Y.C.; et al. Electrochemical upgrade of CO2 from amine capture solution. Nat. Energy 2021, 6, 46–53. [Google Scholar] [CrossRef]
- Pérez-Gallent, E.; Vankani, C.; Sánchez-Martínez, C.; Anastasopol, A.; Goetheer, E. Integrating CO2 Capture with Electrochemical Conversion Using Amine-Based Capture Solvents as Electrolytes. Ind. Eng. Chem. Res. 2021, 60, 4269–4278. [Google Scholar] [CrossRef]
- Ahmad, N.; Wang, X.; Sun, P.; Chen, Y.; Rehman, F.; Xu, J.; Xu, X. Electrochemical CO2 reduction to CO facilitated by MDEA-based deep eutectic solvent in aqueous solution. Renew. Energy 2021, 177, 23–33. [Google Scholar] [CrossRef]
- Wang, M.; Herzog, H.J.; Hatton, T.A. CO2 Capture Using Electrochemically Mediated Amine Regeneration. Ind. Eng. Chem. Res. 2020, 59, 7087–7096. [Google Scholar] [CrossRef]
- O’rear, D.J. Process for Conversion of Lpg and CH4 to syNgas and Higher Valued Products. Patent No. US6774148, 10 August 2004. Also published as GB0426607, US20030236312. [Google Scholar]
- Johnston, D.A. Carbon Dioxide Absorbed from Air and Hydrogen from Electrolysis of Water, for Production of Carbon Monoxide, Alcohols, Fischer-Tropsch Hydrocarbons & Fuels. Patent No. GB2448685, 29 October 2008. [Google Scholar]
- Olah, G.A.; Prakash, G.K.S. Electrolysis of Carbon Dioxide in Aqueous Media to Carbon Monoxide and Hydrogen for Production of Methanol. Patent No. US7704369, 27 April 2010. Also published as CN101743343, AU2008276180, CA2690980, EP2167706, ES2659978, JP2010533784, JP5144755, KR20100036317, US2009014336, WO2009012154. [Google Scholar]
- Olah, G.A.; Prakash, G.K.S. Electrolysis of Carbon Dioxide in Aqueous Media to Carbon Monoxide and Hydrogen for Production of Methanol. Patent No. US8138380, 20 March 2012. Also published as US2010193370. [Google Scholar]
- Sivasankar, N.; Cole, E.B.; Teamey, K. Electrochemical Production of Synthesis Gas from Carbon Dioxide. Patent No. US8721866, 13 May 2014. Also published as AU2011282783, BR112013002217, CA2805852, CN103119204, EP2598670, JP2013532774, KR20140012018, US8721866, US10119196, US2011114504, US2014238871, WO2012015921. [Google Scholar]
- Masel, R.I.; Salehi-Khojin, A. Electrocatalytic Process for Carbon Dioxide Conversion. Patent No. US9012345, 21 April 2015. Also published as US2013157174. [Google Scholar]
- Masel, R.I.; Qingmei, C.H.E.N.; Liu, Z.; Kutz, R. Electrochemical Device for Converting Carbon Dioxide to a Reaction Product. Patent No. US9481939, 1 November 2016. Also published as US2016108530. [Google Scholar]
- Kuhl, K.P.; Cave, E.R.; Leonard, G. Reactor with Advanced Architecture for the Electrochemical Reaction of CO2, CO and Other Chemical Compounds. Patent No. US2017321333, 9 November 2017. Also published as WO2017192787. [Google Scholar]
- Masel, R.I.; Salehi-Khojin, A.; Kutz, R. Electrocatalytic Process for Carbon Dioxide Conversion. Patent No. US9815021, 14 September 2017. Also published as US2017259206, WO2017176600. [Google Scholar]
- Masel, R.I.; Salehi-Khojin, A. Electrocatalytic Process for Carbon Dioxide Conversion. Patent No. US9555367, 31 January 2017. Also published as US2015209722. [Google Scholar]
- Krishnamurthy, K.; Gärtner, L.-E.; Rupieper, A.; Bostick, D. Process and Apparatus for Manufacturing Carbon Monoxide. Patent No. WO2018228723, 20 December 2018. [Google Scholar]
- Masel, R.I. Devices for Electrocatalytic Conversion of Carbon Dioxide. Patent No. US10173169, 26 April 2018. Also published as US2018111083. [Google Scholar]
- Rüger, D. Synthesis Gas Production from CO2 and H2O in a CO-Electrolysis. Patent No. WO2018228643, 20 December 2018. [Google Scholar]
- Sivasankar, N.; Cole, E.B.; Teamey, K. Electrochemical Production of Synthesis Gas from Carbon Dioxide. Patent No. US10119196, 6 November 2018. Also published as AU2011282783, BR112013002217, CA2805852, CN103119204, EP2598670, JP2013532774, KR20140012018, US8721866, US2011114504, US2014238871, WO2012015921. [Google Scholar]
- Lister, T.E.; Dufek, E.J.; Wilson, A.D.; Diaz Aldana, L.A.; Adhikari, B.; Gao, N. Methods and Systems for the Electrochemical Reduction of Carbon Dioxide Using Switchable Polarity Materials. Patent No. WO2019070526, 11 April 2019. [Google Scholar]
- Masel, R.I. Electrocatalytic Process for Carbon Dioxide Conversion. Patent No. US2019211463, 11 July 2019. [Google Scholar]
- Kuhl, K.P.; Cave, E.R.; Leonard, G. Reactor with Advanced Architecture for the Electrochemical Reaction of CO2, CO and Other Chemical Compounds. Patent No. US10822709, 3 November 2020. Also published as CA3022807, CA3022812, CA3124239, CN109643813, CN109643816, CN115198294, CN116231017, DK3453064, DK3453065, EP3453064, EP3828315, EP3954807, ES2879900, ES2885066, HUE055019, HUE056900, JP2019515142, JP2019520474, JP2021059788, JP6784776, JP6816165, JP7194368, PL3453064, PL3453065, PT3453064, PT3453065, US10648091, US11124886, US11680327, US2017321333, US2017321334, US2020354843, US2021164116, US2022010437, WO2017192787, WO2017192788. [Google Scholar]
- Kuhl, K.P.; Cave, E.R.; Leonard, G. Reactor with Advanced Architecture for the Electrochemical Reaction of CO2, CO and Other Chemical Compounds. Patent No. US11124886, 21 September 2021. Also published as CA3022807, CA3022812, CA3124239, CN109643813, CN109643816, CN115198294, CN116231017, DK3453064, DK3453065, EP3453064, EP3828315, EP3954807, ES2879900, ES2885066, HUE055019, HUE056900, JP2019515142, JP2019520474, JP2021059788, JP6784776, JP6816165, JP7194368, PL3453064, PL3453065, PT3453064, PT3453065, US10648091, US10822709, US11680327, US2017321333, US2017321334, US2020354843, US2021164116, US2022010437, WO2017192787, WO2017192788. [Google Scholar]
- Lister, T.E.; Dufek, E.J.; Wilson, A.D.; Diaz Aldana, L.A.; Adhikari, B.; Gao, N. Methods and Systems for the Electrochemical Reduction of Carbon Dioxide Using Switchable Polarity Materials. Patent No. US10975477, 13 April 2021. Also published as US2020255958, WO2019070526. [Google Scholar]
- Omersa, K. Carbon Dioxide Conversion Using Combined Fuel Cell and Electrolysis Cell. Patent No. US2021313608, 7 October 2021. [Google Scholar]
- Fontecave, M.; Mougel, V.; Tran, N.H.; Wakerley, D.; Lamaison, S. Method for Converting Carbon Dioxide (CO2) into Syngas by an Electrolysis Reaction. Patent No. US2022064804, 3 March 2022. Also published as CA3123971, DK3670701, EP3670700, EP3670701, ES2929181, JP2022515169, US2022056602, WO2020127772, WO2020127821. [Google Scholar]
- Rüger, D. Synthesis Gas Production from CO2 and H2O in a CO-Electrolysis. Patent No. US11214488, 4 January 2022. Also published as CA3067061, DK3472370, EP3415661, EP3472370, US2020095124, WO2018228643. [Google Scholar]
- Zhang, C.; Pan, L.; Guo, H.; Xu, X.; Wang, J. System for Preparing Synthesis Gas by Reducing Electrolytic Urea-Carbon Dioxide. Patent No. CN218115613, 23 December 2022. [Google Scholar]
- Kuhl, K.P.; Cave, E.R.; Leonard, G. Reactor with Advanced Architecture for the Electrochemical Reaction of CO2, CO and Other Chemical Compounds. Patent No. US11680327, 20 June 2023. Also published as CA3022807, CA3022812, CA3124239, CN109643813, CN109643816, CN115198294, CN116231017, DK3453064, DK3453065, EP3453064, EP3828315, EP3954807, ES2879900, ES2885066, HUE055019, HUE056900, JP2019515142, JP2019520474, JP2021059788, JP6784776, JP6816165, JP7194368, PL3453064, PL3453065, PT3453064, PT3453065, US10648091, US10822709, US11124886, US2017321333, US2017321334, US2020354843, US2021164116, US2022010437, WO2017192787, WO2017192788. [Google Scholar]
- Schreiber, A.; Peschel, A.; Hentschel, B.; Zapp, P. Life cycle assessment of power-to-syngas: Comparing high temperature co-electrolysis and steam methane reforming. Front. Energy Res. 2020, 8, 533850. [Google Scholar] [CrossRef]
- Harteck, P.; Dondes, S. Decomposition of carbon dioxide by ionizing radiation. Part I. J. Chem. Phys. 1955, 23, 902–908. [Google Scholar] [CrossRef]
- Hirschfelder, J.O.; Taylor, H.S. The alpha-particle reactions in carbon monoxide, oxygen and carbon dioxide systems. J. Chem. Phys. 1938, 6, 783–790. [Google Scholar] [CrossRef]
- Lind, S.C.; Bardwell, D.C. The chemical action of gaseous ions produced by alpha particles. VI. Reactions of the oxides of carbon. J. Am. Chem. Soc. 1925, 47, 2675–2697. [Google Scholar] [CrossRef]
- Kalashnikov, N.A.; Kalinichenko, B.S.; Shvetsov, I.K. Influence of reagent phase state, form and dose rate irradiation on efficiency of radiolytic decomposition of water and carbon dioxide. At. Energiya 1992, 72, 47–53. [Google Scholar]
- Avdonina, E.N. Hot hydrogen atoms reactions and the mechanisms of liquid normal alkanes radiolysis. Radiat. Eff. 1977, 31, 241–248. [Google Scholar] [CrossRef]
- Huerta Parajon, M.; Rajesh, P.; Mu, T.; Pimblott, S.M.; LaVerne, J. H atom yields in the radiolysis of water. Radiat. Phys. Chem. 2008, 77, 1203–1207. [Google Scholar] [CrossRef]
- Schofield, J.; Reiff, S.C.; Pimblott, S.M.; LaVerne, J.A. Radiolytic hydrogen generation at silicon carbide-water interfaces. J. Nucl. Mater. 2016, 469, 43–50. [Google Scholar] [CrossRef]
- Laverne, J.A.; Pimblott, S.M. New mechanism for H2 formation in water. J. Phys. Chem. A 2000, 104, 9820–9822. [Google Scholar] [CrossRef]
- Pimblott, S.M.; La Verne, J.A. Molecular product formation in the electron radiolysis of water. Radiat. Res. 1992, 129, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Liu, C.; Liu, W.; Zhang, D. Factors influencing hydrogen yield in water radiolysis and implications for hydrocarbon generation: A review. Arab. J. Geosci. 2018, 11, 542. [Google Scholar] [CrossRef]
- Southworth, J.S.; Pimblott, S.M.; Orr, R.M.; Koehler, S.P.K. A novel method for measuring the radiolysis yields of water adsorbed on ZrO2 nanoparticles. Radiat. Phys. Chem. 2020, 174, 108924. [Google Scholar] [CrossRef]
- Watanabe, D.; Yoshida, T.; Allen, C.; Tanabe, T. Enhancement of gamma-ray radiolysis of carbon dioxide with the assistance of solid materials. J. Radioanal. Nucl. Chem. 2007, 272, 461–465. [Google Scholar] [CrossRef]
- Ikezoe, Y.; Sato, S.; Shimizu, S.; Matsuoka, S. Effect of water on the radiolysis of carbon dioxide. Radiat. Phys. Chem. 1981, 17, 69–70. [Google Scholar] [CrossRef]
- Ryazantsev, S.V.; Feldman, V.I. Matrix-isolation studies on the radiation-induced chemistry in H2O/CO2 systems: Reactions of oxygen atoms and formation of HOCO radical. J. Phys. Chem. A 2015, 119, 2578–2586. [Google Scholar] [CrossRef]
- Wu, X.Z.; Hatashita, M.; Enokido, Y.; Kakihana, H. Reduction of carbon dioxide in γ ray irradiated carbon dioxide: Water system containing cu2+ and so32. Chem. Lett. 2000, 5, 572–573. [Google Scholar] [CrossRef]
- Kalashnikov, N.A.; Kalinichenko, B.S.; Shvetsov, I.K. Effect of the reagent phase and the radiation power and type on the radiolysis efficiency of water and carbon dioxide. Sov. At. Energy 1992, 72, 43–48. [Google Scholar] [CrossRef]
- Kalashnikov, N.A.; Kalinichenko, B.S.; Shvetsov, I.K. Effect of the temperature, pressure, and inorganic gas impurities on the radiolysis efficiency of water and carbon dioxide. Sov. At. Energy 1992, 72, 49–53. [Google Scholar] [CrossRef]
- Kalinichenko, B.S.; Kalashnikov, N.A.; Kulazhko, V.G.; Shvetsov, I.K. Method of Producing Carbon Monoxide. Patent No. SU1426943, 30 September 1988. [Google Scholar]
- Kalinichenko, B.S.; Kalashnikov, N.A.; Kulazhko, V.G.; Shvetsov, I.K. Method of Producing Carbon Monoxide. Patent No. SU1490074, 30 June 1989. [Google Scholar]
- Kalinichenko, B.S.; Kalashnikov, N.A.; Kulazhko, V.G.; Shvetsov, I.K. Method of Producing Hydrogen by Radiolysis of Water Vapor. Patent No. RU1824375, 30 June 1993. [Google Scholar]
- Ikezoe, Y.; Sato, S. Radiation chemical reactions in carbon dioxide-propane system formation of carbon monoxide by fission fragments. J. Nucl. Sci. Technol. 1976, 13, 503–507. [Google Scholar] [CrossRef]
- Ikezoe, Y.; Sato, S. Radiation chemical reactions in carbon dioxide–propane system. II. Reoxidation of carbon monoxide at low propane concentration. Bull. Chem. Soc. Jpn. 1978, 51, 33–36. [Google Scholar] [CrossRef]
- Dyer, A.; Moorse, G.E. The radiolysis of simple gas mixtures-I. Rates of production and destruction of methane in mixtures with carbon dioxide as a major constituent. Radiat. Phys. Chem. 1982, 20, 315–321. [Google Scholar] [CrossRef]
- Lind, S.C.; Bardwell, D.C. The chemical action of gaseous ions produced by alpha particles. IX. Saturated hydrocarbons. J. Am. Chem. Soc. 1926, 48, 2335–2351. [Google Scholar] [CrossRef]
- Honig, R.E.; Sheppard, C.W. An experimental comparison of the chemical effects of deuterons and of alpha particles on methane and n-butane. J. Phys. Chem. 1946, 50, 119–143. [Google Scholar] [CrossRef]
- Camps, J.A.; Turnbull, D.M. Synthetic Gas Production for Methanol—Current and Future Trends. Hydrog. Prod. Mark. 1980, 116, 123–146. [Google Scholar]
- Sehested, J. Industrial and scientific directions of methanol catalyst development. J. Catal. 2019, 371, 368–375. [Google Scholar] [CrossRef]
- Herman, R.G.; Klier, K.; Simmons, G.W.; Finn, B.P.; Bulko, J.B.; Kobylinski, T.P. Catalytic synthesis of methanol from COH2. I. Phase composition, electronic properties, and activities of the Cu/ZnO/M2O3 catalysts. J. Catal. 1979, 56, 407–429. [Google Scholar] [CrossRef]
- Mehta, S.; Simmons, G.W.; Klier, K.; Herman, R.G. Catalytic synthesis of methanol from COH2. II. Electron microscopy (TEM, STEM, microdiffraction, and energy dispersive analysis) of the Cu ZnO and Cu/ZnO/Cr2O3 catalysts. J. Catal. 1979, 57, 339–360. [Google Scholar] [CrossRef]
- Herman, R.G.; Simmons, G.W.; Klier, K. Catalytic synthesis of methanol from CO/H2 III. the role of alumina and ceria in the cu/zno system. In Studies in Surface Science and Catalysis; Elsevier: Amsterdam, The Netherlands, 1981; Volume 7, pp. 475–489. [Google Scholar] [CrossRef]
- Klier, K.; Chatikavanij, V.; Herman, R.G.; Simmons, G.W. Catalytic synthesis of methanol from COH2. IV. The effects of carbon dioxide. J. Catal. 1982, 74, 343–360. [Google Scholar] [CrossRef]
- Bozzano, G.; Manenti, F. Efficient methanol synthesis: Perspectives, technologies and optimization strategies. Prog. Energy Combust. Sci. 2016, 56, 71–105. [Google Scholar] [CrossRef]
- Ali, K.A.; Abdullah, A.Z.; Mohamed, A.R. Recent development in catalytic technologies for methanol synthesis from renewable sources: A critical review. Renew. Sustain. Energy Rev. 2015, 44, 508–518. [Google Scholar] [CrossRef]
- Jadhav, S.G.; Vaidya, P.D.; Bhanage, B.M.; Joshi, J.B. Catalytic carbon dioxide hydrogenation to methanol: A review of recent studies. Chem. Eng. Res. Des. 2014, 92, 2557–2567. [Google Scholar] [CrossRef]
- Ramos, F.S.; Farias, A.M.D.D.; Borges, L.E.P.; Monteiro, J.L.; Fraga, M.A.; Sousa-Aguiar, E.F.; Appel, L.G. Role of dehydration catalyst acid properties on one-step DME synthesis over physical mixtures. Gas-Liq. Technol. Pap. Present. 12th Braz. Congr. Catal. 2005, 101, 39–44. [Google Scholar] [CrossRef]
- Tian, P.; Wei, Y.; Ye, M.; Liu, Z. Methanol to olefins (MTO): From fundamentals to commercialization. ACS Catal. 2015, 5, 1922–1938. [Google Scholar] [CrossRef]
- Ye, M.; Li, H.; Zhao, Y.; Zhang, T.; Liu, Z. MTO Processes Development: The Key of Mesoscale Studies. In Mesoscale Modeling in Chemical Engineering Part II, 2015; Li, J., Marin, G.B., Eds.; Academic Press Inc.: Cambridge, MA, USA, 2015; Volume 47, pp. 279–335. [Google Scholar] [CrossRef]
- Gogate, M.R. Methanol-to-olefins process technology: Current status and future prospects. Pet. Sci. Technol. 2019, 37, 559–565. [Google Scholar] [CrossRef]
- Carbon Recycling International (CRI); HS Orka. George Olah CO2 to Renewable Methanol Plant. Available online: https://www.chemicals-technology.com/projects/george-olah-renewable-methanol-plant-iceland/ (accessed on 17 August 2024).
- Carbon Recycling International (CRI). Emission to Liquid (ETL) Technology. Available online: https://www.carbonrecycling.is/george-olah (accessed on 17 August 2023).
- Carbon Recycling International (CRI). First large-Scale CO2 to Methanol Plant Inaugurated. Available online: https://carbonrecycling.com/about/news/first-large-scale-CO2-to-methanol-plant-inaugurated (accessed on 1 May 2024).
- Goehna, H.; Koenig, P. Producing methanol from CO2. CHEMTECH 1994, 24, 36–39. [Google Scholar]
- Konig, P.; Gohna, H. Process of Producing Methanol. Patent No. US5631302, 20 May 1997. [Google Scholar]
- Konig, P.; Gohna, H. Process of Producing Methanol. Patent No. US5827901, 27 October 1998. [Google Scholar]
- Müller, D.; Bormann, A. Process and Plant for Producing Methanol. Patent No. US8471076, 25 June 2013. Also published as ATE528274, BRPI0816071, CN101790501, DE102007040707, EG25675, EP2181083, MX2010002206, MY183147, US2011065966, WO2009030353. [Google Scholar]
- Kopetsch, H. Process and Plant for Producing Methanol. Patent No. US8629190, 14 January 2014. Also published as CN102171170, CN105732313, DE102008049622, EP2328854, US2011178187, WO2010037441. [Google Scholar]
- Brehm, L.; Göhna, H.; König, P. Process and Reactor System for Synthesis of Methanol with Cycle Gas and Purge Gas Recycling. Patent No. EP3016924, 11 May 2016. Also published as WO2014173452. [Google Scholar]
- Rodrigues, M.T.; Zonetti, P.C.; Alves, O.C.; Soua-Aguiar, E.F.; Borges, L.P.; Appel, L.E.G. RWGS reaction employing Ni/Mg(Al,Ni)O—The role of the O vacancies. Appl. Catal. A Gen. 2017, 543, 98–103. [Google Scholar] [CrossRef]
- Uhm, S.J.; Han, S.H.; Song, S.M.; Joo, O.S. Process for the Production of Methanol from Waste Gas. Patent No. KR0138587, 1 May 1998. Also published as WO9606064, JPH09510734, KR960007519. [Google Scholar]
- Joo, O.-S.; Jung, K.-D.; Moon, I.; Rozovskii, A.Y.; Lin, G.I.; Han, S.-H.; Uhm, S.-J. Carbon dioxide hydrogenation to form methanol via a reverse-water-gas-shift reaction (the camere process). Ind. Eng. Chem. Res. 1999, 38, 1808–1812. [Google Scholar] [CrossRef]
- Joo, O.S.; Jung, K.D.; Jung, Y. CAMERE process for methanol synthesis from CO2 hydrogenation. In Studies in Surface Science and Catalysis; Elsevier Inc.: Amsterdam, The Netherlands, 2004; Volume 153, pp. 67–72. [Google Scholar]
- Anicic, B.; Trop, P.; Goricanec, D. Comparison between two methods of methanol production from carbon dioxide. Energy 2014, 77, 279–289. [Google Scholar] [CrossRef]
- Toro, C.; Sciubba, E. Sabatier based power-to-gas system: Heat exchange network design and thermoeconomic analysis. Appl. Energy 2018, 229, 1181–1190. [Google Scholar] [CrossRef]
- Vogt, C.; Monai, M.; Kramer, G.J.; Weckhuysen, B.M. The renaissance of the Sabatier reaction and its applications on Earth and in space. Nat. Catal. 2019, 2, 188–197. [Google Scholar] [CrossRef]
- Rihko-Struckmann, L.K.; Peschel, A.; Hanke-Rauschenbach, R.; Sundmacher, K. Assessment of methanol synthesis utilizing exhaust CO2 for chemical storage of electrical energy. Proc. Ind. Eng. Chem. Res. 2010, 49, 11073–11078. [Google Scholar] [CrossRef]
- Liang, X.L.; Xie, J.R.; Liu, Z.M. A Novel Pd-decorated Carbon Nanotubes-promoted Pd-ZnO Catalyst for CO2 Hydrogenation to Methanol. Catal. Lett. 2015, 145, 1138–1147. [Google Scholar] [CrossRef]
- Kiatphuengporn, S.; Donphai, W.; Jantaratana, P.; Yigit, N.; Föttinger, K.; Rupprechter, G.; Chareonpanich, M. Cleaner production of methanol from carbon dioxide over copper and iron supported MCM-41 catalysts using innovative integrated magnetic field-packed bed reactor. J. Clean. Prod. 2017, 142, 1222–1233. [Google Scholar] [CrossRef]
- Wang, J.; Li, G.; Li, Z.; Tang, C.; Feng, Z.; An, H.; Liu, H.; Liu, T.; Li, C. A highly selective and stable ZnO-ZrO2 solid solution catalyst for CO2 hydrogenation to methanol. Sci. Adv. 2017, 3, e1701290. [Google Scholar] [CrossRef]
- Kar, S.; Kothandaraman, J.; Goeppert, A.; Prakash, G.K.S. Advances in catalytic homogeneous hydrogenation of carbon dioxide to methanol. J. CO2 Util. 2018, 23, 212–218. [Google Scholar] [CrossRef]
- Duyar, M.S.; Tsai, C.; Snider, J.L.; Singh, J.A.; Gallo, A.; Yoo, J.S.; Medford, A.J.; Abild-Pedersen, F.; Studt, F.; Kibsgaard, J.; et al. A Highly Active Molybdenum Phosphide Catalyst for Methanol Synthesis from CO and CO2. Angew. Chem.-Int. Ed. 2018, 57, 15045–15050. [Google Scholar] [CrossRef] [PubMed]
- Pacholik, G.; Enzlberger, L.; Benzer, A.; Rameshan, R.; Latschka, M.; Rameshan, C.; Föttinger, K. In situ XPS studies of MoS2-based CO2 hydrogenation catalysts. J. Phys. D Appl. Phys. 2021, 54, 324002. [Google Scholar] [CrossRef]
- Schrenk, F.; Lindenthal, L.; Pacholik, G.; Navratil, T.; Berger, T.M.; Drexler, H.; Rameshan, R.; Ruh, T.; Föttinger, K.; Rameshan, C. Perovskite-Type Oxide Catalysts in CO2 Utilization: A Principal Study of Novel Cu-Doped Perovskites for Methanol Synthesis. Compounds 2022, 2, 378–387. [Google Scholar] [CrossRef]
- Alves, G.A.S.; Pacholik, G.; Pollitt, S.; Wagner, T.; Rameshan, R.; Rameshan, C.; Föttinger, K. Mn-promoted MoS2 catalysts for CO2 hydrogenation: Enhanced methanol selectivity due to MoS2/MnOx interfaces. Catal. Sci. Technol. 2024, 14, 1138–1147. [Google Scholar] [CrossRef]
- Xu, D.; Cheng, B.; Wang, W.; Jiang, C.; Yu, J. Ag2cro4/g-c3n4/graphene oxide ternary nanocomposite Z-scheme photocatalyst with enhanced CO2 reduction activity. Appl. Catal. B Environ. 2018, 231, 368–380. [Google Scholar] [CrossRef]
- Schlager, S.; Dumitru, L.M.; Haberbauer, M.; Fuchsbauer, A.; Neugebauer, H.; Hiemetsberger, D.; Wagner, A.; Portenkirchner, E.; Sariciftci, N.S. Electrochemical reduction of carbon dioxide to methanol by direct injection of electrons into immobilized enzymes on a modified electrode. ChemSusChem 2016, 9, 631–635. [Google Scholar] [CrossRef]
- Schlager, S.; Dibenedetto, A.; Aresta, M.; Apaydin, D.H.; Dumitru, L.M.; Neugebauer, H.; Sariciftci, N.S. Biocatalytic and bioelectrocatalytic approaches for the reduction of carbon dioxide using enzymes. Energy Technol. 2017, 5, 812–821. [Google Scholar] [CrossRef]
- Bansode, A.; Urakawa, A. Towards full one-pass conversion of carbon dioxide to methanol and methanol-derived products. J. Catal. 2014, 309, 66–70. [Google Scholar] [CrossRef]
- Ganesh, I. Conversion of carbon dioxide into methanol—A potential liquid fuel: Fundamental challenges and opportunities (a review). Renew. Sustain. Energy Rev. 2014, 31, 221–257. [Google Scholar] [CrossRef]
- Kommoß, B.; Klemenz, S.; Schmitt, F.; Hocke, E.; Vogel, K.; Drochner, A.; Albert, B.; Etzold, B.; Vogel, H.G. Heterogeneously Catalyzed Hydrogenation of Supercritical CO2 to Methanol. Chem. Eng. Technol. 2017, 40, 1907–1915. [Google Scholar] [CrossRef]
- Álvarez, A.; Bansode, A.; Urakawa, A.; Bavykina, A.V.; Wezendonk, T.A.; Makkee, M.; Gascon, J.; Kapteijn, F. Challenges in the Greener Production of Formates/Formic Acid, Methanol, and DME by Heterogeneously Catalyzed CO2 Hydrogenation Processes. Chem. Rev. 2017, 117, 9804–9838. [Google Scholar] [CrossRef] [PubMed]
- Szima, S.; Cormos, C.-C. Improving methanol synthesis from carbon-free H2 and captured CO2: A techno-economic and environmental evaluation. J. CO2 Util. 2018, 24, 555–563. [Google Scholar] [CrossRef]
- Al-Saydeh, S.A.; Zaidi, S.J. Carbon dioxide conversion to methanol: Opportunities and fundamental challenges. In Carbon Dioxide Chemistry, Capture and Oil Recovery; Iyad, K., Janah, S., Hassan, S., Eds.; IntechOpen: Rijeka, Croatia, 2018; pp. 42–61. [Google Scholar] [CrossRef]
- Marlin, D.S.; Sarron, E.; Sigurbjörnsson, Ó. Process advantages of direct CO2 to methanol synthesis. Front. Chem. 2018, 6, 446. [Google Scholar] [CrossRef] [PubMed]
- Bayomie, O.S.; Bouallou, C. Energy and conversion investigations of different process configurations for hydrogenation of CO2 into methanol from industrial flue gases. Chem. Eng. Trans. 2018, 70, 1345–1350. [Google Scholar] [CrossRef]
- Meunier, N.; Chauvy, R.; Mouhoubi, S.; Thomas, D.; De Weireld, G. Alternative production of methanol from industrial CO2. Renew. Energy 2020, 146, 1192–1203. [Google Scholar] [CrossRef]
- Din, I.U.; Usman, M.; Khan, S.; Helal, A.; Alotaibi, M.A.; Alharthi, A.I.; Centi, G. Prospects for a green methanol thermo-catalytic process from CO2 by using MOFs based materials: A mini-review. J. CO2 Util. 2021, 43, 101361. [Google Scholar] [CrossRef]
- Pacholik, G.; Föttinger, K. Process for Preparing Methanol. Patent No. US2023365481, 16 November 2023. [Google Scholar]
- Hamedi, H.; Brinkmann, T.; Shishatskiy, S. Membrane-assisted methanol synthesis processes and the required permselectivity. Membranes 2021, 11, 596. [Google Scholar] [CrossRef]
- Milani, D.; Khalilpour, R.; Zahedi, G.; Abbas, A. A model-based analysis of CO2 utilization in methanol synthesis plant. J. CO2 Util. 2015, 10, 12–22. [Google Scholar] [CrossRef]
- Abdelaziz, O.Y.; Hosny, W.M.; Gadalla, M.A.; Ashour, F.H.; Ashour, I.A.; Hulteberg, C.P. Novel process technologies for conversion of carbon dioxide from industrial flue gas streams into methanol. J. CO2 Util. 2017, 21, 52–63. [Google Scholar] [CrossRef]
- Simonov, A. Electrolysis Breakthrough Could Solve the Hydrogen Conundrum. 2019. Available online: https://phys.org/news/2019-09-electrolysis-breakthrough-hydrogen-conundrum.html (accessed on 22 January 2022).
- Haldor Topsoe. Electrify Methanol Production for a Sustainable Business. 2019. Available online: https://info.topsoe.com/emethanol (accessed on 5 December 2021).
- Masel, R.I.; Ni, Z.R.; Chen, Q.; Rosen, B.A. Hydrogenation of Formic Acid to Formaldehyde. Patent No. US9193593, 24 November 2015. Also published as AU2014218628, CN105339336, CN107557086, EP2958882, US2014239231, WO2014130962. [Google Scholar]
- Masel, R.I.; Rosen, B.A.; Zhu, W. Devices and Processes for Carbon Dioxide Conversion into Useful Fuels and Chemicals. Patent No. US9181625, 10 November 2015. Also published as CN104822861, EP2898120, US2014093799, WO2014047661, WO201404766. [Google Scholar]
- Masel, R.I. Process for the Sustainable Production of Acrylic Acid. Patent No. US9790161, 17 October 2017. Also published as US2019135726 (9 May 2019), US2018057439 (1 March 2018), US20160207866 (21 July 2016), AU2017200539. [Google Scholar]
- Masel, R.I. System and Process for the Production of Renewable Fuels and Chemicals. Patent No. EP3504359, 3 July 2019. Also published as CN109642332, KR20190043156, WO2018044720. [Google Scholar]
- Kaczur, J.J.; Yang, H.; Sajjad, S.D.; Masel, R.I. Method and System for Electrochemical Production of Formic Acid from Carbon Dioxide. Patent No. US2019010620, 1 October 2019. [Google Scholar]
- Olah, G.A.; Prakash, G.K.S. Efficient and Selective Conversion of Carbon Dioxide to Methanol, Dimethyl Ether and Derived Products. Patent No. US7605293, 20 October 2009. Also published as US20060235091. [Google Scholar]
- Kuhl, K.P.; Hatsukade, T.; Cave, E.R.; Abram, D.N.; Kibsgaard, J.; Jaramillo, T.F. Electrocatalytic conversion of carbon dioxide to methane and methanol on transition metal surfaces. J. Am. Chem. Soc. 2014, 136, 14107–14113. [Google Scholar] [CrossRef]
- Wiranarongkorn, K.; Eamsiri, K.; Chen, Y.S.; Arpornwichanop, A. A comprehensive review of electrochemical reduction of CO2 to methanol: Technical and design aspects. J. CO2 Util. 2023, 71, 102477. [Google Scholar] [CrossRef]
- Ampelli, C.; Genovese, C.; Perathoner, S.; Centi, G.; Errahali, M.; Gatti, G.; Marchese, L. An electrochemical reactor for the CO2 reduction in gas phase by using conductive polymer based electrocatalysts. Chem. Eng. Trans. 2014, 41, 13–18. [Google Scholar] [CrossRef]
- ElMekawy, A.; Hegab, H.M.; Mohanakrishna, G.; Elbaz, A.F.; Bulut, M.; Pant, D. Technological advances in CO2 conversion electro-biorefinery: A step toward commercialization. Bioresour. Technol. 2016, 215, 357–370. [Google Scholar] [CrossRef] [PubMed]
- Masel, R.I.; Liu, Z.; Rosen, B.A. Electrochemical Device Comprising Novel Catalyst Mixtures. Patent No. US9957624, 1 May 2018. Also published as US20170022618. [Google Scholar]
- Hatsukade, T.; Kuhl, K.P.; Cave, E.R.; Abram, D.N.; Jaramillo, T.F. Insights into the electrocatalytic reduction of CO2 on metallic silver surfaces. Phys. Chem. Chem. Phys. 2014, 16, 13814–13819. [Google Scholar] [CrossRef]
- Díez-Ramírez, J.; Sánchez, P.; Valverde, J.L.; Dorado, F. Electrochemical promotion and characterization of PdZn alloy catalysts with K and Na ionic conductors for pure gaseous CO2 hydrogenation. J. CO2 Util. 2016, 16, 375–383. [Google Scholar] [CrossRef]
- Prakash, G.K.S.; Olah, G.A.; Goeppert, A. Beyond oil and gas: The methanol economy. In Proceedings of the Electrochemistry and Climate Change—219th ECS Meeting, Montreal, QC, Canada, 1–6 May 2011; pp. 31–40. [Google Scholar] [CrossRef]
- Liu, W.C.; Baek, J.; Somorjai, G.A. The Methanol Economy: Methane and Carbon Dioxide Conversion. Top. Catal. 2018, 61, 530–541. [Google Scholar] [CrossRef]
- Goeppert, A.; Czaun, M.; Jones, J.P.; Surya Prakash, G.K.; Olah, G.A. Recycling of carbon dioxide to methanol and derived products-closing the loop. Chem. Soc. Rev. 2014, 43, 7995–8048. [Google Scholar] [CrossRef]
- Shulenberger, A.M.; Jonsson, F.R.; Ingolfsson, O.; Tran, K.-C. Process for Producing Liquid Fuel from Carbon Dioxide and Water. Patent No. US8198338, 12 June 2012. Also published as US2012201717, US8506910, WO2007108014. [Google Scholar]
- Shulenberger, A.M.; Jonsson, F.R.; Ingolfsson, O.; Tran, K.-C. Process and System for Producing Liquid Fuel from Carbon Dioxide and Water. Patent No. US8506910, 13 August 2013. Also published as US2012201717, US8198338, WO2007108014. [Google Scholar]
- Zaromb, S. Apparatus and Methods for Carbon Dioxide Capture and Conversion. Patent No. US8413420, 9 April 2013. [Google Scholar]
- Sigurbjornsson, O.F.; Singh, S.; Eythorsson, D. System and Process to Capture Industrial Emissions and Recycle for the Production of Chemicals. Patent No. WO2014087433, 12 June 2014. [Google Scholar]
- Simmons, W.W.; White, S.P.; Perkins, C. Various Methods and Apparatuses for Multi-Stage Synthesis Gas Generation. Patent No. US9663363, 30 May 2017. Also published as WO2013158343, US2012181483. [Google Scholar]
- Bowker, M.; Hayward, J. Catalyst Suitable for Methanol Synthesis. Patent No. WO2018138512, 2 August 2018. [Google Scholar]
- Li, X.; Xu, Y.; Ru, J.; Yang, K.; Zhang, T. Comprehensive Treatment Device for Carbon Dioxide Generated by Waste Incineration. Patent No. CN218154221, 27 December 2022. [Google Scholar]
- Wix, C.; Stummann, T.D. Conversion of Carbon Dioxide and Water to Synthesis Gas for Producing Methanol and Hydrocarbon Products. Patent No. CA3203055, 30 June 2022. Also published as AU2021405257 CN116601335 TW202241835 WO2022136374. [Google Scholar]
- Al-Rowaili, F.N.; Khaled, M.; Jamal, A.; Onaizi, S.A.; Zahid, U. Electrochemical Reduction of Carbon Dioxide. Patent No. US11512400, 29 November 2022. Also published as US2022186385 WO2022125831. [Google Scholar]
- Hou, C.; Lv, H.; Ding, L.A.; Zhang, X. Peak Regulation Power Generation System and Method Coupled with Carbon Dioxide Capture and Utilization. Patent No. CN114899884, 12 August 2022. [Google Scholar]
- Roustaei, A.H.; Djemai, A. System for Production, Conversion and Reproduction of Gas-Liquid-Gas Hydrogen Cycle with Absorption of Carbon Dioxide, Comprises Water Electrolyzer Device with Electrolysis Chamber, Electrodes and Electrolyte, and Gas Transformation Device. Patent No. FR2939450, 11 June 2010. [Google Scholar]
- Hoppe, W.; Bringezu, S.; Wachter, N. Economic assessment of CO2-based methane, methanol and polyoxymethylene production. J. CO2 Util. 2018, 27, 170–178. [Google Scholar] [CrossRef]
- Ghasemzadeh, K.; Sadati Tilebon, S.M.; Nasirinezhad, M.; Basile, A. Economic Assessment of Methanol Production. In Methanol: Science and Engineering; Elsevier: Amsterdam, The Netherlands, 2018; pp. 613–632. [Google Scholar] [CrossRef]
- Nguyen, T.B.H.; Leonzio, G.; Zondervan, E. Supply chain optimization framework for CO2 capture, utilization, and storage in Germany. Phys. Sci. Rev. 2023, 8, 1685–1711. [Google Scholar] [CrossRef]
- Pratschner, S.; Skopec, P.; Hrdlicka, J.; Winter, F. Power-to-green methanol via CO2 hydrogenation—A concept study including oxyfuel fluidized bed combustion of biomass. Energies 2021, 14, 4638. [Google Scholar] [CrossRef]
- Gaikwad, A.; Maga, D.; Schlüter, S. Comparative Lifecycle Assessment of Methanol and Urea Produced from Steel Mill Gases. Chem.-Ing.-Tech. 2022, 94, 1476–1488. [Google Scholar] [CrossRef]
- Meessen, J. Urea synthesis. Chem. Ing. Tech. 2014, 86, 2180–2189. [Google Scholar] [CrossRef]
- Ding, J.; Ye, R.; Fu, Y.; He, Y.; Wu, Y.; Zhang, Y.; Zhong, Q.; Kung, H.H.; Fan, M. Direct synthesis of urea from carbon dioxide and ammonia. Nat. Commun. 2023, 14, 4586. [Google Scholar] [CrossRef] [PubMed]
- Berghout, N.; McCulloch, S. Putting CO2 to Use; IEA: Paris, France, 2019; 86p, Available online: https://www.iea.org/reports/putting-CO2-to-use (accessed on 18 April 2022).
- Cheng, K.; Cheng, W. Tripolycyanamide Whole Circulation Production Process and Device. Patent No. CN108558783, 21 September 2018. [Google Scholar]
- Lee, J.M. Process for Manufacturing Melamine from Urea. Patent No. US5384404, 24 January 1995. [Google Scholar]
- Li, D. One-Step Method Used for Production of Melamine by Circulating Ammonia Gas as Carrying Gas. Patent No. CN103601693, 2 June 2012. [Google Scholar]
- Mennen, J.H. Integrated Production of Urea and Melamine. Patent No. US9981924, 29 May 2018. [Google Scholar]
- Sioli, G. Process for the Production of High Purity Melamine from Urea. Patent No. US9045439, 2 June 2012. [Google Scholar]
- Brueske, S.; Kramer, C.; Fisher, A. Chemical Bandwidth Study; US Department of Energy, Office of Energy Efficiency & Renewable Energy: Washington, DC, USA, 2015. Available online: https://www.energy.gov/eere/iedo/articles/bandwidth-study-us-chemical-manufacturing (accessed on 21 May 2020).
- Hosono, H.; Kitano, M. Advances in materials and applications of inorganic electrides. Chem. Rev. 2021, 121, 3121–3185. [Google Scholar] [CrossRef] [PubMed]
- Kyriakou, V.; Garagounis, I.; Vourros, A.; Vasileiou, E.; Stoukides, M. An electrochemical haber-bosch process. Joule 2020, 4, 142–158. [Google Scholar] [CrossRef]
- Liu, H.; Wei, L.; Liu, F.; Pei, Z.; Shi, J.; Wang, Z.J.; He, D.; Chen, Y. Homogeneous, heterogeneous, and biological catalysts for electrochemical N2 reduction toward NH3 under ambient conditions. ACS Catal. 2019, 9, 5245–5267. [Google Scholar] [CrossRef]
- Wang, Z.; Li, Y.; Yu, H.; Xu, Y.; Xue, H.; Li, X.; Wang, H.; Wang, L. Ambient Electrochemical Synthesis of Ammonia from Nitrogen and Water Catalyzed by Flower-Like Gold Microstructures. ChemSusChem 2018, 11, 3480–3485. [Google Scholar] [CrossRef]
- Shipman, M.A.; Symes, M.D. Recent progress towards the electrosynthesis of ammonia from sustainable resources. Catal. Today 2017, 286, 57–68. [Google Scholar] [CrossRef]
- Kyriakou, V.; Garagounis, I.; Vasileiou, E.; Vourros, A.; Stoukides, M. Progress in the Electrochemical Synthesis of Ammonia. Catal. Today 2017, 286, 2–13. [Google Scholar] [CrossRef]
- Topsoe and First Ammonia Launch Zero Emission Ammonia Production with the World’s Largest Reservation of Electrolyzer Capacity. Available online: https://www.topsoe.com/press-releases/topsoe-and-first-ammonia?utm_medium=email&_hsmi=225986923&_hsenc=p2ANqtz-9hzpaCDUilA4TCFqDA1kUOcPkVabbU%E2%80%A6 (accessed on 16 September 2022).
- Knudsen, J.N. Green Ammonia: Brighten Two Horizons at Once. Available online: https://www.topsoe.com/processes/green-ammonia (accessed on 3 December 2023).
- Boyce, J.; Sacchi, R.; Goetheer, E.; Steubing, B. A prospective life cycle assessment of global ammonia decarbonisation scenarios. Heliyon 2024, 10, e27547. [Google Scholar] [CrossRef]
- Shi, L.; Liu, L.; Yang, B.; Sheng, G.; Xu, T. Evaluation of industrial urea energy consumption (EC) based on life cycle assessment (LCA). Sustainability 2020, 12, 3793. [Google Scholar] [CrossRef]
- Shirmohammadi, R.; Aslani, A.; Ghasempour, R.; Romeo, L.M.; Petrakopoulou, F. Exergoenvironmental analysis and thermoeconomic optimization of an industrial post-combustion CO2 capture and utilization installation. J. CO2 Util. 2022, 59, 101927. [Google Scholar] [CrossRef]
- Yang, M.; Rosentrater, K.A. Life Cycle Assessment of Urea-Formaldehyde Adhesive and Phenol-Formaldehyde Adhesives. Environ. Process. 2020, 7, 553–561. [Google Scholar] [CrossRef]
- Litskas, V.D. Environmental Impact Assessment for Animal Waste, Organic and Synthetic Fertilizers. Nitrogen 2023, 4, 16–25. [Google Scholar] [CrossRef]
- Pulselli, F.M.; Patrizi, N.; Focardi, S. Calculation of the unit emergy value of water in an Italian watershed. Ecol. Model. 2011, 222, 2929–2938. [Google Scholar] [CrossRef]
- Lyu, Y.; Raugei, M.; Zhang, X.; Mellino, S.; Ulgiati, S. Environmental cost and impacts of chemicals used in agriculture: An integration of emergy and Life Cycle Assessment. Renew. Sustain. Energy Rev. 2021, 151, 111604. [Google Scholar] [CrossRef]
- Rivas-Aybar, D.; John, M.; Biswas, W. Environmental Life Cycle Assessment of a Novel Hemp-Based Building Material. Materials 2023, 16, 7208. [Google Scholar] [CrossRef]
- Zhang, L.; Liang, Z.; Hu, Y.; Schmidhalter, U.; Zhang, W.; Ruan, S.; Chen, X. Integrated assessment of agronomic, environmental and ecosystem economic benefits of blending use of controlled-release and common urea in wheat production. J. Clean. Prod. 2021, 287, 125572. [Google Scholar] [CrossRef]
- Masjedi, S.K.; Kazemi, A.; Moeinnadini, M.; Khaki, E.; Olsen, S.I. Urea production: An absolute environmental sustainability assessment. Sci. Total Environ. 2024, 908, 168225. [Google Scholar] [CrossRef]
- Marvel, C.S.; Elliott, J.R.; Boettner, F.E.; Yuska, H. The Structure of Urea-Formaldehyde Resins1. J. Am. Chem. Soc. 1946, 68, 1681–1686. [Google Scholar] [CrossRef]
- Nguon, O.; Lagugné-Labarthet, F.; Brandys, F.A.; Li, J.; Gillies, E.R. Microencapsulation by in situ polymerization of amino resins. Polym. Rev 2017, 58, 326–375. [Google Scholar] [CrossRef]
- Pizzi, A. Urea-formaldehyde adhesives. In Advanced Wood Adhesives Technology; CRC Press: Boca Raton, FL, USA, 1994; pp. 19–66. [Google Scholar]
- Rammon, R.M.; Johns, W.E.; Magnuson, J.; Dunker, A.K. The Chemical Structure of UF Resins. J. Adhes. 1986, 19, 115–135. [Google Scholar] [CrossRef]
- Antov, P.; Savov, V.; Krišt’ák, L.; Réh, R.; Mantanis, G.I. Eco-friendly, high-density fiberboards bonded with urea-formaldehyde and ammonium lignosulfonate. Polymers 2021, 13, 220. [Google Scholar] [CrossRef] [PubMed]
- Ormondroyd, G.A. Adhesives for wood composites. In Wood Composites; Ansell, M.P., Ed.; Woodhead Publishing: Sawston, UK, 2015; pp. 47–66. [Google Scholar] [CrossRef]
- Rowell, R.M. Handbook of Wood Chemistry and Wood Composites, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2012. [Google Scholar] [CrossRef]
- Meyer, B. Urea-Formaldehyde Resins; Addison-Wesley Publishing Co.: London, UK, 1979; 142p. [Google Scholar]
- Myers, G.E.; Koutsky, J.A. Formaldehyde Liberation and Cure Behavior of Urea-Formaldehyde Resins. Holzforschung 1990, 44, 117–126. [Google Scholar] [CrossRef]
- Myers, G.E. New technologies and materials for bonding wood products. Adhes. Age 1988, 31, 31–36. [Google Scholar]
- Myers, G.E.; Koutsky, J.A. Procedure for measuring formaldehyde liberation from formaldehyde-based resins. For. Prod. J. 1987, 37, 56–60. [Google Scholar]
- Myers, G.E. Effect of ventilation rate and board loading on formaldehyde concentration: A critical review of the literature. For. Prod. J. 1984, 34, 59–68. [Google Scholar]
- Myers, G.E. How mole ratio of UF resin affects formaldehyde emission and other properties: A literature critique. For. Prod. J. 1984, 34, 35–41. [Google Scholar]
- Myers, G.E. Formaldehyde emission from particleboard and plywood paneling: Measurement, mechanism, and product standards. For. Prod. J. 1983, 33, 27–37. [Google Scholar]
- Myers, G.E.; Nagaoka, M. Emission of formaldehyde by particleboard: Effect of ventilation rate and loading on air-contamination levels. For. Prod. J. 1981, 31, 39–44. [Google Scholar]
- River, B.H.; Ebewele, R.O.; Myers, G.E. Failure mechanisms in wood joints bonded with urea-formaldehyde adhesives. Holz Als Roh- Und Werkst. 1994, 52, 179–184. [Google Scholar] [CrossRef]
- Ebewele, R.O.; River, B.H.; Myers, G.E. Behavior of amine-modified urea–formaldehyde-bonded wood joints at low formaldehyde/urea molar ratios. J. Appl. Polym. Sci. 1994, 52, 689–700. [Google Scholar] [CrossRef]
- Ebewele, R.O.; River, B.H.; Myers, G.E. Polyamine-modified urea–formaldehyde-bonded wood joints. III. Fracture toughness and cyclic stress and hydrolysis resistance. J. Appl. Polym. Sci. 1993, 49, 229–245. [Google Scholar] [CrossRef]
- Ebewele, R.O.; Myers, G.E.; River, B.H.; Koutsky, J.A. Polyamine-modified urea-formaldehyde resins. I. Synthesis, structure, and properties. J. Appl. Polym. Sci. 1991, 42, 2997–3012. [Google Scholar] [CrossRef]
- Ganesh, I. Conversion of carbon dioxide into several potential chemical commodities following different pathways—A review. Mater. Sci. Forum 2013, 764, 1–82. [Google Scholar] [CrossRef]
- Chan, F.L.; Altinkaya, G.; Fung, N.; Tanksale, A. Low temperature hydrogenation of carbon dioxide into formaldehyde in liquid media. Catal. Today 2018, 309, 242–247. [Google Scholar] [CrossRef]
- Roffael, E. Significance of wood extractives for wood bonding. Appl. Microbiol. Biotechnol. 2016, 100, 1589–1596. [Google Scholar] [CrossRef]
- Dessbesell, L.; Yuan, Z.; Leitch, M.; Paleologou, M.; Pulkki, R.; Xu, C.C. Capacity design of a kraft lignin biorefinery for production of biophenol via a proprietary low-temperature/low-pressure lignin depolymerization process. ACS Sustain. Chem. Eng. 2018, 6, 9293–9303. [Google Scholar] [CrossRef]
- Starace, A.K.; Black, B.A.; Lee, D.D.; Palmiotti, E.C.; Orton, K.A.; Michener, W.E.; Ten Dam, J.; Watson, M.J.; Beckham, G.T.; Magrini, K.A.; et al. Characterization and Catalytic Upgrading of Aqueous Stream Carbon from Catalytic Fast Pyrolysis of Biomass. ACS Sustain. Chem. Eng. 2017, 5, 11761–11769. [Google Scholar] [CrossRef]
- Liu, M.; Wang, Y.; Wu, Y.; He, Z.; Wan, H. “Greener” adhesives composed of urea-formaldehyde resin and cottonseed meal for wood-based composites. J. Clean. Prod. 2018, 187, 361–371. [Google Scholar] [CrossRef]
- Zhan, B.; Zhang, L.; Deng, Y.; Fan, M.; Yan, L. Sustainable adhesives for ultra-composites from biomass powder. Chem. Eng. J. 2024, 485, 149984. [Google Scholar] [CrossRef]
- Al-gheethi, A.A.; Memon, Z.A.; Balasbaneh, A.T.; Al-kutti, W.A.; Mokhtar, N.; Othman, N.; Juki, M.I.; Noman, E.A.; Algaifi, H.A. Critical Analysis for Life Cycle Assessment of Bio-Cementitious Materials Production and Sustainable Solutions. Sustainability 2022, 14, 1920. [Google Scholar] [CrossRef]
- Lee, S.M.; Park, J.Y.; Park, S.B.; Han, S.T.; Kang, E.C. Comparative study of the storage stability between a melamine-urea- formaldehyde and a urea-formaldehyde resin. For. Prod. J. 2012, 62, 146–149. [Google Scholar] [CrossRef]
- Priha, E. Formaldehyde release from resin-containing wood board dusts: Evaluation of methods to determine formaldehyde. Appl. Occup. Environ. Hyg. 1996, 11, 465–470. [Google Scholar] [CrossRef]
- Park, B.D.; Lee, S.M.; Roh, J.K. Effects of formaldehyde/urea mole ratio and melamine content on the hydrolytic stability of cured urea-melamine-formaldehyde resin. Eur. J. Wood Wood Prod. 2009, 67, 121–123. [Google Scholar] [CrossRef]
- Park, B.D.; Lee, S.M. Hydrolytic stability of cured urea-melamine-formaldehyde resins depending on hydrolysis conditions and hardener types. J. Korean Wood Sci. Technol. 2015, 43, 672–681. [Google Scholar] [CrossRef]
- Jeong, B.; Park, B.D.; Causin, V. Influence of synthesis method and melamine content of urea-melamine-formaldehyde resins to their features in cohesion, interphase, and adhesion performance. J. Ind. Eng. Chem. 2019, 79, 87–96. [Google Scholar] [CrossRef]
- Suchý, P.; Straková, E.; Herzig, I.; Staňa, J.; Kalusová, R.; Pospíchalová, M. Toxicological risk of melamine and cyanuric acid in food and feed. Interdiscip. Toxicol. 2009, 2, 55–59. [Google Scholar] [CrossRef]
- Bann, B.; Miller, S.A. Melamine And Derivatives Of Melamine. Chem. Rev. 1958, 58, 131–172. [Google Scholar] [CrossRef]
- Braun, D.; Ritzert, H.J. Properties and processing behaviour of urea-melamine-formaldehyde resins. Kunststoffe Ger. Plast. 1987, 77, 29–31. [Google Scholar]
- No, B.Y.; Kim, M.G. Evaluation of melamine-modified urea-formaldehyde resins as particleboard binders. J. Appl. Polym. Sci. 2007, 106, 4148–4156. [Google Scholar] [CrossRef]
- Shi, J.; Ye, S. Effects of addition ammonia modified urea-melamine-formaldehyde resin on the adhesions and formaldehyde emission in plywood. Proc. Adv. Mater. Res. 2010, 113–116, 1226–1229. [Google Scholar] [CrossRef]
- Ranjbaran, S.; Nazerian, M.; Kermanian, H.; Koosha, M.; Garmaroody, E.R. High strength papers impregnated with urea/melamine formaldehyde resin/nanosilica nanocomposite coatings: The effects of paper type, blend ratio and nano-content. Mater. Today Commun. 2020, 25, 101300. [Google Scholar] [CrossRef]
- Li, Z.; Chen, H.; Xu, Q.; Li, X.; Ma, H.; Yuan, Q. Styrene and BPO poly (urea-melamine-formaldehyde) microcapsules prepared via in situ polymerization to promote the self-healing properties of epoxy composites. J. Coat. Technol. Res. 2022, 19, 1837–1850. [Google Scholar] [CrossRef]
- Sokolova, E.G.; Varankina, G.S.; Rusakov, D.S. Urea–melamine formaldehyde resin for the manufacture of water-resistant plywood. Polym. Sci.-Ser. D 2022, 15, 557–561. [Google Scholar] [CrossRef]
- Silva, D.A.L.; Lahr, F.A.R.; Varanda, L.D.; Christoforo, A.L.; Ometto, A.R. Environmental performance assessment of the melamine-urea-formaldehyde (MUF) resin manufacture: A case study in Brazil. J. Clean. Prod. 2015, 96, 299–307. [Google Scholar] [CrossRef]
- Yılmaz, E. Environmental impact assessment of melamine coated medium density fiberboard (MDF-LAM) production and cumulative energy demand: A case study in Türkiye. Case Stud. Constr. Mater. 2024, 20, e02733. [Google Scholar] [CrossRef]
- Lao, W.L.; Ma, L.P.; Wang, C.; Liu, C.W.; Li, Y. Environmental impacts evaluation and promotion measures of wood-based composite doors. J. Build. Eng. 2023, 80, 108164. [Google Scholar] [CrossRef]
- Tang, Y.; Yuan, Z.; Gong, Y.; Yin, M.; Yang, X.; Chen, D.; Yi, J.; Lei, L.; Liu, C.; Li, X.; et al. System and Process for Melamine Production by Gas-Phase Quenching Method of Energy Efficient and Cost Saving Type. Patent No. US9114371, 25 August 2015. [Google Scholar]
- Solmi, M.V.; Schmitz, M.; Leitner, W. CO2 as a Building Block for the Catalytic Synthesis of Carboxylic Acids. In Studies in Surface Science and Catalysis; Elsevier Inc.: Amsterdam, The Netherlands, 2019; Volume 178, pp. 105–124. [Google Scholar] [CrossRef]
- Hölscher, M.; Gürtler, C.; Keim, W.; Müller, T.E.; Peters, M.; Leitner, W. Carbon dioxide as a carbon resource—Recent trends and perspectives. Z. Fur Naturforschung-Sect. B J. Chem. Sci. 2012, 67, 961–975. [Google Scholar] [CrossRef]
- Scott, M.; Westhues, C.G.; Kaiser, T.; Baums, J.C.; Jupke, A.; Franciò, G.; Leitner, W. Methylformate from CO2: An integrated process combining catalytic hydrogenation and reactive distillation. Green Chem. 2019, 21, 6307–6317. [Google Scholar] [CrossRef]
- Schmitz, M.; Solmi, M.V.; Leitner, W. Catalytic processes combining CO2 and alkenes into value-added chemicals: Synthesis of cyclic carbonates, lactones, carboxylic acids, esters, aldehydes, alcohols, and amines. Top. Organomet. Chem. 2019, 63, 17–38. [Google Scholar] [CrossRef]
- Peters, M.; Köhler, B.; Kuckshinrichs, W.; Leitner, W.; Markewitz, P.; Müller, T.E. Chemical technologies for exploiting and recycling carbon dioxide into the value chain. ChemSusChem 2011, 4, 1216–1240. [Google Scholar] [CrossRef] [PubMed]
- Ostapowicz, T.G.; Schmitz, M.; Krystof, M.; Klankermayer, J.; Leitner, W. Carbon dioxide as a C1 building block for the formation of carboxylic acids by formal catalytic hydrocarboxylation. Angew. Chem.-Int. Ed. 2013, 52, 12119–12123. [Google Scholar] [CrossRef] [PubMed]
- Leitner, W.; Franciò, G.; Scott, M.; Westhues, C.; Langanke, J.; Lansing, M.; Hussong, C.; Erdkamp, E. Carbon2Polymer—Chemical Utilization of CO2 in the Production of Isocyanates. Chem.-Ing.-Tech. 2018, 90, 1504–1512. [Google Scholar] [CrossRef]
- Suh, H.S.; Ha, J.Y.; Yoon, J.H.; Ha, C.-S.; Suh, H.; Kim, I. Polyester polyol synthesis by alternating copolymerization of propylene oxide with cyclic acid anhydrides by using double metal cyanide catalyst. React. Funct. Polym. 2010, 70, 288–293. [Google Scholar] [CrossRef]
- Ballivet-Tkatchenko, D.; Dos Santos, J.H.Z.; Philippot, K.; Vasireddy, S. Carbon dioxide conversion to dimethyl carbonate: The effect of silica as support for SnO2 and ZrO2 catalysts. C. R. Chim. 2011, 14, 780–785. [Google Scholar] [CrossRef]
- Kizlink, J.; Pastucha, I. Preparation of dimethyl carbonate from methanol and carbon dioxide in the presence of Sn(IV) and Ti(IV) alkoxides and metal acetates. Collect. Czech. Chem. Commun. 1995, 60, 687–692. [Google Scholar] [CrossRef]
- Sun, W.; Zheng, L.; Wang, Y.; Li, D.; Liu, Z.; Wu, L.; Fang, T.; Wu, J. Study of thermodynamics and experiment on direct synthesis of dimethyl carbonate from carbon dioxide and methanol over yttrium oxide. Ind. Eng. Chem. Res. 2020, 59, 4281–4290. [Google Scholar] [CrossRef]
- Hu, X.; Cheng, H.; Kang, X.; Chen, L.; Yuan, X.; Qi, Z. Analysis of direct synthesis of dimethyl carbonate from methanol and CO2 intensified by in-situ hydration-assisted reactive distillation with side reactor. Chem. Eng. Process.-Process Intensif. 2018, 129, 109–117. [Google Scholar] [CrossRef]
- Guo, C.; Liu, X.; Wang, F.; Cao, Y.; Zheng, S.; He, G. Economic Analysis and Life Cycle Environmental Assessment of Imidazolium-Based Ionic Liquids for Separation of the Methanol/Dimethyl Carbonate Azeotrope. ACS Sustain. Chem. Eng. 2023, 11, 10482–10495. [Google Scholar] [CrossRef]
- Garcia-Herrero, I.; Cuéllar-Franca, R.M.; Enríquez-Gutiérrez, V.M.; Alvarez-Guerra, M.; Irabien, A.; Azapagic, A. Environmental Assessment of Dimethyl Carbonate Production: Comparison of a Novel Electrosynthesis Route Utilizing CO2 with a Commercial Oxidative Carbonylation Process. ACS Sustain. Chem. Eng. 2016, 4, 2088–2097. [Google Scholar] [CrossRef]
- Zhao, S.Y.; Wang, S.P.; Zhao, Y.J.; Ma, X.B. An in situ infrared study of dimethyl carbonate synthesis from carbon dioxide and methanol over well-shaped CeO2. Chin. Chem. Lett. 2017, 28, 65–69. [Google Scholar] [CrossRef]
- Tomishige, K.; Sakaihori, T.; Ikeda, Y.; Fujimoto, K. A novel method of direct synthesis of dimethyl carbonate from methanol and carbon dioxide catalyzed by zirconia. Catal. Lett. 1999, 58, 225–229. [Google Scholar] [CrossRef]
- Stoian, D.C.; Taboada, E.; Llorca, J.; Molins, E.; Medina, F.; Segarra, A.M. Boosted CO2 reaction with methanol to yield dimethyl carbonate over Mg–Al hydrotalcite-silica lyogels. Chem. Commun. 2013, 49, 5489–5491. [Google Scholar] [CrossRef] [PubMed]
- Aresta, M.; Dibenedetto, A.; Pastore, C.; Pápai, I.; Schubert, G. Reaction mechanism of the direct carboxylation of methanol to dimethylcarbonate: Experimental and theoretical studies. Top. Catal. 2006, 40, 71–81. [Google Scholar] [CrossRef]
- Tundo, P.; Selva, M. The chemistry of dimethyl carbonate. Acc. Chem. Res. 2002, 35, 706–716. [Google Scholar] [CrossRef]
- Tundo, P.; Perosa, A. Green organic syntheses: Organic carbonates as methylating agents. Chem. Rec. 2002, 2, 13–23. [Google Scholar] [CrossRef]
- Lui, Y.W.; Chan, B.; Lui, M.Y. Methylation with Dimethyl Carbonate/Dimethyl Sulfide Mixtures: An Integrated Process without Addition of Acid/Base and Formation of Residual Salts. ChemSusChem 2022, 15, e202102538. [Google Scholar] [CrossRef]
- Delledonne, D.; Rivetti, F.; Romano, U. Developments in the production and application of dimethylcarbonate. Appl. Catal. A Gen. 2001, 221, 241–251. [Google Scholar] [CrossRef]
- Astruc, L.; Miranda, J.; Donis, I.R.; Vialle, C.; Sablayrolles, C. Dimethyl Carbonate Production by Urea Transesterification, Process Simulation and Environmental Assessment. In Computer Aided Chemical Engineering; Elsevier B.V.: Amsterdam, The Netherlands, 2020; Volume 48, pp. 97–102. [Google Scholar] [CrossRef]
- Huang, S.; Yan, B.; Wang, S.; Ma, X. Recent advances in dialkyl carbonates synthesis and applications. Chem. Soc. Rev. 2015, 44, 3079–3116. [Google Scholar] [CrossRef]
- Monteiro, J.G.M.S.; De Queiroz Fernandes Araújo, O.; De Medeiros, J.L. Sustainability metrics for eco-technologies assessment, part I: Preliminary screening. Clean Technol. Environ. Policy 2009, 11, 209–214. [Google Scholar] [CrossRef]
- Monteiro, J.G.M.S.; de Queiroz Fernandes Araújo, O.; de Medeiros, J.L. Sustainability metrics for eco-technologies assessment, Part II. Life cycle analysis. Clean Technol. Environ. Policy 2009, 11, 459–472. [Google Scholar] [CrossRef]
- Lee, Y.G.; Lee, H.U.; Lee, J.M.; Kim, N.Y.; Jeong, D.H. Design of Dimethyl Carbonate (DMC) Synthesis Process Using CO2, Techno-economic Analysis, and Life Cycle Assessment. Korean J. Chem. Eng. 2024, 41, 117–133. [Google Scholar] [CrossRef]
- Aresta, M.; Quaranta, E.; Dibenedetto, A.; Tommasi, I.; Marciniec, B. CO2-catalysed carbamation of aminofunctional silanes. Appl. Organomet. Chem. 2000, 14, 871–873. [Google Scholar] [CrossRef]
- Tundo, P.; McElroy, C.R.; Aricò, F. Synthesis of carbamates from amines and dialkyl carbonates: Influence of leaving and entering groups. Synlett 2010, 2010, 1567–1571. [Google Scholar] [CrossRef]
- Sun, J.; Aly, K.I.; Kuckling, D. A novel one-pot process for the preparation of linear and hyperbranched polycarbonates of various diols and triols using dimethyl carbonate. RSC Adv. 2017, 7, 12550–12560. [Google Scholar] [CrossRef]
- Liu, T.; Wang, G.; Yang, X. Controlled synthesis of aliphatic polycarbonate diols using dimethyl carbonate and various diols. J. Chin. Chem. Soc. 2022, 69, 995–1001. [Google Scholar] [CrossRef]
- Tundo, P.; Aricò, F.; McElroy, C.R. The Greening of Chemistry. In The Chemical Element: Chemistry’s Contribution to Our Global Future, 1st ed.; Wiley-VCH: Hoboken, NJ, USA, 2011; pp. 189–233. [Google Scholar] [CrossRef]
- Dernehl, C.U. Health hazards associated with polyurethane foams. J. Occup. Med. 1966, 8, 59–62. [Google Scholar]
- González, S.G.; Bravo, A.R.; Carpintero, V.C.; Cuenca-Romero, L.A. Towards an ecological transition in the construction sector through the production of new eco-efficient products. Proc. E3S Web Conf. 2023, 379, 04004. [Google Scholar] [CrossRef]
- Rodrigo-Bravo, A.; Alameda Cuenca-Romero, L.; Calderón, V.; Rodríguez, Á.; Gutiérrez-González, S. Comparative Life Cycle Assessment (LCA) between standard gypsum ceiling tile and polyurethane gypsum ceiling tile. Energy Build. 2022, 259, 111867. [Google Scholar] [CrossRef]
- Rey-Álvarez, B.; Silvestre, J.; García-Martínez, A.; Sánchez-Montañés, B. A comparative approach to evaluate the toxicity of building materials through life cycle assessment. Sci. Total Environ. 2024, 912, 168897. [Google Scholar] [CrossRef] [PubMed]
- Llantoy, N.; Chàfer, M.; Cabeza, L.F. A comparative life cycle assessment (LCA) of different insulation materials for buildings in the continental Mediterranean climate. Energy Build. 2020, 225, 110323. [Google Scholar] [CrossRef]
- Klug, V.; Schöggl, J.P.; Dallinger, D.; Hiebler, K.; Stueckler, C.; Steiner, A.; Arzt, A.; Kappe, C.O.; Baumgartner, R.J. Comparative Life Cycle Assessment of Different Production Processes for Waterborne Polyurethane Dispersions. ACS Sustain. Chem. Eng. 2021, 9, 8980–8989. [Google Scholar] [CrossRef]
- Mohamed, N.A.; El-Dash, K.M.; Attia, T.M.; Abdel-Monem, M. Analysis of alternative building envelope solutions to improve energy efficiency. In Proceedings of the 2023 2nd International Conference on Smart Cities 4.0, Smart Cities 4.0 2023, Cairo, Egypt, 22–24 October 2023; pp. 138–145. [Google Scholar] [CrossRef]
- Liang, S.; Liu, H.; Jiang, T.; Song, J.; Yang, G.; Han, B. Highly efficient synthesis of cyclic carbonates from CO2 and epoxides over cellulose/KI. Chem. Commun. 2011, 47, 2131–2133. [Google Scholar] [CrossRef]
- Nodehi, M.; Aguayo, F.; Madey, N.; Zhou, L. A Comparative Review of Polymer, Bacterial-based, and Alkali-Activated (also Geopolymer) Binders: Production, Mechanical, Durability, and Environmental impacts (life cycle assessment (LCA)). Constr. Build. Mater. 2024, 422, 135816. [Google Scholar] [CrossRef]
- Fridrihsone, A.; Romagnoli, F.; Kirsanovs, V.; Cabulis, U. Life Cycle Assessment of vegetable oil based polyols for polyurethane production. J. Clean. Prod. 2020, 266, 121403. [Google Scholar] [CrossRef]
- Bachmann, M.; Kätelhön, A.; Winter, B.; Meys, R.; Müller, L.J.; Bardow, A. Renewable carbon feedstock for polymers: Environmental benefits from synergistic use of biomass and CO2. Faraday Discuss. 2021, 230, 227–246. [Google Scholar] [CrossRef]
- Polo Fonseca, L.; Duval, A.; Luna, E.; Ximenis, M.; De Meester, S.; Avérous, L.; Sardon, H. Reducing the carbon footprint of polyurethanes by chemical and biological depolymerization: Fact or fiction? Curr. Opin. Green Sustain. Chem. 2023, 41, 100802. [Google Scholar] [CrossRef]
- Recupido, F.; Lama, G.C.; Ammendola, M.; Bossa, F.D.L.; Minigher, A.; Campaner, P.; Morena, A.G.; Tzanov, T.; Ornelas, M.; Barros, A.; et al. Rigid composite bio-based polyurethane foams: From synthesis to LCA analysis. Polymer 2023, 267, 125674. [Google Scholar] [CrossRef]
- Silva, R.; Barros-Timmons, A.; Quinteiro, P. Life cycle assessment of fossil- and bio-based polyurethane foams: A review. J. Clean. Prod. 2023, 430, 139697. [Google Scholar] [CrossRef]
- Wiatrowski, M.; Tan, E.C.D.; Davis, R. TEA and LCA of bio-based polyurethanes. In Rethinking Polyester Polyurethanes: Algae Based Renewable, Sustainable, Biodegradable and Recyclable Materials; Elsevier: Amsterdam, The Netherlands, 2023; pp. 153–176. [Google Scholar] [CrossRef]
- Zhou, C.H.; Beltramini, J.N.; Lu, G.Q. Chemoselective catalytic conversion of glycerol as a biorenewable source to valuable commodity chemicals. Chem. Soc. Rev. 2008, 37, 527–549. [Google Scholar] [CrossRef] [PubMed]
- Pagliaro, M.; Rossi, M. The Future of Glycerol: New Usages for a Versatile Raw Material; RSC Publishing: Cambridge, UK, 2008; 127p. [Google Scholar] [CrossRef]
- Zheng, Y.; Chen, X.; Shen, Y. Commodity Chemicals Derived from Glycerol, an Important Biorefinery Feedstock. Chem. Rev. 2008, 110, 1807. [Google Scholar] [CrossRef] [PubMed]
- Ozorio, L.P.; Pianzolli, R.; Da Cruz MacHado, L.; Miranda, J.L.; Turci, C.C.; Guerra, A.C.O.; Souza-Aguiar, E.F.; Mota, C.J.A. Metal-impregnated zeolite Y as efficient catalyst for the direct carbonation of glycerol with CO2. Appl. Catal. A Gen. 2015, 504, 187–191. [Google Scholar] [CrossRef]
- Dibenedetto, A.; Pastore, C.; Aresta, M. Direct carboxylation of alcohols to organic carbonates: Comparison of the Group 5 element alkoxides catalytic activity. An insight into the reaction mechanism and its key steps. Catal. Today 2006, 115, 88–94. [Google Scholar] [CrossRef]
- Aresta, M.; Dibenedetto, A.; Nocito, F.; Pastore, C. A study on the carboxylation of glycerol to glycerol carbonate with carbon dioxide: The role of the catalyst, solvent and reaction conditions. J. Mol. Catal. A Chem. 2006, 257, 149–153. [Google Scholar] [CrossRef]
- Hammond, C.; Lopez-Sanchez, J.A.; Hasbi Ab Rahim, M.; Dimitratos, N.; Jenkins, R.L.; Carley, A.F.; He, Q.; Kiely, C.J.; Knight, D.W.; Hutchings, G.J. Synthesis of glycerol carbonate from glycerol and urea with gold-based catalysts. Dalton Trans. 2011, 40, 3927–3937. [Google Scholar] [CrossRef]
- Yoo, J.W.; Mouloungui, Z. Catalytic carbonylation of glycerin by urea in the presence of zinc mesoporous system for the synthesis of glycerol carbonate. In Studies in Surface Science and Catalysis; Elsevier Inc.: Amsterdam, The Netherlands, 2003; Volume 146, pp. 757–760. [Google Scholar] [CrossRef]
- Wang, L.; Ma, Y.; Wang, Y.; Liu, S.; Deng, Y. Efficient synthesis of glycerol carbonate from glycerol and urea with lanthanum oxide as a solid base catalyst. Catal. Commun. 2011, 12, 1458–1462. [Google Scholar] [CrossRef]
- Christy, S.; Noschese, A.; Lomelí-Rodriguez, M.; Greeves, N.; Lopez-Sanchez, J.A. Recent progress in the synthesis and applications of glycerol carbonate. Curr. Opin. Green Sustain. Chem. 2018, 14, 99–107. [Google Scholar] [CrossRef]
- Ahamad, A.; Singh, P.; Tiwary, D. Plastic and Microplastic in the Environment: Management and Health Risks; Wiley: Hoboken, NJ, USA, 2022; pp. 1–299. [Google Scholar] [CrossRef]
- Kumar, A.; Thakur, V.K.; Nezhad, H.Y.; Lee, K.S. Prospects of sustainable polymers. Sci. Rep. 2024, 14, 9430. [Google Scholar] [CrossRef]
- Kosloski-Oh, S.C.; Fieser, M.E. From source to disposal: Cracking the code for sustainable polymers. One Earth 2023, 6, 587–590. [Google Scholar] [CrossRef]
- Zeng, X.; Chen, Q.; Zhao, C.; Xie, S.; Xie, H.; Huang, C. Eugenol-derived Organic Liquids as an In-Situ CO2 Capturing and Conversion System for Eugenol-based Polycarbonate Synthesis. Chem.-Asian J. 2022, 17, e202200503. [Google Scholar] [CrossRef] [PubMed]
- Torres-Giner, S. Sustainable Polymer Technologies for a Circular Economy. Appl. Sci. 2023, 13, 5864. [Google Scholar] [CrossRef]
- Sadeghi, K.; Jeon, Y.; Seo, J. Roadmap to the sustainable synthesis of polymers: From the perspective of CO2 upcycling. Prog. Mater. Sci. 2023, 135, 101103. [Google Scholar] [CrossRef]
- Mohanty, A.K.; Wu, F.; Mincheva, R.; Hakkarainen, M.; Raquez, J.M.; Mielewski, D.F.; Narayan, R.; Netravali, A.N.; Misra, M. Sustainable polymers. Nat. Rev. Methods Primers 2022, 2, 46. [Google Scholar] [CrossRef]
- Okolie, O.; Kumar, A.; Edwards, C.; Lawton, L.A.; Oke, A.; McDonald, S.; Thakur, V.K.; Njuguna, J. Bio-Based Sustainable Polymers and Materials: From Processing to Biodegradation. J. Compos. Sci. 2023, 7, 213. [Google Scholar] [CrossRef]
- Alper, E.; Yuksel Orhan, O. CO2 utilization: Developments in conversion processes. Petroleum 2017, 3, 109–126. [Google Scholar] [CrossRef]
- Elmas, S.; Subhani, M.A.; Leitner, W.; Müller, T.E. Anion effect controlling the selectivity in the zinc-catalysed copolymerisation of CO2 and cyclohexene oxide. Beilstein J. Org. Chem. 2015, 11, 42–49. [Google Scholar] [CrossRef]
- Leitner, W.; Müller, T.E.; Gürtler, C. How can we put the climate killer CO2 to good use? In Annual Report: Science for a Better Life; Bayer: Leverkusen, Germany, 2010; pp. 32–37. [Google Scholar]
- Gütlbauer, F.; Getoff, N. Radiation chemical carboxylation of hydroxycompounds. Int. J. Appl. Radiat. Isot. 1965, 16, 673–680. [Google Scholar] [CrossRef]
- Krapfenbauer, K.F.; Getoff, N. Radiation-and photo-induced formation of salicylic acid from phenol and CO2 in aqueous solution. World Resour. Rev. 1997, 9, 421–433. [Google Scholar]
- Getoff, N. Radiation chemistry and the environment. Radiat. Phys. Chem. 1999, 54, 377–384. [Google Scholar] [CrossRef]
- Getoff, N.; Solar, S.; Quint, R.M. One-electron oxidation of mitomycin C and its corresponding peroxyl radicals. A steady-state and pulse radiolysis study. Radiat. Phys. Chem. 1997, 50, 575–583. [Google Scholar] [CrossRef]
- Getoff, N.; Solar, S.; Richter, U.B.; Haenel, M.W. Pulse radiolysis of pyrene in aprotic polar organic solvents: Simultaneous formation of pyrene radical cations and radical anions. Radiat. Phys. Chem. 2003, 66, 207–214. [Google Scholar] [CrossRef]
- Getoff, N.; Schenck, G.O. On the formation of amino acids by gamma-ray-induced carboxylation of amines in aqueous solutions. Radiat. Res. 1967, 31, 486–505. [Google Scholar] [CrossRef] [PubMed]
- Fjodorov, V.V.; Getoff, N. Radiation induced carboxylation of methanol under elevated CO2-pressure. Radiat. Phys. Chem. 1983, 22, 841–848. [Google Scholar] [CrossRef]
- Getoff, N.; Gütlbauer, F.; Schenck, G.O. Radiation-chemical carboxylation of formic acid and methanol in aqueous solution. Int. J. Appl. Radiat. Isot. 1966, 17, 341–349. [Google Scholar] [CrossRef]
- Caputo, G.; Galia, A.; Scrò, F.; Spadaro, G.; Filardo, G. Gamma radiation induced polymerization of vinyl monomers in dense CO2. Radiat. Phys. Chem. 2002, 63, 45–51. [Google Scholar] [CrossRef]
- Hayashi, K. Radiation and photo-induced ionic polymerization. Polym. J. 1980, 12, 571–581. [Google Scholar] [CrossRef][Green Version]
- Bartoníček, B. Gamma radiolysis of solid succinic acid. Int. J. Radiat. Phys. Chem. 1973, 5, 361–369. [Google Scholar] [CrossRef]
- Wang, W.; Chang, Z.; Wang, M.; Zhang, Z. Effect of carboxyl on vulcanization and mechanical properties of carboxylated acrylic rubber prepared by 60Co-γ-ray-induced polymerization. J. Appl. Polym. Sci. 2006, 102, 5587–5594. [Google Scholar] [CrossRef]
- Dispenza, C.; Filardo, G.; Silvestri, G.; Spadaro, G. Carboxylation of linear low density polyethylene through gamma irradiation in presence of supercritical carbon dioxide. Grafted groups analysis via derivatization procedures. Colloid Polym. Sci. 1997, 275, 390–395. [Google Scholar] [CrossRef]
- Spadaro, G.; De Gregorio, R.; Galia, A.; Valenza, A.; Filardo, G. Gamma radiation induced maleation of polypropylene using supercritical CO2: Preliminary results. Polymer 2000, 41, 3491–3494. [Google Scholar] [CrossRef]
- Clean Energy Technologies. Canada’s CO2 Capture & Storage Technology Roadmap; CANMET Energy Technology Centre: Devon, AL, Canada, 2006; 89p, Available online: https://natural-resources.canada.ca/our-natural-resources/energy-sources-distribution/carbon-management/canadas-co2-capture-and-storage-technology-roadmap/4327 (accessed on 29 June 2022).
- Zhang, Y. Progress in carbon dioxide sequestration by mineral carbonation. CIESC J. 2007, 58, 1. [Google Scholar]
- Sanna, A.; Uibu, M.; Caramanna, G.; Kuusik, R.; Maroto-Valer, M.M. A review of mineral carbonation technologies to sequester CO2. Chem. Soc. Rev. 2014, 43, 8049–8080. [Google Scholar] [CrossRef] [PubMed]
- Delgado Torróntegui, M. Assessing the Mineral Carbonation Science and Technology; Swiss Federal Institute of Technology, ETH: Zurich, Switzerland, 2010. [Google Scholar]
- Romanov, V.; Soong, Y.; Carney, C.; Rush, G.E.; Nielsen, B.; O’Connor, W. Mineralization of Carbon Dioxide: A Literature Review. ChemBioEng Rev. 2015, 2, 231–256. [Google Scholar] [CrossRef]
- Aminu, M.D.; Nabavi, S.A.; Rochelle, C.A.; Manovic, V. A review of developments in carbon dioxide storage. Appl. Energy 2017, 208, 1389–1419. [Google Scholar] [CrossRef]
- Wang, B.; Pan, Z.; Cheng, H.; Zhang, Z.; Cheng, F. A review of carbon dioxide sequestration by mineral carbonation of industrial byproduct gypsum. J. Clean. Prod. 2021, 302, 126930. [Google Scholar] [CrossRef]
- Bocin Dumitriu, A.; Perez Fortes, M.D.M.; Tzimas, E.; Sveen, T. Carbon Capture and Utilisation Workshop: Background and Proceedings; JRC Publications: Luxembourg, 2013; 77p, Available online: https://publications.jrc.ec.europa.eu/repository/handle/JRC86324 (accessed on 22 July 2021).
- Romão, I.S.S. Production of Magnesium Carbonates from Serpentinites for CO2 Mineral Sequestration-Optimisation towards Industrial Application. Ph.D. Thesis, Abo Akademi University, Turku, Findland, 2015. [Google Scholar]
- Romão, I.S.; Gando-Ferreira, L.M.; Da Silva, M.M.V.G.; Zevenhoven, R. CO2 sequestration with serpentinite and metaperidotite from Northeast Portugal. Miner. Eng. 2016, 94, 104–114. [Google Scholar] [CrossRef]
- Zevenhoven, R.; Fagerlund, J.; Nduagu, E.; Romão, I.; Jie, B.; Highfield, J. Carbon storage by mineralisation (CSM): Serpentinite rock carbonation via Mg(OH)2 reaction intermediate without CO2 pre-separation. In Proceedings of the 11th International Conference on Greenhouse Gas Control Technologies, GHGT 2012, Kyoto, Japan, 18–22 November 2012; pp. 5945–5954. [Google Scholar] [CrossRef]
- Huijgen, W.J.J.; Ruijg, G.J.; Comans, R.N.J.; Witkamp, G.J. Energy consumption and net CO2 sequestration of aqueous mineral carbonation. Ind. Eng. Chem. Res. 2006, 45, 9184–9194. [Google Scholar] [CrossRef]
- Santos, R.M.; Verbeeck, W.; Knops, P.; Rijnsburger, K.; Pontikes, Y.; Van Gerven, T. Integrated mineral carbonation reactor technology for sustainable carbon dioxide sequestration: ‘CO2 Energy Reactor’. In Proceedings of the 11th International Conference on Greenhouse Gas Control Technologies, GHGT 2012, Kyoto, Japan, 18–22 November 2012; pp. 5884–5891. [Google Scholar] [CrossRef]
- Lackner, K.S.; Wendt, C.H.; Butt, D.P.; Joyce Jr, E.L.; Sharp, D.H. Carbon dioxide disposal in carbonate minerals. Energy 1995, 20, 1153–1170. [Google Scholar] [CrossRef]
- Chen, Z.Y.; O’Connor, W.K.; Gerdemann, S.J. Chemistry of aqueous mineral carbonation for carbon sequestration and explanation of experimental results. Environ. Prog. 2006, 25, 161–166. [Google Scholar] [CrossRef]
- O’Connor, W.K.; Dahlin, D.C.; Rush, G.E.; Gerdemann, S.J.; Penner, L.R. Energy and economic evaluation of ex situ aqueous mineral carbonation. In Greenhouse Gas Control Technologies; Elsevier Ltd.: Amsterdam, The Netherlands, 2005; pp. 2011–2015. [Google Scholar] [CrossRef]
- Gerdemann, S.J.; O’Connor, W.K.; Dahlin, D.C.; Penner, L.R.; Rush, H. Ex situ aqueous mineral carbonation. Environ. Sci. Technol. 2007, 41, 2587–2593. [Google Scholar] [CrossRef] [PubMed]
- Benhelal, E.; Rashid, M.I.; Rayson, M.S.; Brent, G.F.; Oliver, T.; Stockenhuber, M.; Kennedy, E.M. Direct aqueous carbonation of heat activated serpentine: Discovery of undesirable side reactions reducing process efficiency. Appl. Energy 2019, 242, 1369–1382. [Google Scholar] [CrossRef]
- Fernandez, A.; West, K. Technology Roadmap—Low-Carbon Transition in the Cement Industry; International Energy Agency: Paris, France, 2018; 62p. [Google Scholar]
- Olajire, A.A. A review of mineral carbonation technology in sequestration of CO2. J. Pet. Sci. Eng. 2013, 109, 364–392. [Google Scholar] [CrossRef]
- Zevenhoven, R.; Romão, I.; Slotte, M. Mineralisation of CO2 using serpentinite rock—Towards industrial application. In Proceedings of the IEAGHG/IETS Iron & Steel Industry CCUS & Process Integration Workshop, Tokyo, Japan, 5–7 November 2013; 26p. Available online: https://ieaghg.org/docs/General_Docs/Iron%20and%20Steel%202%220Secured%220presentations/203_1520%1520Mikko%1520Helle.pdf (accessed on 13 July 2024).
- Doucet, F.J. Scoping Study on CO2 Mineralization Technologies; CGS-2011-007 Report; CGS: Pretoria, South Africa, 2001; 88p, Available online: https://www.academia.edu/4061042/Scoping_study_on_CO2_mineralization_technologies (accessed on 13 July 2024).
- Hemmati, A.; Shayegan, J.; Bu, J.; Yeo, T.Y.; Sharratt, P. Process optimization for mineral carbonation in aqueous phase. Int. J. Miner. Process. 2014, 130, 20–27. [Google Scholar] [CrossRef]
- Priestnall, M. Making Money from Mineralisation of CO2. Carbon Capture J. 3 February 2013. 3p. Available online: https://www.carboncapturejournal.com/news/making-money-from-mineralisation-of-co2/3251.aspx?Category=a#:~:text=Mineral%20carbonation%20brings%20opportunities%20particularly%20to%20the,wider%20industrial%20and%20power%20generation%20CCS%20markets.d (accessed on 29 June 2022).
- Zevenhoven, R. Metals production, CO2 mineralization and LCA. Metals 2020, 10, 16. [Google Scholar] [CrossRef]
- Priestnall, M. Method and System of Sequestrating Carbon Dioxide. Patent No. GB2515995, 14 January 2015. [Google Scholar]
- Priestnall, M. Method and System of Activation of Mineral Silicate Minerals. Patent No. US9963351, 8 May 2018. Also published as CN106573197, DK3129125, EP3129125, ES2824676, GB2516141, PL3129125, US2017029284, WO2015154887. [Google Scholar]
- Priestnall, M. Can Mineral Carbonation Be Used for Industrial Carbon Dioxide Sequestration? Environmental Chemistry Group Buletin. 2014. 3p. Available online: https://www.envchemgroup.com/can-mineral-carbonation-be-used-for-industrial-co2-sequestration.html (accessed on 29 June 2022).
- Priestnall, M. Decarbonising flue gas using CO2 mineralisation—Project experience on ships. In Proceedings of the Keeping the Momentum; Geological Society: London, UK, 2014; 9p. [Google Scholar]
- Priestnall, M. CO2 mineralisation via fuel cells. How to make CCS profitable. In Proceedings of the Finding Petroleum CCS Forum; Geological Society: London, UK, 2010; 14p; Available online: http://www.findingpetroleum.com/files/event15/ccc.pdf (accessed on 29 June 2022).
- Evans, M. Programme and Business Plan. In Direct Air CO2 Capture & Mineralisation; Cambridge Carbon Capture: Cambridge, UK, 2022; Chapter 4; pp. 14–18. Available online: https://assets.publishing.service.gov.uk/media/6281f0cd8fa8f5561cd9b1b7/cambridge-carbon-cam202029.pdf (accessed on 29 June 2022).
- Fernandez Pales, A.; Remme, U.; Berly, T.; McCulloch, S.; Stanley, T.; Vass, T.; Teter, J.; Abergel, T. Exploring Clean Energy Pathways: The Role of CO2 Storage; International Energy Agency (IEA): Paris, France, 2019; 105p. [Google Scholar] [CrossRef]
- Zhang, J.; Shi, C.; Li, Y.; Pan, X.; Poon, C.-S.; Xie, Z. Influence of carbonated recycled concrete aggregate on properties of cement mortar. Constr. Build. Mater. 2015, 98, 1–7. [Google Scholar] [CrossRef]
- Alberici, S.; Noothout, P.; Mir, G.U.R.; Stork, M.; Wiersma, F.; Mac Dowell, N.; Shah, N.; Fennell, P. Assessing the Potential of CO2 Utilisation in the UK; Gardiner, A., Ed.; Final Report SISUK17099; 2017; 138p. Available online: https://assets.publishing.service.gov.uk/media/5cc9bd9c40f0b64c1e187664/SISUK17099AssessingCO2_utilisationUK_ReportFinal_260517v2__1_.pdf (accessed on 25 May 2024).
- O’Connor, W.K.; Dahlin, D.C.; Rush, G.E.; Dahlin, C.L.; Collins, W.K. Carbon dioxide sequestration by direct mineral carbonation: Process mineralogy of feed and products. Miner. Metall. Process. 2002, 19, 95–101. [Google Scholar] [CrossRef]
- O’Connor, W.K.; Dahlin, D.C.; Rush, G.E.; Gedermann, S.J.; Penner, L.R.; Nilsen, D.N. Aqueous Mineral Carbonation; DOE/ARC-TR-04-002; National Energy Technology Laboratory: Albany, OR, USA, 2005; 463p.
- O’Connor, W.K.; Dahlin, D.C.; Turner, P.C.; Walters, R.P. Carbon dioxide sequestration by ex-situ mineral carbonation. Technology 1999, 7, 115–123. [Google Scholar]
- Teir, S.; Eloneva, S.; Revitzer, H.; Zevenhoven, R.; Salminen, J.; Fogelholm, C.-J.; Poylio, E. Method of Producing Calcium Carbonate from Waste and by-Products. Patent No. US8603428, 10 December 2013. [Google Scholar]
- Batuecas, E.; Liendo, F.; Tommasi, T.; Bensaid, S.; Deorsola, F.A.; Fino, D. Recycling CO2 from flue gas for CaCO3 nanoparticles production as cement filler: A Life Cycle Assessment. J. CO2 Util. 2021, 45, 101446. [Google Scholar] [CrossRef]
- Khoo, Z.Y.; Ho, E.H.Z.; Li, Y.; Yeo, Z.; Low, J.S.C.; Bu, J.; Chia, L.S.O. Life cycle assessment of a CO2 mineralisation technology for carbon capture and utilisation in Singapore. J. CO2 Util. 2021, 44, 101378. [Google Scholar] [CrossRef]
- Zhuang, Q.; Clements, B.; Li, Y. From ammonium bicarbonate fertilizer production process to power plant CO2 capture. Int. J. Greenh. Gas Control 2012, 10, 56–63. [Google Scholar] [CrossRef]
- Dowsett, M.R.; Lewis, C.M.; North, M.; Parkin, A. Exploring the scope of capacitance-assisted electrochemical carbon dioxide capture. Dalton Trans. 2018, 47, 10447–10452. [Google Scholar] [CrossRef] [PubMed]
- Lamb, K.J.; Dowsett, M.R.; Chatzipanagis, K.; Scullion, Z.W.; Kröger, R.; Lee, J.D.; Aguiar, P.M.; North, M.; Parkin, A. Capacitance-Assisted Sustainable Electrochemical Carbon Dioxide Mineralisation. ChemSusChem 2018, 11, 137–148. [Google Scholar] [CrossRef]
- Balucan, R.D.; Dlugogorski, B.Z.; Kennedy, E.M.; Belova, I.V.; Murch, G.E. Energy cost of heat activating serpentinites for CO2 storage by mineralisation. Int. J. Greenh. Gas Control 2013, 17, 225–239. [Google Scholar] [CrossRef]
- Britten, R. System for Halting the Increase in Atmospheric Carbon Dioxide and Method of Operation Thereof. Patent No. US2011171107, 14 July 2011. Abandoned. [Google Scholar]
- Jones, J.D.; St Angelo, D. Removing Carbon Dioxide from Waste Streams through Co-Generation of Carbonate and/or Bicarbonate Minerals. Patent No. US9205375, 8 December 2015. Also published as AT543562, AU2008302064, BRPI0817116, CA2930096, CA2699572, CN101970084, EP2205341, ES2386060, GEP20135757, HK1143107, JP2010540214, JP2013066887, JP2015186801, JP6110103, JP6145129, KR101485854, KR20100101069, PL2205341, RU2010115481, RU2012144036, RU2477168, RU2531827, US2009127127, WO2009039445, ZA201001932. [Google Scholar]
- Sano, K.; Saito, H.; Miyamoto, H.; Imada, T. Carbon Dioxide Fixation Method and Carbon Dioxide Fixation System. Patent No. US2022297058, 22 September 2022. Also published as JP2022145080. [Google Scholar]
- Sano, K.; Saito, H.; Miyamoto, H.; Suzuki, A. Carbon Dioxide Fixation System and Method Using Seawater Electrolysis. Patent No. US2023051291, 16 February 2023. Also published as JP2023025798. [Google Scholar]
- Idir, R.; Cyr, M.; Tagnit-Hamou, A. Use of waste glass in cement-based materials. Environ. Ingénierie Développement (Déchets Sci. Et Tech.) 2015, 57, 7. [Google Scholar] [CrossRef]
- Glavind, M. Sustainability of cement, concrete and cement replacement materials in construction. In Sustainability of Construction Materials; Elsevier Ltd.: Amsterdam, The Netherlands, 2009; pp. 120–147. [Google Scholar] [CrossRef]
- Riley, B.J.; McFarlane, J.; DelCul, G.D.; Vienna, J.D.; Contescu, C.I.; Forsberg, C.W. Molten salt reactor waste and effluent management strategies: A review. Nucl. Eng. Des. 2019, 345, 94–109. [Google Scholar] [CrossRef]
- Was, G.S.; Petti, D.; Ukai, S.; Zinkle, S. Materials for future nuclear energy systems. J. Nucl. Mater. 2019, 527, 151837. [Google Scholar] [CrossRef]
- Mignacca, B.; Locatelli, G. Economics and finance of Molten Salt Reactors. Prog. Nucl. Energy 2020, 129, 103503. [Google Scholar] [CrossRef]
- Roper, R.; Harkema, M.; Sabharwall, P.; Riddle, C.; Chisholm, B.; Day, B.; Marotta, P. Molten salt for advanced energy applications: A review. Ann. Nucl. Energy 2022, 169, 108924. [Google Scholar] [CrossRef]
- Wu, J.; Chen, J.; Cai, X.; Zou, C.; Yu, C.; Cui, Y.; Zhang, A.; Zhao, H. A Review of Molten Salt Reactor Multi-Physics Coupling Models and Development Prospects. Energies 2022, 15, 8296. [Google Scholar] [CrossRef]
- Rehm, T.E. Advanced nuclear energy: The safest and most renewable clean energy. Curr. Opin. Chem. Eng. 2023, 39, 100878. [Google Scholar] [CrossRef]
- Wilson, J.F.; Srinivasan, S.S.; Moore, B.M.; Henderson, L.; Iii, S.E.; Sharma, P.C. Hydrogen Production Using Nuclear Energy; NP-T-4.2; International Atomic Energy Agency: Vienna, Austria, 2013; 400p. [Google Scholar]
- Kugeler, K.; Schulten, R.; Kugeler, M.; Niessen, H.F.; Roeth-Kamat, M.; Hohn, H.; Woike, O.; Germer, J.H. The Pebble-Bed High-Temperature Reactor as a Source of Nuclear Process Heat. Julich, Germany. 1974, Volume 4. 84p. Available online: http://inis.iaea.org/search/search.aspx?orig_q=RN:07275833 (accessed on 8 December 2018).
- Singh, J.; Niessen, H.F.; Harth, R.; Fedders, H.; Reutler, H.; Panknin, W.; Müller, W.D.; Harms, H.G. The nuclear heated steam reformer—Design and semitechnical operating experiences. Nucl. Eng. Des. 1984, 78, 179–194. [Google Scholar] [CrossRef]
- Niessen, H.F.; Bhattacharyya, A.T.; Busch, M.; Hesse, K.; Zentis, A. Erprobung und Versuchsergebnisse des PNP-Teströhrenspaltofens in der EVA II-Anlage [Test and Results of the Test Tubes, Chemical Heat Pipe PNP in Eva II-Anlage]; Juel-2231/FZJ-2014-04914; Kernforschungsanlage Jülich GmbH; Zentralbiliothek Verlag: Jülich, Germany, 1988. [Google Scholar]
- Harth, R.; Jansing, W.; Teubner, H. Experience gained from the EVA II and KVK operation. Nucl. Eng. Des. 1990, 121, 173–182. [Google Scholar] [CrossRef]
- Kugeler, K.; Kugeler, M.; Niessen, H.F.; Hammelmann, K.H. Steam reformers heated by helium from high temperature reactors. Nucl. Eng. Des. 1975, 34, 129–145. [Google Scholar] [CrossRef]
- Fedders, H.; Harth, R.; Höhlein, B. Experiments for combining nuclear heat with the methane steam-reforming process. Nucl. Eng. Des. 1975, 34, 119–127. [Google Scholar] [CrossRef]
- Harth, R.; Jansing, W.; Mauersberger, R.; Teubner, H. Status of High-Temperature Reactor Component Development for Process Heat Generation. Atomkernenerg. Kerntech. 1985, 47, 176–183. [Google Scholar]
- Dumm, K.; Theymann, W.; Der Decken, C.B.V. Design and qualification of components for high-temperature reactors. Nucl. Eng. Des. 1990, 121, 193–198. [Google Scholar] [CrossRef]
- Höhlein, B.; Niessen, H.; Range, J.; Schiebahn, H.J.R.; Vorwerk, M. Methane from synthesis gas and operation of high-temperature methanation. Nucl. Eng. Des. 1984, 78, 241–250. [Google Scholar] [CrossRef]
- Kugeler, K.; Niessen, H.F.; Röth-Kamat, M.; Böcker, D.; Rüter, B.; Theis, K.A. Transport of nuclear heat by means of chemical energy (nuclear long-distance energy). Nucl. Eng. Des. 1975, 34, 65–72. [Google Scholar] [CrossRef]
- Fedders, H.; Höhlein, B. Operating a pilot plant circuit for energy transport with hydrogen-rich gas. Int. J. Hydrogen Energy 1982, 7, 793–800. [Google Scholar] [CrossRef]
- Boltendahl, U.; Harth, R. Wärmetransport auf kaltem Wege [Transport of Heat Energy using Cold Gases]. Nukleare Fernenergie, Projekt ADAM und EVA. Bild Der Wiss. 1980, 17, 44–55. [Google Scholar]
- Kesten, A.S.; Boedeker, L.R.; Couch, H.T. A self-driven chemical heat pipe. J. Heat Recovery Syst. 1983, 3, 311–319. [Google Scholar] [CrossRef]
- Nuernberg, H.W.; Wolff, G. Energy Conversion Method. Patent No. US3558047, 26 January 1971. [Google Scholar]
- Kugeler, K.; Kugeler, M.; Niessen, H.F.; Hohn, H. Considerations on high temperature reactors for process heat applications. Nucl. Eng. Des. 1975, 34, 15–32. [Google Scholar] [CrossRef]
- Inagaki, Y.; Hino, R.; Hada, K.; Haga, K.; Nishihara, T.; Takeda, T.; Shiozawa, S. Out-of-pile demonstration test program of HTTR hydrogen production system by steam reforming of natural gas. In Proceedings of the 5th International Conference on Nuclear Engineering, ICONE5, Nice, France, 26–30 May 1997; p. 172. [Google Scholar]
- Takeda, T.; Iwatsuki, J. Counter-permeation of deuterium and hydrogen through INCONEL 600®. Nucl. Technol. 2004, 146, 83–95. [Google Scholar] [CrossRef]
- Hishida, M.; Nekoya, S.; Takizuka, T.; Emori, K.; Ogawa, M.; Ouchi, M.; Okamoto, Y.; Sanokawa, K.; Nakano, T.; Hagiwara, T.; et al. Construction and Performance Tests of Hydrogen Secondary Cooling System, (I) Facility and Performance of Components. Nippon Genshiryoku Gakkaishi/J. At. Energy Soc. Jpn. 1980, 22, 181–188. [Google Scholar] [CrossRef]
- Hishida, M.; Takizuka, T.; Ogawa, M.; Nekoya, S.; Emori, K.; Ouchi, M.; Sanokawa, K. Construction and Performance Tests of Hydrogen Secondary Cooling System, (II) Results of Performance Tests. Nippon Genshiryoku Gakkaishi/J. At. Energy Soc. Jpn. 1980, 22, 326–334. [Google Scholar] [CrossRef]
- Tochio, D.; Shimizu, A.; Hamamoto, S.; Sakaba, N. Helium leak and chemical impurities control technology in HTTR. J. Nucl. Sci. Technol. 2014, 51, 1407–1412. [Google Scholar] [CrossRef][Green Version]
- Nishihara, T.; Sakaki, A.; Inagaki, Y.; Takami, K. Development of high temperature isolation valve for the HTTR hydrogen production system. Trans. At. Energy Soc. Jpn. 2004, 3, 381–387. [Google Scholar] [CrossRef]
- Inaba, Y.; Nishihara, T.; Nitta, Y. Analytical study on fire and explosion accidents assumed in HTGR hydrogen production system. Nucl. Technol. 2004, 146, 49–57. [Google Scholar] [CrossRef]
- Inaba, Y.; Nishihara, T.; Groethe, M.A.; Nitta, Y. Study on explosion characteristics of natural gas and methane in semi-open space for the HTTR hydrogen production system. Nucl. Eng. Des. 2004, 232, 111–119. [Google Scholar] [CrossRef]
- Verfondern, K.; Nishihara, T. Safety aspects of the combined HTTR/steam reforming complex for nuclear hyrogen production. Prog. Nucl. Energy 2005, 47, 527–534. [Google Scholar] [CrossRef]
- Inaba, Y.; Ohashi, H.; Nishihara, T.; Sato, H.; Inagaki, Y.; Takeda, T.; Hayashi, K.; Takada, S. Study on control characteristics for HTTR hydrogen production system with mock-up test facility System controllability test for fluctuation of chemical reaction. Nucl. Eng. Des. 2005, 235, 111–121. [Google Scholar] [CrossRef]
- Hada, K.; Nishihara, T.; Shibata, T.; Shiozawa, S. Design of a steam reforming system to be connected to the HTTR. In Proceedings of the Third JAERI Symposium on HTGR Technologies, Oarai, Japan, 15–16 February 1996; pp. 229–240. [Google Scholar]
- Nishihara, T.; Shimizu, A.; Inagaki, Y.; Tanihira, M. Flow Scheme and Controllability of the HTTR Hydrogen Production System. Trans. At. Energy Soc. Jpn. 2003, 2, 517–524. [Google Scholar] [CrossRef][Green Version]
- Tanaka, T.; Baba, O.; Shiozawa, S.; Okubo, M.; Tobioka, T. Present status of HTTR construction and its testing program. In Proceedings of the JAERI seminar on HTGR technologies, Tokai, Japan, 7–8 November 1994. 26p. [Google Scholar]
- Ohashi, H.; Inaba, Y.; Nishihara, T.; Inagaki, Y.; Takeda, T.; Hayashi, K.; Katanishi, S.; Takada, S.; Ogawa, M.; Shiozawa, S. Performance test results of mock-up test facility of httr hydrogen production system. J. Nucl. Sci. Technol. 2004, 41, 385–392. [Google Scholar] [CrossRef]
- Ohashi, H.; Inaba, Y.; Nishihara, T.; Takeda, T.; Hayashi, K.; Takada, S.; Inagaki, Y. Development of control technology for HTTR hydrogen production system with mock-up test facility. System controllability test for loss of chemical reaction. Nucl. Eng. Des. 2006, 236, 1396–1410. [Google Scholar] [CrossRef]
- Inagaki, Y.; Ohashi, H.; Inaba, Y.; Sato, H.; Nishihara, T.; Takeda, T.; Hayashi, K.; Ogawa, M. Research and development on system integration technology for connection of hydrogen production systems to an HTGR. Nucl. Technol. 2007, 157, 111–119. [Google Scholar] [CrossRef]
- Onuki, K.; Inagaki, Y.; Hino, R.; Tachibana, Y. Research and development on nuclear hydrogen production using HTGR at JAERI. Prog. Nucl. Energy 2005, 47, 496–503. [Google Scholar] [CrossRef]
- Karasawa, H.; Shiina, K.; Sasahira, A.; Hoshino, K. Storage and hydrogen production usage of recycling material in the flexible fuel cycle initiative. In Proceedings of the International Conference on Nuclear Energy Systems for Future Generation and Global Sustainability, Tsukuba, Japan, 9–13 October 2005. 6p. [Google Scholar]
- Icerman, L. Open-loop chemical heat pipes. Energy 1979, 4, 1187–1188. [Google Scholar] [CrossRef]
- Lee, M.-Y.; Park, K.T.; Lee, W.; Lim, H.; Kwon, Y.; Kang, S. Current achievements and the future direction of electrochemical CO2 reduction: A short review. Crit. Rev. Environ. Sci. Technol. 2020, 50, 769–815. [Google Scholar] [CrossRef]
- Burdyny, T.; Smith, W.A. CO2 reduction on gas-diffusion electrodes and why catalytic performance must be assessed at commercially-relevant conditions. Energy Environ. Sci. 2019, 12, 1442–1453. [Google Scholar] [CrossRef]
- Jensen, S.H.; Larsen, P.H.; Mogensen, M. Hydrogen and synthetic fuel production from renewable energy sources. Int. J. Hydrogen Energy 2007, 32, 3253–3257. [Google Scholar] [CrossRef]
- Fujiwara, S.; Kasai, S.; Yamauchi, H.; Yamada, K.; Makino, S.; Matsunaga, K.; Yoshino, M.; Kameda, T.; Ogawa, T.; Momma, S.; et al. Hydrogen production by high temperature electrolysis with nuclear reactor. Prog. Nucl. Energy 2008, 50, 422–426. [Google Scholar] [CrossRef]
- Kasai, S.; Fujiwara, S.; Yamada, K.; Ogawa, T.; Matsunaga, K.; Yoshino, M.; Hoashp, E.; Makino, S. Nuclear hydrogen production by high-temperature electrolysis. Trans. At. Energy Soc. Jpn. 2009, 8, 122–141. [Google Scholar] [CrossRef]
- Chang, J. Status of nuclear hydrogen production technology development project in Korea. In Proceedings of the International Conference on Non-Electric Applications of Nuclear Power: Seawater Desalination, Hydrogen Production and Other Industrial Applications, Oarai, Japan, 16–19 April 2007; pp. 130–132. [Google Scholar]
- International Atomic Energy Agency. Non-electric applications of nuclear power: Seawater desalination, hydrogen production and other industrial applications. In Proceedings of the International Conference on Non-Electric Applications of Nuclear Power, Oarai, Japan, 16–19 April 2007. [Google Scholar]
- Bents, D.J.; Scullin, V.J.; Chang, B.J.; Johnson, D.W.; Garcia, C.P.; Jakupca, I.J. Hydrogen-oxygen PEM regenerative fuel cell development at Nasa Glenn Research Center. Fuel Cells Bull. 2006, 2006, 12–14. [Google Scholar] [CrossRef]
- Sohal, M.S.; O’Brien, J.E.; Stoots, C.M.; Sharma, V.I.; Yildiz, B.; Virkar, A. Degradation issues in solid oxide cells during high temperature electrolysis. J. Fuel Cell Sci. Technol. 2012, 9, 011017. [Google Scholar] [CrossRef]
- Zhang, X.; O’Brien, J.E.; O’Brien, R.C.; Housley, G.K. Durability evaluation of reversible solid oxide cells. J. Power Sources 2013, 242, 566–574. [Google Scholar] [CrossRef]
- O’Brien, J.E.; O’Brien, R.C.; Zhang, X.; Tao, G.G.; Butler, B.J. Long-term performance of solid oxide stacks with electrode-supported cells operating in the steam electrolysis mode. In Proceedings of the ASME International Mechanical Engineering Congress and Exposition, IMECE 2011, Denver, CO, USA, 11–17 November 2011; pp. 495–503. [Google Scholar]
- Harvego, E.A.; O’Brien, J.E.; McKellar, M.G. System evaluation and life-cycle cost analysis of a commercial scale high-temperature electrolysis hydrogen production plant. In Proceedings of the ASME International Mechanical Engineering Congress and Exposition, IMECE 2012, Houston, TX, USA, 9–15 November 2012; pp. 875–884. [Google Scholar] [CrossRef]
- Schmeda-Lopez, D.; McConnaughy, T.B.; McFarland, E.W. Radiation enhanced chemical production: Improving the value proposition of nuclear power. Energy 2018, 162, 491–504. [Google Scholar] [CrossRef]
- Livingston, P. Radiation Reduction of Carbon Dioxide: A New Chemical Industry? ChemRxiv 2020, 1, 14. [Google Scholar] [CrossRef]
- LaTourrette, T.; Light, T.; Knopman, D.; Bartis, J.T. Managing Spent Nuclear Fuel. Strategy Alternatives and Policy Implications. RAND Corporation Monograph Series 2010. 95p. Available online: https://www.rand.org/content/dam/rand/pubs/monographs/2010/RAND_MG970.pdf (accessed on 23 January 2023).
- Mccullum, R. Integrated Used Fuel Management: Industry Perspectives; Rod McCullum, Nuclear Energy Institute, Nuclear Waste Technical Review Board: Las Vegas, NV, USA, 2009; 15p. Available online: https://www.nwtrb.gov/docs/default-source/meetings/2009/june/mccullum.pdf?sfvrsn=5 (accessed on 23 January 2023).
- Office of Civilian Radioactive Waste Management. The Report to the President and the Congress by the Secretary of Energy on the Need for a Second Repository; DOE/RW-0595; U.S. Department of Energy: Las Vegas, NV, USA, 10 December 2008; 15p.
- Chu, M.S.Y. Spent nuclear fuel recycling practices, technologies and impact on geological repository systems. In Geological Repository Systems for Safe Disposal of Spent Nuclear Fuels and Radioactive Waste; Elsevier Inc.: Amsterdam, The Netherlands, 2010; pp. 29–42. [Google Scholar] [CrossRef]
- Paviet, P.; Simpson, M.F. Nuclear fuel recycling. In Nuclear Engineering Handbook, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2016; pp. 387–469. [Google Scholar] [CrossRef]
- Buckner, M.R.; Burchill, W.E. The Case for Nuclear Fuel Recycling. Nuclear News. February 2016, pp. 42–48. Available online: https://www.ans.org/pubs/magazines/download/article-1011/ (accessed on 23 January 2023).
- Perkins, C.; Jovanovic, Z.; Laska, T.E. Various Methods and Apparatuses for a Radiant-Heat Driven Chemical Reactor. Patent No. US8814961, 26 August 2014. Also published as US20120241677. [Google Scholar]
- Simmons, W.W.; Perkins, C.; Jovanovic, Z.; Hilton, C.; Popp, P.; Schramm, B.J.; Turner, J.T. Various Methods and Apparatuses for Ultra-High Heat Flux Chemical Reactor. Patent No. US9011560, 21 April 2015. Also published as US2012145965, CN102985195, WO2011155962, CA2799827, AU2010355257. [Google Scholar]
- Wickham, A.J.; Best, J.V.; Wood, C.J. Recent advances in the ories of carbon dioxide radiolysis and radiolytic graphite corrosion. Radiat. Phys. Chem. 1977, 10, 107–117. [Google Scholar] [CrossRef]
- Ramirez-Corredores, M.M. Sustainable production of CO2-derived materials. NPJ Mater. Sustain. 2024, in press.





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).