
Diminishing Performance of Pt/CNT in Ethanol Oxidation after High-Potential Scanning



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental Section
3. Findings and Analysis
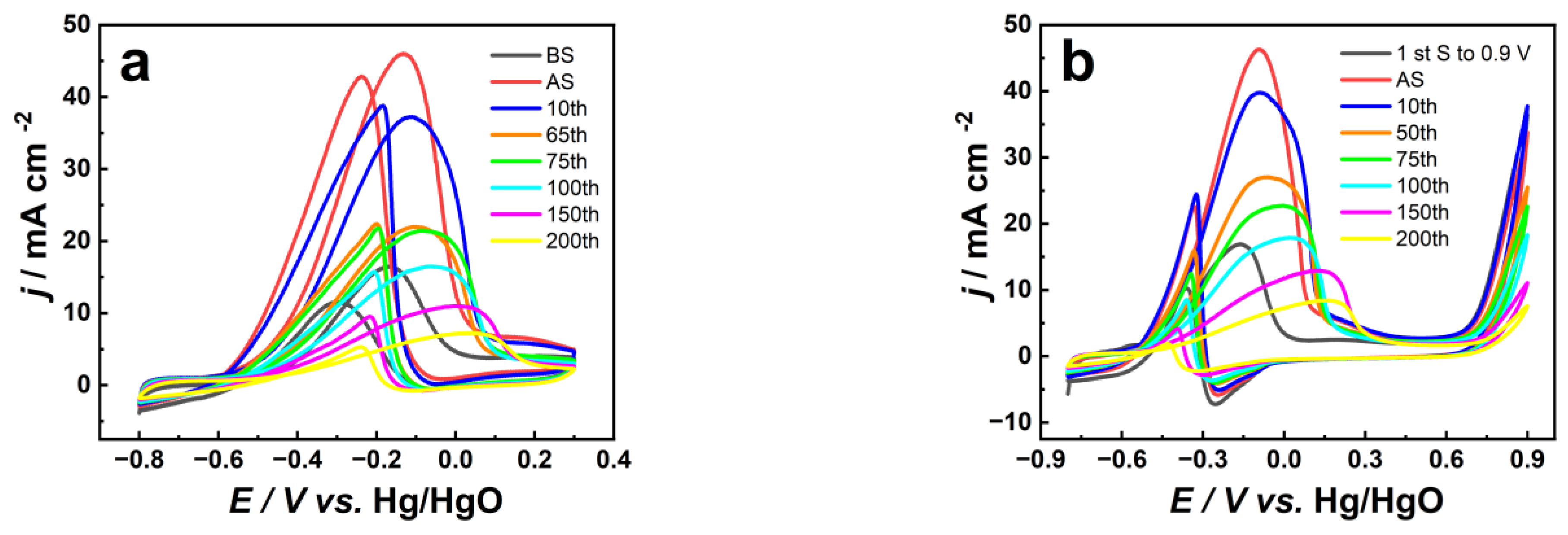
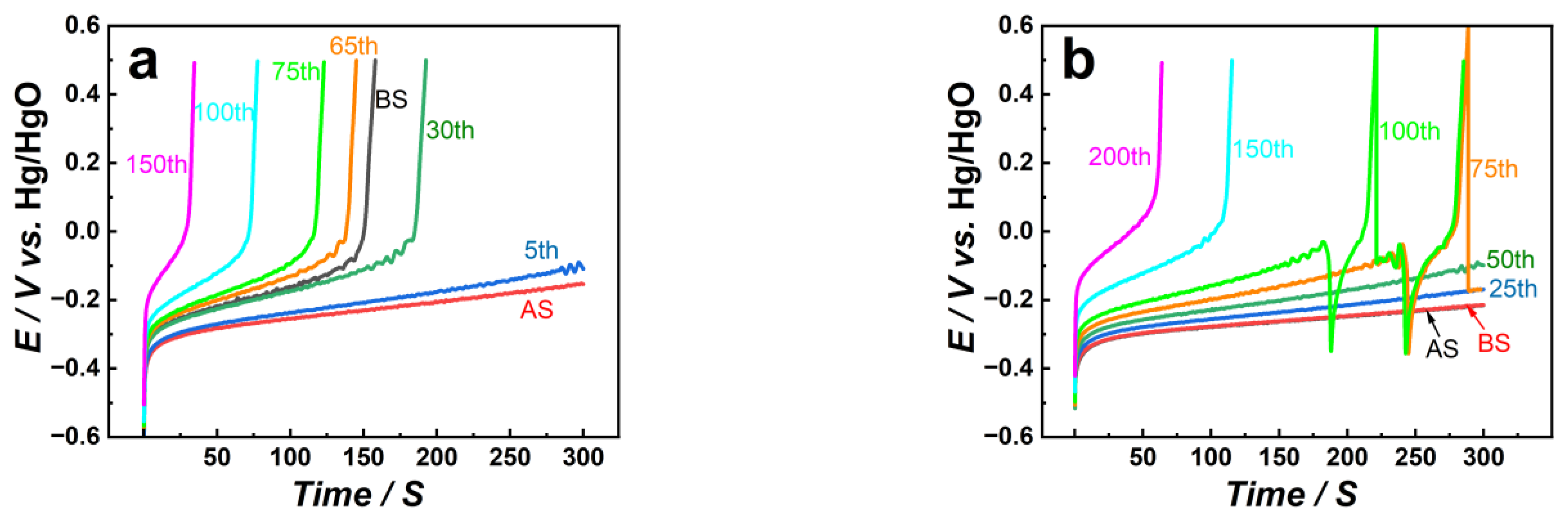
3.1. Electrochemical Behavior of Pt/CNT Catalysts
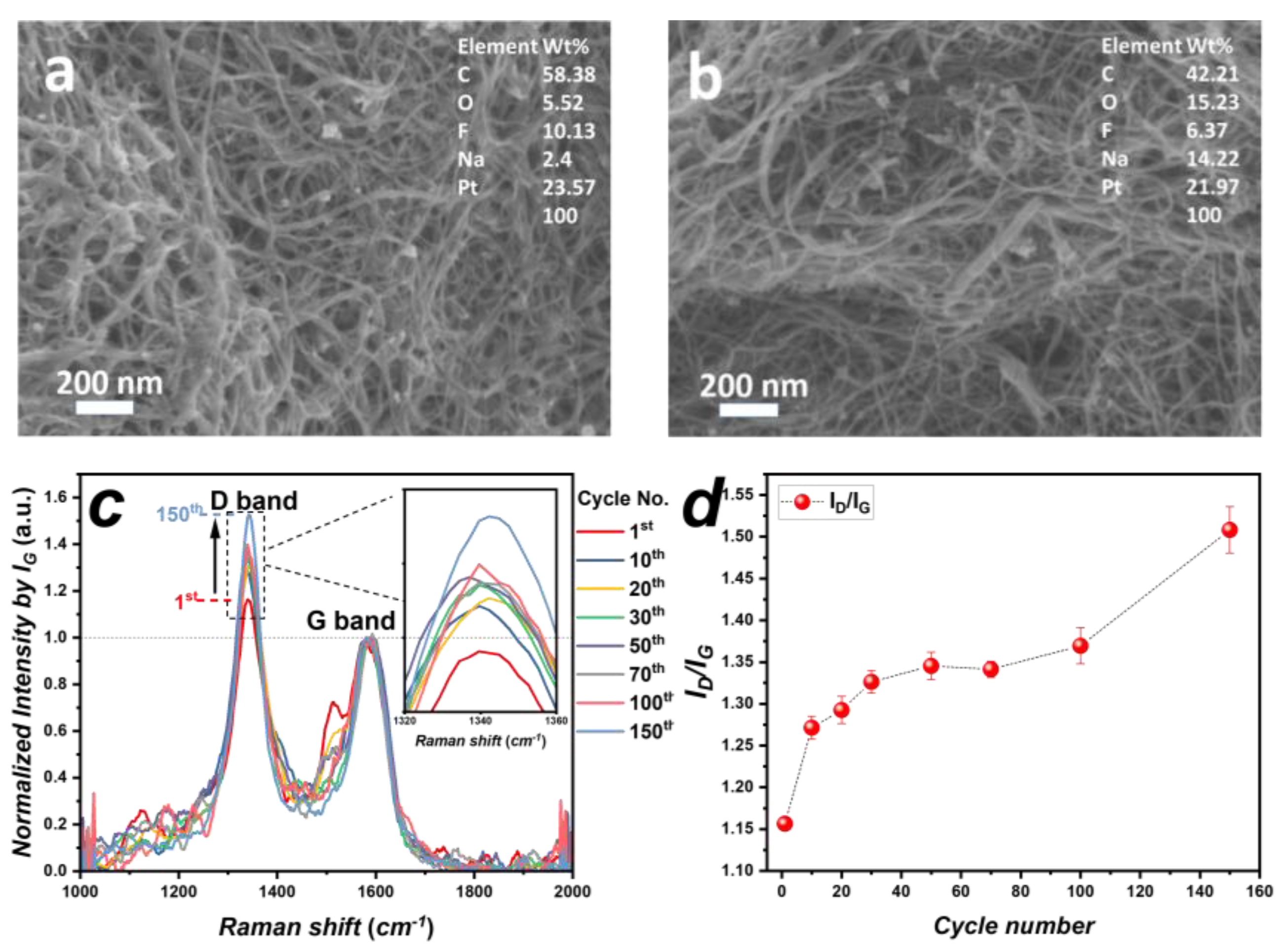
3.2. Characteristics of Pt/CNT Catalysts
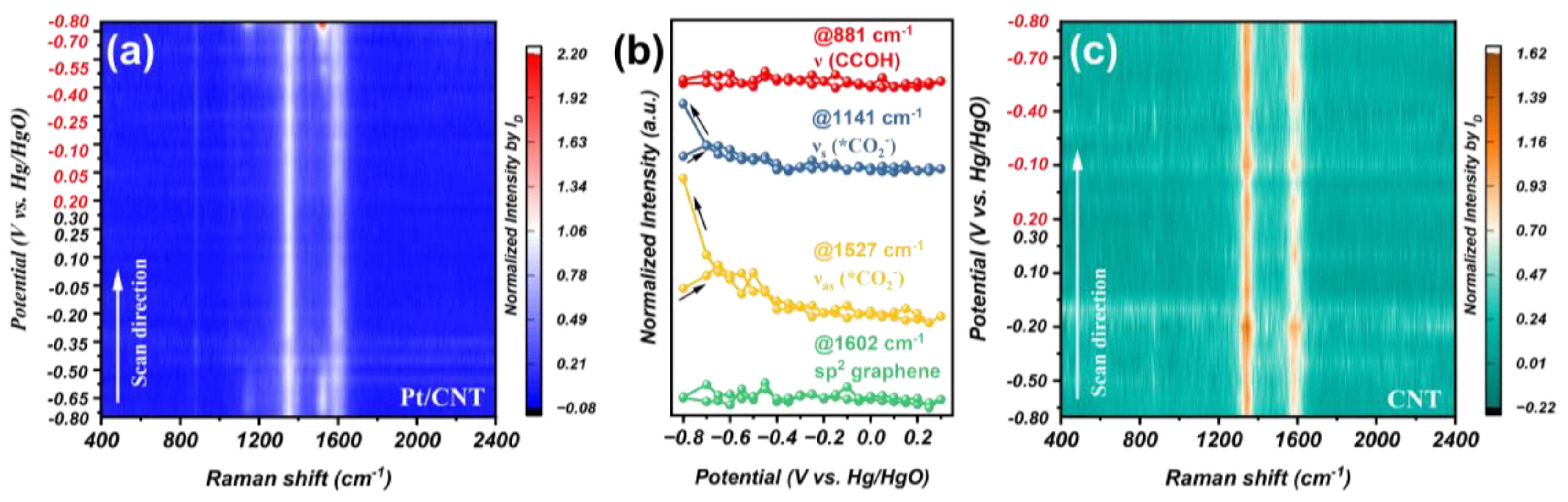
3.3. Reaction Intermediates Analyzed through In Situ Raman
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jahromi, M.M.; Heidary, H. Automotive Applications of PEM Technology; INC: New York, NY, USA, 2022; pp. 347–405. [Google Scholar] [CrossRef]
- Parekh, A. Recent developments of proton exchange membranes for PEMFC: A review. Front. Energy Res. 2022, 10, 956132. [Google Scholar] [CrossRef]
- Hong, B.K.; Kim, S.H.; Kim, C.M. Powering the future through hydrogen and polymer electrolyte membrane fuel cells current commercialisation and key challenges with focus on work at Hyundai. Johns. Matthey Technol. Rev. 2020, 64, 236–251. [Google Scholar] [CrossRef]
- Tang, A.; Yang, Y.; Yu, Q.; Zhang, Z.; Yang, L. A Review of Life Prediction Methods for PEMFCs in Electric Vehicles. Sustainability 2022, 14, 9842. [Google Scholar] [CrossRef]
- Lee, S.W.; Lee, B.H.; Kim, T.Y.; Baik, C.; Kim, M.S.; Chai, G.S.; Pak, C. Multifunctional non-Pt ternary catalyst for the hydrogen oxidation and oxygen evolution reactions in reversal-tolerant anode. Catal. Commun. 2019, 130, 105758. [Google Scholar] [CrossRef]
- You, E.; Lee, S.W.; You, D.; Lee, B.; Pak, C. Effect of metal composition and carbon support on the durability of the reversal-tolerant anode with irru alloy catalyst. Catalysts 2020, 10, 932. [Google Scholar] [CrossRef]
- Gao, Z.; Liu, F.; Su, J.; Guo, L.; Liu, H. Experimental investigation of reverse voltage phenomenon during galvanostatic start-up of a proton exchange membrane fuel cell. Energy Convers. Manag. 2022, 258, 115386. [Google Scholar] [CrossRef]
- Qin, C.; Wang, J.; Yang, D.; Li, B.; Zhang, C. Proton exchange membrane fuel cell reversal: A review. Catalysts 2016, 6, 197. [Google Scholar] [CrossRef]
- Cai, C.; Wan, Z.; Rao, Y.; Chen, W.; Zhou, J.; Tan, J.; Pan, M. Water electrolysis plateau in voltage reversal process for proton exchange membrane fuel cells. J. Power Sources 2020, 455, 227952. [Google Scholar] [CrossRef]
- Chen, H.; Zhao, X.; Zhang, T.; Pei, P. The reactant starvation of the proton exchange membrane fuel cells for vehicular applications: A review. Energy Convers. Manag. 2019, 182, 282–298. [Google Scholar] [CrossRef]
- Dou, M.; Hou, M.; Liang, D.; Shen, Q.; Zhang, H.; Lu, W.; Shao, Z.; Yi, B. Behaviors of proton exchange membrane fuel cells under oxidant starvation. J. Power Sources 2011, 196, 2759–2762. [Google Scholar] [CrossRef]
- Wang, Y.; Xie, X.; Zhou, C.; Feng, Q.; Zhou, Y.; Yuan, X.Z.; Xu, J.; Fan, J.; Zeng, L.; Li, H.; et al. Study of relative humidity on durability of the reversal tolerant proton exchange membrane fuel cell anode using a segmented cell. J. Power Sources 2020, 449, 227542. [Google Scholar] [CrossRef]
- Cai, C.; Rao, Y.; Zhou, J.; Zhang, L.; Chen, W.; Wan, Z.; Tan, J.; Pan, M. Carbon corrosion: A novel termination mechanism of the water electrolysis plateau during voltage reversal. J. Power Sources 2020, 473, 228542. [Google Scholar] [CrossRef]
- Wang, Y.; Liao, J.; Li, Z.; Wu, B.; Lou, J.; Zeng, L.; Zhao, T. Ir-Pt/C composite with high metal loading as a high-performance anti-reversal anode catalyst for proton exchange membrane fuel cells. Int. J. Hydrogen Energy 2022, 47, 13101–13111. [Google Scholar] [CrossRef]
- Labi, T.; Van Schalkwyk, F.; Andersen, S.M.; Morgen, P.; Ray, S.C.; Chamier, J. Increasing fuel cell durability during prolonged and intermittent fuel starvation using supported IrOx. J. Power Sources 2021, 490, 229568. [Google Scholar] [CrossRef]
- Zhou, X.; Ji, H.; Li, B.; Zhang, C. High-Repetitive Reversal Tolerant Performance of Proton-Exchange Membrane Fuel Cell by Designing a Suitable Anode. ACS Omega 2020, 5, 10099–10105. [Google Scholar] [CrossRef]
- Wang, Y.; Leung, D.Y.C.; Xuan, J.; Wang, H. A review on unitized regenerative fuel cell technologies, part-A: Unitized regenerative proton exchange membrane fuel cells. Renew. Sustain. Energy Rev. 2016, 65, 961–977. [Google Scholar] [CrossRef]
- Rocha, A.; Ferreira, R.B.; Falcão, D.S.; Pinto, A.M.F.R. Experimental study on a unitized regenerative fuel cell operated in constant electrode mode: Effect of cell design and operating conditions. Renew. Energy 2023, 215, 118870. [Google Scholar] [CrossRef]
- Gayen, P.; Saha, S.; Liu, X.; Sharma, K.; Ramani, V.K. High-performance AEM unitized regenerative fuel cell using Pt-pyrochlore as bifunctional oxygen electrocatalyst. Proc. Natl. Acad. Sci. USA 2021, 118, e2107205118. [Google Scholar] [CrossRef]
- Rivera-Gavidia, L.M.; Fernández de la Puente, I.; Hernández-Rodríguez, M.A.; Celorrio, V.; Sebastián, D.; Lázaro, M.J.; Pastor, E.; García, G. Bi-functional carbon-based catalysts for unitized regenerative fuel cells. J. Catal. 2020, 387, 138–144. [Google Scholar] [CrossRef]
- Wang, Y.J.; Fang, B.; Wang, X.; Ignaszak, A.; Liu, Y.; Li, A.; Zhang, L.; Zhang, J. Recent advancements in the development of bifunctional electrocatalysts for oxygen electrodes in unitized regenerative fuel cells (URFCs). Prog. Mater. Sci. 2018, 98, 108–167. [Google Scholar] [CrossRef]
- Moore, C.E.; Eastcott, J.; Cimenti, M.; Kremliakova, N.; Gyenge, E.L. Novel methodology for ex situ characterization of iridium oxide catalysts in voltage reversal tolerant proton exchange membrane fuel cell anodes. J. Power Sources 2019, 417, 53–60. [Google Scholar] [CrossRef]
- Roh, C.W.; Kim, H.E.; Choi, J.; Lim, J.; Lee, H. Monodisperse IrOx deposited on Pt/C for reversal tolerant anode in proton exchange membrane fuel cell. J. Power Sources 2019, 443, 227270. [Google Scholar] [CrossRef]
- Mandal, P.; Hong, B.K.; Oh, J.G.; Litster, S. Understanding the voltage reversal behavior of automotive fuel cells. J. Power Sources 2018, 397, 397–404. [Google Scholar] [CrossRef]
- Chang, J.; Wang, G.; Chang, X.; Yang, Z.; Wang, H.; Li, B.; Zhang, W.; Kovarik, L.; Du, Y.; Orlovskaya, N.; et al. Interface synergism and engineering of Pd/Co@N-C for direct ethanol fuel cells. Nat. Commun. 2023, 14, 1346. [Google Scholar] [CrossRef] [PubMed]
- Begum, H.; Ahmed, M.S.; Lee, D.W.; Kim, Y.B. Carbon nanotubes-based PdM bimetallic catalysts through N4-system for efficient ethanol oxidation and hydrogen evolution reaction. Sci. Rep. 2019, 9, 11051. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Chen, S.; Qian, G.; Song, L.; Chen, D.; Zhou, X.; Duan, X. On the nature of Pt-carbon interactions for enhanced hydrogen generation. J. Catal. 2020, 389, 492–501. [Google Scholar] [CrossRef]
- Azcoaga Chort, M.F.; Nagel, P.A.; Veizaga, N.S.; Rodríguez, V.I.; de Miguel, S.R. Effect of Sn content on Pt/CNT electrocatalysts for direct ethanol fuel cell application. Can. J. Chem. Eng. 2022, 100, 1848–1857. [Google Scholar] [CrossRef]
- Tian, M.H.; Yang, Y.; Desmond, C.; Liu, F.; Zhu, Z.Q.; Li, Q.X. Pt-Surface-Enriched Platinum–Tungsten Bimetallic Nanoparticles Catalysts on Different Carbon Supports for Electro-Oxidation of Ethanol. Catal. Lett. 2020, 150, 3386–3393. [Google Scholar] [CrossRef]
- Huang, H.; Zhang, W.; Fu, Y.; Wang, X. Controlled growth of nanostructured MnO2 on carbon nanotubes for high-performance electrochemical capacitors. Electrochim. Acta 2015, 152, 480–488. [Google Scholar] [CrossRef]
- Ning, L.; Liu, X.; Deng, M.; Huang, Z.; Zhu, A.; Zhang, Q.; Liu, Q. Palladium-based nanocatalysts anchored on CNT with high activity and durability for ethanol electro-oxidation. Electrochim. Acta 2019, 297, 206–214. [Google Scholar] [CrossRef]
- Hu, F.; Chen, W. End-opened carbon nanotube array supported Pd as anode for alkaline fuel cell. Electrochem. Commun. 2011, 13, 955–958. [Google Scholar] [CrossRef]
- Ramachandran, K.; Vinothkannan, M.; Kim, A.R.; Ramakrishnan, S.; Yoo, D.J. Ultrafine bimetallic alloy supported on nitrogen doped reduced graphene oxide toward liquid-fuel oxidation: Profile of improved performance and extended durability. Int. J. Hydrogen Energy 2019, 44, 21769–21780. [Google Scholar] [CrossRef]
- Raj Kumar, T.; Jin Yoo, D.; Kim, A.R.; Gnana Kumar, G. Green synthesis of Pt-Pd bimetallic nanoparticle decorated reduced graphene oxide and its robust catalytic activity for efficient ethylene glycol electrooxidation. New J. Chem. 2018, 42, 14386–14393. [Google Scholar] [CrossRef]
- Zhang, W.; Sherrell, P.; Minett, A.I.; Razal, J.M.; Chen, J. Carbon nanotube architectures as catalyst supports for proton exchange membrane fuel cells. Energy Environ. Sci. 2010, 3, 1286–1293. [Google Scholar] [CrossRef]
- Kim, M.J.; Lee, K.B.; Seo, M.G.; Lee, K.Y. Porous MnO2/CNT catalysts with a large specific surface area for the decomposition of hydrogen peroxide. Korean J. Chem. Eng. 2017, 34, 2147–2153. [Google Scholar] [CrossRef]
- Tang, T.; Gan, Q.; Guo, X.; Dong, H.; Zhang, J.; Zhao, Y.; Tian, J.; Yang, X. A hybrid catalyst of Pt/CoNiO2 on carbon nanotubes and its synergetic effect towards remarkable ethanol electro-oxidation in alkaline media. Sustain. Energy Fuels 2018, 2, 229–236. [Google Scholar] [CrossRef]
- Yin, S.; Zhu, Q.; Yang, L.; Qiang, Y.; Huang, F.; Luo, L. Carbon nanotubes supported Pt catalysts for ethanol oxidation in. Adv. Mater. Res. 2013, 662, 132–135. [Google Scholar] [CrossRef]
- Hu, F.; Cui, X.; Chen, W. Ultralong-CNTA-supported Pd-based anodes for ethanol oxidation. J. Phys. Chem. C 2010, 114, 20284–20289. [Google Scholar] [CrossRef]
- Dong, J.C.; Zhang, X.G.; Briega-Martos, V.; Jin, X.; Yang, J.; Chen, S.; Yang, Z.L.; Wu, D.Y.; Feliu, J.M.; Williams, C.T.; et al. In situ Raman spectroscopic evidence for oxygen reduction reaction intermediates at platinum single-crystal surfaces. Nat. Energy 2019, 4, 60–67. [Google Scholar] [CrossRef]
- Xu, J.; Wang, B.-X.; Lyu, D.; Wang, T.; Wang, Z. In situ Raman investigation of the femtosecond laser irradiated NiFeOOH for enhanced electrochemical ethanol oxidation reaction. Int. J. Hydrogen Energy 2023, 48, 10724–10736. [Google Scholar] [CrossRef]
- Tiwari, J.N.; Dang, N.K.; Park, H.J.; Sultan, S.; Kim, M.G.; Haiyan, J.; Lee, Z.; Kim, K.S. Remarkably enhanced catalytic activity by the synergistic effect of palladium single atoms and palladium–cobalt phosphide nanoparticles. Nano Energy 2020, 78, 105166. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Zhu, Y.Z.; Li, J.F. Application of raman spectroscopy in fuel cell. Wuli Huaxue Xuebao/Acta Phys.-Chim. Sin. 2021, 37, 2004052. [Google Scholar] [CrossRef]
- Dong, J.C.; Su, M.; Briega-Martos, V.; Li, L.; Le, J.B.; Radjenovic, P.; Zhou, X.S.; Feliu, J.M.; Tian, Z.Q.; Li, J.F. Direct in Situ Raman Spectroscopic Evidence of Oxygen Reduction Reaction Intermediates at High-Index Pt(hkl) Surfaces. J. Am. Chem. Soc. 2020, 142, 715–719. [Google Scholar] [CrossRef]
- Wang, Y.H.; Zheng, S.; Yang, W.M.; Zhou, R.Y.; He, Q.F.; Radjenovic, P.; Dong, J.C.; Li, S.; Zheng, J.; Yang, Z.L.; et al. In situ Raman spectroscopy reveals the structure and dissociation of interfacial water. Nature 2021, 600, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Hu, F.; Xu, J.; Wei, L.; Wang, Z.; Jiang, F. In situ Raman study of high potential scan enhanced Pt/C and Pd/C catalyst performance for ethanol oxidation. Electrochem. Commun. 2023, 154, 107561. [Google Scholar] [CrossRef]
- Wang, C.; Van Der Vliet, D.; More, K.L.; Zaluzec, N.J.; Peng, S.; Sun, S.; Daimon, H.; Wang, G.; Greeley, J.; Pearson, J.; et al. Multimetallic Au/FePt3 nanoparticles as highly durable electrocatalyst. Nano Lett. 2011, 11, 919–926. [Google Scholar] [CrossRef]
- Chen, T.W.; Huang, W.F.; Kang, J.X.; Zhang, D.F.; Guo, L. Cycling potential engineering surface configuration of sandwich Au@Ni@PtNiAu for superior catalytic durability. Nano Energy 2018, 52, 22–28. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, F.; Xu, J.; Wei, L.; Wang, Z.; Jiang, F. Diminishing Performance of Pt/CNT in Ethanol Oxidation after High-Potential Scanning. Energies 2024, 17, 2122. https://doi.org/10.3390/en17092122
Hu F, Xu J, Wei L, Wang Z, Jiang F. Diminishing Performance of Pt/CNT in Ethanol Oxidation after High-Potential Scanning. Energies. 2024; 17(9):2122. https://doi.org/10.3390/en17092122
Chicago/Turabian StyleHu, Fengping, Jinchang Xu, Lin Wei, Zhenyou Wang, and Fangming Jiang. 2024. "Diminishing Performance of Pt/CNT in Ethanol Oxidation after High-Potential Scanning" Energies 17, no. 9: 2122. https://doi.org/10.3390/en17092122
APA StyleHu, F., Xu, J., Wei, L., Wang, Z., & Jiang, F. (2024). Diminishing Performance of Pt/CNT in Ethanol Oxidation after High-Potential Scanning. Energies, 17(9), 2122. https://doi.org/10.3390/en17092122