One-Pot Hydrothermal Synthesis and Electrochemical Performance of Subspheroidal Core–Shell Structure MoS2/C Composite as Anode Material for Lithium-Ion Batteries

Abstract
:1. Introduction
2. Experimental
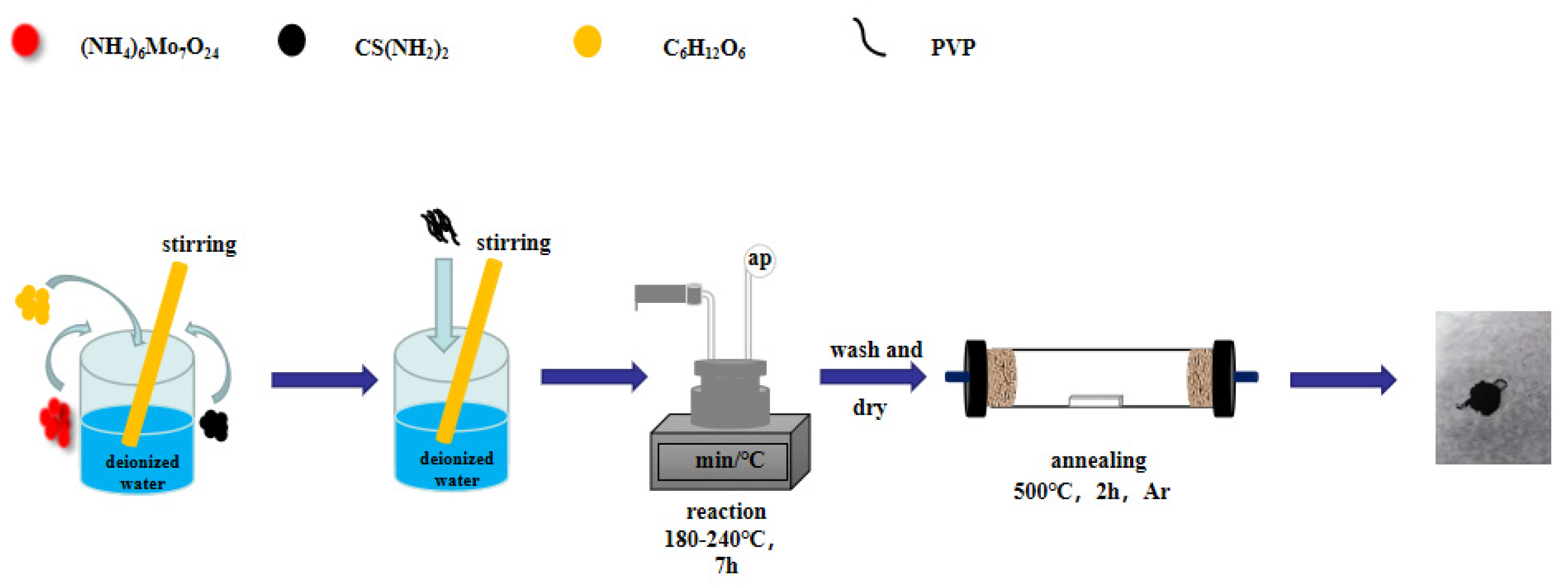
2.1. Synthesis of the MoS2/C Composite
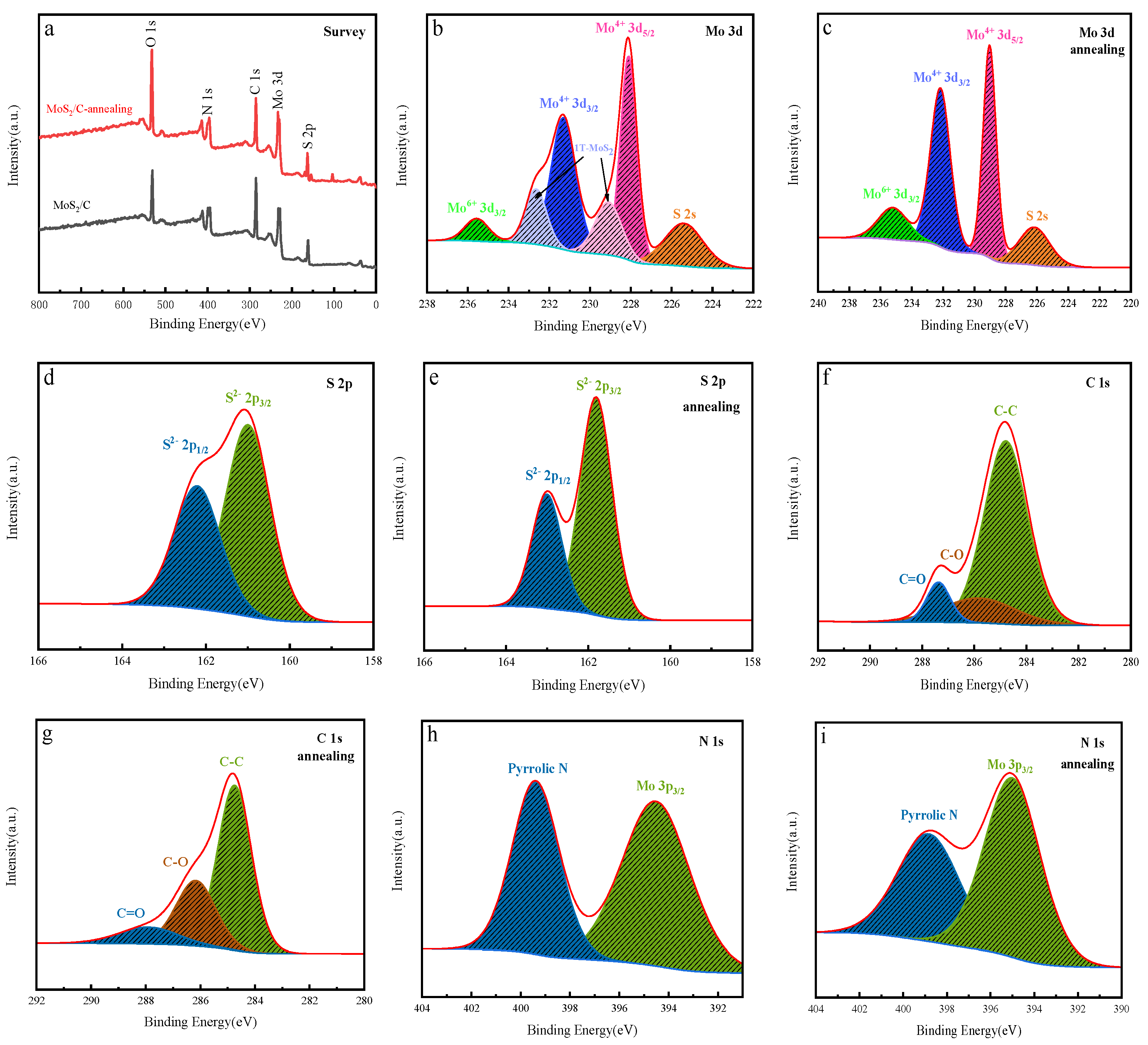
2.2. Characterization of the MoS2/C Composite
2.3. Electrochemical Performance Test
3. Results and Discussion
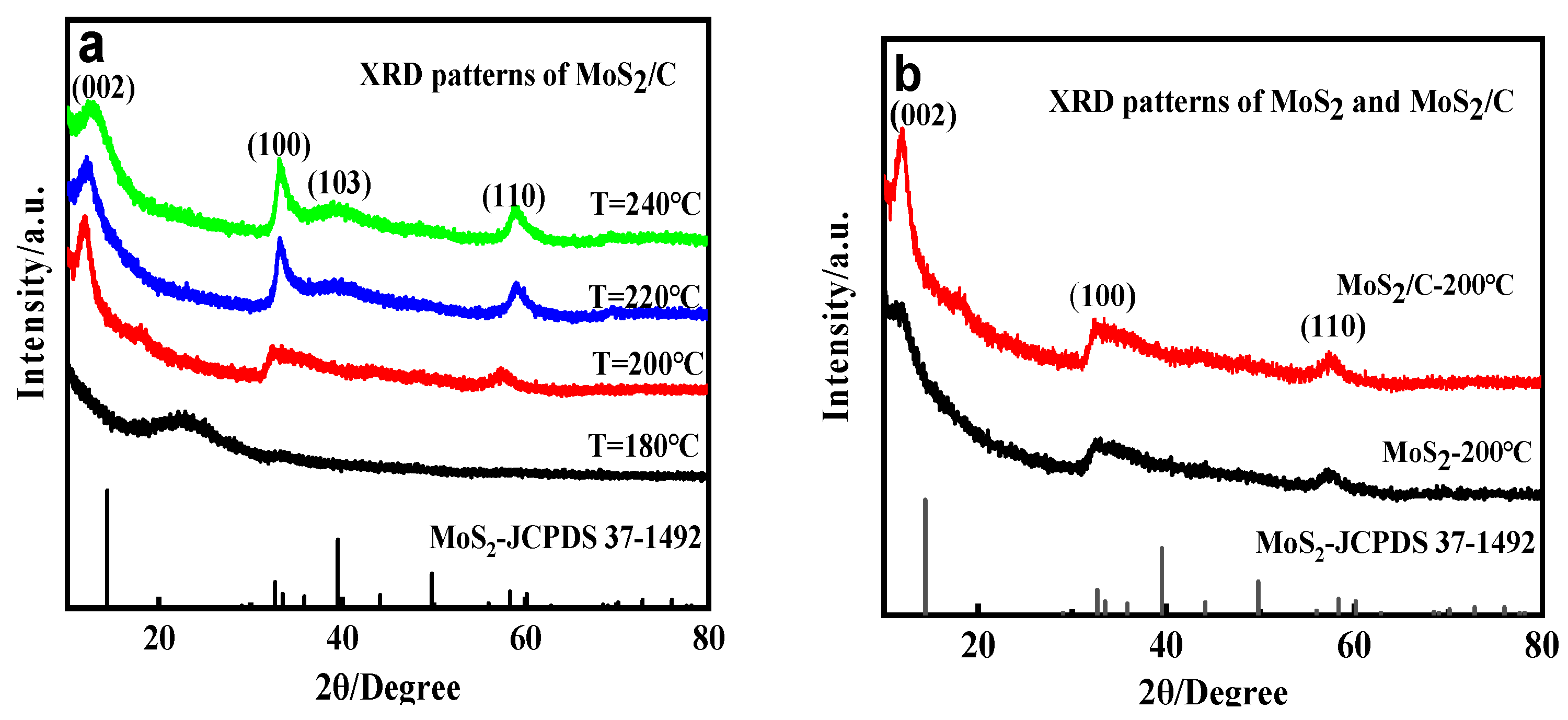
3.1. Effect of Temperature on the Crystal Structure and Morphology
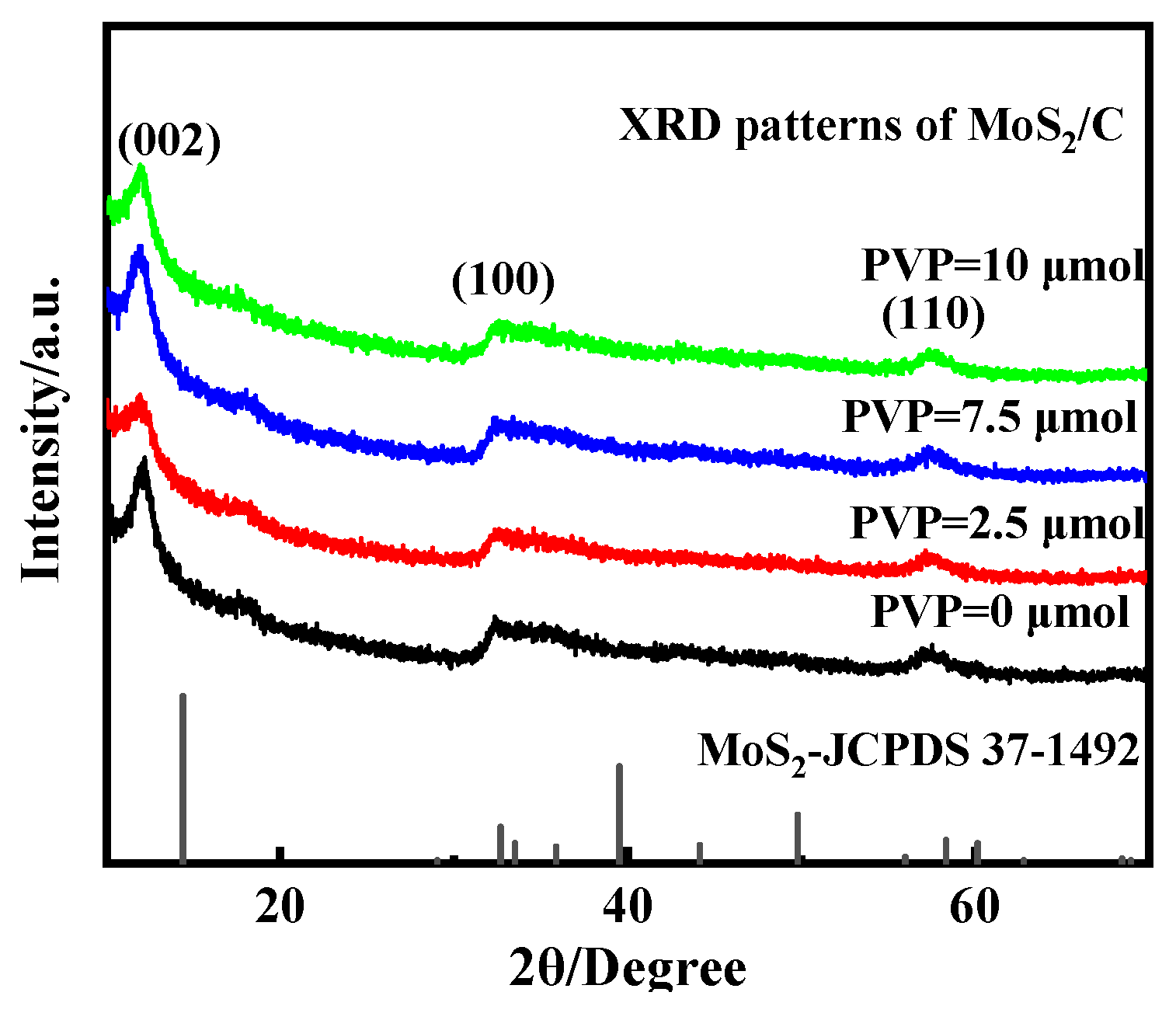
3.2. Effect of PVP on the Crystal Structure and Morphology
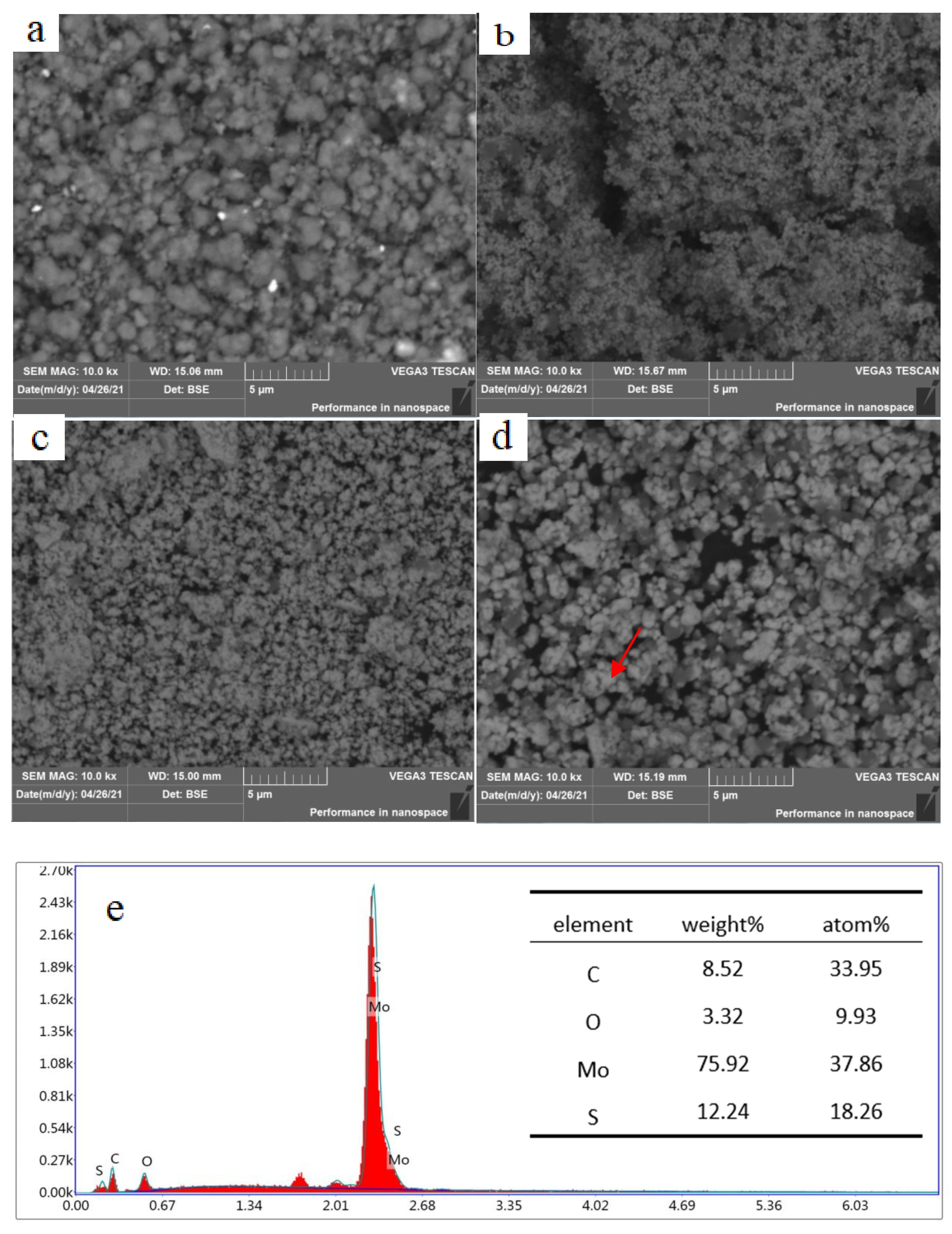
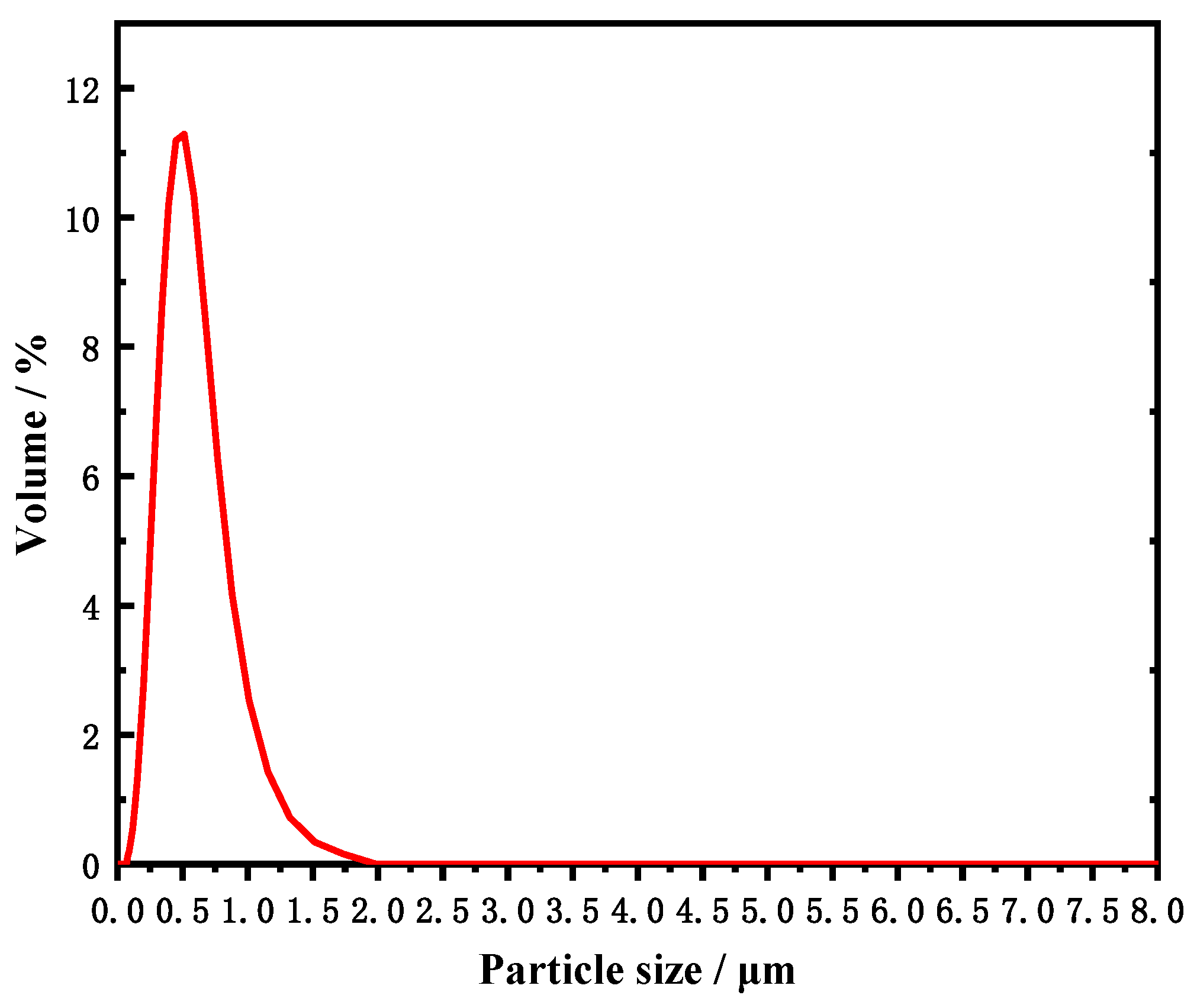
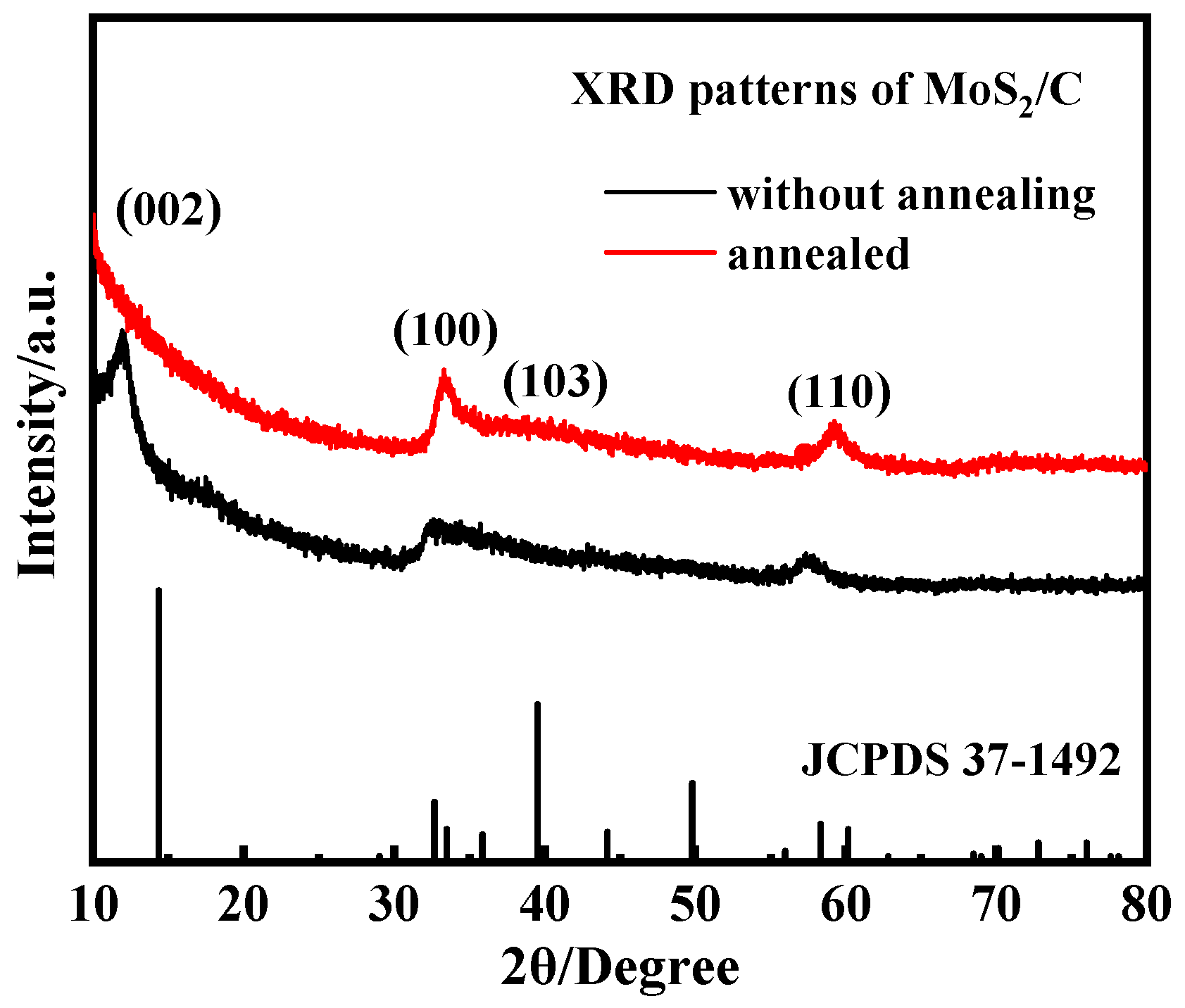
3.3. Effect of Annealing on the Crystal Structure and Morphology
3.4. Synthesis Mechanism
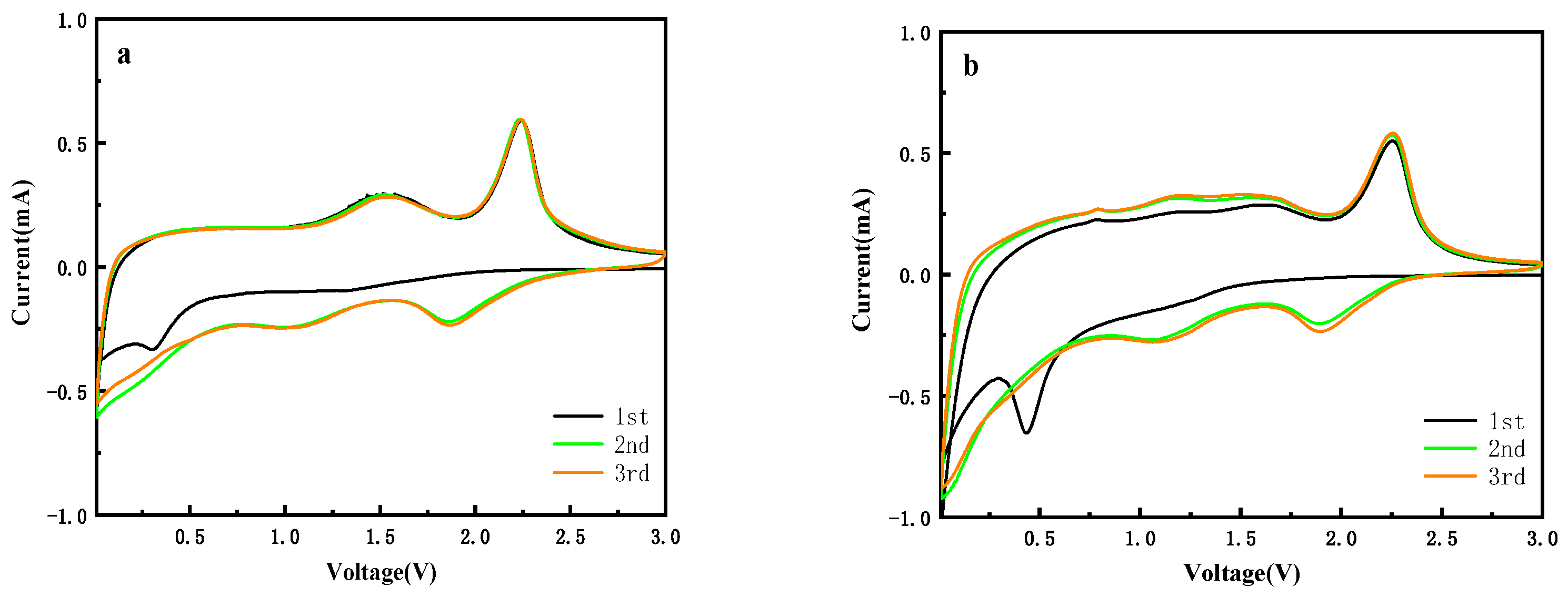
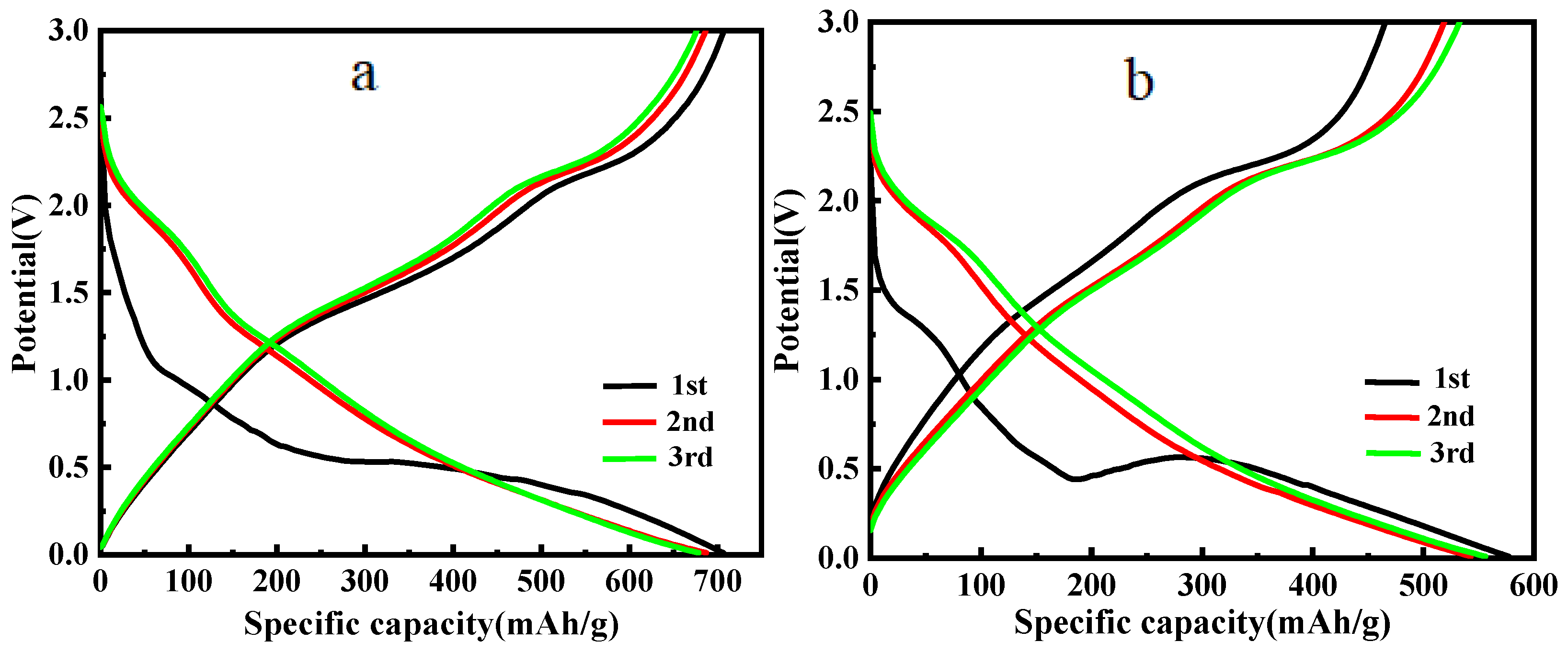
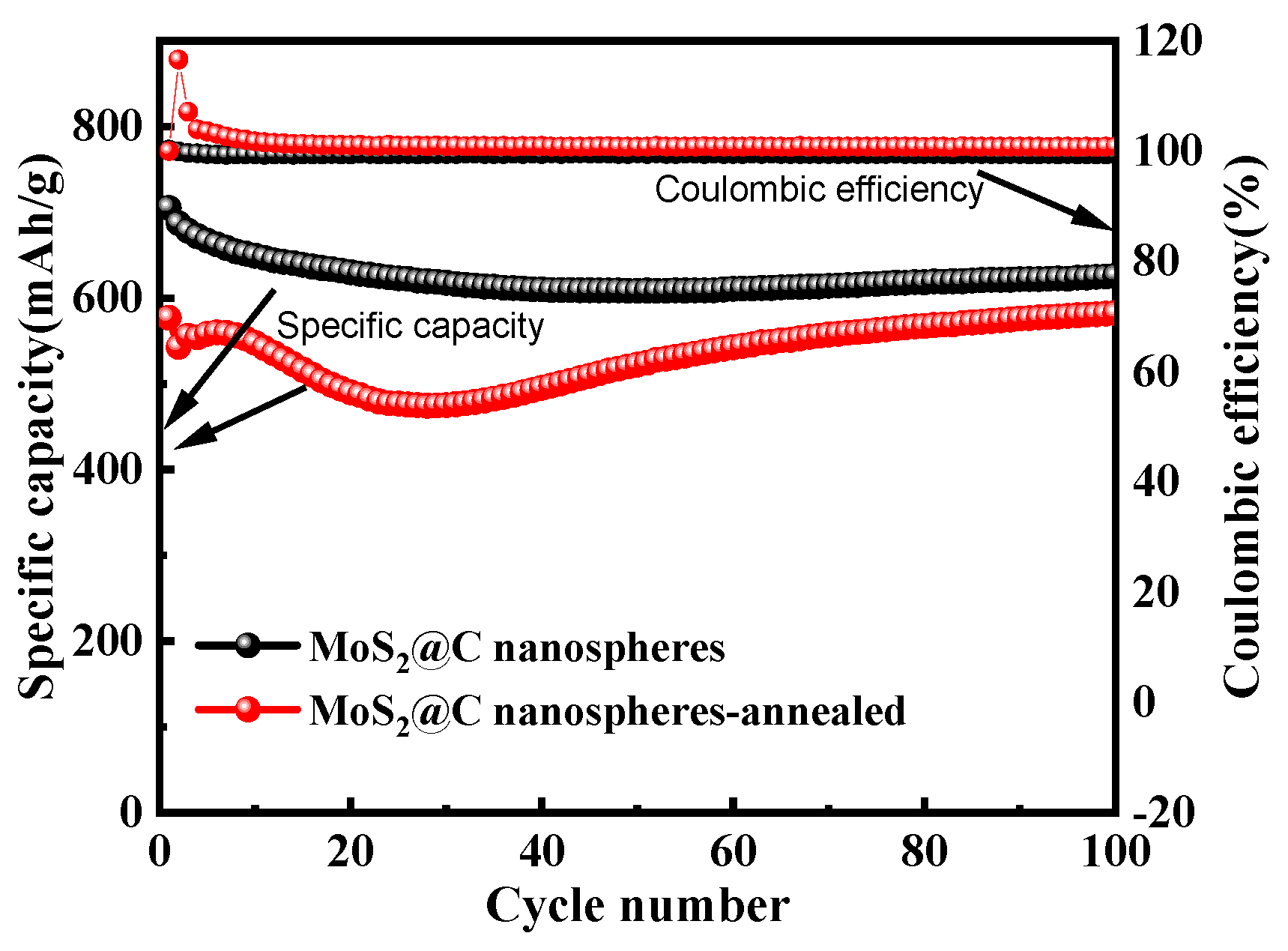
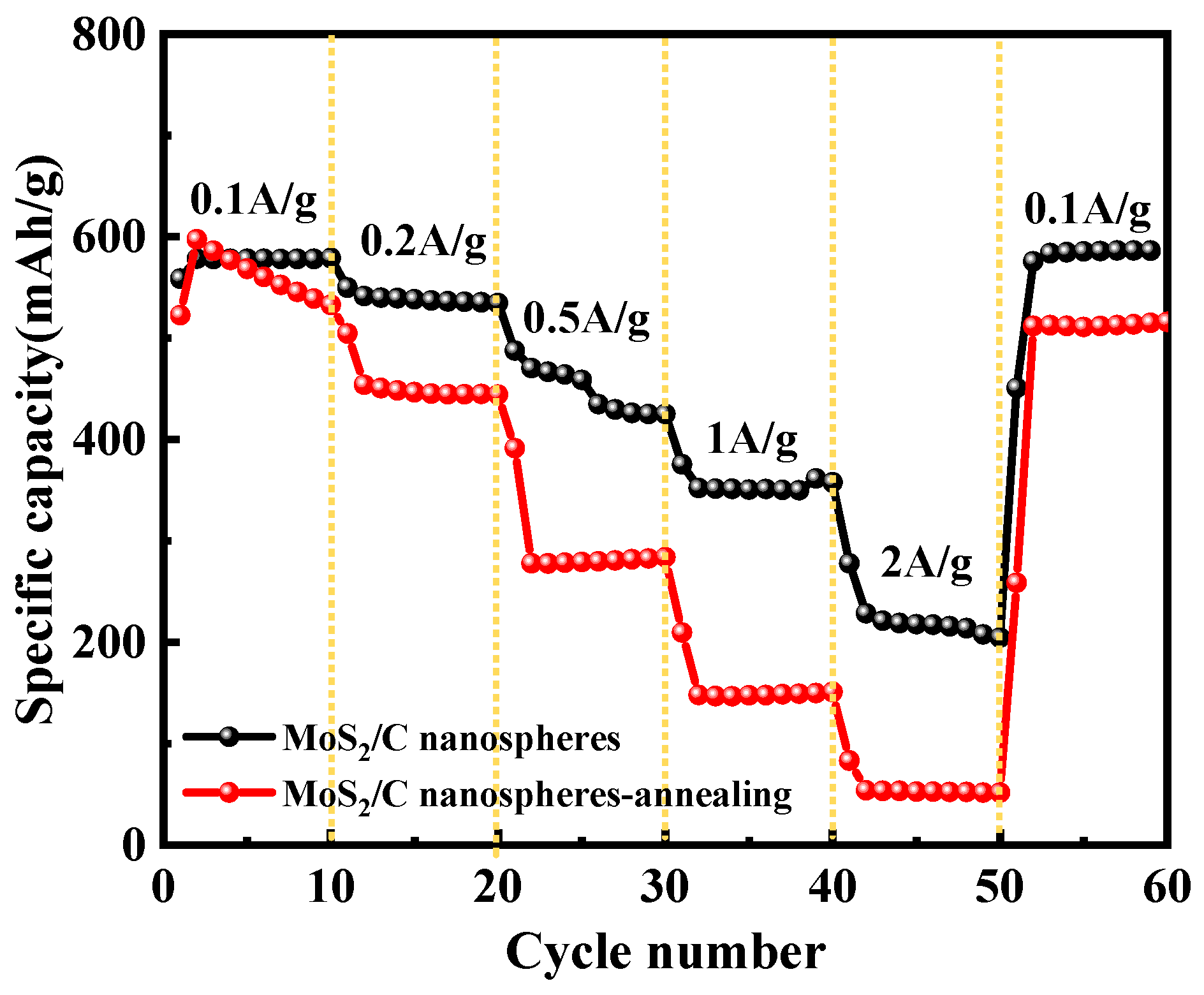
3.5. Electrochemical Performance of MoS2/C
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, Q.; Yao, T.; He, Q.; Wang, H.; Liu, Z.; Wang, D.; Wang, H.; Meng, L. Enhancing lithium-ion storage performance of hollow CoS2/MoS2 nanospheres via N-doped carbon-coating. J. Energy Storage 2023, 72, 108639. [Google Scholar] [CrossRef]
- Wu, L.; Zheng, J.; Wang, L.; Xiong, X.; Shao, Y.; Wang, G.; Wang, J.H.; Zhong, S.; Wu, M. PPy-encapsulated SnS2 nanosheets stabilized by defects on a TiO2 support as a durable anode material for lithium-ion batteries. Angew. Chem. 2019, 131, 821–825. [Google Scholar] [CrossRef]
- Wang, Z.; Feng, G.; Zhen, D.; Gu, F.; Ball, A. A review on online state of charge and state of health estimation for lithium-ion batteries in electric vehicles. Energy Rep. 2021, 7, 5141–5161. [Google Scholar] [CrossRef]
- Yao, T.; Wang, H.; Wang, J.; Liu, T.; Li, C.; Han, X.; Cheng, Y. Metal-organic framework derived Ge/TiO2@ C nanotablets as high-performance anode for lithium-ion batteries. ChemistrySelect 2019, 4, 10576–10580. [Google Scholar] [CrossRef]
- Huang, L.; Li, T.; Liu, Q.; Gu, J. Fluorine-free Ti3C2Tx as anode materials for Li-ion batteries. Electrochem. Commun. 2019, 104, 106472. [Google Scholar] [CrossRef]
- IPCC Climate Change. Mitigation of climate change. In Contribution of Working Group III to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change; United Nations. Economic Commission for Africa: Addis Ababa, Ethiopia, 2014; Volume 1454, p. 147. [Google Scholar]
- Xu, H.; Li, H.; Wang, X. The Anode Materials for Lithium-Ion and Sodium-Ion Batteries Based on Conversion Reactions: A Review. ChemElectroChem 2023, 10, e202201151. [Google Scholar] [CrossRef]
- Kumar, R.; Sahoo, S.; Joanni, E.; Singh, R.K.; Tan, W.K.; Kar, K.K.; Matsuda, A. Recent progress in the synthesis of graphene and derived materials for next generation electrodes of high performance lithium ion batteries. Prog. Energy Combust. Sci. 2019, 75, 100786. [Google Scholar] [CrossRef]
- Arshad, F.; Lin, J.; Manurkar, N.; Fan, E.; Ahmad, A.; Wu, F.; Chen, R.; Li, L. Life cycle assessment of lithium-ion batteries: A critical review. Resour. Conserv. Recycl. 2022, 180, 106164. [Google Scholar] [CrossRef]
- Zahoor, A.; Yu, Y.; Zhang, H.; Nihed, B.; Afrane, S.; Peng, S.; Sápi, A.; Lin, C.J.; Mao, G. Can the new energy vehicles (NEVs) and power battery industry help China to meet the carbon neutrality goal before 2060? J. Environ. Manag. 2023, 336, 117663. [Google Scholar] [CrossRef]
- Nitta, N.; Wu, F.; Lee, J.T.; Yushin, G. Li-ion battery materials: Present and future. Mater. Today 2015, 18, 252–264. [Google Scholar] [CrossRef]
- Cao, B.; Liu, Z.; Xu, C.; Huang, J.; Fang, H.; Chen, Y. High-rate-induced capacity evolution of mesoporous C@ SnO2@ C hollow nanospheres for ultra-long cycle lithium-ion batteries. J. Power Sources 2019, 414, 233–241. [Google Scholar] [CrossRef]
- Cheng, H.; Shapter, J.G.; Li, Y.; Gao, G. Recent progress of advanced anode materials of lithium-ion batteries. J. Energy Chem. 2021, 57, 451–468. [Google Scholar] [CrossRef]
- Wang, W.; Zeng, X.; Warner, J.H.; Guo, Z.; Hu, Y.; Zeng, Y.; Lu, J.; Jin, W.; Wang, S.; Lu, J. Photoresponse-bias modulation of a high-performance MoS2 photodetector with a unique vertically stacked 2H-MoS2/1T@ 2H-MoS2 structure. ACS Appl. Mater. Interfaces 2020, 12, 33325–33335. [Google Scholar] [CrossRef] [PubMed]
- Lei, Z.; Zhan, J.; Tang, L.; Zhang, Y.; Wang, Y. Recent development of metallic (1T) phase of molybdenum disulfide for energy conversion and storage. Adv. Energy Mater. 2018, 8, 1703482. [Google Scholar] [CrossRef]
- Zhang, R.; Qin, Y.; Liu, P.; Jia, C.; Tang, Y.; Wang, H. How does molybdenum disulfide store charge: A minireview. ChemSusChem 2020, 13, 1354–1365. [Google Scholar] [CrossRef] [PubMed]
- Navas, J.; Martínez-Merino, P.; Sánchez-Coronilla, A.; Gallardo, J.J.; Alcántara, R.; Martín, E.I.; Piñero, J.C.; León, J.R.; Aguilar, T.; Toledo, J.H. MoS2 nanosheets vs. nanowires: Preparation and a theoretical study of highly stable and efficient nanofluids for concentrating solar power. J. Mater. Chem. A 2018, 6, 14919–14929. [Google Scholar] [CrossRef]
- Radhakrishnan, S.; KA, S.R.; Kumar, S.R.; Johari, P.; Rout, C.S. Energy storage performance of 2D MoS2 and carbon nanotube heterojunctions in symmetric and asymmetric configuration. Nanotechnology 2021, 32, 155403. [Google Scholar] [CrossRef]
- Li, Y.; Jiang, S.; Qian, Y.; Yan, X.; Zhou, J.; Yi, Z.; Lin, N.; Qian, Y. 2D interspace confined growth of ultrathin MoS2-intercalated graphite hetero-layers for high-rate Li/K storage. Nano Res. 2021, 14, 1061–1068. [Google Scholar] [CrossRef]
- Chen, R.; Lin, C.; Yu, H.; Tang, Y.; Song, C.; Yuwen, L.; Li, H.; Xie, X.; Wang, L.; Huang, W. Templating C60 on MoS2 Nanosheets for 2D Hybrid van der Waals p–n Nanoheterojunctions. Chem. Mater. 2016, 28, 4300–4306. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, Z.; Zhang, H.; Zhou, Y.; Weng, Y.; Xiong, S. Three-Dimensional PrGO-Based Sandwich Composites With MoS2 Flowers as Stuffings for Superior Lithium Storage. Front. Chem. 2020, 8, 94. [Google Scholar] [CrossRef]
- Lou, X.; Liu, Z.; Hou, J.; Zhou, Y.; Chen, W.; Xing, X.; Li, Y.; Liao, Q.; Zhu, X. Modification of the anodes using MoS2 nanoflowers for improving microbial fuel cells performance. Catal. Today 2021, 364, 111–117. [Google Scholar] [CrossRef]
- Liu, Y.; Hu, K.; Hu, E.; Guo, J.; Han, C.; Hu, X. Double hollow MoS2 nano-spheres: Synthesis, tribological properties, and functional conversion from lubrication to photocatalysis. Appl. Surf. Sci. 2017, 392, 1144–1152. [Google Scholar] [CrossRef]
- Liu, M.; Li, N.; Wang, S.; Li, Y.; Liang, C.; Yu, K. 3D nanoflower-like MoS2 grown on wheat straw cellulose carbon for lithium-ion battery anode material. J. Alloys Compd. 2023, 933, 167689. [Google Scholar] [CrossRef]
- Liu, H.; Lin, Y.; Zhang, L. Hierarchical porous MoS2/C nanospheres self-assembled by nanosheets with high electrochemical energy storage performance. Nanoscale Res. Lett. 2020, 15, 199. [Google Scholar] [CrossRef] [PubMed]
- Long, F.; Chen, Y.; Wu, C.; Wang, J.; Mo, S.; Zou, Z.; Zheng, G. Unique three-dimensional hierarchical heterogeneous MoS2/graphene structures as a high-performance anode material for lithium-ion batteries. Ionics 2021, 27, 1977–1986. [Google Scholar] [CrossRef]
- Ye, R.; Bao, Y.; Huang, J.; Zhao, Y.; Fan, Z.; Chen, H.; Hu, X.; Zhuang, N. Enhanced Hydrogen Evolution Activity and Lithium-Ion Storage of Three-Dimensional Porous MoS2 Hybridized with Graphene Aerogel. Catal. Lett. 2020, 150, 3402–3408. [Google Scholar] [CrossRef]
- Zhang, J.; Li, Y.; Gao, T.; Sun, X.; Cao, P.; Zhou, G. Flower-shaped TiO2/C microspheres embedded with fish-scale-like MoS2 as anodes for lithium-ion batteries. Ceram. Int. 2018, 44, 8550–8555. [Google Scholar] [CrossRef]
- Jiao, J.; Du, K.; Wang, Y.; Sun, P.; Zhao, H.; Tang, P.; Fan, Q.; Tian, H.; Li, Q.; Xu, Q. N plasma treatment on graphene oxide-MoS2 composites for improved performance in lithium ion batteries. Mater. Chem. Phys. 2020, 240, 122169. [Google Scholar] [CrossRef]
- Liu, H.; Lei, Y.; Lin, Y.; Zhang, L.; Lu, Y. One-pot Hydrothermal Synthesis of MoS2 Porous Nanospheres and Their Electrochemical Properties. Int. J. Electrochem. Sci. 2020, 15, 1942–1948. [Google Scholar] [CrossRef]
- Liu, Y.; Tang, D.; Zhong, H.; Zhang, Q.; Yang, J.; Zhang, L. A novel MoS2/C nanocomposite as an anode material for lithium-ion batteries. J. Alloys Compd. 2017, 729, 583–589. [Google Scholar] [CrossRef]
- Liu, B.; Li, F.; Li, H.; Zhang, S.; Liu, J.; He, X.; Sun, Z.; Yu, Z.; Zhang, Y.; Huang, X. Monodisperse MoS2/graphite composite anode materials for advanced lithium ion batteries. Molecules 2023, 28, 2775. [Google Scholar] [CrossRef] [PubMed]
- Suchanek, W.L.; Riman, R.E. Hydrothermal synthesis of advanced ceramic powders. Adv. Sci. Technol. 2006, 45, 184–193. [Google Scholar]
- Shi, E.W.; Chen, Z.Z.; Yuan, R.L.; Zheng, Y.Q. Hydrothemal Crystallgraphy, 1st ed.; Science Press: Beijing, China, 2004; pp. 218–219. [Google Scholar]
- Wu, Y.; Zhang, L.L.; Wang, W.; Fan, D.S.; Yang, S.S.; Bai, Y.H.; Li, J.W.; Liu, W. Enhanced hydrothermal synthesis and electrochemi-cal performance of subsphaeroidal MoS2 used as anode material for lithium-ion batterie. Rare Met. Mater. Eng. 2023, 52, 2893–2900. [Google Scholar]
- Ma, B.-B.; Chen, S.-J.; Huang, Y.-W.; Nie, Z.-Z.; Qiu, X.-B.; Xie, X.-Q.; Wu, Z.-J. Electrochemical lithium storage performance of three-dimensional foam-like biocarbon/MoS2 composites. Trans. Nonferrous Met. Soc. China 2021, 31, 255–264. [Google Scholar] [CrossRef]
- Chang, K.; Chen, W.; Ma, L.; Li, H.; Li, H.; Huang, F.; Xu, Z.; Zhang, Q.; Lee, J.-Y. Graphene-like MoS2/amorphous carbon composites with high capacity and excellent stability as anode materials for lithium ion batteries. J. Mater. Chem. 2011, 21, 6251–6257. [Google Scholar] [CrossRef]
- Luo, F.; Feng, X.; Zeng, L.; Lin, L.; Li, X.; Kang, B.; Xiao, L.; Chen, Q.; Wei, M.; Qian, Q. In situ simultaneous encapsulation of defective MoS2 nanolayers and sulfur nanodots into SPAN fibers for high rate sodium-ion batteries. Chem. Eng. J. 2021, 404, 126430. [Google Scholar] [CrossRef]
- Jia, P.; Liang, J.; Yue, L.; Liu, M.; Wang, X.; Liang, C.; Yu, K. Effect of MoS2 decoration on energy storage of wheat straw-derived porous carbon for lithium-ion batteries. Ionics 2023, 29, 5085–5097. [Google Scholar] [CrossRef]
- Teng, Y.; Liu, H.; Liu, D.; He, H.; Chen, Y. Pitaya-like carbon-coated ZnS/carbon nanospheres with inner three-dimensional nanostructure as high-performance anode for lithium-ion battery. J. Colloid Interface Sci. 2019, 554, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Hui, K.; Fu, J.; Liu, J.; Chen, Y.; Gao, X.; Gao, T.; Wei, Q.; Li, C.; Zhang, H.; Tang, M. Yolk–shell nanoarchitecture for stabilizing a Ce2S3 anode. Carbon Energy 2021, 3, 709–720. [Google Scholar] [CrossRef]
- Bai, J.; Zhao, B.; Wang, X.; Ma, H.; Li, K.; Fang, Z.; Li, H.; Dai, J.; Zhu, X.; Sun, Y. Yarn ball-like MoS2 nanospheres coated by nitrogen-doped carbon for enhanced lithium and sodium storage performance. J. Power Sources 2020, 465, 228282. [Google Scholar] [CrossRef]
- Zhu, W.; Shi, C.; Zhao, J.; Wang, Y.; Hu, Y. Structure and electrochemical performance of MoS2 based on different molybdenum-sulfur mole ratios. J. Phys. Chem. Solids 2022, 167, 110749. [Google Scholar] [CrossRef]
- Ramohlola, K.E.; Iwuoha, E.I.; Hato, M.J.; Modibane, K.D. Instrumental techniques for characterization of molybdenum disulphide nanostructures. J. Anal. Methods Chem. 2020, 2020, 8896698. [Google Scholar] [CrossRef]
- Yin, M.; Song, H.; Tang, A.; Xu, G.; Liu, L.; Pan, Y. Effect of carbon sources on morphologies and electrochemical properties of MoS 2/carbon composites. Ionics 2019, 25, 2087–2092. [Google Scholar] [CrossRef]
- Wang, J.; Fang, W.; Hu, Y.; Zhang, Y.; Dang, J.; Wu, Y.; Zhao, H.; Li, Z. Different phases of few-layer MoS2 and their silver/gold nanocomposites for efficient hydrogen evolution reaction. Catal. Sci. Technol. 2020, 10, 154–163. [Google Scholar] [CrossRef]
- Eda, G.; Yamaguchi, H.; Voiry, D.; Fujita, T.; Chen, M.; Chhowalla, M. Photoluminescence from chemically exfoliated MoS2. Nano Lett. 2011, 11, 5111–5116. [Google Scholar] [CrossRef]
- Liu, M.-C.; Xu, Y.; Hu, Y.-X.; Yang, Q.-Q.; Kong, L.-B.; Liu, W.-W.; Niu, W.-J.; Chueh, Y.-L. Electrostatically charged MoS2/graphene oxide hybrid composites for excellent electrochemical energy storage devices. ACS Appl. Mater. Interfaces 2018, 10, 35571–35579. [Google Scholar] [CrossRef]
- Zhang, X.; Huang, L.; Zeng, P.; Wu, L.; Shen, Q.; Gao, Z.; Chen, Y. Hierarchical MoS2 anchored on core-shell Si@C with increased active-sites and charge transfer for superior cycling and rate capability in lithium-ion batteries. Chem. Eng. J. 2019, 357, 625–632. [Google Scholar] [CrossRef]
- Xiao, Z.; Sheng, L.; Jiang, L.; Zhao, Y.; Jiang, M.; Zhang, X.; Zhang, M.; Shi, J.; Lin, Y.; Fan, Z. Nitrogen-doped graphene ribbons/MoS2 with ultrafast electron and ion transport for high-rate Li-ion batteries. Chem. Eng. J. 2021, 408, 127269. [Google Scholar] [CrossRef]
- Gong, C.; Long, M.; Xiao, J.; Li, J.; Chen, J.; Xiao, Y.; Zhang, G.; Gao, H.; Liu, H. One-step hydrothermal reaction induced nitrogen-doped MoS2/MXene composites with superior lithium-ion storage. Batteries 2022, 8, 156. [Google Scholar] [CrossRef]
- Cui, Y.; He, J.; Yuan, F.; Xue, J.; Li, X.; Wang, J. Preparation of MoS2 microspheres through surfactants-assisted hydrothermal synthesis using thioacetamide as reducing agent. Hydrometallurgy 2016, 164, 184–188. [Google Scholar] [CrossRef]
- Zeng, X.; Qin, W. Synthesis of MoS2 nanoparticles using MoO3 nanobelts as precursor via a PVP-assisted hydrothermal method. Mater. Lett. 2016, 182, 347–350. [Google Scholar] [CrossRef]
- Xue, H.; Yue, S.; Wang, J.; Zhao, Y.; Li, Q.; Yin, M.; Wang, S.; Feng, C.; Wu, Q.; Li, H. MoS2 microsphere@ N-doped carbon composites as high performance anode materials for lithium-ion batteries. J. Electroanal. Chem. 2019, 840, 230–236. [Google Scholar] [CrossRef]
- Xu, W.; Wang, T.; Wu, S.; Wang, S. N-doped carbon-coated MoS2 nanosheets on hollow carbon microspheres for high-performance lithium-ion batteries. J. Alloys Compd. 2017, 698, 68–76. [Google Scholar] [CrossRef]
- Choi, J.-H.; Kim, M.-C.; Moon, S.-H.; Kim, H.; Kim, Y.-S.; Park, K.-W. Enhanced electrochemical performance of MoS2/graphene nanosheet nanocomposites. RSC Adv. 2020, 10, 19077–19082. [Google Scholar] [CrossRef] [PubMed]
- Xue, H.; Jiao, Q.; Du, J.; Wang, S.; Feng, C.; Wu, Q.; Li, H.; Lu, Q.; Shi, D.; Zhao, Y. Hollow MoS2/rGO composites as high-performance anode materials for lithium-ion batteries. Ionics 2019, 25, 4659–4666. [Google Scholar] [CrossRef]
- Chen, C.; Xie, X.; Anasori, B.; Sarycheva, A.; Makaryan, T.; Zhao, M.; Urbankowski, P.; Miao, L.; Jiang, J.; Gogotsi, Y. MoS2-on-MXene heterostructures as highly reversible anode materials for lithium-ion batteries. Angew. Chem. Int. Ed. 2018, 57, 1846–1850. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Han, L.; Zhang, X.; Sun, H.; Liu, X.; Lu, T.; Yao, Y.; Pan, L. Multi-role TiO2 layer coated carbon@ few-layered MoS2 nanotubes for durable lithium storage. Chem. Eng. J. 2021, 406, 126873. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, S.; Zeng, X.; Xiao, J.; Dong, P.; Zhao, J.; Sun, S.; Huang, L.; Li, X. TiO2–MoS2 hybrid nano composites with 3D network architecture as binder-free flexible electrodes for lithium ion batteries. J. Mater. Sci. Mater. Electron. 2017, 28, 9519–9527. [Google Scholar] [CrossRef]
- Zhao, J.; Ren, H.; Gu, C.; Guan, W.; Song, X.; Huang, J. Synthesis of hierarchical molybdenum disulfide microplates consisting of numerous crosslinked nanosheets for lithium-ion batteries. J. Alloys Compd. 2019, 781, 174–185. [Google Scholar] [CrossRef]
- Li, H.; Su, Q.; Kang, J.; Feng, H.; Huang, P.; Huang, M.; Feng, M.; Tu, G.; Du, G. MoS2 nanosheets attached on carbon microspheres used as anodes for lithium ion batteries. Ceram. Int. 2018, 44, 5311–5318. [Google Scholar] [CrossRef]
- Chao, Y. Molybdenum Disulfide/Carbon Composites for Lithium Ion Batteries. Ph.D. Thesis, University of Wollongong, Wollongong, Australia, 2020. [Google Scholar]
- Sun, D.; Huang, D.; Wang, H.; Xu, G.-L.; Zhang, X.; Zhang, R.; Tang, Y.; Abd EI-Hady, D.; Alshitari, W.; AL-Bogami, A.S. 1T MoS2 nanosheets with extraordinary sodium storage properties via thermal-driven ion intercalation assisted exfoliation of bulky MoS2. Nano Energy 2019, 61, 361–369. [Google Scholar] [CrossRef]
- Zhu, S.; Dai, Y.; Li, J.; Ye, C.; Zhou, W.; Yu, R.; Liao, X.; Li, J.; Zhang, W.; Zong, W. Cathodic Zn underpotential deposition: An evitable degradation mechanism in aqueous zinc-ion batteries. Sci. Bull. 2022, 67, 1882–1889. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Hao, H.; Liu, J.; Huang, T.; Yu, A. A novel method for preparation of macroposous lithium nickel manganese oxygen as cathode material for lithium ion batteries. Electrochim. Acta 2011, 56, 4065–4069. [Google Scholar] [CrossRef]
- Liu, X.; Ma, X.; Liu, G.; Zhang, X.; Tang, X.; Li, C.; Zang, X.; Cao, N.; Shao, Q. Polyaniline spaced MoS2 nanosheets with increased interlayer distances for constructing high-rate dual-ion batteries. J. Mater. Sci. Technol. 2024, 182, 220–230. [Google Scholar] [CrossRef]
- Mao, Z.-Y.; Sun, Y.-P.; Scott, K. Evaluation of apparent lithium-ion diffusion coefficients in FePO4/LiFePO4 cathode material particles from linear non-equilibrium thermodynamics and principle of electroneutrality. J. Electroanal. Chem. 2016, 766, 107–119. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Material | Morphology | Method | Time | Initial Discharge Specific Capacity | Cyclic Stability | Ref. |
|---|---|---|---|---|---|---|
| MoS2/C | Nuclear crust structure | hydrothermal | 7 h | 705.2 mAh·g−1 | Specific capacity was 625.7 mAh·g−1 after 100 weeks of cycling | This work |
| (0.5 A·g−1) | ||||||
| MoS2/GNS | Nano | hydrothermal | 12 h | 732 mAh·g−1 | Specific capacity less than 100 mAh·g−1 after 100 weeks of cycling | [56] |
| sheet | (0.1 A·g−1) | |||||
| MoS2/rGO | Hollow microspheres | hydrothermal | 8 h | 760 mAh·g−1 | Specific capacity retention was 99.15% after 100 weeks of cycling | [57] |
| (0.5 A·g−1) | ||||||
| MoS2/Mo2TiC2Tx | Sheet | liquid phase mixing | 72 h | 646 mAh·g−1 | Specific capacity retention was 86% after 500 weeks of cycling | [58] |
| (0.1 A·g−1) | ||||||
| C@MoS2@ | Nanotube | template method-agent heat method | / | 455.2 mAh·g−1 | Specific capacity was 455.2 mAh·g−1 after 1000 cycles | [59] |
| TiO2 | (2 A·g−1) | |||||
| MoS2/TiO2 | Micrometer florid | hydrothermal | 16 h | 410.8 mAh·g−1 | Specific capacity retention was 88% after 300 weeks of cycling | [60] |
| (0.8 A·g−1) | ||||||
| MoS2-TiN | Striated | magnetron sputtering | / | 700 mAh·g−1 | Specific capacity retention was 89% after 300 weeks of cycling | [31] |
| (0.1 A·g−1) | ||||||
| MoS2 | Ultra- | thermal drive stripping method | 12 h | 450 mAh·g−1 (0.5 A·g−1) | Specific capacity retention was 94% after 200 weeks of cycling | [2] |
| thin nanosheets | ||||||
| MoS2-PVP@NC | Ball of wool | hydrothermal | 29 h | 607.1 mAh· g−1 | Specific capacity was 356 mAh·g−1 after 300 weeks of cycling | [4] |
| (1 A·g−1) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, W.; Fan, D.; Wang, W.; Yang, S.; Lu, Y.; Fu, L.; Zhang, J.; Wu, Y. One-Pot Hydrothermal Synthesis and Electrochemical Performance of Subspheroidal Core–Shell Structure MoS2/C Composite as Anode Material for Lithium-Ion Batteries. Energies 2024, 17, 1678. https://doi.org/10.3390/en17071678
Liu W, Fan D, Wang W, Yang S, Lu Y, Fu L, Zhang J, Wu Y. One-Pot Hydrothermal Synthesis and Electrochemical Performance of Subspheroidal Core–Shell Structure MoS2/C Composite as Anode Material for Lithium-Ion Batteries. Energies. 2024; 17(7):1678. https://doi.org/10.3390/en17071678
Chicago/Turabian StyleLiu, Wei, Dongsheng Fan, Wei Wang, Shenshen Yang, Yaozong Lu, Lingping Fu, Jingbo Zhang, and Yang Wu. 2024. "One-Pot Hydrothermal Synthesis and Electrochemical Performance of Subspheroidal Core–Shell Structure MoS2/C Composite as Anode Material for Lithium-Ion Batteries" Energies 17, no. 7: 1678. https://doi.org/10.3390/en17071678