Abstract
The development of a compact mechanism has made a great contribution to work on the combustion of hydrocarbon species and facilitates the investigations on chemical kinetics and computational fluid dynamics (CFD) studies. N-propylcyclohexane (NPCH) is one of the important components for jet, diesel, and gasoline fuels which needs a reliable compact reaction kinetics mechanism. This study aims to investigate the construction of a well-validated mechanism for NPCH with a simplified chemical kinetics model that delivers a good prediction ability for the key combustion parameters in a wide range of conditions (temperatures, pressures, and equivalence rates). The NPCH reaction kinetic mechanism was constructed with the aid of a coupling process, simplification process, rate modification, and a combination of standard reduction methods. The model includes a simplified sub-mechanism with 16 species and 58 reactions and a semi-detailed core mechanism with 56 species and 390 reactions. Two key parameters including ignition delay time and laminar flame speed are simulated by the use of ANSYS Chemkin-Pro. The simulation results for these parameters are validated against the available data in the literature, and the results show a good agreement compared to the experimental data over a wide range of conditions covering low to high temperatures at different pressures and equivalence ratios.
1. Introduction
Combustion is one of the phenomena which have had significant effects on the life of humankind. To fully understand the process and improve its efficiency, this phenomenon should be extensively studied. However, it is expensive and time-demanding to just investigate it empirically. So, computational combustion was utilized as a way of investigating combustion besides experimental works. Kinetic modelling is one of the significant aspects of computational combustion that has provided the opportunity of gaining a deeper understanding and knowledge about the combustion phenomena in different media. It has a great role in the improvement in the functionality of practical combustion applications, such as the engines of vehicles. With the aid of kinetic modelling, researchers can survey and analyse the fuel structure and the fundamental chemistry coupled with direct kinetic measurements of intermediate and products species and the rate constants. Fuel modelling can provide an opportunity for academic and industrial investigators to rapidly conduct their intended scientific works over a broad range of conditions while releasing them from time-consuming and expensive changes in the design of a prototype. Due to the complexity, stiffness, and the huge computational cost of using a detailed chemical reaction mechanism for large hydrocarbon fuel species, researchers utilize the approach of developing a reduced form of the detailed mechanisms. The simplification process of a detailed mechanism (reduction in the species and reactions) is based on the numbers and types of the target combustion parameters. However, the process needs to be conducted with scrutiny in order to avoid a significant loss of accuracy.
NPCH is an important hydrocarbon species that is less understood in terms of combustion chemistry, compared to other cycloalkanes. There are a limited number of investigations dedicated to NPCH chemical kinetics. Ristori et al. [1] measured species mole fractions for the oxidation of NPCH in a Jet-Stirred Reactor (JSR) at 1 bar, 950–1250 K, and an equivalence ratio range of 0.5–1.5 and developed a kinetic model for the simulation of the species concentrations measured. The proposed kinetic mechanism generally could simulate the mole fraction profile of the measured species in the oxidation of n-propylcyclohexane, and the main reactions of the oxidation of n-propylcyclohexane were determined. They reported that H-atom abstraction occurs on the saturated cycle and on the side-chain which generates seven distinct radicals, and it is responsible for the production of the major intermediate species measured in the experiments. Dubois et al. [2] investigated the high-temperature ignition characteristics of NPCH in a shock tube at 10–20 bar for a range of equivalence ratios over 1250–1800 K. The ignition delay time (IDT) measurement was based on OH* and CH* radical chemiluminescence.
The experimental data were simulated by the kinetic mechanism that they developed in the work and the work of Ristori et al. [1]. They reported that their model has better performance for the simulation of ignition delay. They also conducted an experiment to measure flame speeds in a constant volume spherical bomb at 1 bar, 403 K, and ϕ = 0.6–1.75. The result disclosed that the peak of laminar flame speed occurs around the equivalence ratio of 1.08 and decreases before and after the equivalence ratio. The simulation result for laminar flame speed showed an overestimation against the experimental data. The autoignition chemistry of NPCH has been experimentally investigated [3] in a rapid compression machine in just lean conditions over a temperature range of 620–930 K and a pressure range of 0.45–1.34 MPa. A high-temperature NPCH mechanism including 545 species and 3105 reactions was constructed by Guo et al. [4]. They extracted a semi-detailed and a skeletal mechanism from the detailed mechanism, and the models were used to simulate the combustion properties of NPCH in comparison with the experimental data and to delineate the major reaction pathways and the important reactions of NPCH combustion with reaction pathway analysis and sensitivity analysis. Based on the simulation results, they suggested that the mechanisms are reliable for predicting the autoignition behaviour of NPCH at high temperatures and can be used for CFD studies and the construction of mechanisms for the high-temperature combustion of other cycloalkanes with one ring. Tian et al. [5] conducted a comparative investigation on the high-temperature autoignition behaviour of cyclohexane, ethylcyclohexane, and n-propylcyclohexane over 1110–1650 K in a shock tube, at atmospheric pressure and equivalence ratios of 0.5, 1.0, and 2.0. They observed that the ignition delay time is affected by the molecular structure of each fuel and followed an order of n-propylcyclohexane < ethylcyclohexane ≈ cyclohexane. A faster H-radical generation of n-propylcyclohexane makes shorter and simpler pathways for the production of the H atoms compared to ethylcyclohexane and cyclohexane. Cyclohexane faces difficulties in the fast generation of H atoms due to the complex pathways of H production, which causes a longer ignition delay time than that of n-propylcyclohexane. Cyclohexane showed a shorter ignition delay time at temperatures higher than 1450 K compared to ethylcyclohexane and n-propylcyclohexane, and they related it to the branching chain favouring the unimolecular reactions dominating the fuel decomposition. Ji et al. [6] conducted an investigation on the laminar flame speeds of cyclohexane, methylcyclohexane, ethylcyclohexane, n-propylcyclohexane, and n-butylcyclohexane. The experiments were conducted in a counterflow configuration for a mixture of the species with air at atmospheric pressure, an equivalence ratio range from 0.7 to 1.5, and an unburned temperature of 353 K. Cyclohexane/air flames demonstrated a relatively faster propagation compared to mono-alkylated cyclohexane/air flames because of the greater amounts of 1,3-butadiene produced in cyclohexane/air flames. The laminar flame speeds of mono-alkylated cyclohexanes were approximately similar to each other. It was reported that the greater production of propene and allyl radical causes the lower flame propagation rates for mono-alkylated cyclohexanes. The experimental results were reproduced by the JetSurF (version 1.1) mechanism with good agreement. Recently, in 2021, an investigation on the combustion chemistry of NPCH was conducted by Ahmed et al. [7] that helps to better the understanding of combustion aspects of this species by providing comprehensive experimental data, detailed kinetic modelling, and quantum chemistry investigation of n-propylcyclohexane. They studied the autoignition behaviour of NPCH in lean and stoichiometric conditions over low and high temperatures in a shock tube and RCM at 20 and 40 bar and also the species mole fractions in a JSR and pressurized flow reactor (PFR), in a range of conditions. They developed a comprehensive detailed chemical kinetic mechanism including estimated reaction rates based on analogies with existing and analogous species mechanisms and estimated thermochemistry with quantum chemistry methods and a group additivity approach. In general, the model predicted the overall autoignition behaviour of NPCH and could reproduce the mole fraction experimental results with an acceptable agreement, but it was concluded that the model needs more improvements for the simulation of some measured intermediate species in the work and to fix the over-predictions observed for the ignition delay in high-temperature and negative-temperature coefficient (NTC) regions.
Apart from the issue that there are very few investigations dedicated to NPCH chemical kinetics, which are cited above, the mentioned studies are mostly limited to high temperatures or the development of a detailed mechanism with a relatively large size, and some of them even suffer from the lack of delivering a close simulation for more than one key combustion parameter in a wide range of conditions, such as covering both low- and high-temperature regions in the IDT or the lack of enough accuracy for the prediction of key combustion parameters over a wide range of conditions. Therefore, developing a compact mechanism that facilitates the usage of the NPCH model for practical applications and CFD studies and that can model the important combustion properties of NPCH with good agreement in a wide range of conditions is of great importance. In this regard, a theoretical investigation was conducted to provide a small-sized mechanism through coupling a simplified sub-mechanism to a core detailed mechanism, simulating the laminar flame speed and the autoignition behaviour of NPCH in varying conditions with good agreement against the experimental data.
2. Theoretical Framework
The mechanism construction methodology applies a C0-Cn mechanism as the core and a simplified sub-mechanism of Cn-Cm, where m is the carbon number of the intended heavy hydrocarbon, and this methodology is an effective approach to building a compact mechanism for heavy hydrocarbons [8,9,10]. The methodology procedure includes the incorporation of a detailed/semi-detailed core mechanism into a simplified sub-mechanism with a reduced number of species and reactions for the intended heavy fuel species, so that one representative species of the hydrocarbon classes which are required for the prediction of a target combustion parameter has been considered just for inclusion in the sub-mechanism. Such a simplified sub-mechanism considerably reduces the number of species and reactions in the final model. The simplified sub-mechanism of this study was constructed by utilizing the reaction classes of a detailed mechanism [7] recently developed for NPCH combustion chemistry. Through an analysis of the effect of the detailed mechanism reaction classes on NPCH combustion properties, a series of important reaction classes were selected to be included in the sub-mechanism. The included reactions in the sub-mechanism are given in Table 1. Then, the sub-mechanism was coupled to a C0-C5 core detailed mechanism [11] that was formerly developed to be used for hydrocarbon fuels. This core mechanism satisfied the requirement to have a lightweight model to couple to the sub-mechanism for the purpose of ignition delay prediction and to have the capability of predicting the laminar flame speed of heavy hydrocarbons.

Table 1.
Reactions in the sub-mechanism.
After the development of the mechanism, rate modifications for the reactions given in Table 1 were applied (which can be observed in the mechanism file appended to this paper), to provide the closest agreement compared to the available experimental data for the ignition and flame speed of NPCH in the literature. A combination of the Directed Relation Graph (DRG), Error Propagation Extension to DRG (DRGEP), and Directed Relation Graph Method with Path Flux Analysis (DRGPFA) methods, which are tools available in ANSYS Chemkin-Pro for reduction purposes [12], was applied to reduce the mechanism and provide a more compact model. The final version of the developed mechanism includes 72 species and 448 reactions. The developed mechanism files is appended to this paper as a Supplementary Material.
3. Results and Discussions
The simulations for ignition delay time and laminar flame speed, as the two key combustion properties, were conducted using the zero-dimensional homogeneous closed reactor model and the one-dimensional freely propagating laminar flame speed model in the ANSYS Chemkin-Pro 2021 R1 software. The developed mechanism could satisfactorily model the experimental results with good agreement.
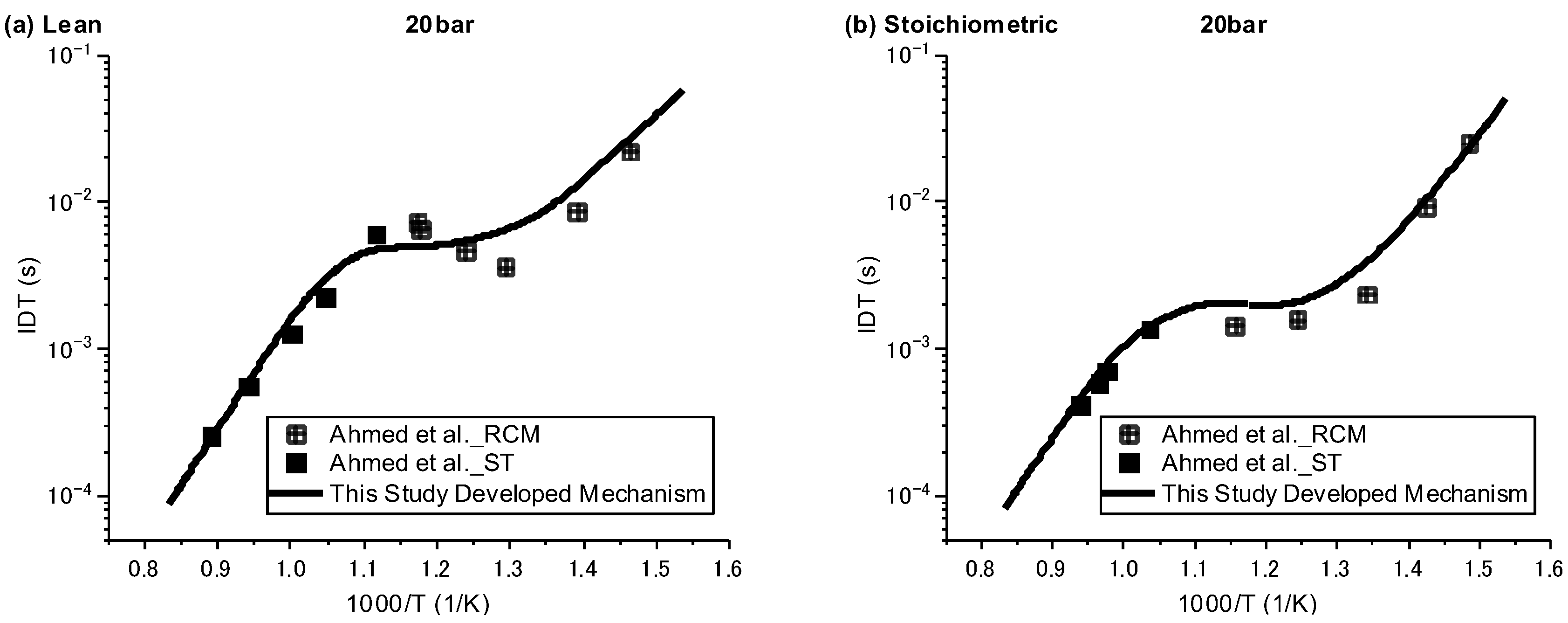
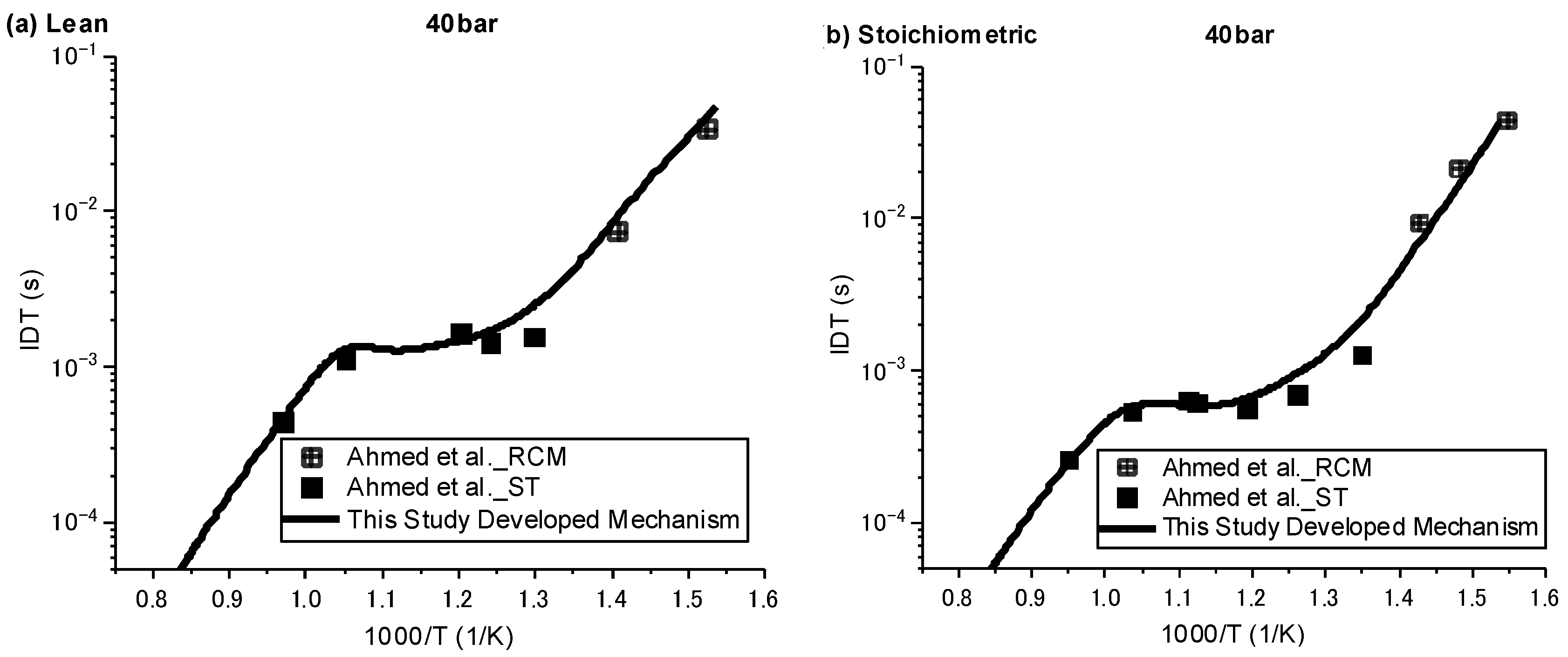
Ignition delay times were simulated against the available experimental data over three regimes of low, intermediate, and high temperature, with pressures of 20 and 40 bar and equivalence ratios of 0.5 and 1. Since the understanding of ignition behaviour for species such as NPCH at low temperatures is of great importance, especially for its usage as a surrogate or a component in a surrogate in engine applications, the experimental data by Ahmed et al. [7], which are the only available data covering both high and low temperatures, were selected for the validation of the results. As can be seen in Figure 1 and Figure 2, in general, the developed mechanism delivers a close emulation of the autoignition behaviour of NPCH in all temperature zones at different pressures and equivalence ratios. The model demonstrates the typical NTC behaviour of hydrocarbon species in accordance with the experimental results. The simulation results also follow the trend of a reduction in ignition delay time at higher pressures. A little discrepancy was observed for the lean condition at 20 bar over the intermediate temperature zone. This issue could be due to the experimental uncertainty or the lack of enough understanding in the combustion chemistry of NPCH, since the discrepancy was observed by using the detailed mechanism [7].
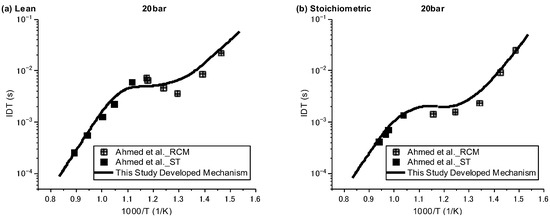
Figure 1.
IDT modelling results against the experimental data at P = 20 bar for lean (φ = 0.5) condition [7].
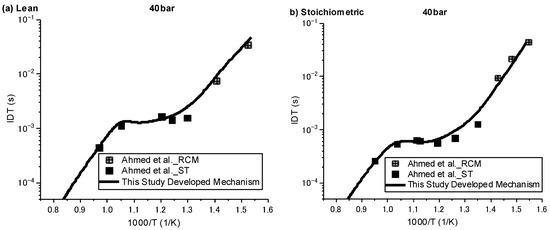
Figure 2.
IDT modelling results against the experimental data at P = 40 bar for stoichiometric (φ = 1) condition [7].
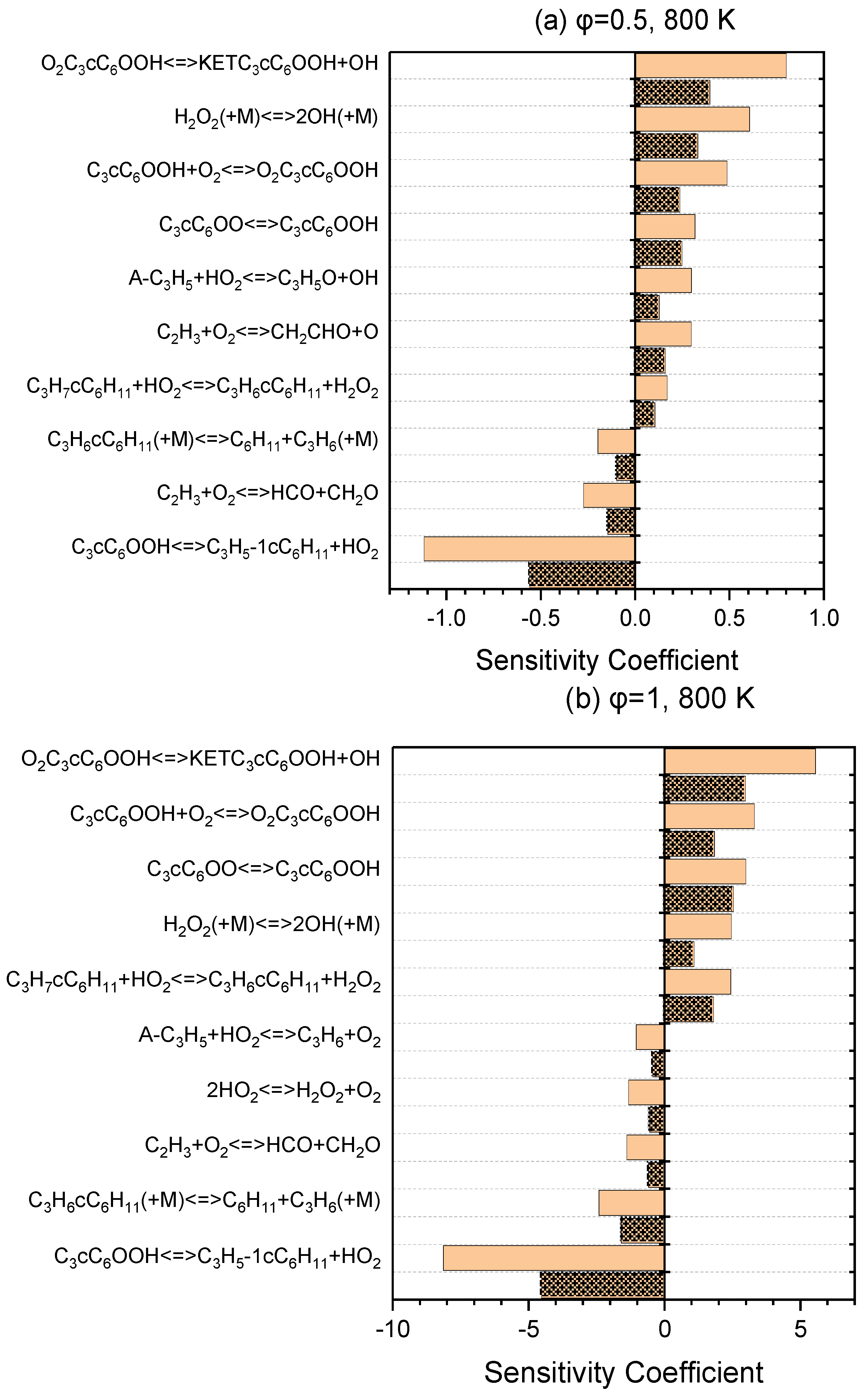
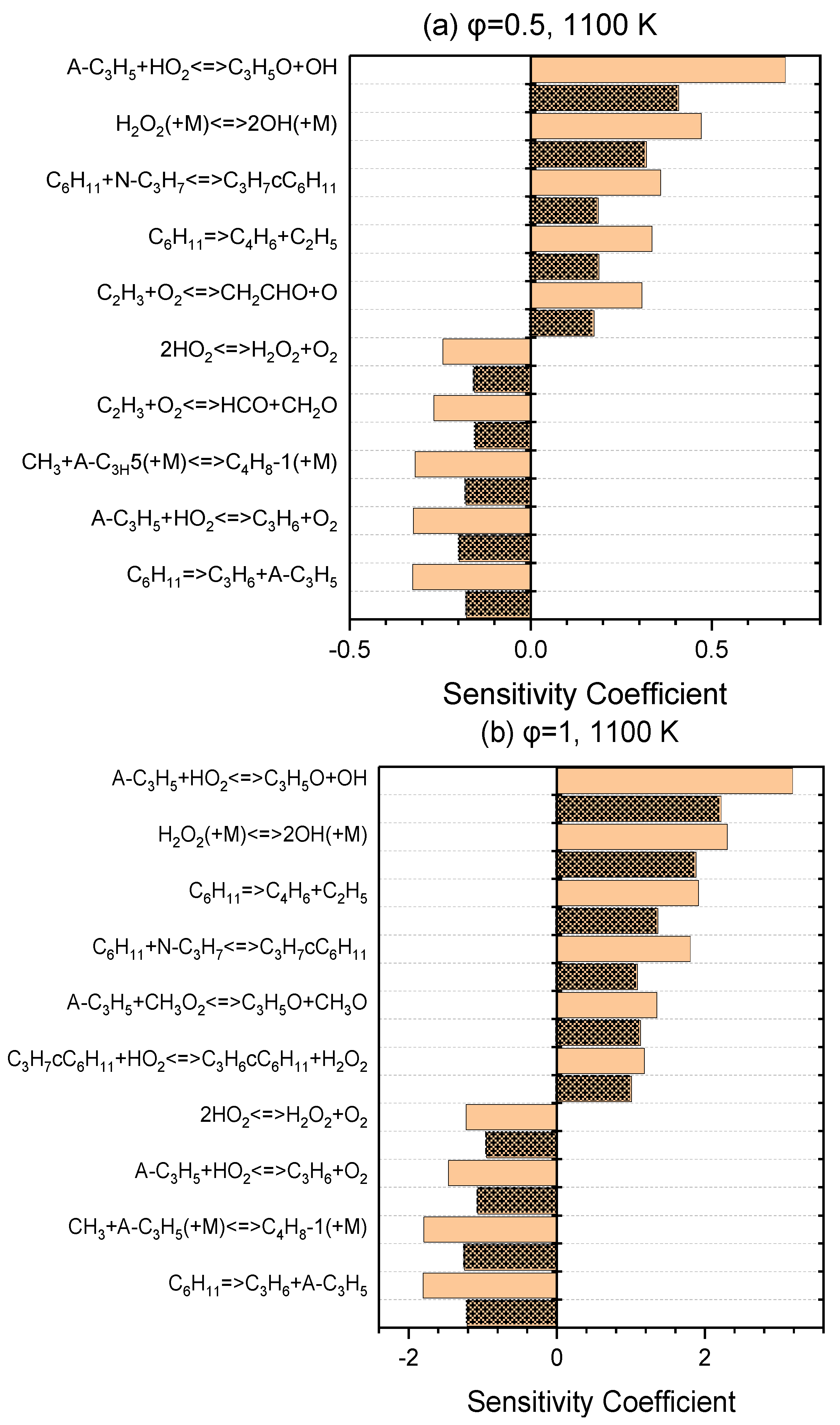
A series of sensitivity analyses were provided using Ansys Chemkin-Pro 2021 R1 in order to identify the key reactions relevant to the ignition of NPCH for the pressures of 20 and 40 bar and equivalence ratios of 0.5 and 1, at 800 K and 1100 K as the representatives of low- and high-temperature regions, respectively. Figure 3 demonstrates the top reactions that have a great impact on the ignition delay time at a low temperature. The isomerization of O2QOOH to the ketohydroperoxides, the second oxygen addition, and the isomerization of alkylperoxy are the top reactions for both equivalence ratios of 0.5 and 1. The H abstraction reaction by the HO2 abstractor is among the top reactions for both equivalence ratios (with a higher rank for φ = 1 than φ = 0.5). The hydrogen peroxide produced via this reaction generates two reactive OH radicals through H2O2 ⟶ 2OH, which has a boosting effect in the combustion process. The reaction of A-C3H5 + HO2 ⇌ C3H5O + OH and the reaction of C2H3 + O2 ⇌ CH2CHO + OH that the C2H3 radicals undergo in the branching reaction and form two kinds of radicals also have a relatively great promoting effect, which can be observed in the ranking for φ = 0.5, but are not in the top ten reactions for φ = 1. The reaction of QOOH ⇌ Alkene + HO2 is the strongest inhibitor and reduces the overall reactivity via the formation of more stable alkenes. As it can be seen in Figure 4, the reactivity at the high temperatures is mainly dominated by the small-species reactions which demonstrate a strong influence in terms of promotion and inhibition. The reactions of small species producing more active radicals are among the top-ranked reactions with a promoting effect. The unimolecular decomposition of fuel also has a strong promoting effect at this temperature. On the other hand, the small-species reactions that produce stable alkenes show a strong inhibiting effect.
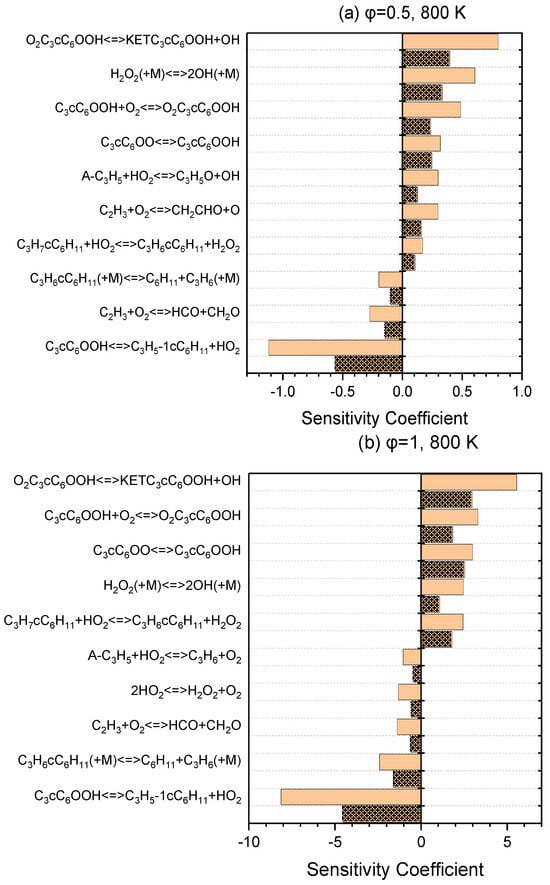
Figure 3.
Sensitivity analysis at 800 K and pressures of 20 bar (simple blocks) and 40 bar (patterned blocks) for lean (a) and stoichiometric (b) conditions.
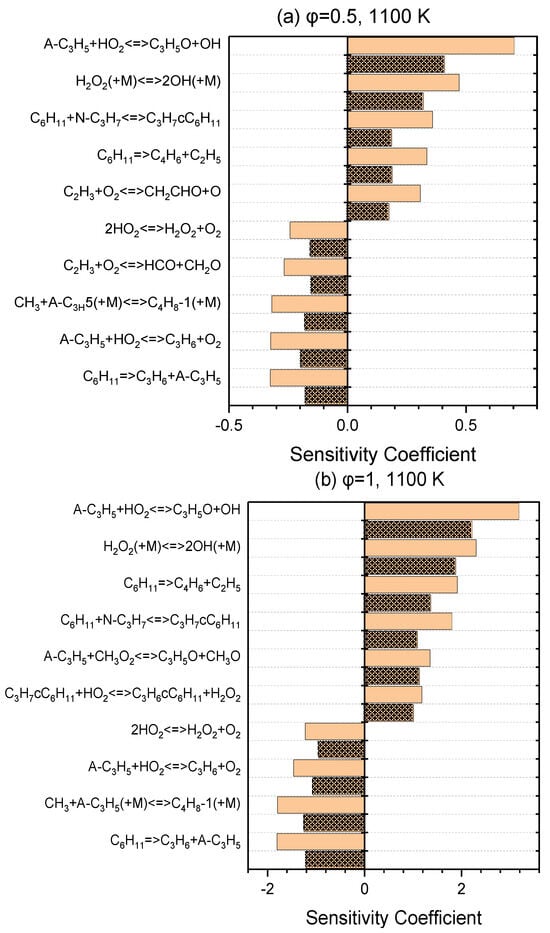
Figure 4.
Sensitivity analysis at 1100 K and pressures of 20 bar (simple blocks) and 40 bar (patterned blocks) for lean (a) and stoichiometric (b) conditions.
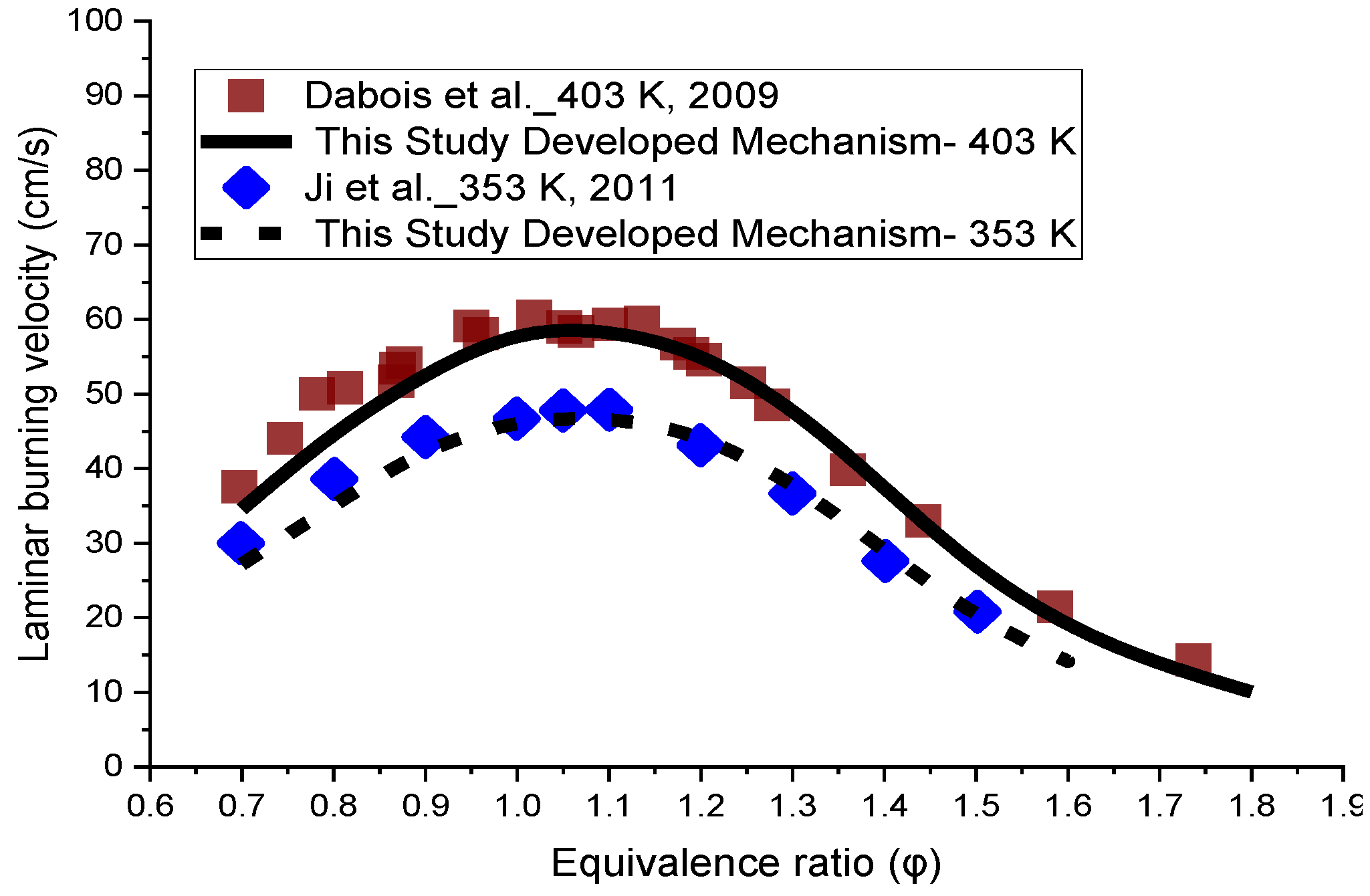
The prediction of laminar flame velocity at atmospheric pressure for inlet gas temperatures of 403 K and 353 K and a range of equivalence ratios is provided in Figure 5. The results were compared to the results of the available experimental data [2,6]. The developed mechanism demonstrates good agreement for the laminar flame speed at both temperatures, compared to the empirical data. A little discrepancy at the lean side is observed between the modelling and experimental results. The slight under-prediction is just seen at the equivalence ratios of 0.7 and 0.8 at both temperatures. However, the mechanism showed a very close agreement for all other conditions at 403 K and 353 K.
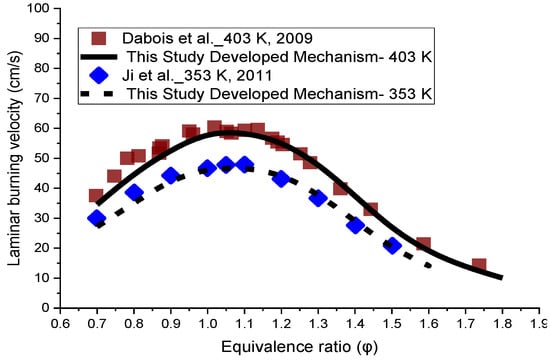
Figure 5.
Laminar flame speed simulation results of the developed mechanism of this study at the unburned temperatures of 403 K and 353 K, against the experimental data at atmospheric pressure [2,6].
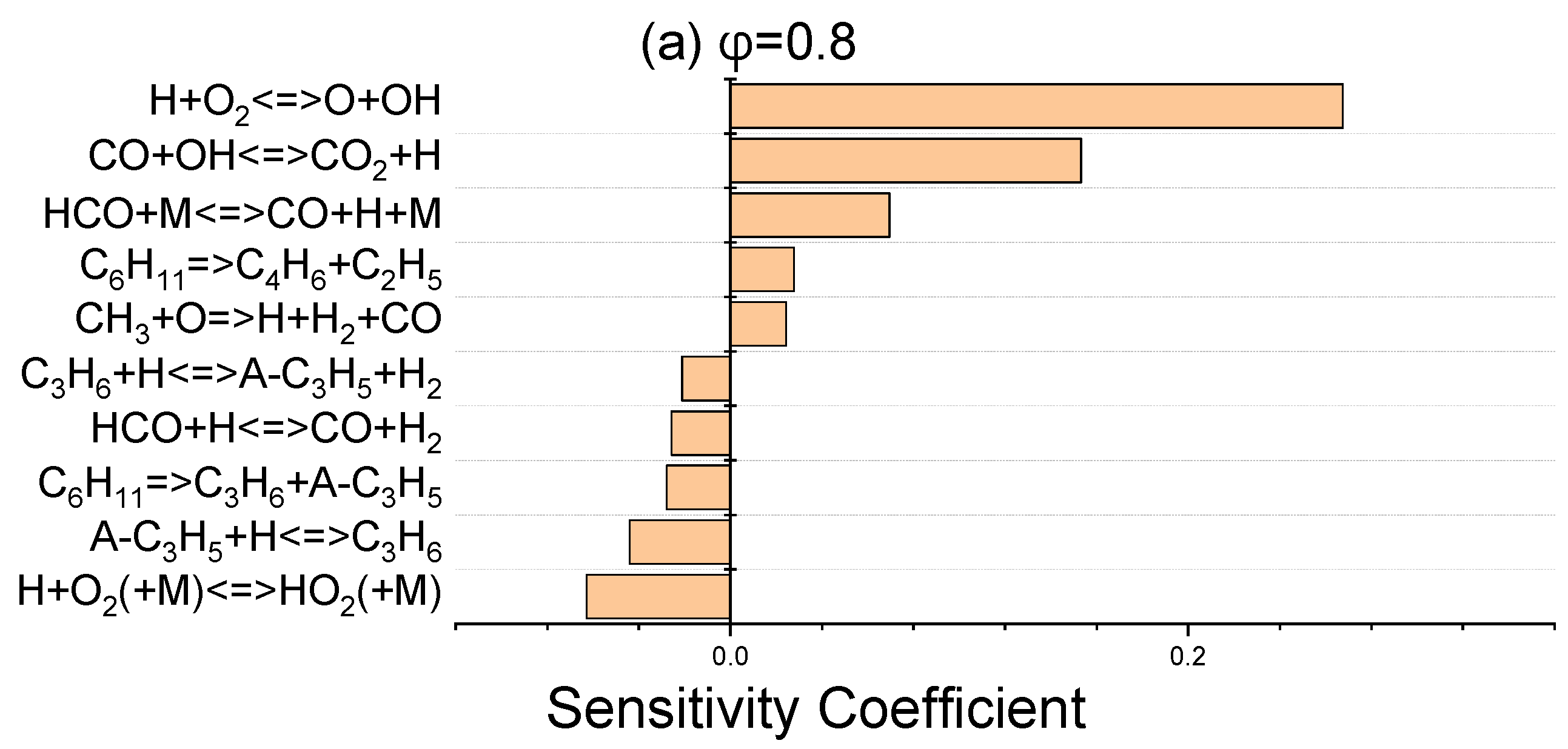
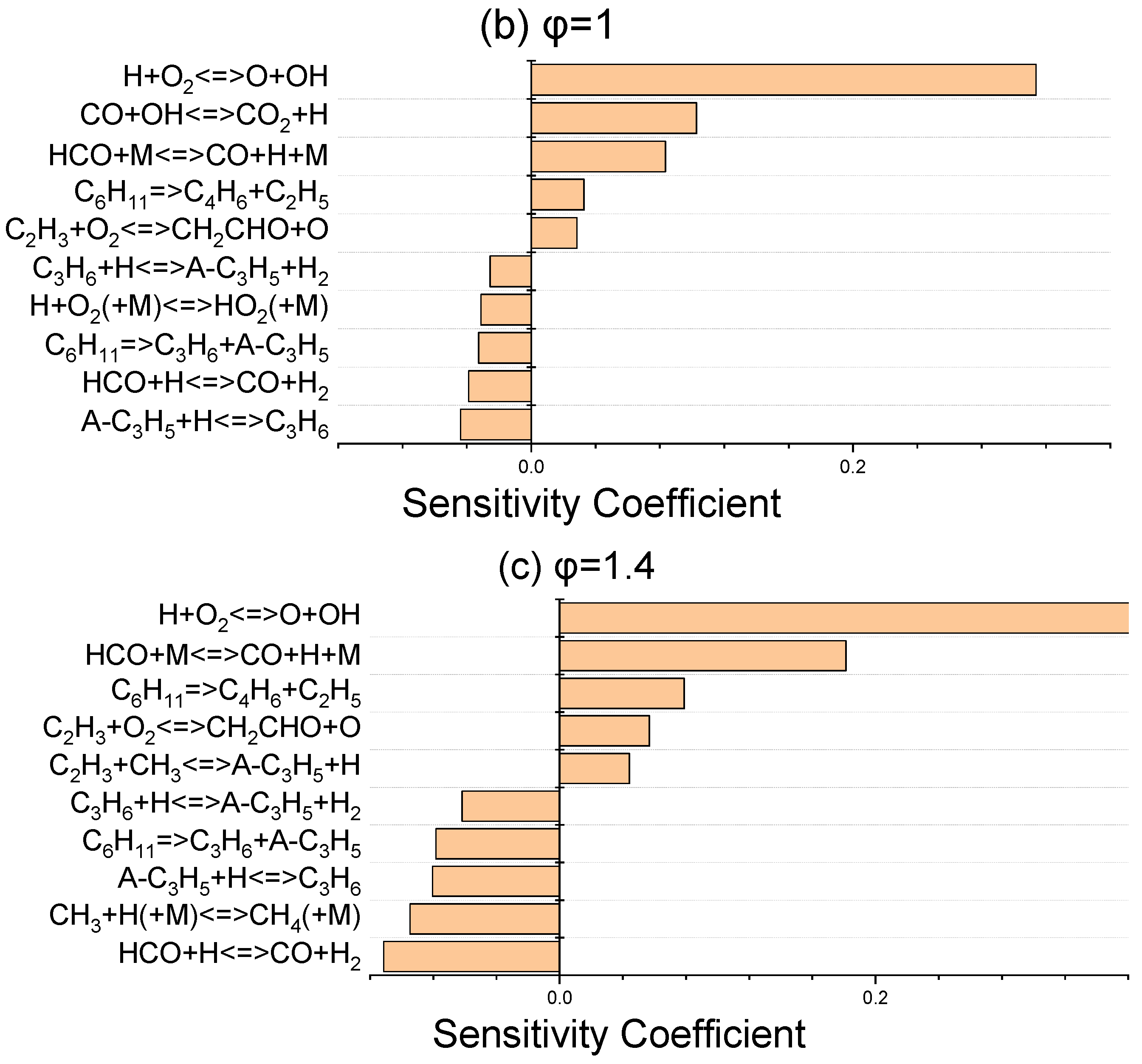
The sensitivity coefficients for laminar flame speed at 403 K for three equivalence ratios of 0.8, 1, and 1.4 were determined using ANSYS Chemkin-Pro 2021 R1 in order to identify the most sensitive reactions which affect the laminar flame speed. Figure 6 discloses that H + O2 ⇌ O + OH, as the dominant chain branching reaction, has the greatest effect on flame speed. The chain propagation reaction of CO + OH ⇌ CO2 + H, as the main oxidation reaction of carbon monoxide, and the chain initiation reaction of HCO + M ⇌ CO + H + M are the next important elementary reactions. These abovementioned three promoter reactions greatly contribute to the laminar flame speed increment via the production of more active radicals of H, O, and OH. In terms of inhibiting reactions, the reactions that have a termination effect on the radical chain process suppress the flame velocity and are given in Figure 6.
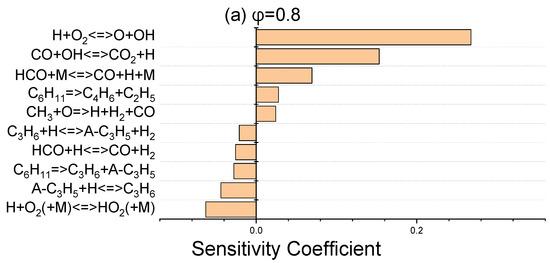
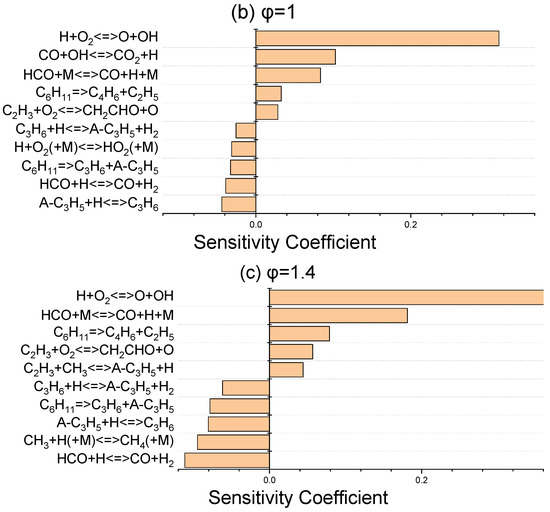
Figure 6.
Sensitivity analysis of laminar flame velocity for NPCH/air mixture at 1 bar and 403 K for equivalence ratios of 0.8 (a), 1 (b), and 1.4 (c).
Regarding the little discrepancy observed in the lean side in Figure 5, there are two issues which prevent further optimization. Firstly, there are common reactions which have an important effect on the flame speed at three equivalence ratios. Any modification of the reactions has a negative effect on the flame speed in stoichiometric and rich conditions. Secondly, some of the important reactions in flame speed have a considerable impact on the ignition delay of NPCH as well, which makes it hard to further optimize the discrepancy observed for the lean condition. Moreover, the availability of more experimental data could help to be certain about the flame speed behaviour of NPCH, then applying the required work for the optimization of the flame speed at the equivalence ratios.
4. Conclusions
A compact reaction kinetic mechanism for n-propylcyclohexane was developed by coupling a simplified sub-mechanism to a core mechanism and utilizing a combination of standard reduction methods so that the developed mechanism includes only 72 species and 448 reactions. Modifications to the reaction rates of the sub-mechanism were applied in order to emulate the empirical data of NPCH key combustion properties. The mechanism could closely predict the ignition delay behaviour of this fuel species against the experimental data over the low-to-high-temperature region with good agreement for lean and stoichiometric conditions at different pressures. Sensitivity analyses were conducted to identify the key reactions of the developed mechanism affecting the ignition delay. The simulations of the laminar flame speed were conducted at 1 bar for the unburned temperatures of 403 K and 353 K and were compared against the experimental data. The developed mechanism could emulate the experimental results with good agreement. The important reactions that have a great effect on laminar flame speed were identified via sensitivity analysis. In conclusion, the good predictive ability of the combustion parameters and the compact size of the simplified developed mechanism make it a useful tool for CFD simulation works and surrogate development.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/en17051103/s1. Supplementary material including mechanism files is appended to this paper.
Author Contributions
Conceptualization, H.S.S.; Methodology, H.S.S.; Software, H.S.S.; Validation, H.S.S.; Investigation, H.S.S.; Writing—original draft, H.S.S.; Writing—review & editing, K.J.H.; Supervision, K.J.H. and M.P. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the University of Sheffield Institutional Open Access Fund. For the purpose of open access, the author has applied a Creative Commons Attribution (CC BY) licence to any Author-Accepted Manuscript version arising.
Data Availability Statement
Data are contained within the article and supplementary materials.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ristori, A.; Dagaut, P.; EL Bakali, A.; Cathonnet, M. The oxidation of n-propylcyclohexane: Experimental results and kinetic modeling. Combust. Sci. Technol. 2001, 165, 197–228. [Google Scholar] [CrossRef]
- Dubois, T.; Chaumeix, N.; Paillard, C.E. Experimental and modeling study of n-propylcyclohexane oxidation under en-gine-relevant conditions. Energy Fuels 2009, 23, 2453–2466. [Google Scholar] [CrossRef]
- Crochet, M.; Minetti, R.; Ribaucour, M.; Vanhove, G. A detailed experimental study of n-propylcyclohexane autoignition in lean conditions. Combust. Flame 2010, 157, 2078–2085. [Google Scholar] [CrossRef]
- Guo, J.; Wang, J.; Hua, X.; Li, Z.; Tan, N.; Li, X. Mechanism construction and simulation for high-temperature combustion of n-propylcyclohexane. Chem. Res. Chin. Univ. 2014, 30, 480–488. [Google Scholar] [CrossRef]
- Tian, Z.; Zhang, Y.; Yang, F.; Pan, L.; Jiang, X.; Huang, Z. Comparative study of experimental and modeling autoignition of cyclohexane, ethylcyclohexane, and n-propylcyclohexane. Energy Fuels 2014, 28, 7159–7167. [Google Scholar] [CrossRef]
- Ji, C.; Dames, E.; Sirjean, B.; Wang, H.; Egolfopoulos, F.N. An experimental and modeling study of the propagation of cy-clohexane and mono-alkylated cyclohexane flames. Proc. Combust. Inst. 2011, 33, 971–978. [Google Scholar] [CrossRef]
- Ahmed, A.; Corrubia, J.A.; Al-Lehaibi, M.; Farid, F.; Wang, H.; Wang, Z.; Chen, B.; Roberts, W.L.; Miller, D.L.; Farooq, A.; et al. A comprehensive combustion chemistry study of n-propylcyclohexane. Combust. Flame 2021, 233, 111576. [Google Scholar] [CrossRef]
- Chang, Y.; Jia, M.; Li, Y.; Liu, Y.; Xie, M.; Wang, H.; Reitz, R.D. Development of a skeletal mechanism for diesel surrogate fuel by using a decoupling methodology. Combust. Flame 2015, 162, 3785–3802. [Google Scholar] [CrossRef]
- Saraee, H.S.; Hughes, K.J.; Shi, S.; Ingham, D.B.; Pourkashanian, M. Skeletal and Compact Validated Mechanisms for Iso-dodecane Using a Decoupling Methodology. Energy Fuels 2023, 37, 2307–2318. [Google Scholar] [CrossRef]
- Saraee, H.S.; Hughes, K.J.; Pourkashanian, M. A compact chemical kinetic mechanism for modelling isocetane. J. Energy Inst. 2023, 108, 101253. [Google Scholar] [CrossRef]
- Narayanaswamy, K.; Pitsch, H.; Pepiot, P. A component library framework for deriving kinetic mechanisms for multi-component fuel surrogates: Application for jet fuel surrogates. Combust. Flame 2016, 165, 288–309. [Google Scholar] [CrossRef]
- Available online: https://www.ansys.com/products/fluids/ansys-chemkin-pro (accessed on 24 February 2024).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).