Methanol Electro-Oxidation on Pt-Ru Alloy Nanoparticles Supported on Carbon Nanotubes

Abstract
:1. Introduction
2. Experimental Section
3. Results and Discussion
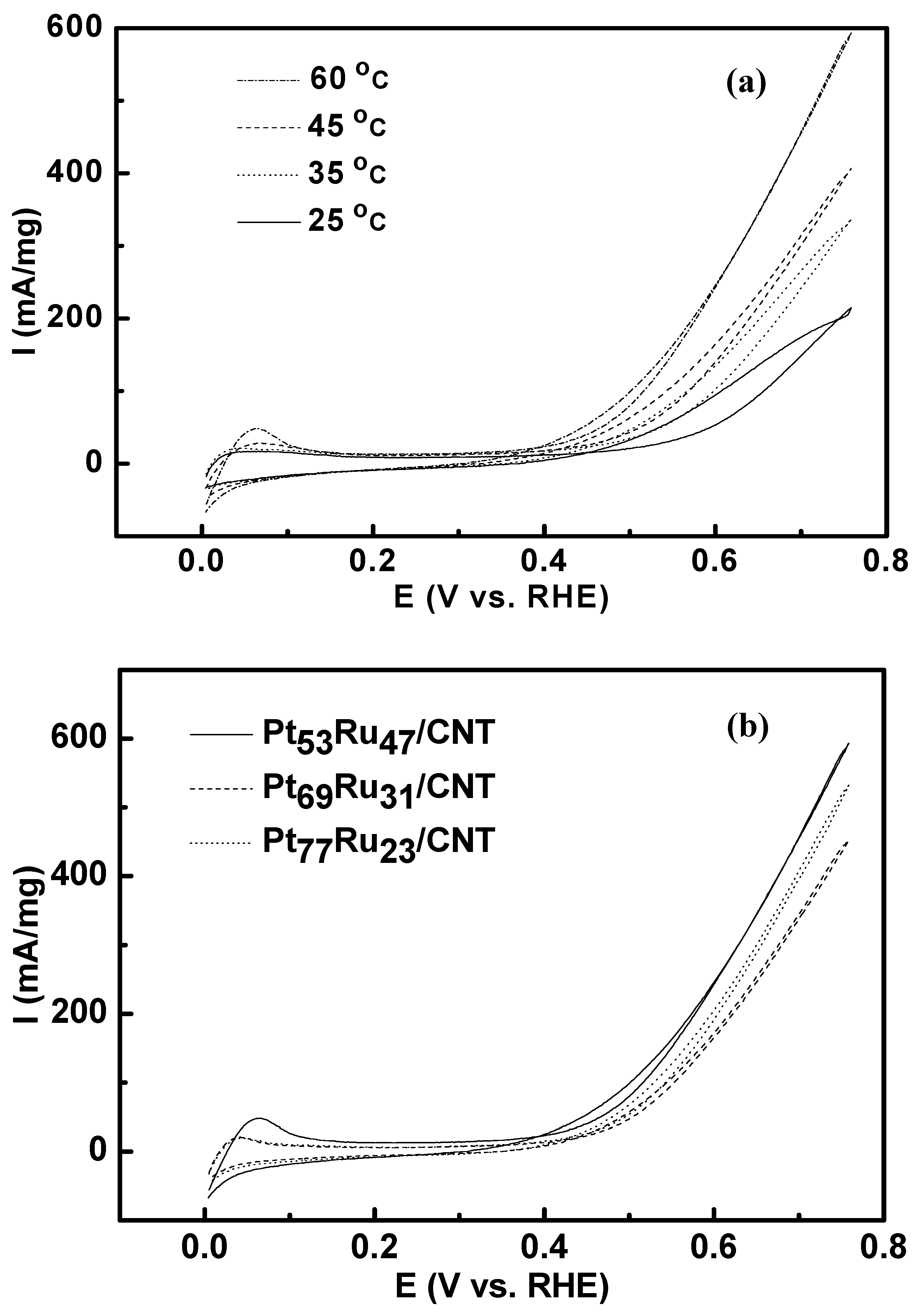
3.1. Cyclic Voltammetry

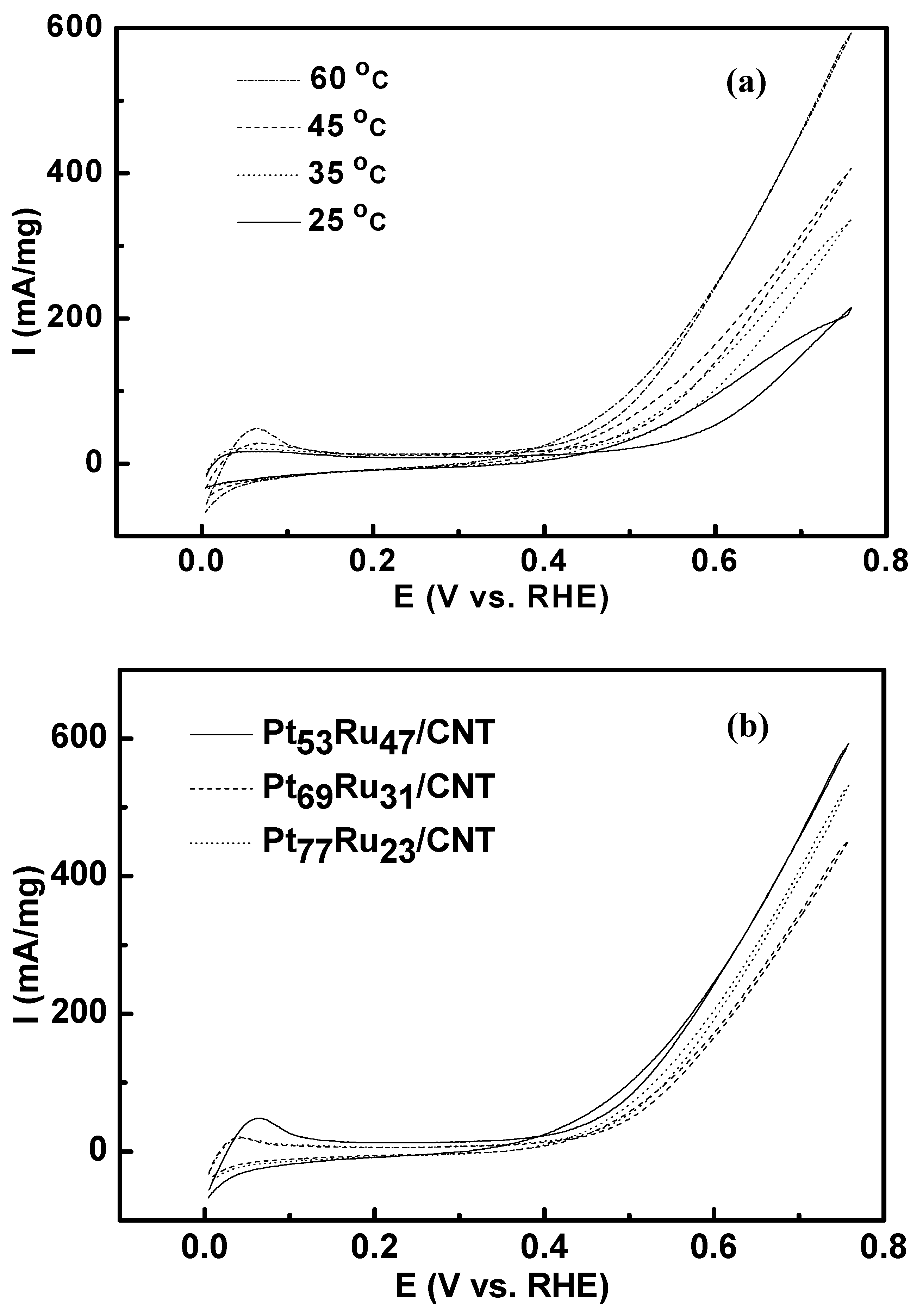{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
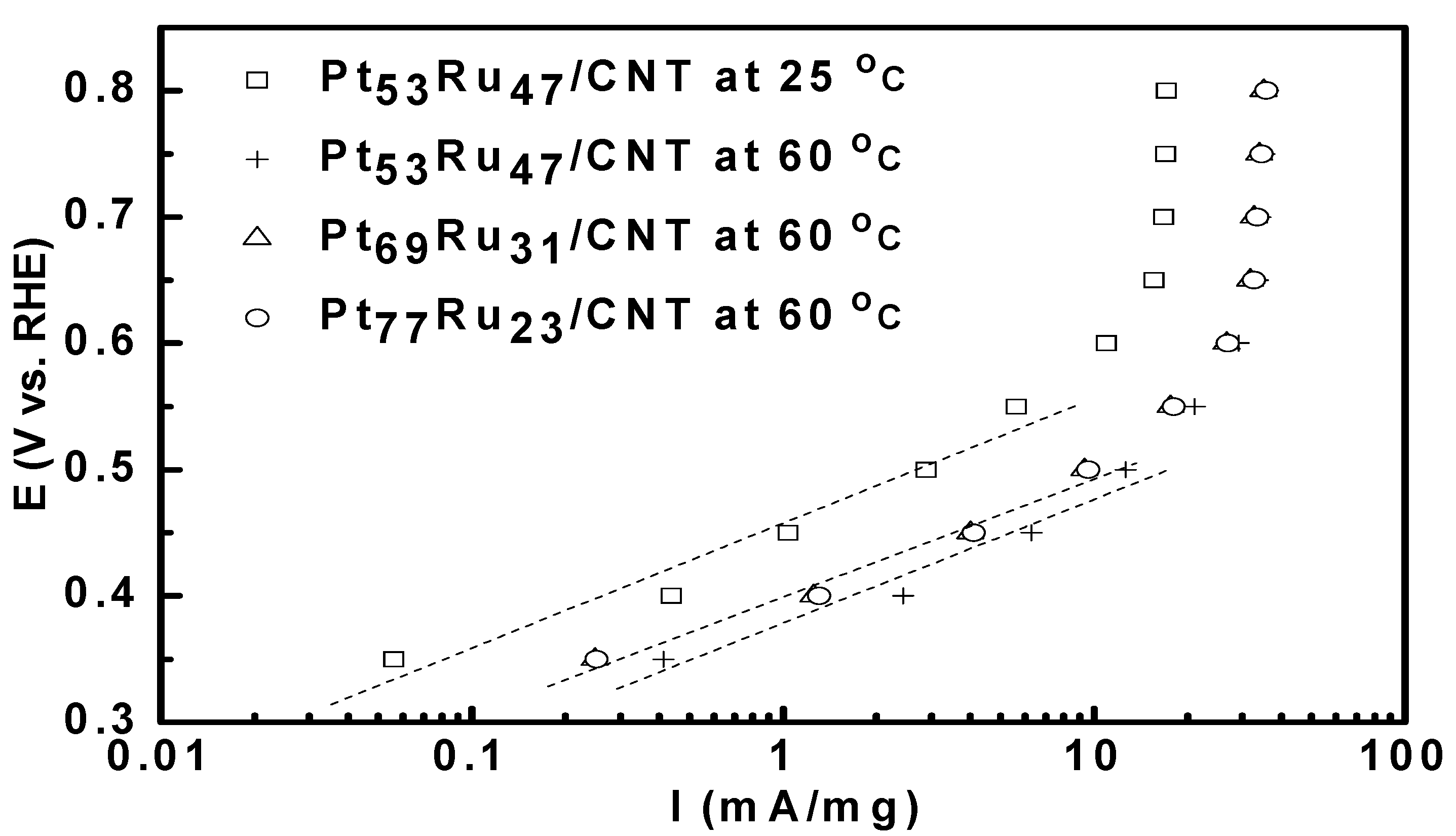
| Catalysts | Onset potential (mV) | Tafel slope (mV dec-1) | ||
|---|---|---|---|---|
| 25 °C | 60 °C | 25 °C | 60 °C | |
| Pt53Ru47/CNT | 253 | 215 | 98.8 | 97.8 |
| Pt69Ru31/CNT | 272 | 240 | 101.3 | 105.7 |
| Pt77Ru23/CNT | 262 | 230 | 101.1 | 105.5 |
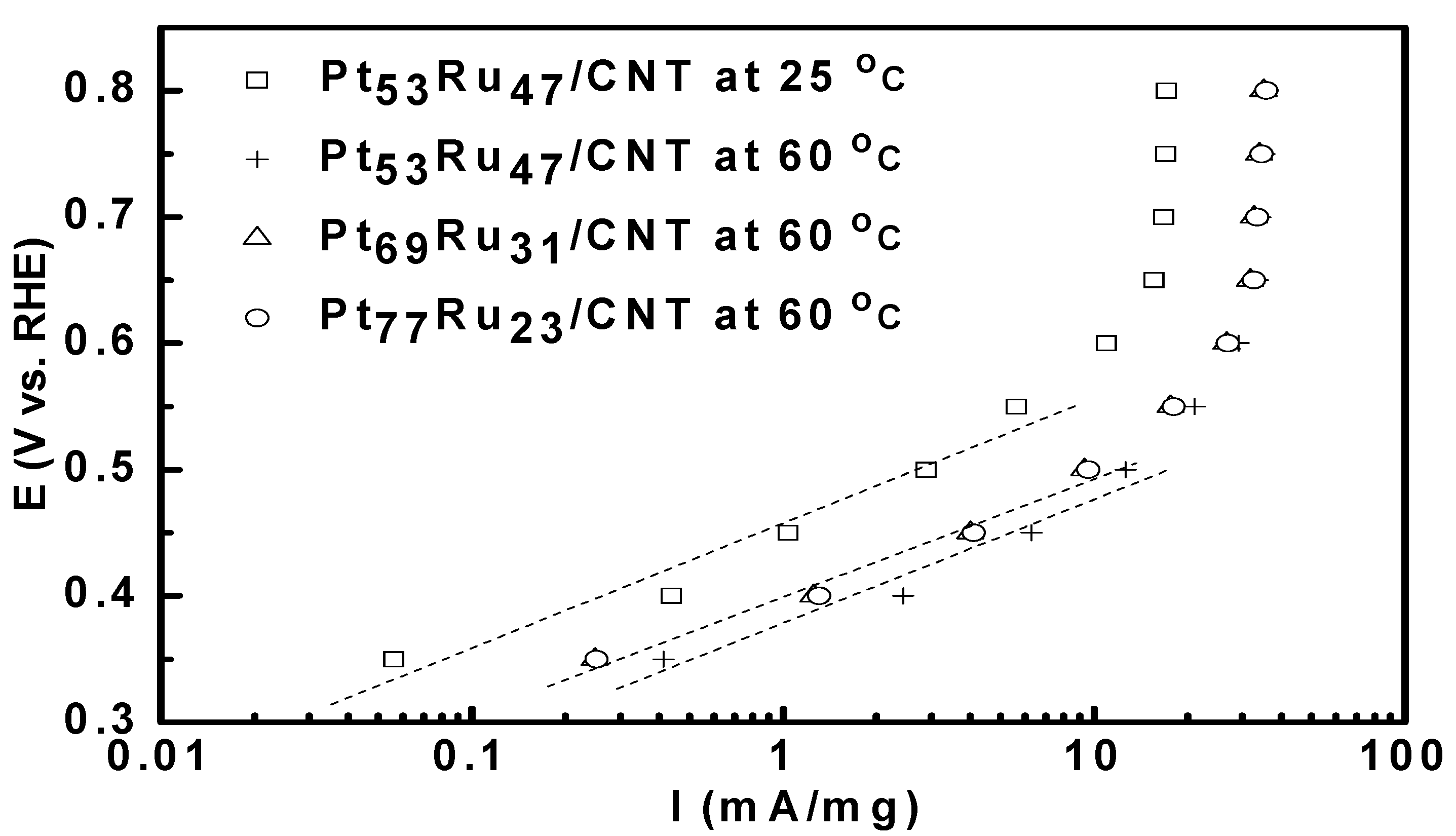
3.2. Bulter-Volmer Plot (Tafel Plot)
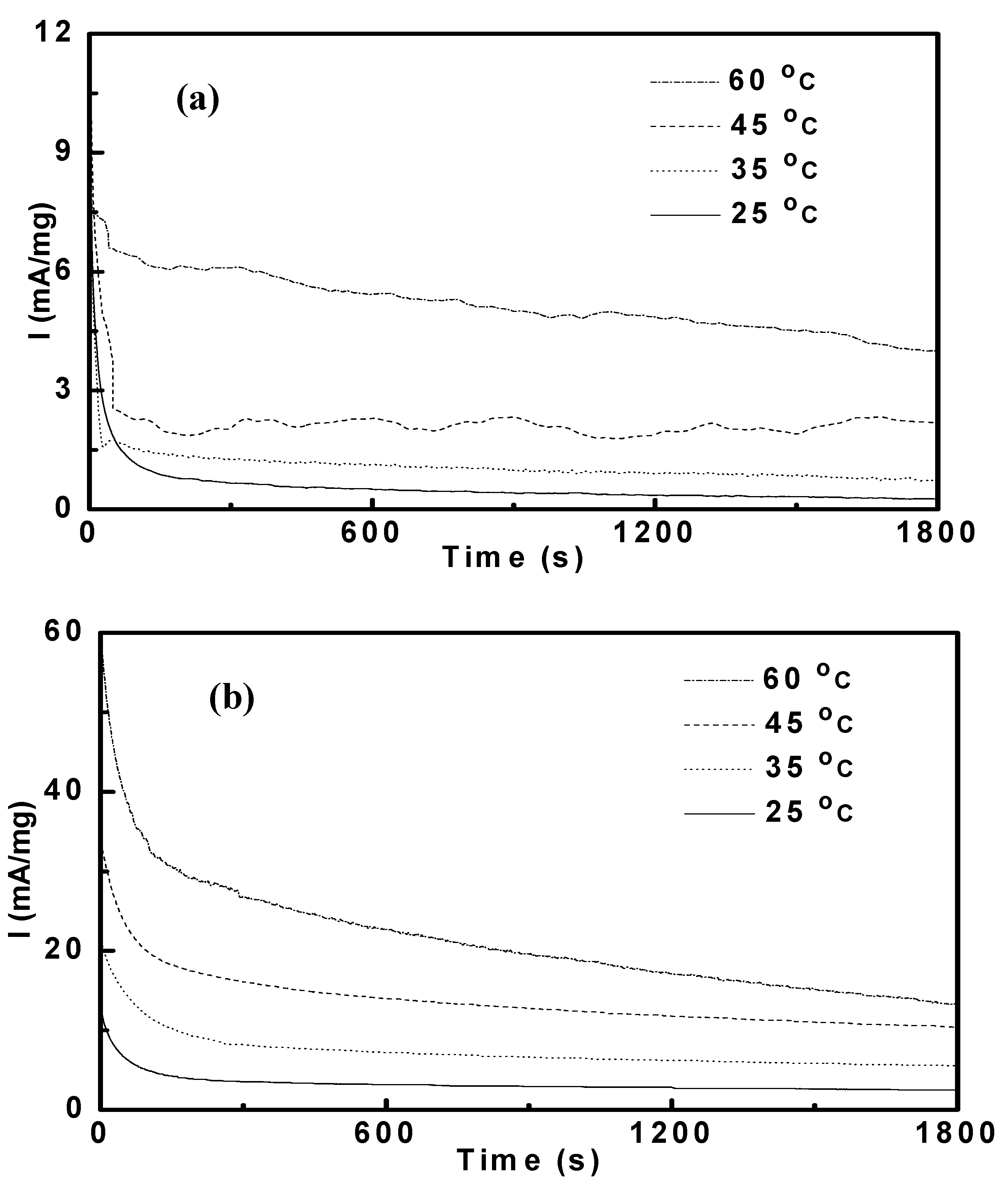
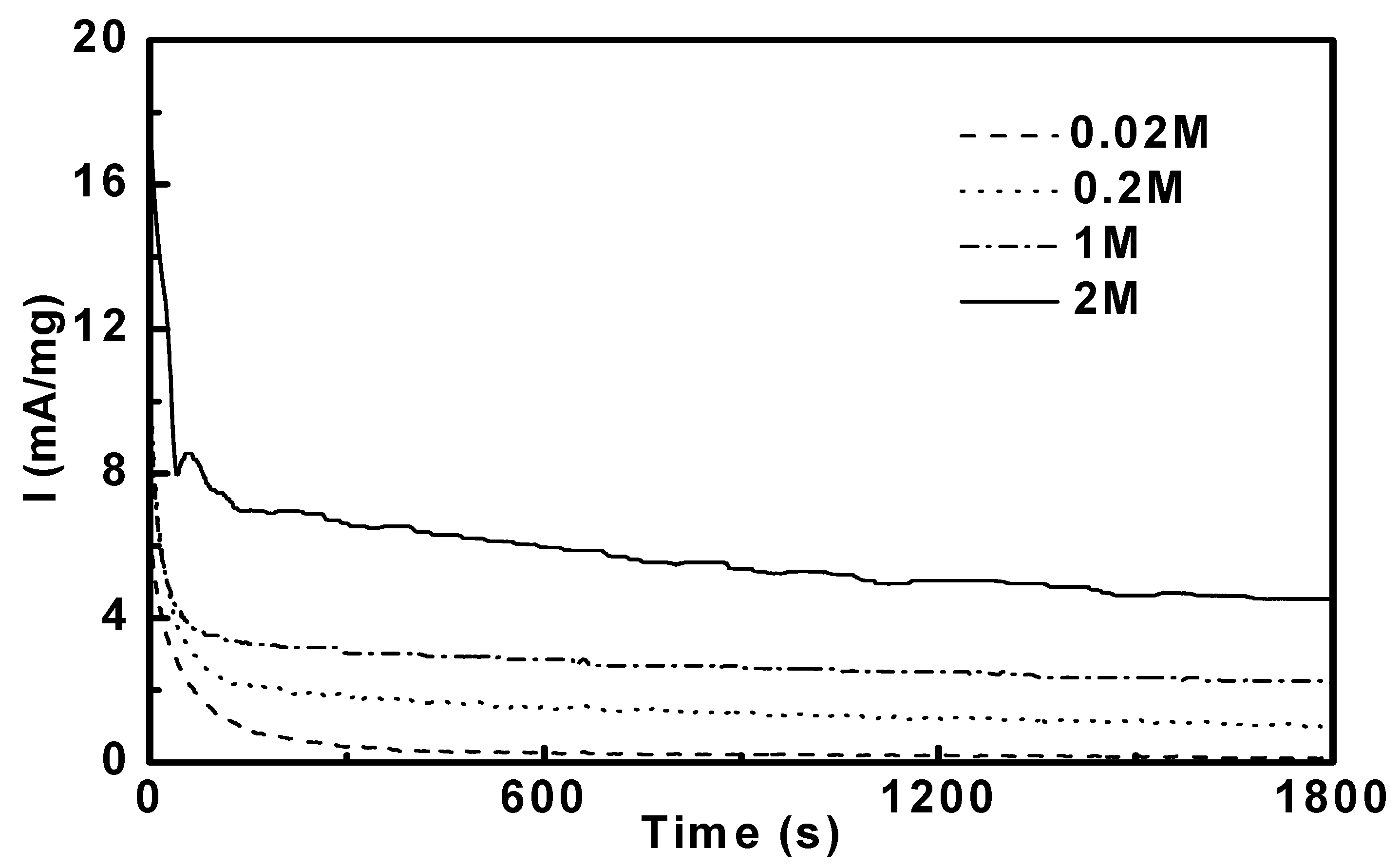
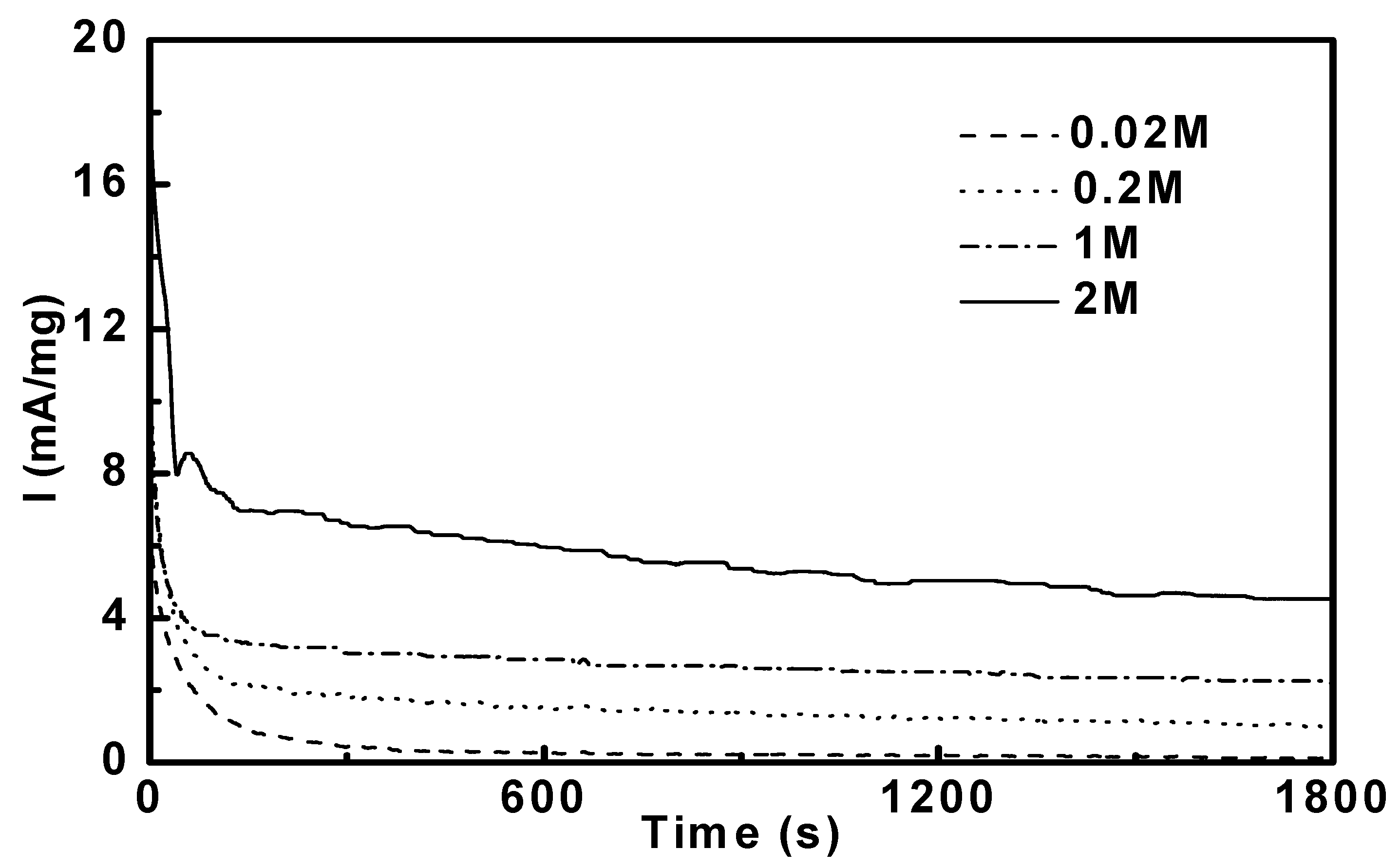
3.3. Potentiostatic Methanol Oxidation on PtRu/CNT
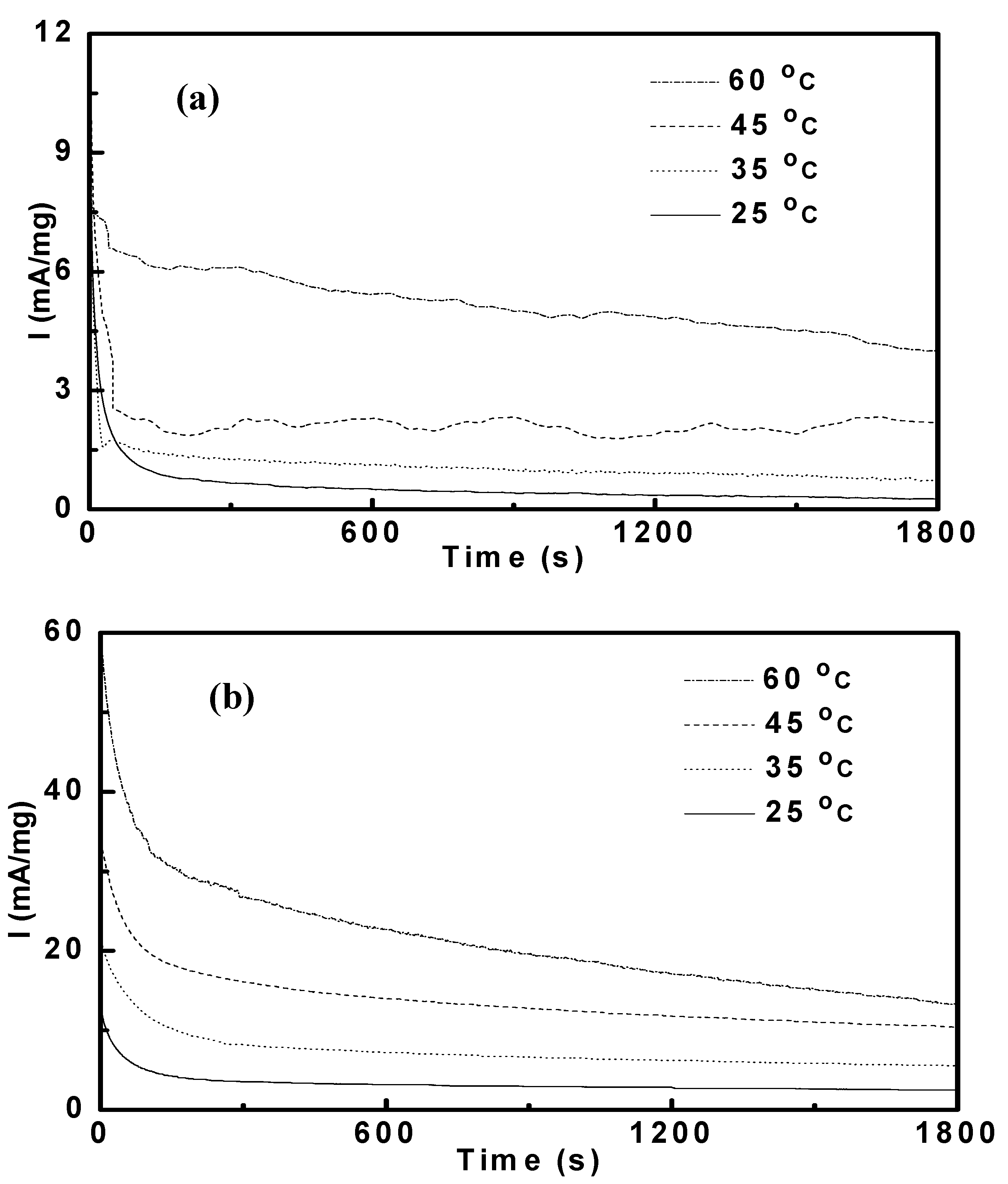
| Catalysts | Applied potentials | |||||||
|---|---|---|---|---|---|---|---|---|
| 0.4 V | 0.5 V | |||||||
| 25 °C | 35 °C | 45 °C | 60 °C | 25 °C | 35 °C | 45 °C | 60 °C | |
| Pt53Ru47/CNT | 0.93 | 1.64 | 2.64 | 5.52 | 4.60 | 6.98 | 13.3 | 26.3 |
| Pt69Ru31/CNT | 0.28 | 0.49 | 0.88 | 1.98 | 2.03 | 3.73 | 6.27 | 14.5 |
| Pt77Ru23/CNT | 0.53 | 0.97 | 1.71 | 3.38 | 3.05 | 5.70 | 9.95 | 19.6 |
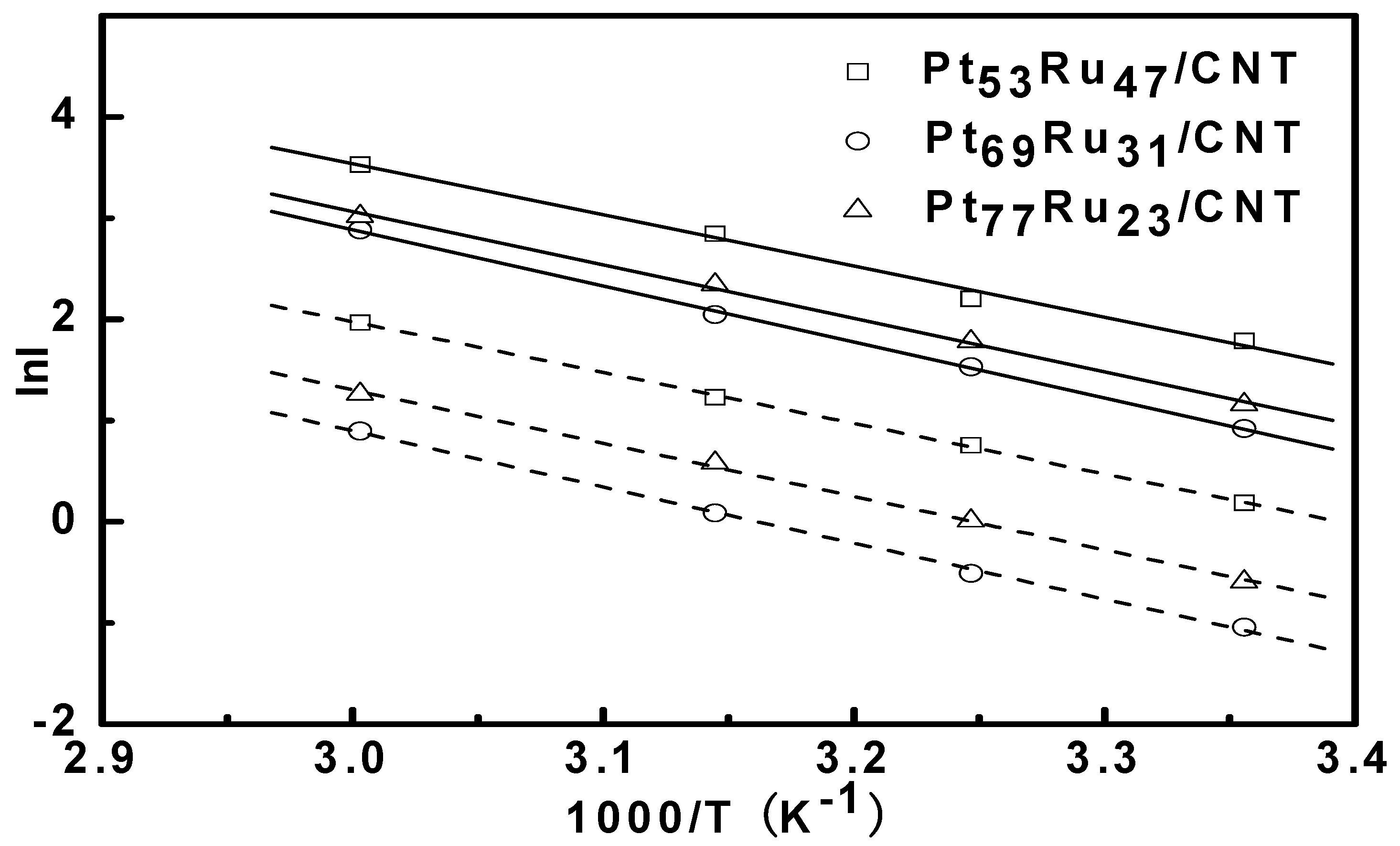
| Catalysts | Activation energy (kJ/mol) |
|---|---|
| Pt53Ru47/CNT | 41.9 ± 0.3 |
| Pt69Ru31/CNT | 46.0 ± 0.1 |
| Pt77Ru23/CNT | 43.8 ± 0.1 |
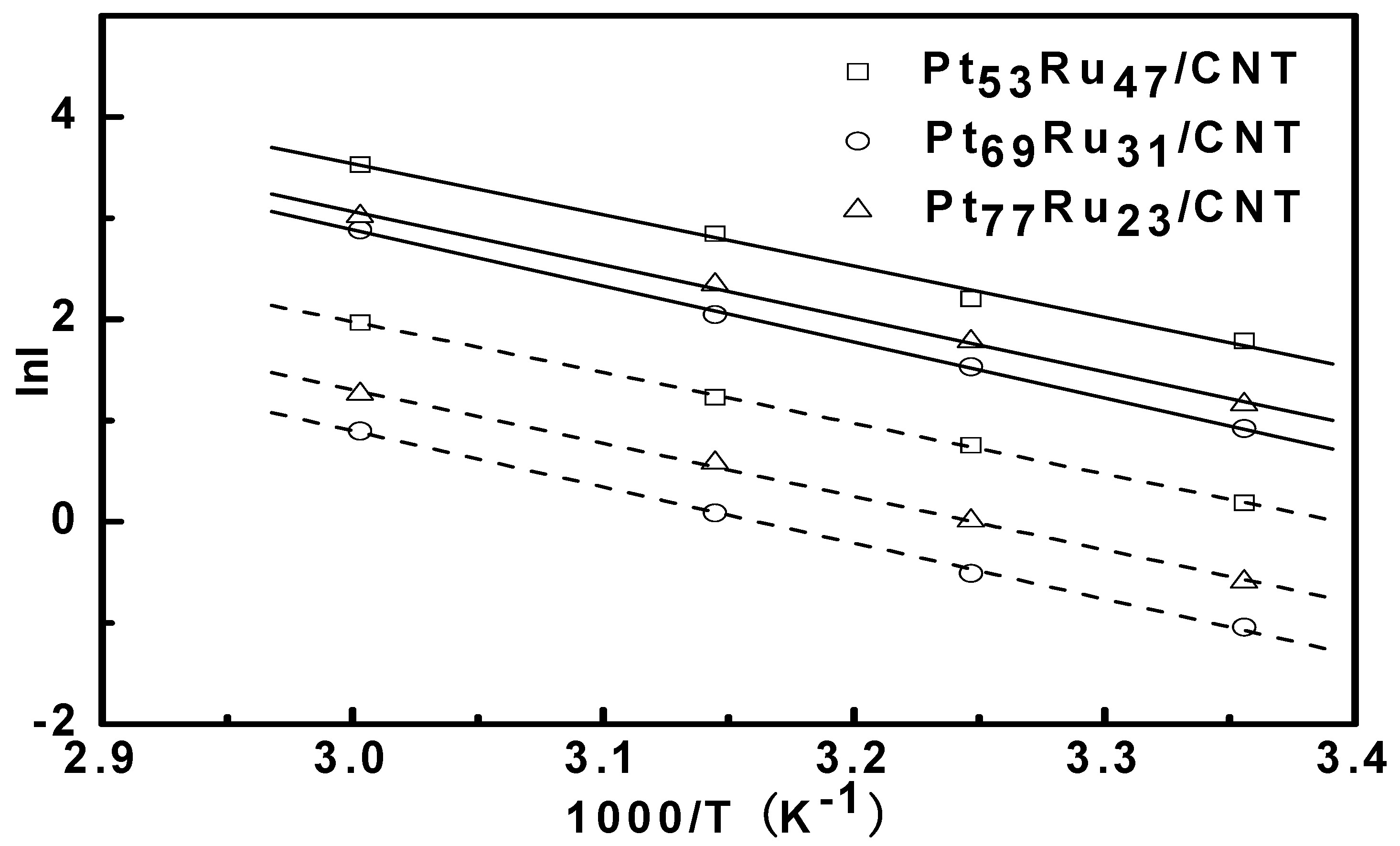
3.4. Reaction Orders in Methanol Electro-Oxidation
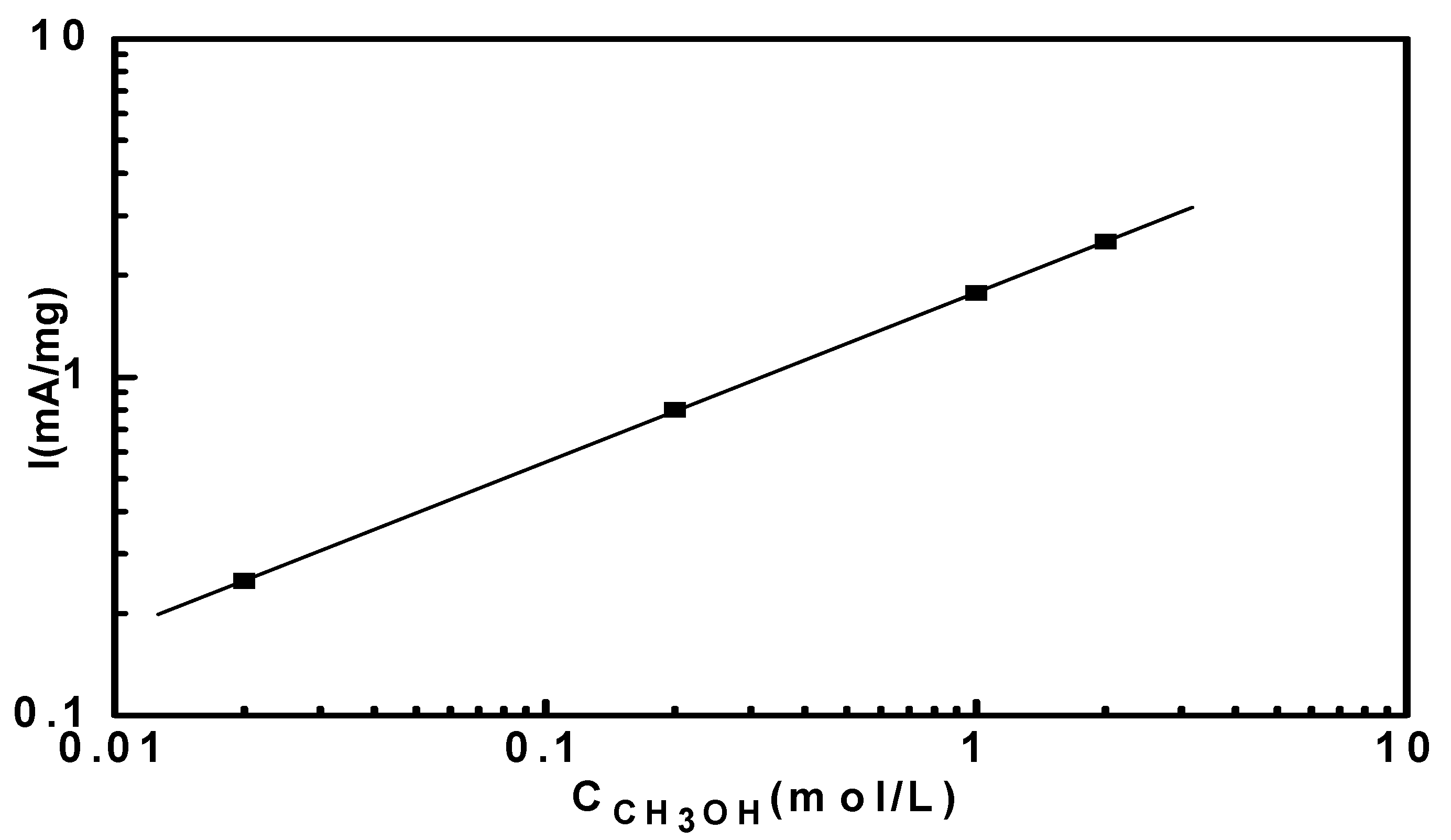
4. Conclusions
Acknowledgements
References and Notes
- Vidakovic, T.; Christov, M.; Sundmacher, K. Rate expression for electrochemical oxidation of methanol on a direct methanol fuel cell anode. J. Electroanal. Chem. 2005, 580, 105–121. [Google Scholar] [CrossRef]
- Kabbabi, A.; Durand, R.; Beden, B.; Hahn, F.; Leger, J.M.; Lamy, C. In situ FTIRS study of the electrocatalytic oxidation of carbon monoxide and methanol at platinum–ruthenium bulk alloy electrodes. J. Electroanal. Chem. 1998, 444, 41–53. [Google Scholar] [CrossRef]
- Jusys, Z.; Kaiser, J.; Behm, R.J. Composition and activity of high surface area PtRu catalysts towards adsorbed CO and methanol electrooxidation: A DEMS study. Electrochim. Acta 2002, 47, 3693–3706. [Google Scholar] [CrossRef]
- Laborde, H.; Leger, J.M.; Lamy, C. Electrocatalytic oxidation of methanol and C1 molecules on highly dispersed electrodes Part II: Platinum-ruthenium in polyaniline. J. Appl. Electrochem. 1994, 24, 1019–1027. [Google Scholar]
- Bagotzsky, V.S.; Vassiliev, Y.B.; Khazova, O.A. Generalized scheme of chemisorption, electrooxidation and electroreduction of simple organic compounds on platinum group metals. J. Electroanal. Chem. 1977, 81, 229. [Google Scholar] [CrossRef]
- Bockris, J.O’M.; Wroblowa, H. Electrocatalysis. J. Electroanal. Chem. Interfacial Electrochem. 1964, 7, 428. [Google Scholar]
- Watanabe, M.; Motoo, S. Electrocatalysis by ad-atoms: Part II. Enhancement of the oxidation of methanol on platinum by ruthenium ad-atoms. J. Electroanal. Chem. Interfacial Electrochem. 1975, 60, 267. [Google Scholar] [CrossRef]
- Gasteiger, H.A.; Markovic, N.; Ross, P.N.; Cairns, E.J. Temperature-dependent methanol electro-oxidation on well-characterized Pt-Ru alloys. J. Electrochem. Soc. 1994, 141, 1795–1803. [Google Scholar] [CrossRef]
- Takasu, Y.; Fujiwara, T.; Murakami, Y.; Sasaki, K.; Oguri, M.; Asaki, T.; Sugimoto, W. Effect of structure of carbon-supported PtRu electrocatalysts on the electrochemical oxidation of methanol. J. Electrochem. Soc. 2000, 147, 4421–4427. [Google Scholar] [CrossRef]
- Goodenough, J.B.; Manohara, R.; Shukla, A.K.; Ramesh, K.V. Intraalloy electron transfer and catalyst performance: a spectroscopic and electrochemical study. Chem. Mater. 1989, 1, 391–398. [Google Scholar] [CrossRef]
- Hamnett, A. Mechanism and electrocatalysis in the direct methanol fuel cell. Catal. Today 1997, 38, 445–457. [Google Scholar] [CrossRef]
- Liu, R.; Iddir, H.; Fan, Q.; Hou, G.; Bo, A.; Ley, K.L.; Smotkin, E.S.; Sung, Y.E.; Kim, H.; Thomas, S.; Wieckowski, A. Potential-dependent infrared absorption spectroscopy of adsorbed CO and X-ray photoelectron spectroscopy of arc-melted single-phase Pt, PtRu, PtOs, PtRuOs, and Ru electrodes. J. Phys. Chem.B. 2000, 104, 3518–3531. [Google Scholar] [CrossRef]
- Waszczuk, P.; Wieckowski, A.; Zelenay, P.; Gottesfeld, S.; Coutanceau, C.; Leger, J.M.; Lamy, C. Adsorption of CO poison on fuel cell nanoparticle electrodes from methanol solutions: a radioactive labeling study. J. Electroanal. Chem. 2001, 511, 55–64. [Google Scholar] [CrossRef]
- Che, G.; Lakshmi, B.B.; Martin, C.R.; Fisher, E.R. Metal-nanocluster-filled carbon nanotubes: catalytic properties and possible applications in electrochemical energy storage and production. Langmuir 1999, 15, 750–758. [Google Scholar] [CrossRef]
- Li, W.; Liang, C.; Qiu, J.; Zhou, W.; Han, H.; Wei, Z.; Sun, G.; Xin, Q. Carbon nanotubes as support for cathode catalyst of a direct methanol fuel cell. Carbon 2002, 40, 791–794. [Google Scholar] [CrossRef]
- Xing, Y. Synthesis and electrochemical characterization of uniformly-dispersed high loading Pt nanoparticles on sonochemically-treated carbon nanotubes. J. Phys. Chem. B 2004, 108, 19255–19259. [Google Scholar] [CrossRef]
- Wang, X.; Waje, M.; Yan, Y. CNT-based electrodes with high efficiency for proton exchange membrane fuel cells. Electrochem. Solid State Lett. 2004, 8, A42–A44. [Google Scholar] [CrossRef]
- Liu, Z.L.; Lee, J.Y.; Chen, W.; Han, M.; Gan, L.M. Physical and electrochemical characterizations of microwave-assisted polyol preparation of carbon-supported PtRu nanoparticles. Langmuir 2004, 20, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Park, K.W.; Sung, Y.E.; Han, S.; Yun, Y.; Hyeon, T. Origin of the enhanced catalytic activity of carbon nanocoil-supported PtRu alloy electrocatalysts. J. Phys. Chem. B 2004, 108, 939–944. [Google Scholar] [CrossRef]
- Lin, Y.; Cui, X.; Yen, C.H.; Wai, C.M. PtRu/Carbon nanotube nanocomposite synthesized in supercritical fluid: a novel electrocatalyst for direct methanol fuel cell. Langmuir 2005, 21, 11474–11479. [Google Scholar] [CrossRef] [PubMed]
- Frackowiak, E.; Lota, G.; Cacciaguerra, T.; Beguin, F. Carbon nanotubes with Pt–Ru catalyst for methanol fuel cell. Electrochem. Comm. 2006, 8, 129–132. [Google Scholar] [CrossRef]
- Girishkumar, G.; Hall, T.D.; Vinodgopal, K.; Kamat, P.V. Single wall carbon nanotube supports for portable direct methanol fuel cells. J. Phys. Chem. B 2006, 110, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wang, X.; Chen, Z.; Waje, M.; Yan, Y. Pt−Ru supported on double-walled carbon nanotubes as high-performance anode catalysts for direct methanol fuel cells. J. Phys. Chem. B 2006, 110, 15353–15358. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Xing, Y. Pt−Ru nanoparticles supported on carbon nanotubes as methanol fuel cell catalysts. J. Phys. Chem. C 2007, 111, 2803–2808. [Google Scholar] [CrossRef]
- Yu, W.Y.; Tu, W.X.; Liu, H.F. Synthesis of nanoscale platinum colloids by microwave dielectric heating. Langmuir 1999, 15, 6–9. [Google Scholar] [CrossRef]
- Li, W.; Liang, C.; Zhou, W.; Qiu, J.; Zhou, Z.; Han, H.; Wei, Z.; Sun, G.; Xin, Q. Preparation and characterization of multiwalled carbon nanotube-supported platinum for cathode catalysts of direct methanol fuel cells. J. Phys. Chem. B 2003, 107, 6292–6299. [Google Scholar] [CrossRef]
- Xing, Y.; Li, L.; Chusuei, C.C.; Hull, R.V. Sonochemical oxidation of multiwalled carbon nanotubes. Langmuir 2005, 21, 4185–4190. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.J.; Gasteiger, H.A.; Stab, G.D.; Urban, P.M.; Kolb, D.M.; Behm, R.J. Characterization of high-surface-area electrocatalysts using a rotating disk electrode configuration. J. Electrochem. Soc. 1998, 145, 2354–2358. [Google Scholar] [CrossRef]
- Arico, A.S.; Baglio, V.; Blasi, A.D.; Modica, E.; Antonucci, P.L.; Antonucci, V. Analysis of the high-temperature methanol oxidation behaviour at carbon-supported Pt–Ru catalysts. J. Electroanal. Chem. 2003, 557, 167–176. [Google Scholar] [CrossRef]
- Gasteiger, H.A.; Markovic, N.; Ross, P.N.; Cairns, E.J. Methanol electrooxidation on well-characterized platinum-ruthenium bulk alloys. J. Phys. Chem. 1993, 97, 12020–12029. [Google Scholar] [CrossRef]
- Kauranen, P.; Skou, E.; Munk, J. Kinetics of methanol oxidation on carbon-supported Pt and Pt + Ru catalysts. J. Electroanal. Chem. 1996, 404, 1–13. [Google Scholar] [CrossRef]
- Ticanelli, E.; Beery, T.G.; Paffett, M.T.; Gottesfeld, S. An electrochemical, ellipsometric, and surface science investigation of the PtRu bulk alloy surface. J. Electroanal. Chem. 1989, 258, 61–77. [Google Scholar] [CrossRef]
- Choi, J.H.; Park, K.W.; Kwon, B.K.; Sung, Y.E. Methanol oxidation on Pt/Ru, Pt/Ni, and Pt/Ru/Ni anode electrocatalysts at different temperatures for DMFCs. J. Electrochem. Soc. 2003, 150, A973–A978. [Google Scholar] [CrossRef]
- Raicheva, S.N.; Christov, M.V.; Sokolova, E.I. Effect of the temperature on the electrochemical behaviour of aliphatic alcohols. Electrochim. Acta 1981, 26, 1669–1676. [Google Scholar] [CrossRef]
- Chu, D.; Gilman, S. Methanol electro-oxidation on unsupported Pt-Ru alloys at different temperatures. J. Electrochem. Soc. 1996, 143, 1685–1690. [Google Scholar] [CrossRef]
- Madey, T.E.; Engelhardt, H.A.; Menzel, D. Adsorption of oxygen and oxidation of CO on the ruthenium (001) surface. Surf. Sci. 1975, 48, 304–328. [Google Scholar] [CrossRef]
- Vidakovic, T.; Christov, M.; Sundmacher, K.; Nagabhushana, K.S.; Fei, W.; Kinge, S.; Bonnemann, H. PtRu colloidal catalysts: Characterisation and determination of kinetics for methanol oxidation. Electrochim. Acta 2007, 52, 2277–2284. [Google Scholar] [CrossRef]
- Roth, C.; Marty, N.; Hahn, F.; Leger, J.M.; Lamy, C.; Fuess, H. Characterization of differently synthesized Pt-Ru fuel cell catalysts by cyclic voltammetry, FTIR spectroscopy, and in single cells. J. Electrochem. Soc. 2002, 149, E433–E439. [Google Scholar] [CrossRef]
- Meli, G.; Leger, J.M.; Lamy, C. Direct electrooxidation of methanol on highly dispersed platinum-based catalyst electrodes: temperature effect. J. Appl. Electrochem. 1993, 23, 197–202. [Google Scholar] [CrossRef]
- Schmidt, T.J.; Gasteiger, H.A.; Behm, R.J. Methanol electrooxidation on a colloidal PtRu-alloy fuel-cell catalyst. Electrochem. Commun. 1999, 1, 1–4. [Google Scholar] [CrossRef]
- Chrzanowski, W.; Wieckowski, A. Surface structure effects in Platinum/Ruthenium methanol oxidation electrocatalysis. Langmuir 1998, 14, 1967–1970. [Google Scholar] [CrossRef]
- Herrero, E.; Franaszczuk, K.; Wieckowski, A. Electrochemistry of methanol at low index crystal planes of platinum: an integrated voltammetric and chronoamperometric study. J. Phys. Chem. 1994, 98, 5074–5083. [Google Scholar] [CrossRef]
- Hoster, H.; Iwasita, T.; Baumgartner, H.; Vielstich, W. Pt–Ru model catalysts for anodic methanol oxidation: Influence of structure and composition on the reactivity. Phys. Chem. Chem. Phys. 2001, 3, 337–346. [Google Scholar] [CrossRef]
- Chrzanowski, W.; Kim, H.; Wieckowski, A. Enhancement in methanol oxidation by spontaneously deposited ruthenium on low-index platinum electrodes. Catal. Lett. 1998, 50, 69–75. [Google Scholar] [CrossRef]
- Entina, V.S.; Petri, O.A. Electrooxidation of methanol on Pt-Ru and Ru electrodes at different temperatures. Elektrokhimiya 1968, 4, 678. [Google Scholar]
- McNicol, B.D. Electrocatalytic problems associated with the development of direct methanol-air fuel cells. J. Electroanal. Chem. 1981, 118, 71–87. [Google Scholar] [CrossRef]
- Hills, C.W.; Nashner, M.S.; Frenkel, A.I.; Shapley, J.R.; Nuzzo, R.G. Carbon support effects on bimetallic Pt−Ru nanoparticles formed from molecular precursors. Langmuir 1999, 15, 690–700. [Google Scholar] [CrossRef]
- Capon, A.; Parsons, R. The oxidation of formic acid at noble metal electrodes Part III. Intermediates and mechanism on platinum electrodes. J. Electroanal. Chem. 1973, 45, 205. [Google Scholar] [CrossRef]
- Tang, D.; Ci, L.; Zhou, W.; Xie, S. Effect of H2O adsorption on the electrical transport properties of double-walled carbon nanotubes. Carbon 2006, 44, 2155–2159. [Google Scholar] [CrossRef]
- Bard, A.J.; Faulkner, L.R. Electrochemical Methods Fundamentals and Applications, 2nd ed.; John Wiley: New York, NY, USA, 2001. [Google Scholar]
- Tripkovic, A.V.; Popovic, K.D.; Lovic, J.D. Kinetic study of methanol oxidation on Pt2Ru3/C catalyst in alkaline media. J. Serb. Chem. Soc. 2007, 72, 1095–1101. [Google Scholar] [CrossRef]
- Gojkovic, S.L.; Vidakovic, T.R.; Durovic, D.R. Kinetic study of methanol oxidation on carbon-supported PtRu electrocatalyst. Electrochim. Acta 2003, 48, 3607–3614. [Google Scholar] [CrossRef]
- Franaszczuk, K.; Herrero, E.; Zelenay, P.; Wieckowski, A.; Wang, J.; Masel, R.I. A comparison of electrochemical and gas-phase decomposition of methanol on platinum surfaces. J. Phys. Chem. 1992, 96, 8509–8516. [Google Scholar] [CrossRef]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Li, L.; Xing, Y. Methanol Electro-Oxidation on Pt-Ru Alloy Nanoparticles Supported on Carbon Nanotubes. Energies 2009, 2, 789-804. https://doi.org/10.3390/en20300789
Li L, Xing Y. Methanol Electro-Oxidation on Pt-Ru Alloy Nanoparticles Supported on Carbon Nanotubes. Energies. 2009; 2(3):789-804. https://doi.org/10.3390/en20300789
Chicago/Turabian StyleLi, Liang, and Yangchuan Xing. 2009. "Methanol Electro-Oxidation on Pt-Ru Alloy Nanoparticles Supported on Carbon Nanotubes" Energies 2, no. 3: 789-804. https://doi.org/10.3390/en20300789
APA StyleLi, L., & Xing, Y. (2009). Methanol Electro-Oxidation on Pt-Ru Alloy Nanoparticles Supported on Carbon Nanotubes. Energies, 2(3), 789-804. https://doi.org/10.3390/en20300789