

An n-Heptane Oxidation Mechanism Suitable for Low- to High-Temperature Combustion


Abstract
:1. Introduction
2. Selection of Comparison Mechanism
3. Construction of a Simplified Mechanism of n-Heptane
3.1. Construction of Skeleton Mechanism
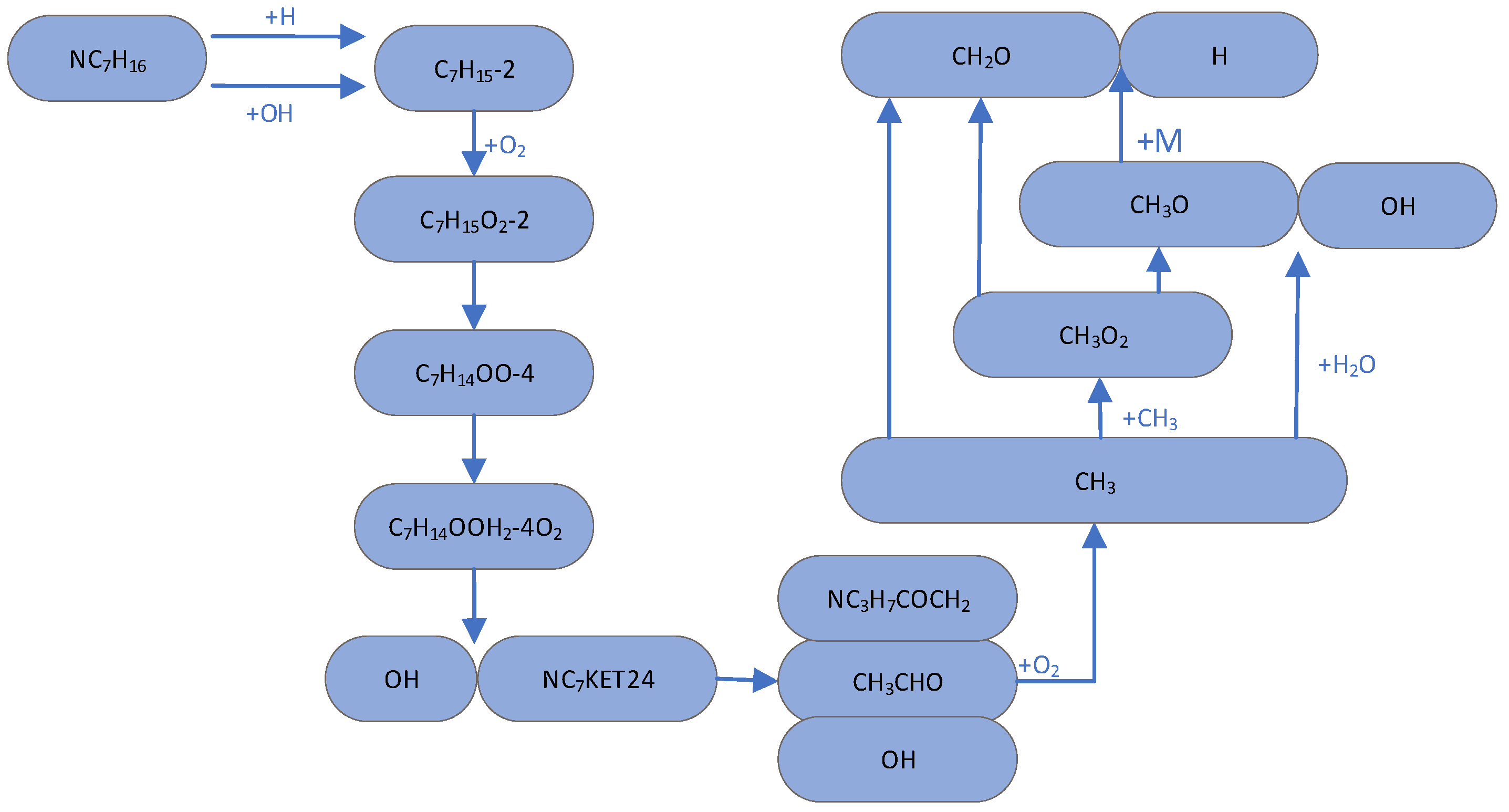
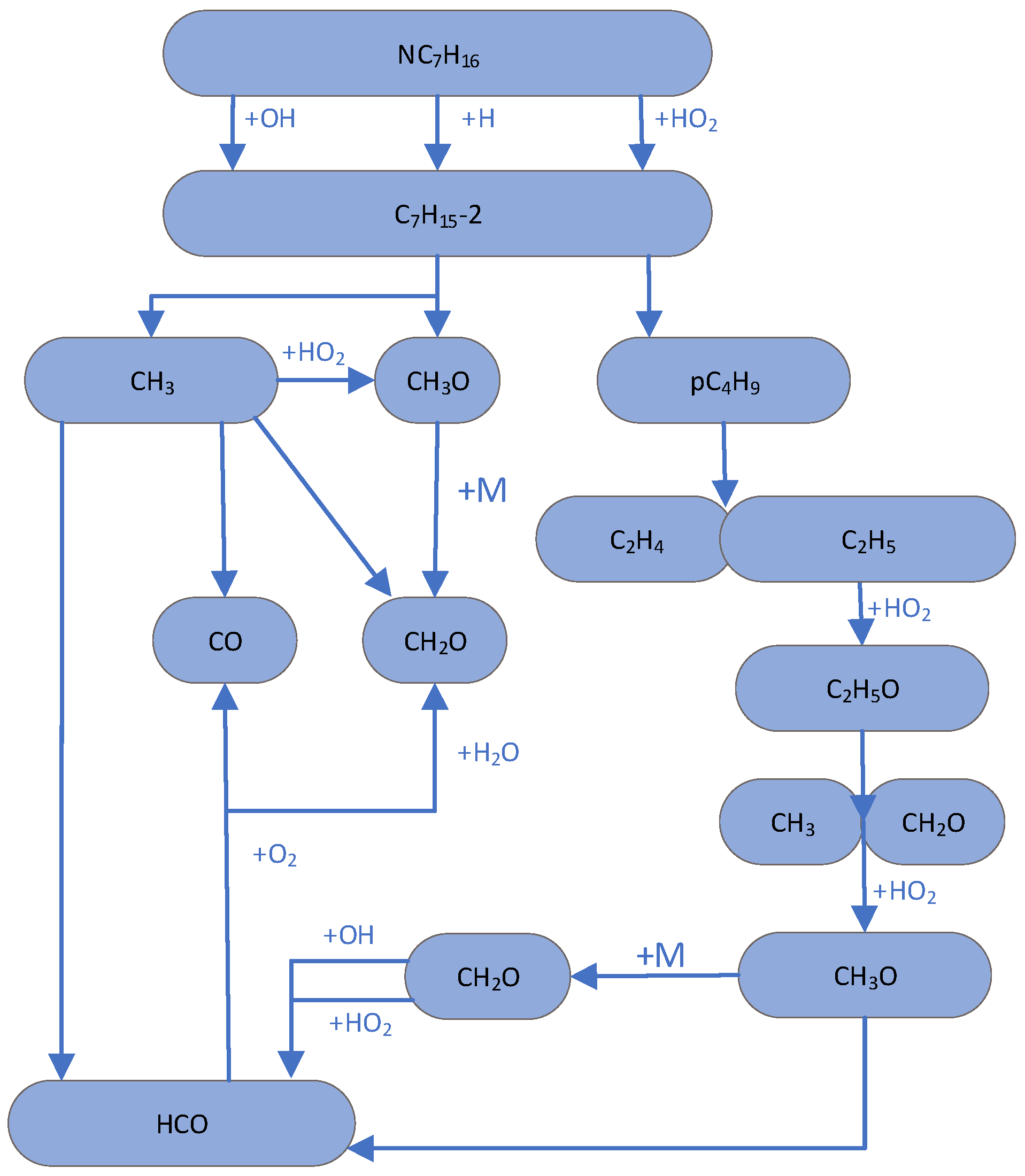
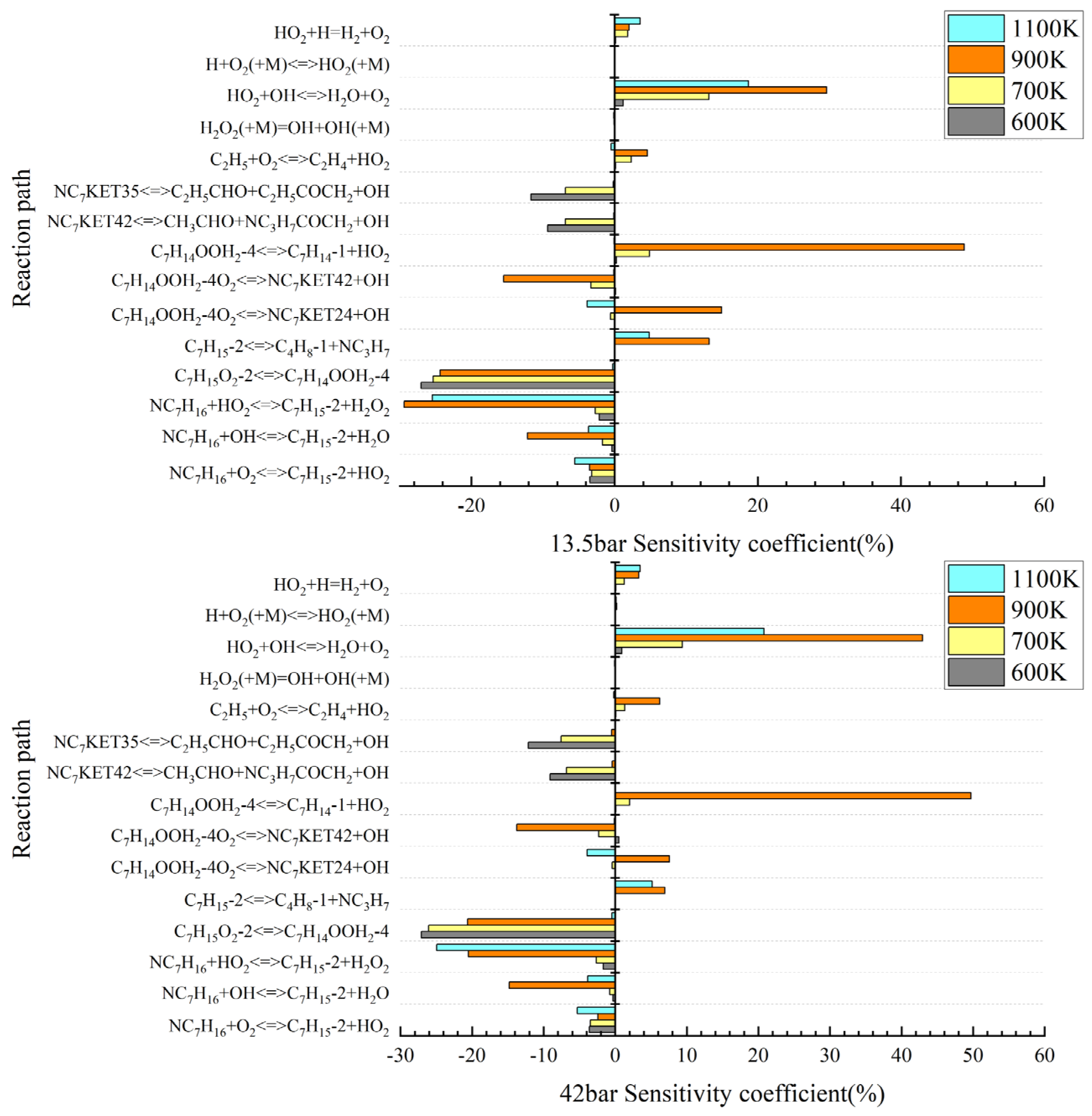
3.2. Important Primitives and Path Supplements in the Initial Stage of the Low-Temperature Reaction
| NC7H16 + O2 => C7H15-2 + HO2 | C7H14OOH2-4O2 => NC7KET35 + OH |
| NC7H16 + H => C7H15-2 + H2 | C7H14OOH2-4O2 => NC7KET42 + OH |
| NC7H16 + OH => C7H15-2 + H2O | C7H14OOH2-4 + O2 => C7H14OOH2-4O2 |
| NC7H16 + HO2 => C7H15-2 + H2O2 | NC7KET42 => CH3CHO + NC3H7COCH2 + OH |
| C7H15-2 + O2 => C7H15O2-2 | NC7KET24 => NC3H7CHO + CH3COCH2 + OH |
| C7H15O2-2 => C7H14OOH2-4 | NC7KET35 => C2H5CHO + C2H5COCH2 + OH |
| C7H15-2 => C7H14-1 + H | C7H14OOH2-4 => C7H14-3 + HO2 |
| C7H15-2 => pC4H9 + C3H6 | C7H14OOH2-4 => C7H14O2-4 + OH |
| C7H15-2 => C4H8-1 + NC3H7 | C7H14OOH2-4 => OH + C2H5CHO + C4H8-1 |
| C7H15-2 => C2H5 + C5H10-1 | C7H14OOH2-4 => OH + CH3CHO + C5H10-1 |
| C7H14OOH2-4O2 => NC7KET24 + OH | C7H14OOH2-4 => OH + NC3H7CHO + C3H6 |
3.3. Study on Reaction Intermediates and Paths
| 1. | NC7H16 + O2 <=> C7H15-2 + HO2 | 4.000 × 1013 => 4.1 × 1013 |
| 2. | NC7H16 + OH <=> C7H15-2 + H2O | 9.400 × 107 => 1.6 × 108 |
| 3. | NC7H16 + HO2 <=> C7H15-2 + H2O2 | 3 × 1013 => 2.1 × 1013 |
| 4. | C7H15O2-2 <=> C7H14OOH2-4 | 2.500 × 1010 => 3 × 1010 |
| 5. | C7H14OOH2-4O2 <=> NC7KET42 + OH | 1.250 × 1010 => 1.5 × 1010 |
| 6. | C7H14OOH2-4 <=> C7H14-1 + HO2 | 6.615 × 1018 => 1.3 × 1019 |
| 7. | HO2 + H = H2 + O2 | 1.660 × 1013 => 0.7 × 1013 |
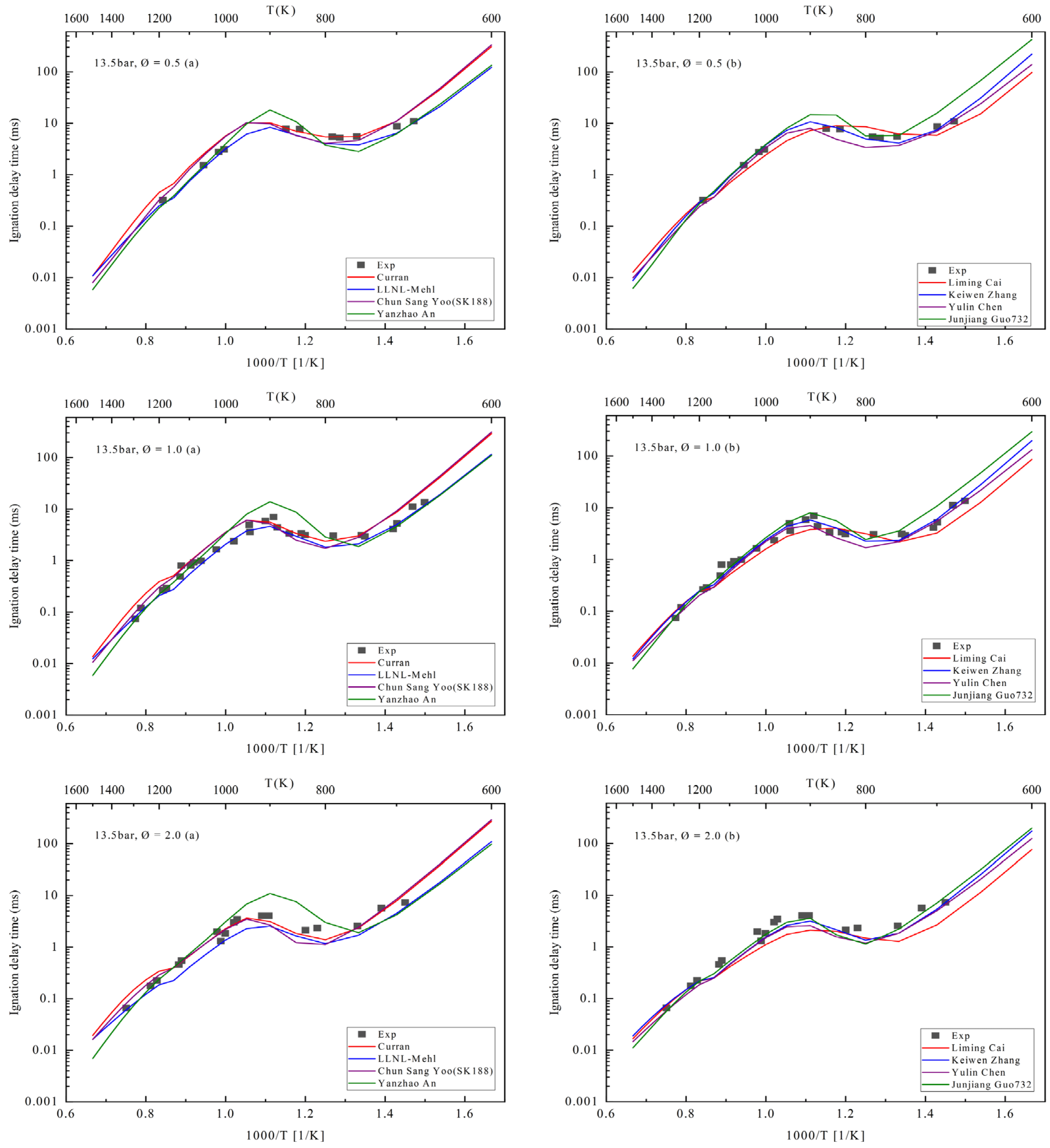
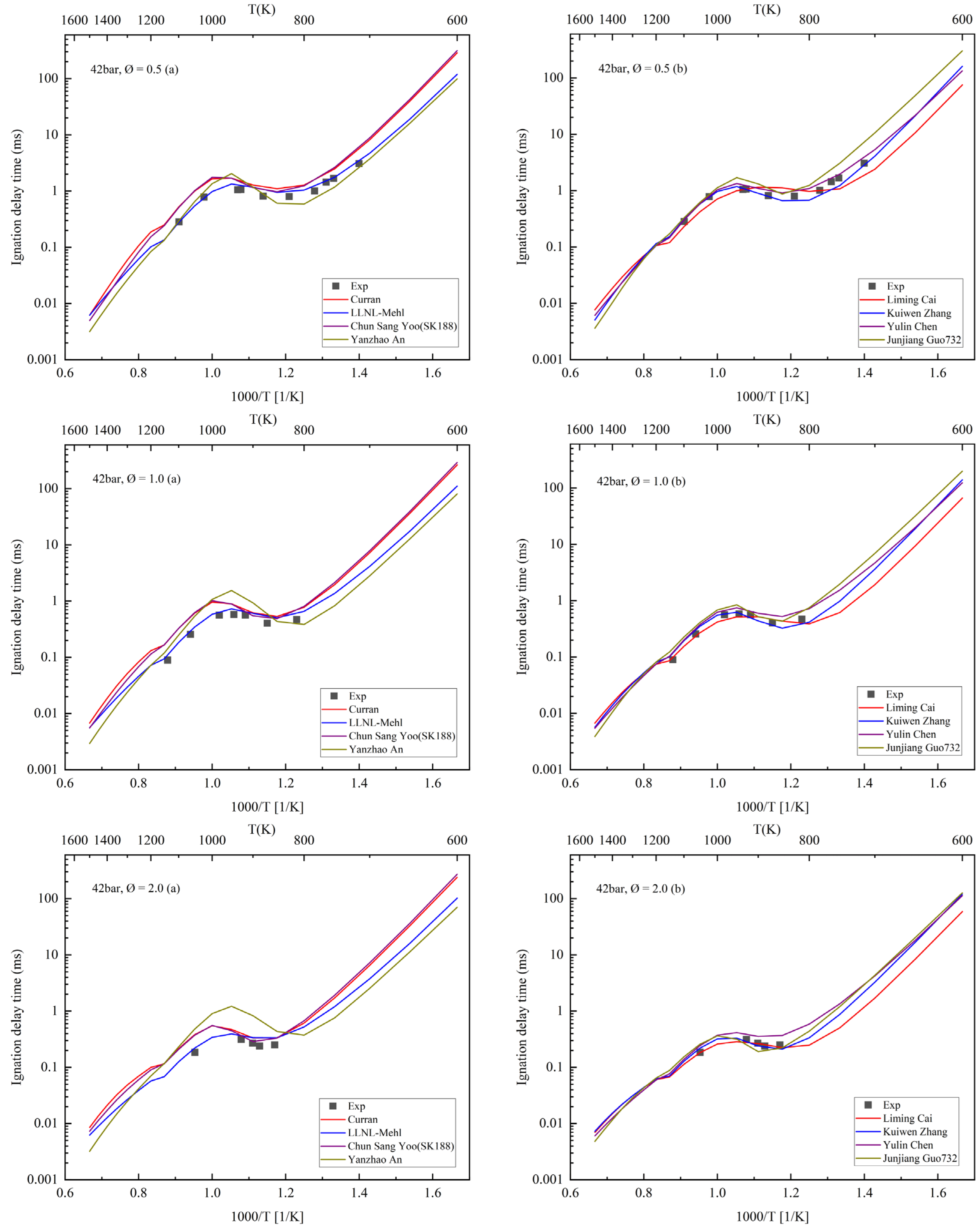
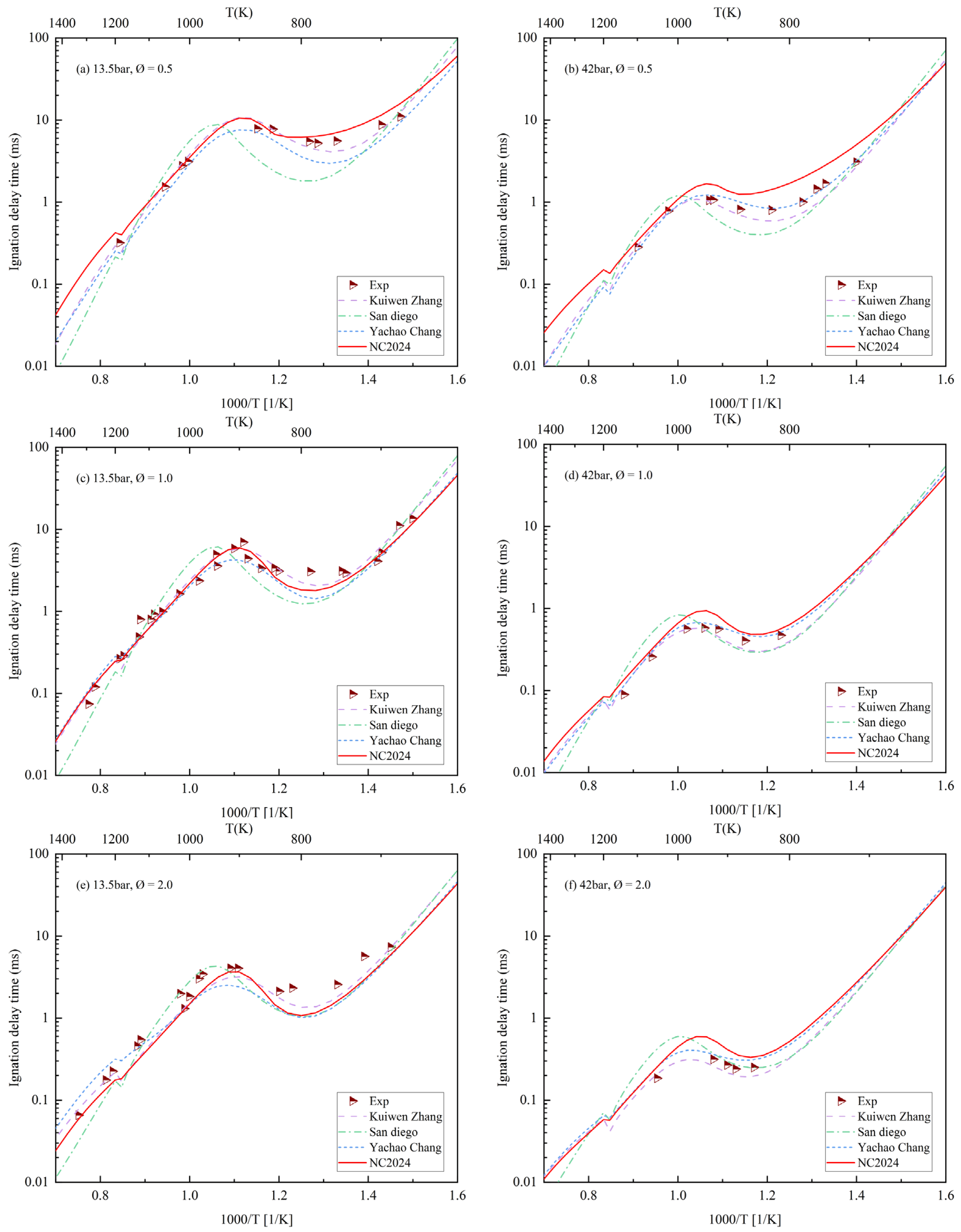
4. Ignition Delay Period Verification
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Wu, Y.; Panigrahy, S.; Sahu, A.B.; Bariki, C.; Beeckmann, J.; Liang, J.; Mohamed, A.A.; Dong, S.; Tang, C.; Pitsch, H.; et al. Understanding the antagonistic effect of methanol as a component in surrogate fuel models: A case study of methanol/-heptane mixtures. Combust. Flame 2021, 226, 229–242. [Google Scholar] [CrossRef]
- Zhu, J.; Zhou, D.; Yu, L.; Qian, Y.; Lu, X. Construction of a skeletal multi-component diesel surrogate model by integrating chemical lumping and genetic algorithm. Fuel 2022, 313, 122711. [Google Scholar] [CrossRef]
- Coats, C.M.; Williams, A. Investigation of the ignition and combustion of n-heptane-oxygen mixtures. Symp. (Int.) Combust. 1979, 17, 611–621. [Google Scholar] [CrossRef]
- Westbrook, C.K.; Dryer, F.L. Chemical kinetics and modeling of combustion processes. Symp. (Int.) Combust. 1981, 18, 749–767. [Google Scholar] [CrossRef]
- Westbrook, C.K.; Warnatz, J.; Pitz, W.J. A detailed chemical kinetic reaction mechanism for the oxidation of iso-octane and n-heptane over an extended temperature range and its application to analysis of engine knock. Symp. (Int.) Combust. 1989, 22, 893–901. [Google Scholar] [CrossRef]
- Curran, H.; Gaffuri, P.; Pitz, W.; Westbrook, C. A Comprehensive Modeling Study of n-Heptane Oxidation. Combust. Flame 1998, 114, 149–177. [Google Scholar] [CrossRef]
- Mehl, M.; Pitz, W.J.; Westbrook, C.K.; Curran, H.J. Kinetic modeling of gasoline surrogate components and mixtures under engine conditions. Proc. Combust. Inst. 2011, 33, 193–200. [Google Scholar] [CrossRef]
- Mehl, M.; Vanhove, G.; Pitz, W.J.; Ranzi, E. Oxidation and combustion of the n-hexene isomers: A wide range kinetic modeling study. Combust. Flame 2008, 155, 756–772. [Google Scholar] [CrossRef]
- Sarathy, S.; Westbrook, C.; Mehl, M.; Pitz, W.; Togbe, C.; Dagaut, P.; Wang, H.; Oehlschlaeger, M.; Niemann, U.; Seshadri, K.; et al. Comprehensive chemical kinetic modeling of the oxidation of 2-methylalkanes from C7 to C20. Combust. Flame 2011, 158, 2338–2357. [Google Scholar] [CrossRef]
- Doute Christine, D.J.-L.; Christian, V. Detailed Reaction Mechanisms for Low Pressure Premixed n-Heptane Flames. Combust. Sci. Technol. 1999, 147, 61–109. [Google Scholar] [CrossRef]
- Seiser, R.; Pitsch, H.; Seshadri, K.; Pitz, W.; Gurran, H. Extinction and autoignition of n-heptane in counterflow configuration. Proc. Combust. Inst. 2000, 28, 2029–2037. [Google Scholar] [CrossRef]
- Curran, H. A comprehensive modeling study of iso-octane oxidation. Combust. Flame 2002, 129, 253–280. [Google Scholar] [CrossRef]
- Chen, Y.L.; Chen, J.Y. Towards improved automatic chemical kinetic model reduction regarding ignition delays and flame speeds. Combust. Flame 2018, 190, 293–301. [Google Scholar] [CrossRef]
- Cai, L.; Pitsch, H.; Mohamed, S.Y.; Raman, V.; Bugler, J.; Curran, H.; Sarathy, S.M. Optimized reaction mechanism rate rules for ignition of normal alkanes. Combust. Flame 2016, 173, 468–482. [Google Scholar] [CrossRef]
- Ranzi, E. A wide-range modeling study of n-heptane oxidation. Combust. Flame 1995, 103, 91–106. [Google Scholar] [CrossRef]
- Ranzi, E. A Wide-Range Kinetic Modeling Study of Oxidation and Combustion of Transportation Fuels and Surrogate Mixtures. Energy Fuels 2006, 20, 1024–1032. [Google Scholar] [CrossRef]
- Ranzi, E.; Frassoldati, A.; Grana, R.; Cuoci, A.; Faravelli, T.; Kelley, A.P.; Law, C.K. Hierarchical and comparative kinetic modeling of laminar flame speeds of hydrocarbon and oxygenated fuels. Prog. Energy Combust. Sci. 2012, 38, 468–501. [Google Scholar] [CrossRef]
- Pelucchi, M.; Bissoli, M.; Cavallotti, C.; Cuoci, A.; Faravelli, T.; Frassoldati, A.; Ranzi, E.; Stagni, A. Improved Kinetic Model of the Low-Temperature Oxidation of n-Heptane. Energy Fuels 2014, 28, 7178–7193. [Google Scholar] [CrossRef]
- Herbinet, O.; Husson, B.; Serinyel, Z.; Cord, M.; Warth, V.; Fournet, R.; Glaude, P.-A.; Sirjean, B.; Battin-Leclerc, F.; Wang, Z.; et al. Experimental and modeling investigation of the low-temperature oxidation of n-heptane. Combust. Flame 2012, 159, 3455–3471. [Google Scholar] [CrossRef]
- Hakka, H.; Cracknell, R.; Pekalski, A.; Glaude, P.-A.; Battin-Leclerc, F. Experimental and modeling study of ultra-rich oxidation of n-heptane. Fuel 2015, 144, 358–368. [Google Scholar] [CrossRef]
- Nehse, M.; Warnat, J.; Chevalier, C. Kinetic modeling of the oxidation of large aliphatic hydrocarbons. Symp. (Int.) Combust. 1996, 26, 773–780. [Google Scholar] [CrossRef]
- Ahmed, S.S.; Mauß, F.; Moréac, G.; Zeuch, T. A comprehensive and compact n-heptane oxidation model derived using chemical lumping. Phys. Chem. Chem. Phys. 2007, 9, 1107–1126. [Google Scholar] [CrossRef]
- Yoo, C.S.; Lu, T.; Chen, J.H.; Law, C.K. Direct numerical simulations of ignition of a lean-heptane/air mixture with temperature inhomogeneities at constant volume: Parametric study. Combust. Flame 2011, 158, 1727–1741. [Google Scholar] [CrossRef]
- Liu, Z.; Yang, L.; Song, E.; Wang, J.; Zare, A.; Bodisco, T.A.; Brown, R.J. Development of a reduced multi-component combustion mechanism for a diesel/natural gas dual fuel engine by cross-reaction analysis. Fuel 2021, 293, 120388. [Google Scholar] [CrossRef]
- Liu, X.L.; Wang, H.; Yao, M.F. Investigation of the chemical kinetics process of diesel combustion in a compression ignition engine using the large eddy simulation approach. Fuel 2020, 270, 117544. [Google Scholar] [CrossRef]
- Kohse-Höinghaus, K. Combustion in the future: The importance of chemistry. Proc. Combust. Inst. 2021, 38, 1–56. [Google Scholar] [CrossRef]
- Jinzhe Zeng, L.C.; Xu, M.; Zhu, T.; John, Z.; Zhang, H. Complex reaction processes in combustion unraveled by neural network-based molecular dynamics simulation. Nat. Commun. 2020, 11, 5713. [Google Scholar] [CrossRef]
- Klippenstein, S.J. From theoretical reaction dynamics to chemical modeling of combustion. Theor. React. Dyn. Chem. Model. Combust. 2017, 36, 77–111. [Google Scholar] [CrossRef]
- Panchigar, D.; Kar, K.; Shukla, S.; Mathew, R.M.; Chadha, U.; Selvaraj, S.K. Machine learning-based CFD simulations: A review, models, open threats, and future tactics. Neural Comput. Appl. 2022, 34, 21677–21700. [Google Scholar] [CrossRef]
- Shigeyuki Tanaka, F.A.; Keck, J.C. A reduced chemical kinetic model for HCCI combustion of primary reference fuels in a rapid compression machine. Combust. Flame 2003, 133, 467–481. [Google Scholar] [CrossRef]
- Patel, A.; Kong, S.C.; Reitz, R.D. Development and Validation of a Reduced Reaction Mechanism for HCCI Engine Simulations; SAE Technical Paper; SAE International: Warrendale, PA, USA, 2004. [Google Scholar] [CrossRef]
- Youngchul Ra, R.D.R. A reduced chemical kinetic model for IC engine combustion simulations with primary reference fuels. Combust. Flame 2008, 155, 713–738. [Google Scholar]
- Zeuch, T.; Moréac, G.; Ahmed, S.S.; Mauss, F. A comprehensive skeletal mechanism for the oxidation of n-heptane generated by chemistry-guided reduction. Combust. Flame 2008, 155, 651–674. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, X.; Song, Q.; Zhao, L.; Su, H.; Li, H.; Zeng, X.; Zhao, D.; Xu, J. The effect of temperature and pressure on n-heptane thermal cracking in regenerative cooling channel. Combust. Flame 2018, 194, 233–244. [Google Scholar] [CrossRef]
- Matthew, F.; Campbell, S.W.; Davidson, D.F.; Hanson, R.K. Shock tube study of normal heptane first-stage ignition near 3.5 atm. Combust. Flame 2018, 198, 376–392. [Google Scholar]
- An, Y.-Z.; Pei, Y.-Q.; Qin, J.; Zhao, H.; Li, X. Kinetic modeling of polycyclic aromatic hydrocarbons formation process for gasoline surrogate fuels. Energy Convers. Manag. 2015, 100, 249–261. [Google Scholar] [CrossRef]
- Zhang, K.; Banyon, C.; Bugler, J.; Curran, H.J.; Rodriguez, A.; Herbinet, O.; Battin-Leclerc, F.; B’Chir, C.; Heufer, K.A. An updated experimental and kinetic modeling study of n-heptane oxidation. Combust. Flame 2016, 172, 116–135. [Google Scholar] [CrossRef]
- Guo, J.; Li, S.; Tang, S.; Xiao, L.; Tan, N. Influence of Different Core Mechanisms on Low-Temperature Combustion Characteristics of Large Hydrocarbon Fuels. Energy Fuels 2019, 33, 7835–7851. [Google Scholar] [CrossRef]
- Ciezki, H.; Adomeit, G. Shock-tube investigation of the ignition delay on n-heptane/air-mixtures in a high pressure shock tube under conditions relevant to diesel-engine combustion. AIP Conf. Proc. 1990, 208, 707–712. [Google Scholar]
- Herzler, J.; Jerig, L.; Roth, P. Shock tube study of the ignition of lean n-heptane/air mixtures at intermediate temperatures and high pressures. Proc. Combust. Inst. 2005, 30, 1147–1153. [Google Scholar] [CrossRef]
- Davidson, D.; Hong, Z.; Pilla, G.; Farooq, A.; Cook, R.; Hanson, R. Multi-species time-history measurements during n-heptane oxidation behind reflected shock waves. Combust. Flame 2010, 157, 1899–1905. [Google Scholar] [CrossRef]
- Shen, H.-P.S.; Vanderover, J.; Oehlschlaeger, M.A. A shock tube study of the auto-ignition of toluene/air mixtures at high pressures. Proc. Combust. Inst. 2009, 32, 165–172. [Google Scholar] [CrossRef]
- Davidson, D.F.; Oehlschlaeger, M.A.; Hanson, R.K. Methyl concentration time-histories during iso-octane and n-heptane oxidation and pyrolysis. Proc. Combust. Inst. 2007, 31, 321–328. [Google Scholar] [CrossRef]
- Akih-Kumgeh, B.; Bergthorson, J.M. Comparative Study of Methyl Butanoate and n-Heptane High Temperature Autoignition. Energy Fuels 2010, 24, 2439–2448. [Google Scholar] [CrossRef]
- Ingemarsson, Å.T.; Pedersen, J.R.; Olsson, J.O. Oxidation of n-Heptane in a Premixed Laminar Flame. J. Phys. Chem. A 1999, 103, 8222–8230. [Google Scholar] [CrossRef]
- Held, T.J.; Marchese, A.J.; Dryer, F.L. A Semi-Empirical Reaction Mechanism for n-Heptane Oxidation and Pyrolysis. Combust. Sci. Technol. 1997, 123, 107–146. [Google Scholar] [CrossRef]
- Tanaka, S. Two-stage ignition in HCCI combustion and HCCI control by fuels and additives. Combust. Flame 2003, 132, 219–239. [Google Scholar] [CrossRef]
- Di Sante, R. Measurements of the auto-ignition of n-heptane/toluene mixtures using a rapid compression machine. Combust. Flame 2012, 159, 55–63. [Google Scholar] [CrossRef]
- Guang, H.; Yang, Z.; Huang, Z.; Lu, X. Experimental study of n-heptane ignition delay with carbon dioxide addition in a rapid compression machine under low-temperature conditions. Chin. Sci. Bull. 2012, 57, 3953–3960. [Google Scholar] [CrossRef]
- Karwat, D.M.; Wagnon, S.W.; Wooldridge, M.S.; Westbrook, C.K. Low-temperature speciation and chemical kinetic studies of n-heptane. Combust. Flame 2013, 160, 2693–2706. [Google Scholar] [CrossRef]
- Di, H.; He, X.; Zhang, P.; Wang, Z.; Wooldridge, M.S.; Law, C.K.; Wang, C.; Shuai, S.; Wang, J. Effects of buffer gas composition on low temperature ignition of iso-octane and n-heptane. Combust. Flame 2014, 161, 2531–2538. [Google Scholar] [CrossRef]
- Yong, Y.-C.; Zhong, J.-J. Impacts of Quorum Sensing on Microbial Metabolism and Human Health. In Future Trends in Biotechnology; Springer: Berlin/Heidelberg, Germany, 2012. [Google Scholar]
- Desantes, J.M.; Bermúdez, V.; López, J.J.; López-Pintor, D. A new method to predict high and low-temperature ignition delays under transient thermodynamic conditions and its experimental validation using a Rapid Compression-Expansion Machine. Energy Convers. Manag. 2016, 123, 512–522. [Google Scholar] [CrossRef]
- Ciezki, H.K.; Adomeit, G. Shock-tube investigation of self-ignition of n-heptane-air mixtures under engine relevant conditions. Combust. Flame 1993, 93, 421–433. [Google Scholar] [CrossRef]
- Côme, G.; Warth, V.; Glaude, P.; Fournet, R.; Battin-Leclerc, F.; Scacchi, G. Computer-aided design of gas-phase oxidation mechanisms—Application to the modeling of n-heptane and iso-octane oxidation. Symp. (Int.) Combust. 1996, 26, 755–762. [Google Scholar] [CrossRef]
- Warth, V.; Stef, N.; Glaude, P.A.; Battin-Leclerc, F.; Scacchi, G.; Côme, G.M. Computer-Aided Derivation of Gas-Phase Oxidation Mechanisms: Application to the Modeling of the Oxidation of n-Butane. Combust. Flame 1998, 114, 81–102. [Google Scholar] [CrossRef]
- Zheng, Z.; Yao, M. Numerical study on the chemical reaction kinetics of n-heptane for HCCI combustion process. Fuel 2006, 85, 2605–2615. [Google Scholar] [CrossRef]
- Lapointe, S.; Zhang, K.; McNenly, M.J. Reduced chemical model for low and high-temperature oxidation of fuel blends relevant to internal combustion engines. Proc. Combust. Inst. 2019, 37, 789–796. [Google Scholar] [CrossRef]
- Lin, S.; Xie, M.; Wang, J.; Liang, W.; Law, C.K.; Zhou, W.; Yang, B. Chemical kinetic model reduction through species-targeted global sensitivity analysis (STGSA). Combust. Flame 2021, 224, 73–82. [Google Scholar] [CrossRef]
- Chang, Y.; Jia, M.; Niu, B.; Xie, M.; Zhou, C. Reduction of Detailed Chemical Mechanisms Using Reaction Class Based Global Sensitivity and Path Sensitivity Analyses. Energy Fuels 2019, 33, 9289–9301. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number | Author | Year | Species | Reaction | Temperature (K) | Pressure (Bar) | Equivalence Ratio |
|---|---|---|---|---|---|---|---|
| 1 | Curran et al. [6] | 1998 | 560 | 2539 | 550–1700 | 1–42 | 0.3–1.5 |
| 2 | Mehl [7,8] | 2011 | 1550 | 6000 | 650–1200 | 3–50 | 0.3–1.0 |
| 3 | Chun Sang Yoo [23] | 2011 | 188 | 939 | / | / | / |
| 4 | Yanzhao An [36] | 2015 | 219 | 1229 | 750–180 K | 15–60 | 0.5–2.0 |
| 5 | Liming Cai [14] | 2016 | 1692 | 11,015 | / | / | 0.5–2 |
| 6 | Kuiwen Zhang [37] | 2016 | 1268 | 5336 | 726–1412 | 15–38 | 0.25–4.0 |
| 7 | Chen Yulin [13] | 2018 | 126 | 680 | 750–1100 | 20–40 | 0.5–2.0 |
| 8 | Junjiang Guo [38] | 2019 | 732 | 3022 | / | / | / |
| Author | Temperature (K) | Pressure (Bar) | Equivalent Ratio | Test Equipment | Year |
|---|---|---|---|---|---|
| Ciezki [54] | 660–1350 | 3.2–42 | 0.5–3.0 | shock tube | 1993 |
| Equation | |
|---|---|
| C7H14O2-4 + OH => CH3CO + C5H10-1 + H2O | C3H5-a <=> C3H4-a + H |
| C7H14-1 <=> pC4H9 + C3H5-a | C3H4-a + OH => C3H3 + H2O |
| C7H14-2 => C4H7 + NC3H7 | C3H3 + H => C3H2 + H2 |
| C5H10-1 => C2H5 + C3H5-a | C3H2 + O2 => HCCO + CO + H |
| C5H10-1 + OH => C5H9 + H2O | C2H3CHO + H => C2H3CO + H2 |
| C5H9 => C2H3 + C3H6 | C2H3CO => C2H3 + CO |
| NC4H9O2 => pC4H9 + O2 | C2H5(+M) <=> H + C2H4(+M) |
| pC4H9 => C2H5 + C2H4 | C2H4 + H => C2H3 + H2 |
| C4H8-1 <=> C3H5-a + CH3 | C2H4 + O => CH3 + HCO |
| C4H8OOH1-2 => C4H8-1 + HO2 | C2H3 + O2 => C2H2 + HO2 |
| C4H8-1 + OH => C4H7 + H2O | C2H3 + O2 => CH2O + HCO |
| C4H7 + HO2 => C4H7O + OH | C2H2 + O => HCCO + H |
| C4H7O => CH3CHO + C2H3 | CH3CHO + H => CH3CO + H2 |
| C4H7O => C2H3CHO + CH3 | CH3CO(+M) => CH3 + CO(+M) |
| C4H7 + NC3H7 => C7H14-2 | HCCO + H => CH2(s) + CO |
| C4H7 => C4H6 + H | CH4 + O => CH3 + OH |
| C4H6 + H => C2H3 + C2H4 | CH3 + O2 => CH2O + OH |
| NC3H7 <=> CH3 + C2H4 | CH3 + O => CH2O + H |
| NC3H7 <=> H + C3H6 | CH2O + OH => HCO + H2O |
| C3H6 + OH => C3H5-a + H2O | CH2O + H => HCO + H2 |
| C3H6 + H => C3H5-a + H2 | CH2(s) + CO => HCCO + H |
| C3H6 + CH3 => C3H5-a + CH4 | CH2(s) + M <=> CH2 + M |
| C2H4 + OH => C2H3 + H2O | CH2(s) + CH4 => 2CH3 |
| C2H3 + CH3 => C3H6 | CH2(s) + O2 => CO + OH + H |
| C3H5-a + HO2 => C3H5O + OH | HCO + M => H + CO + M |
| C3H5O => C2H3CHO + H | CO + OH => CO2 + H |
| C3H5O => C2H3 + CH2O | OH + H2 => H + H2O |
| Number | Author | Year | Species | Reaction | Temperature (K) | Pressure (Bar) | Equivalence Ratio |
|---|---|---|---|---|---|---|---|
| 1 | San Diego | 2016 | 64 | 301 | 600–1500 | 1–40 | 0.5–2.0 |
| 2 | Yachao Chang [60] | 2020 | 56 | 131 | / | / | / |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duan, J.; Yang, A.; Wei, W.; Qin, G. An n-Heptane Oxidation Mechanism Suitable for Low- to High-Temperature Combustion. Energies 2025, 18, 1305. https://doi.org/10.3390/en18051305
Duan J, Yang A, Wei W, Qin G. An n-Heptane Oxidation Mechanism Suitable for Low- to High-Temperature Combustion. Energies. 2025; 18(5):1305. https://doi.org/10.3390/en18051305
Chicago/Turabian StyleDuan, Junfa, Aoqing Yang, Wei Wei, and Gaolin Qin. 2025. "An n-Heptane Oxidation Mechanism Suitable for Low- to High-Temperature Combustion" Energies 18, no. 5: 1305. https://doi.org/10.3390/en18051305
APA StyleDuan, J., Yang, A., Wei, W., & Qin, G. (2025). An n-Heptane Oxidation Mechanism Suitable for Low- to High-Temperature Combustion. Energies, 18(5), 1305. https://doi.org/10.3390/en18051305