Evolution of Akaganeite in Rust Layers Formed on Steel Submitted to Wet/Dry Cyclic Tests


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
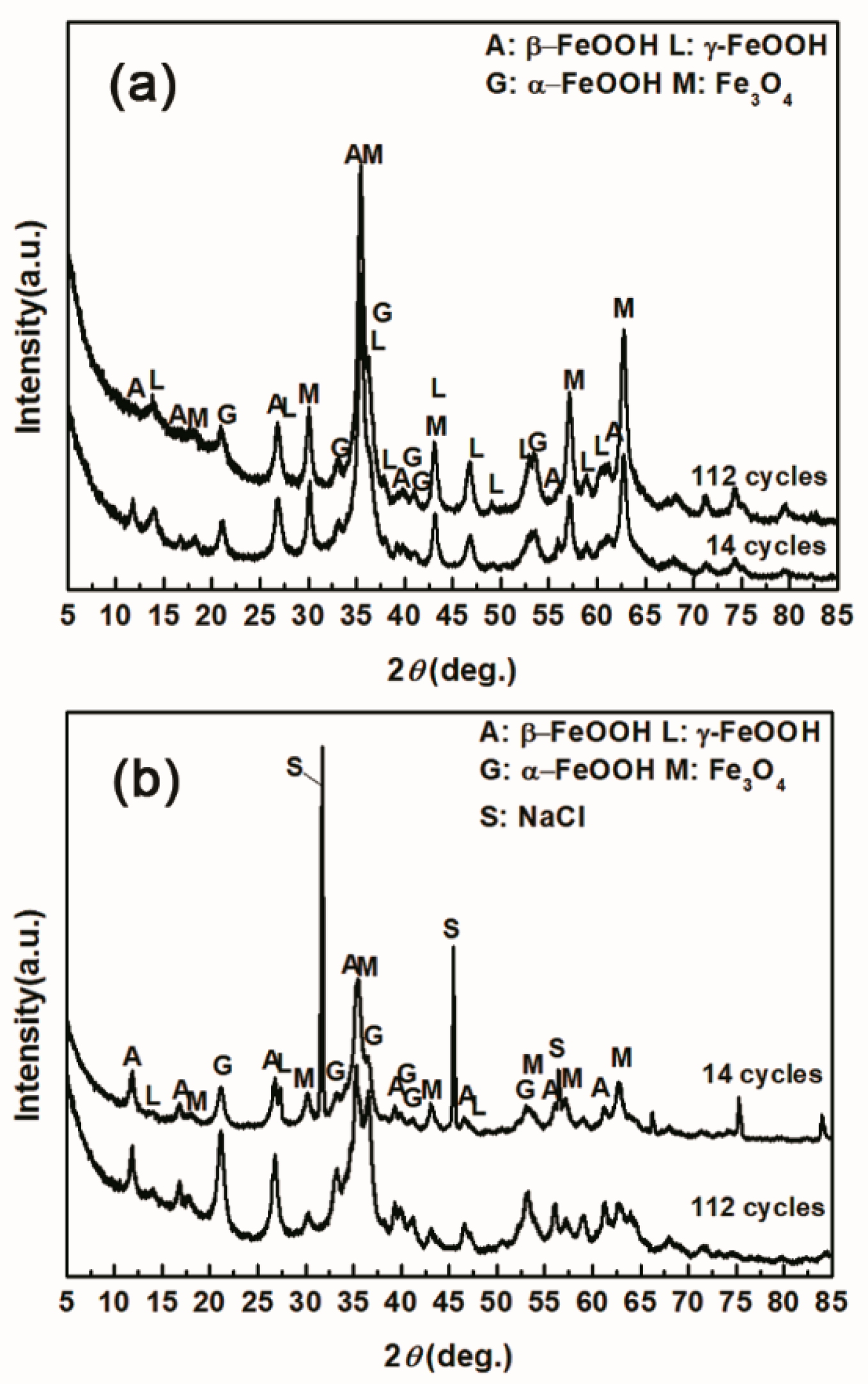
2.2. Characterization of the Iron Rust Phase
2.3. The Wet-Dry Cyclic Corrosion Tests
3. Results and Discussion
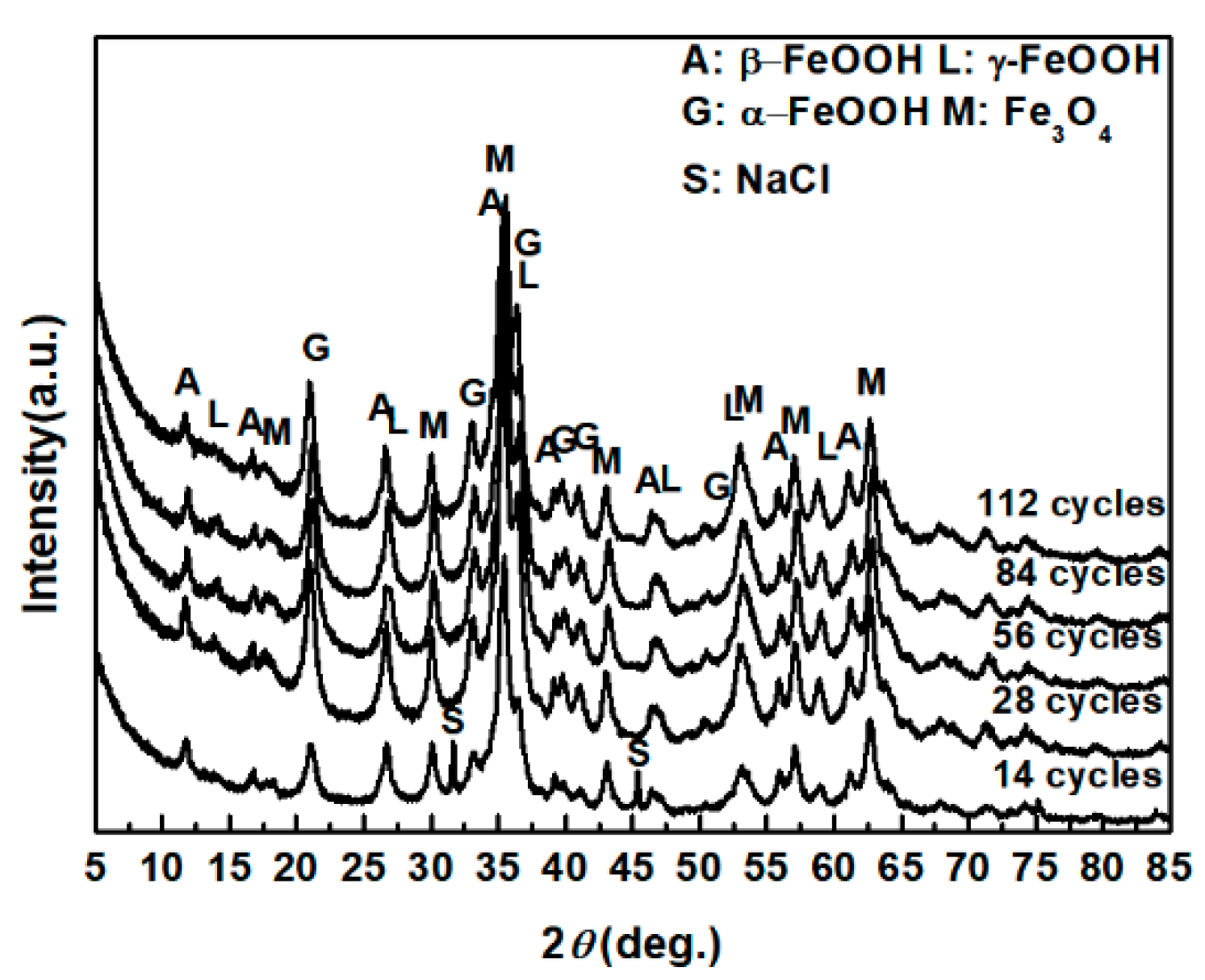
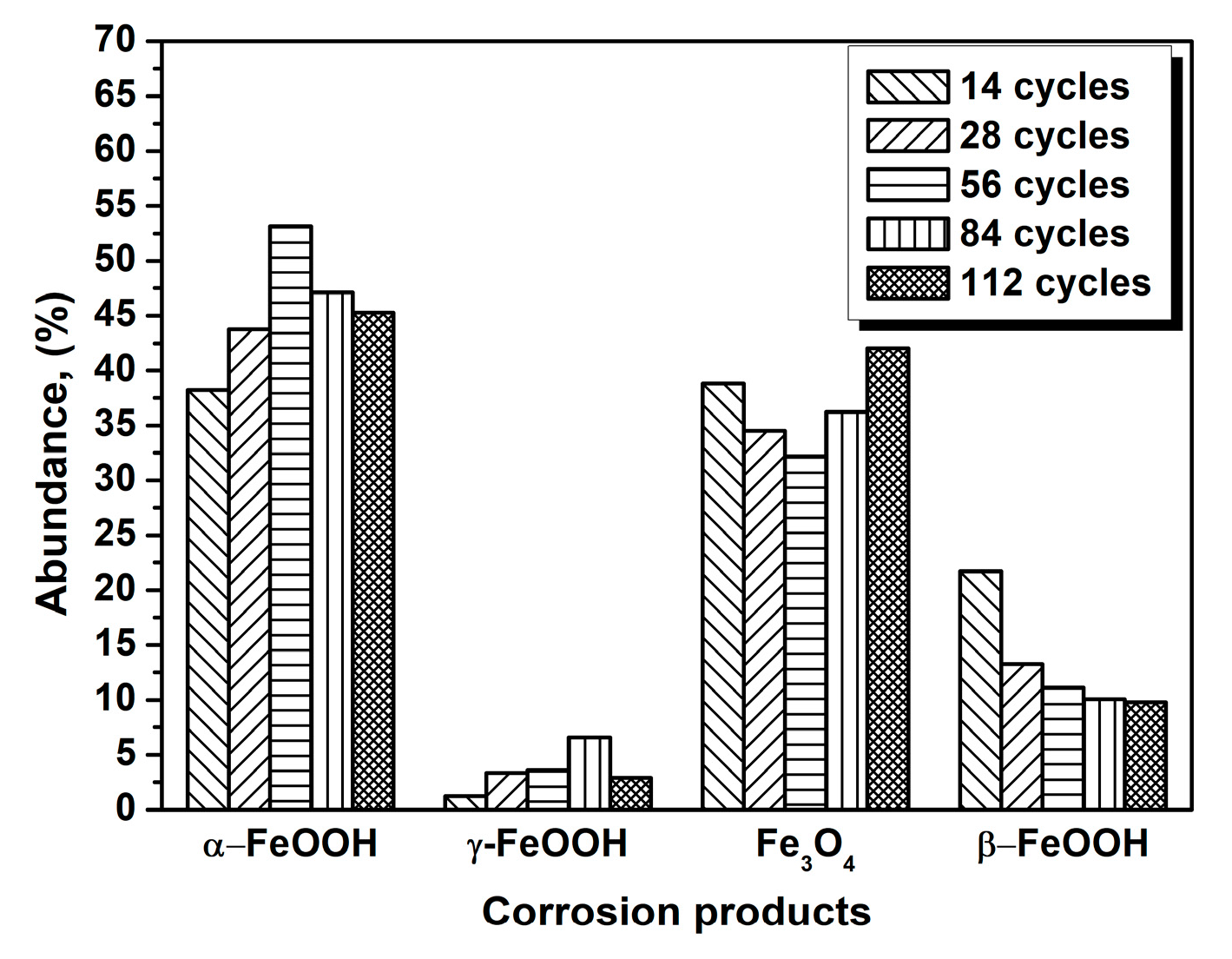
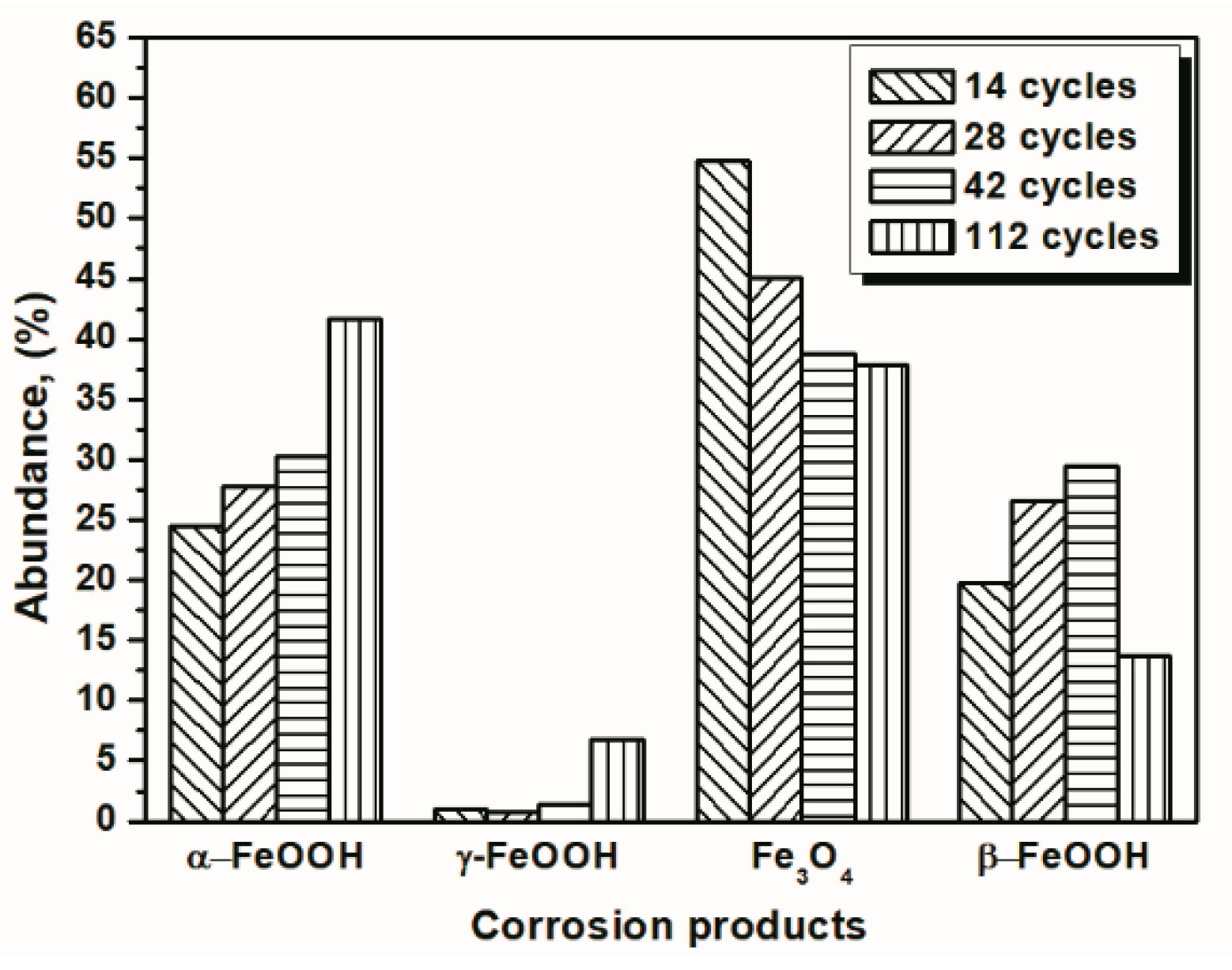
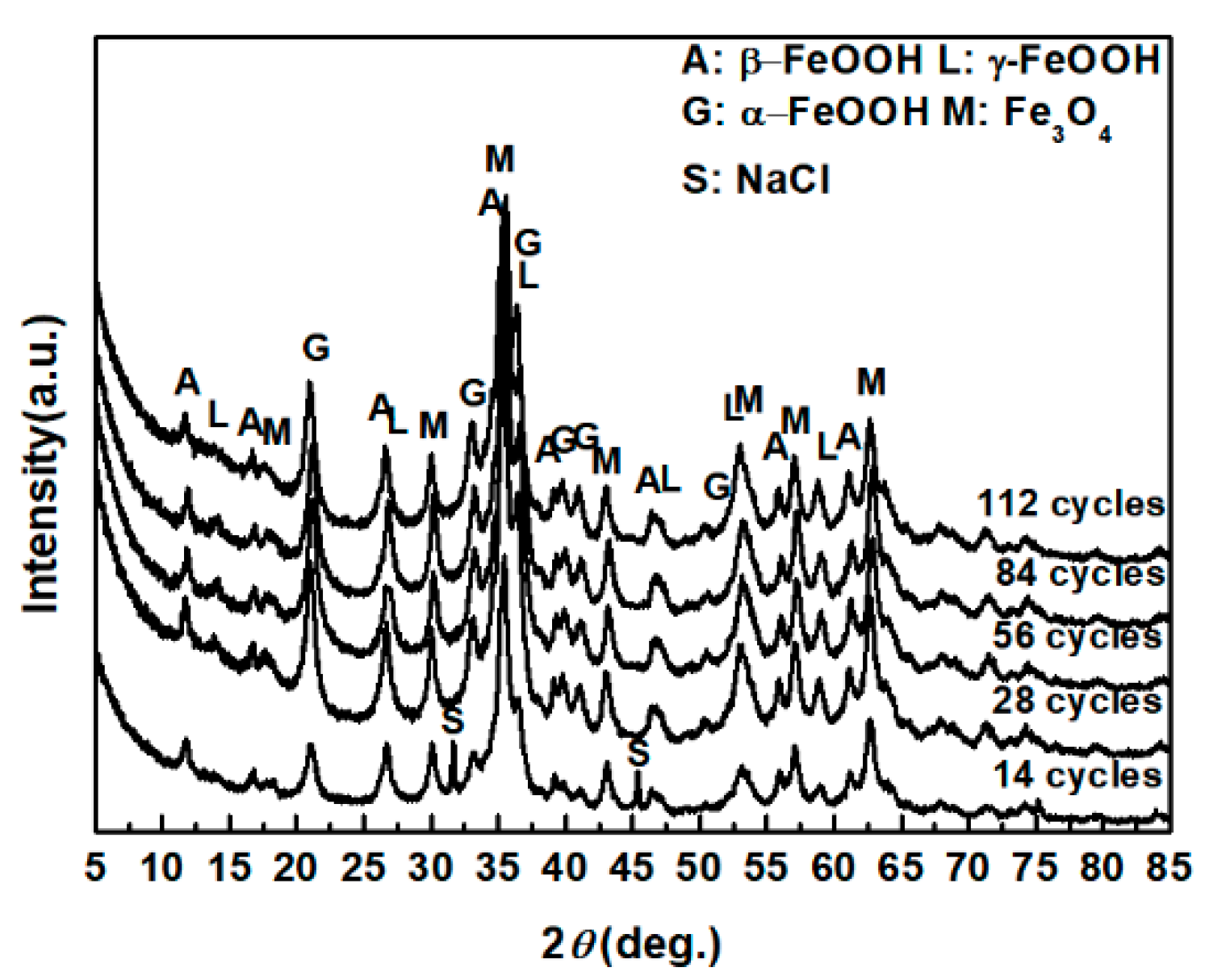
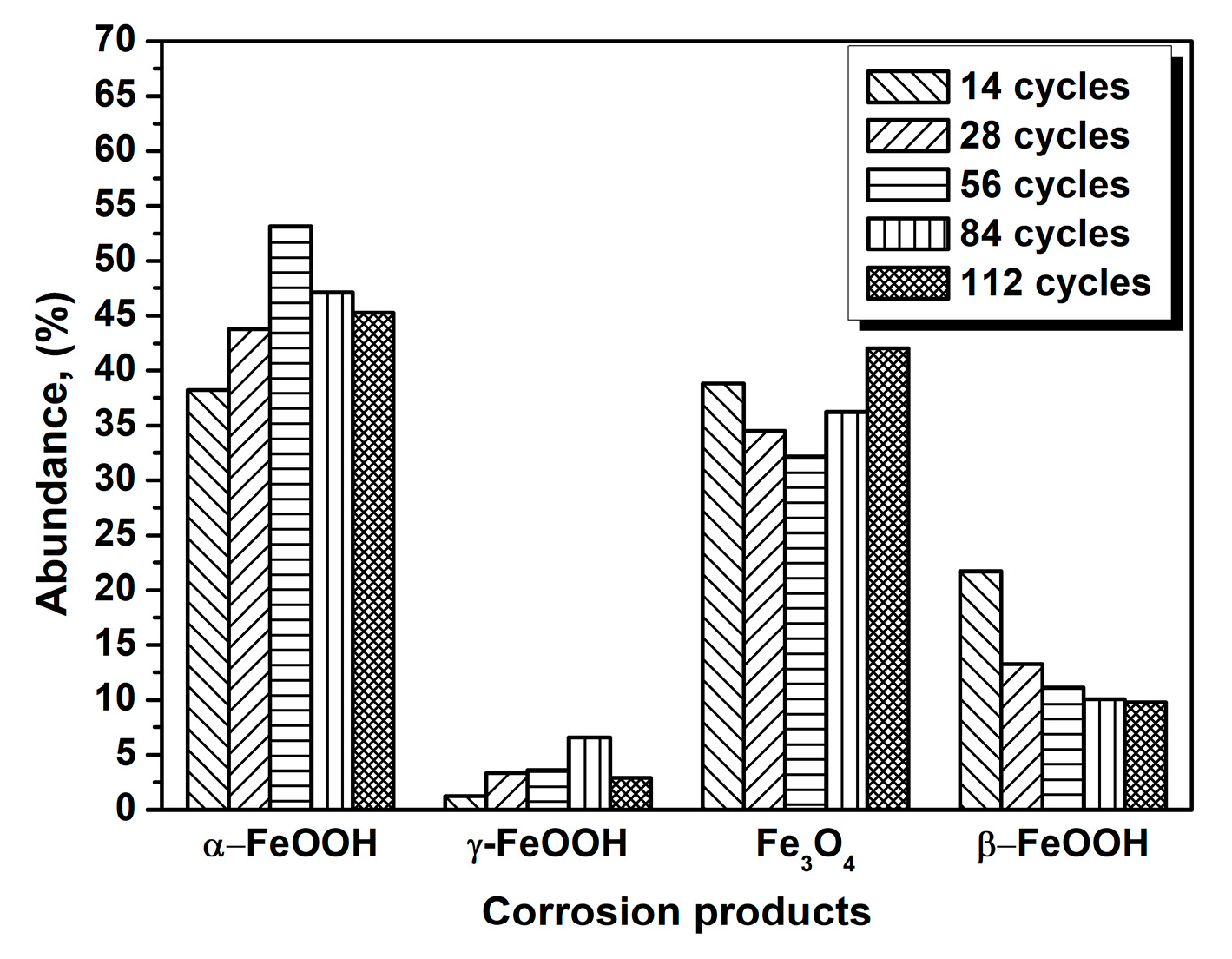
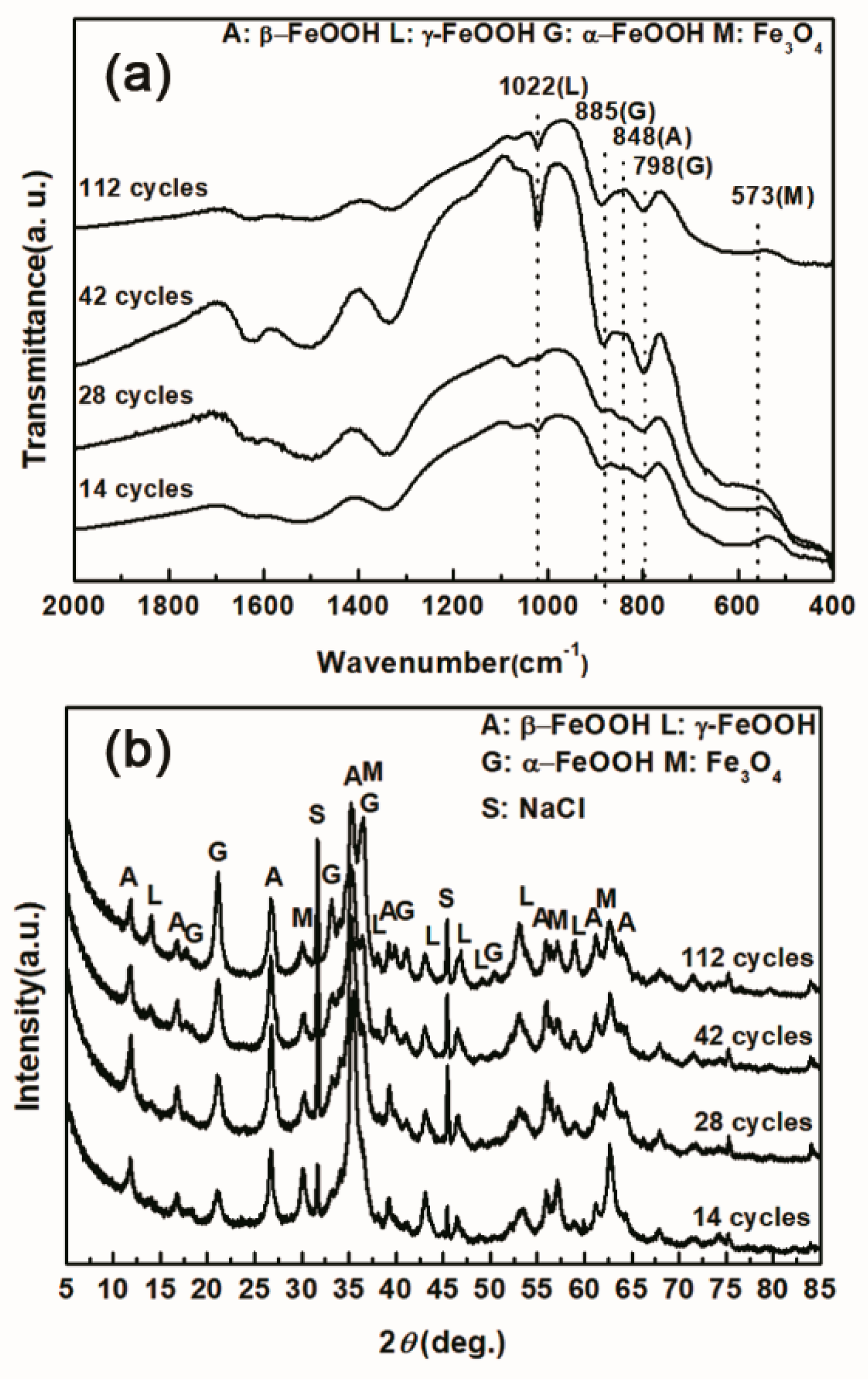
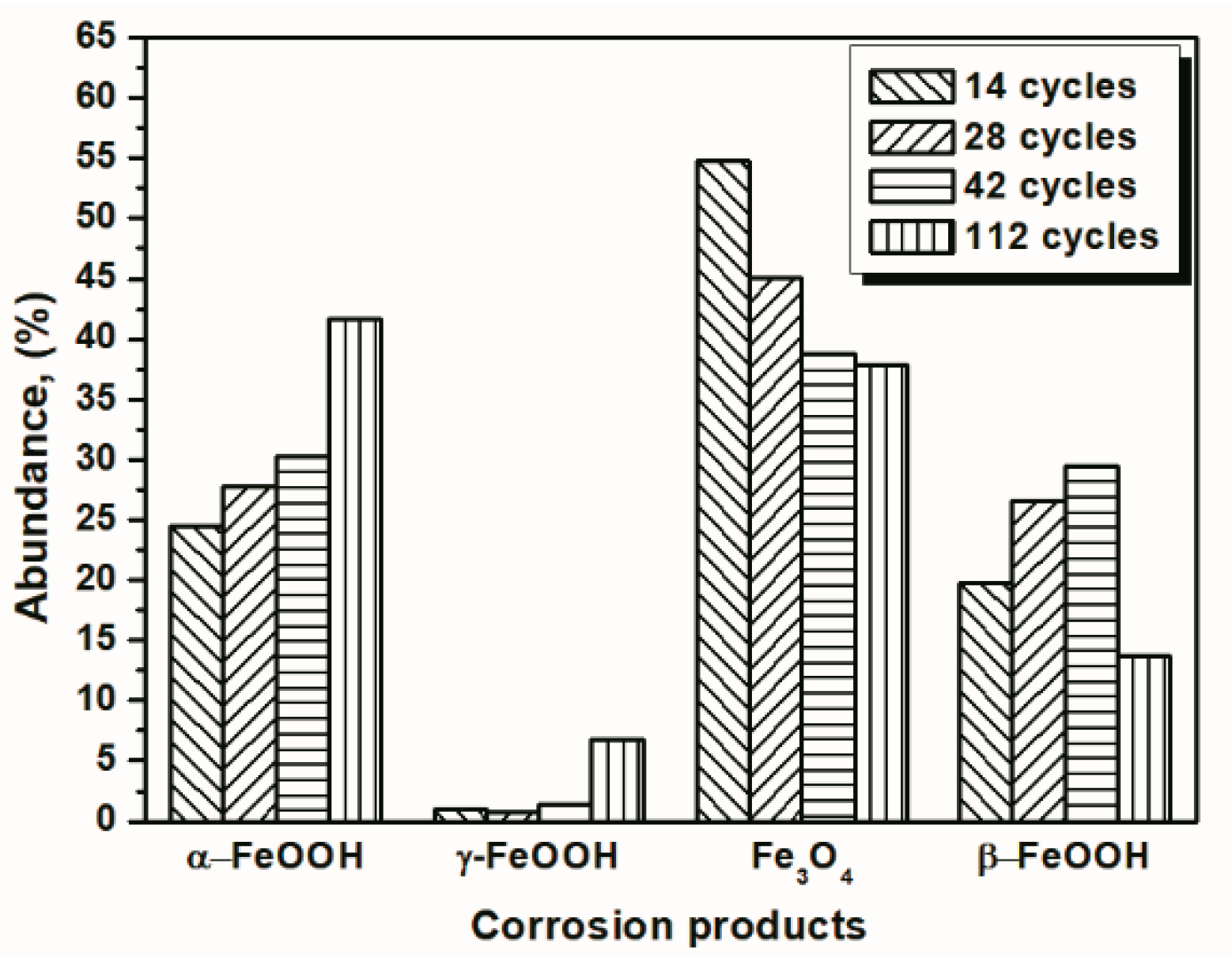
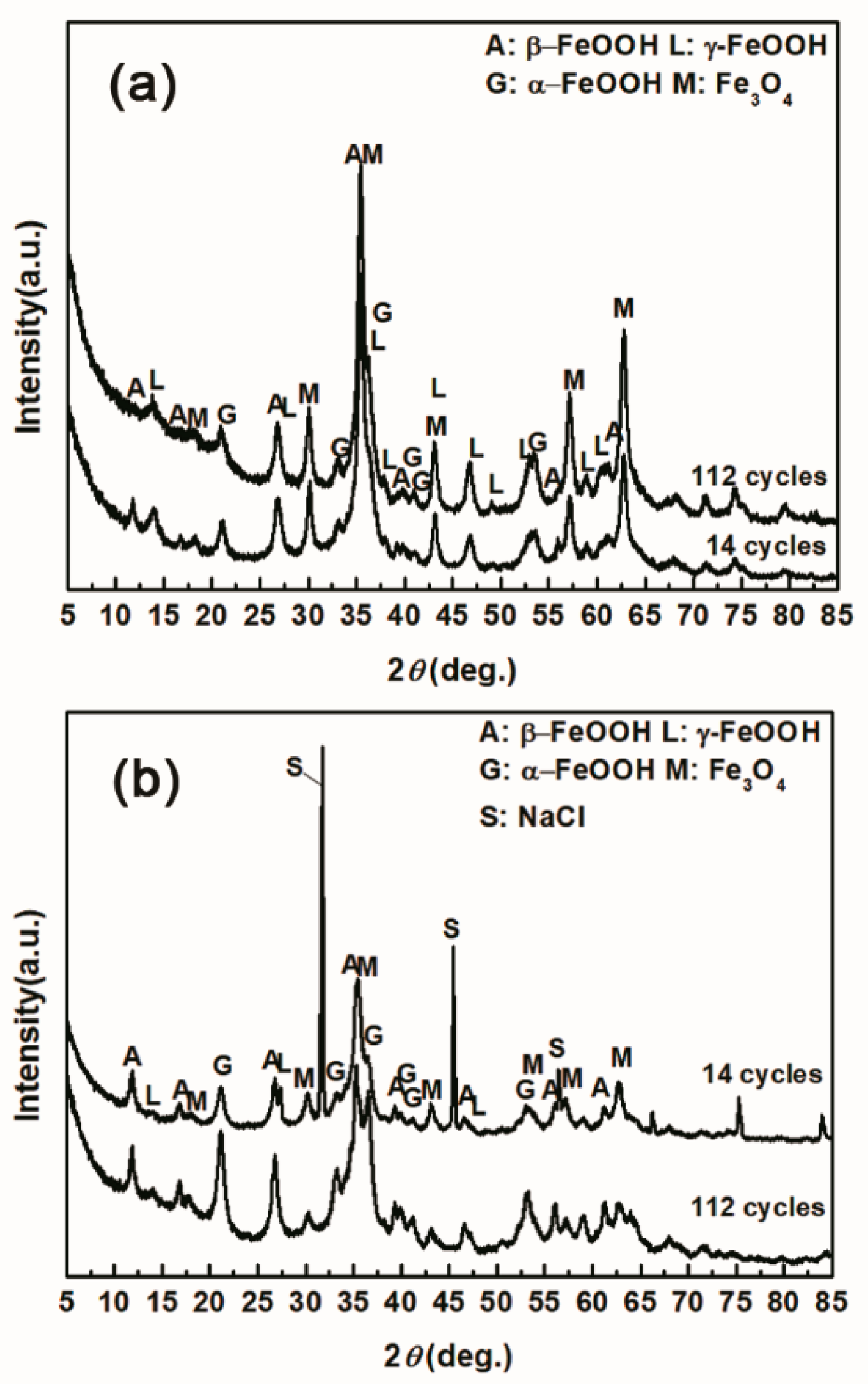
3.1. The Variations in the Relative Amounts of Akaganeite during Repeated Wet-Dry Cycles under the Two Sets of Conditions
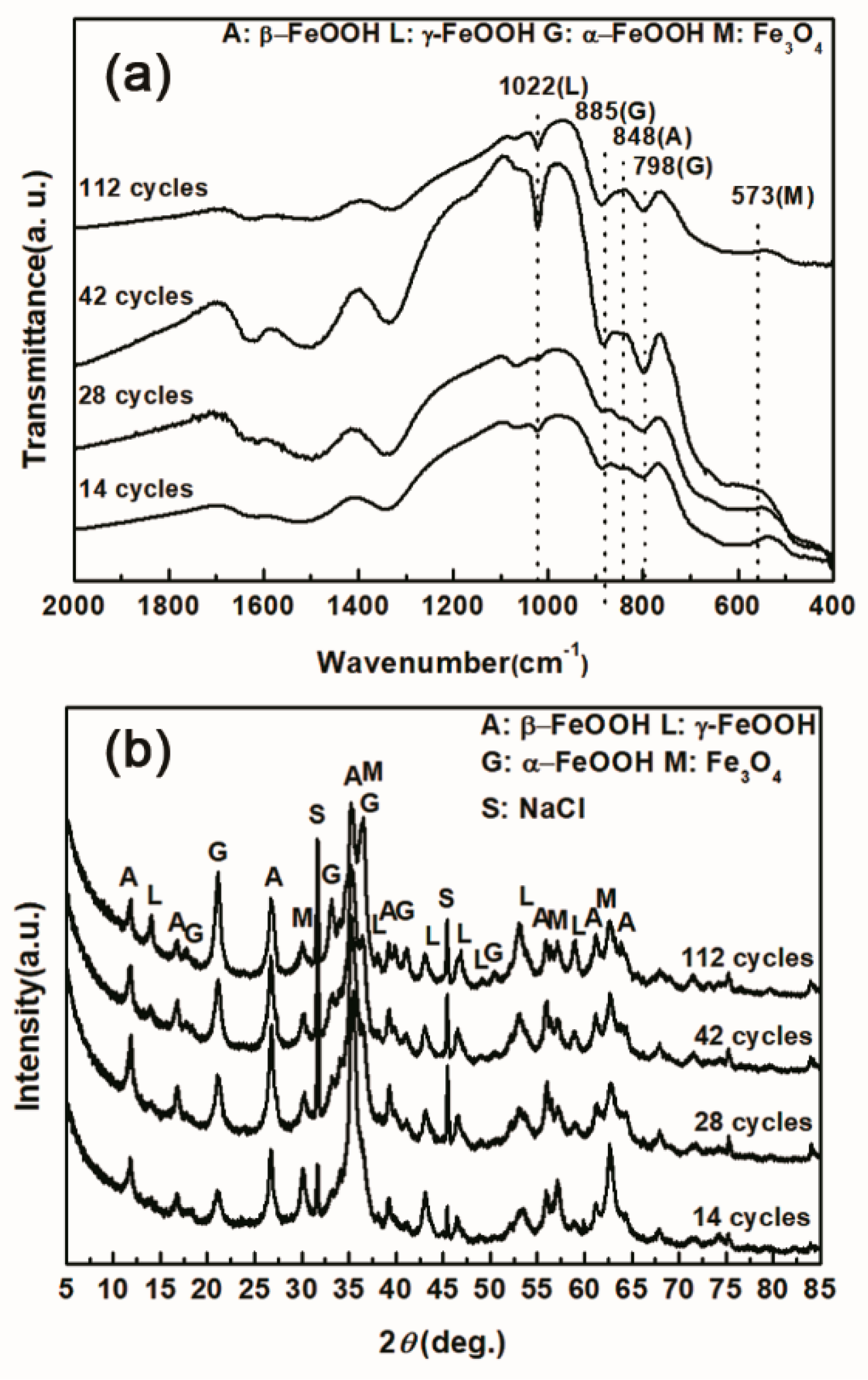
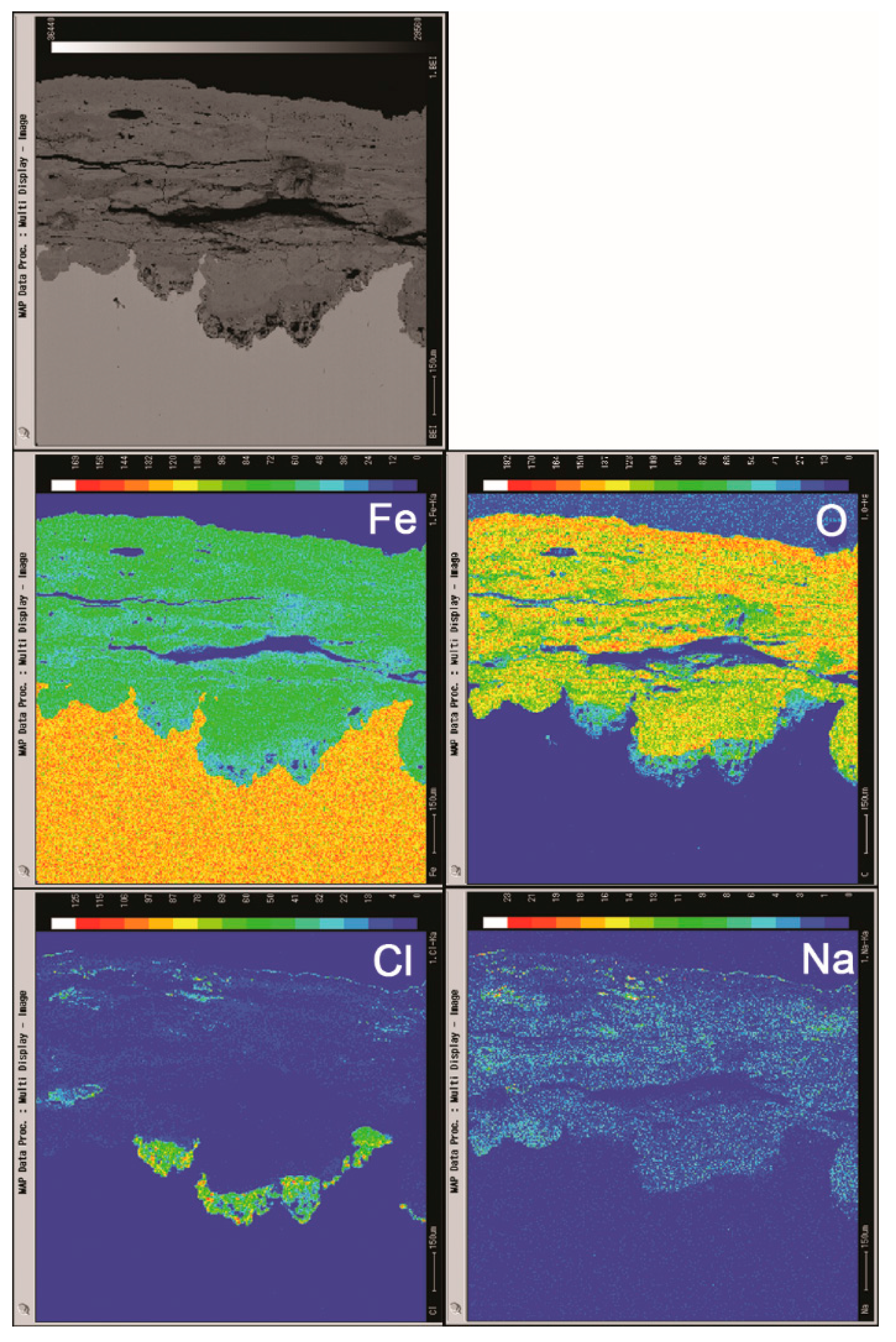
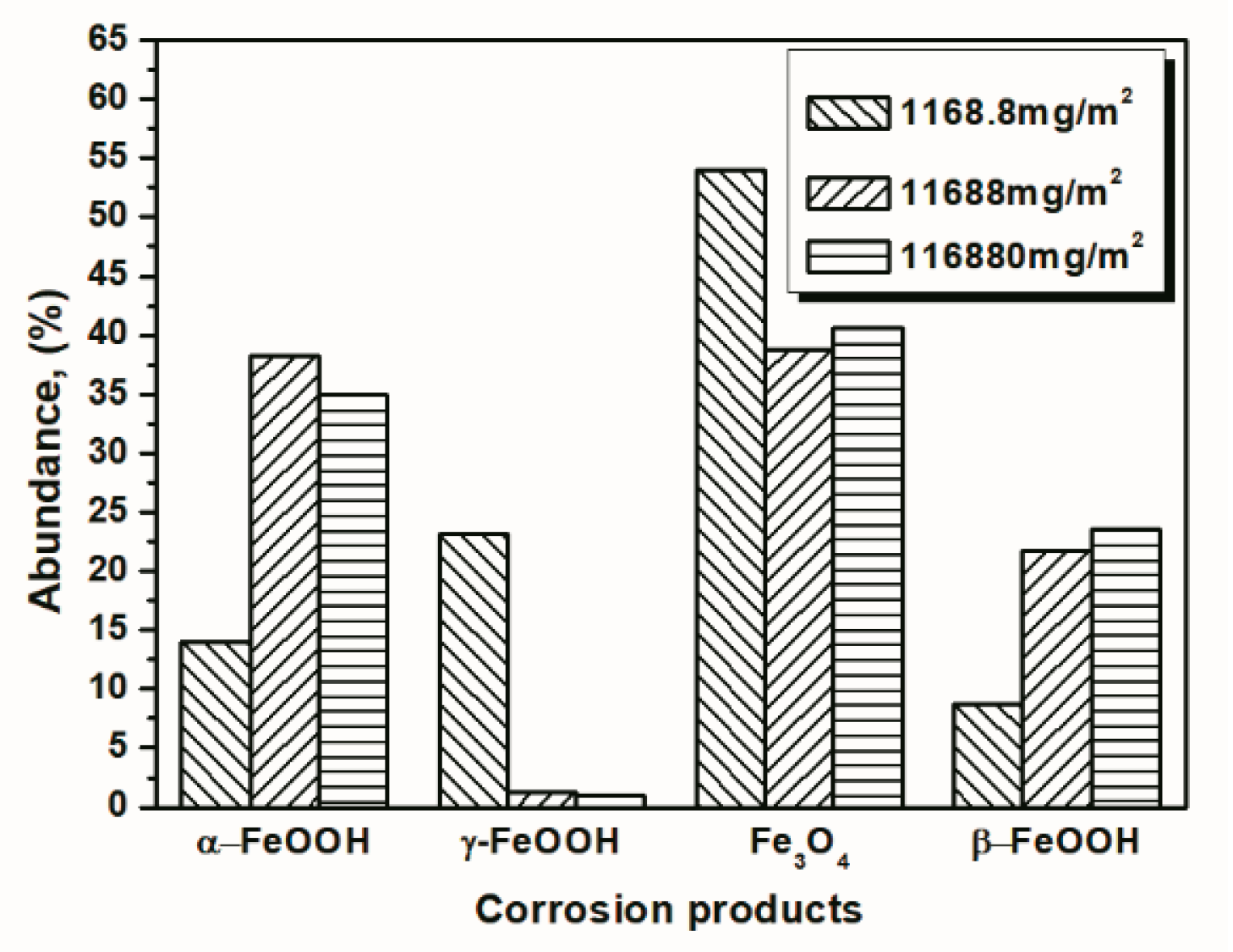
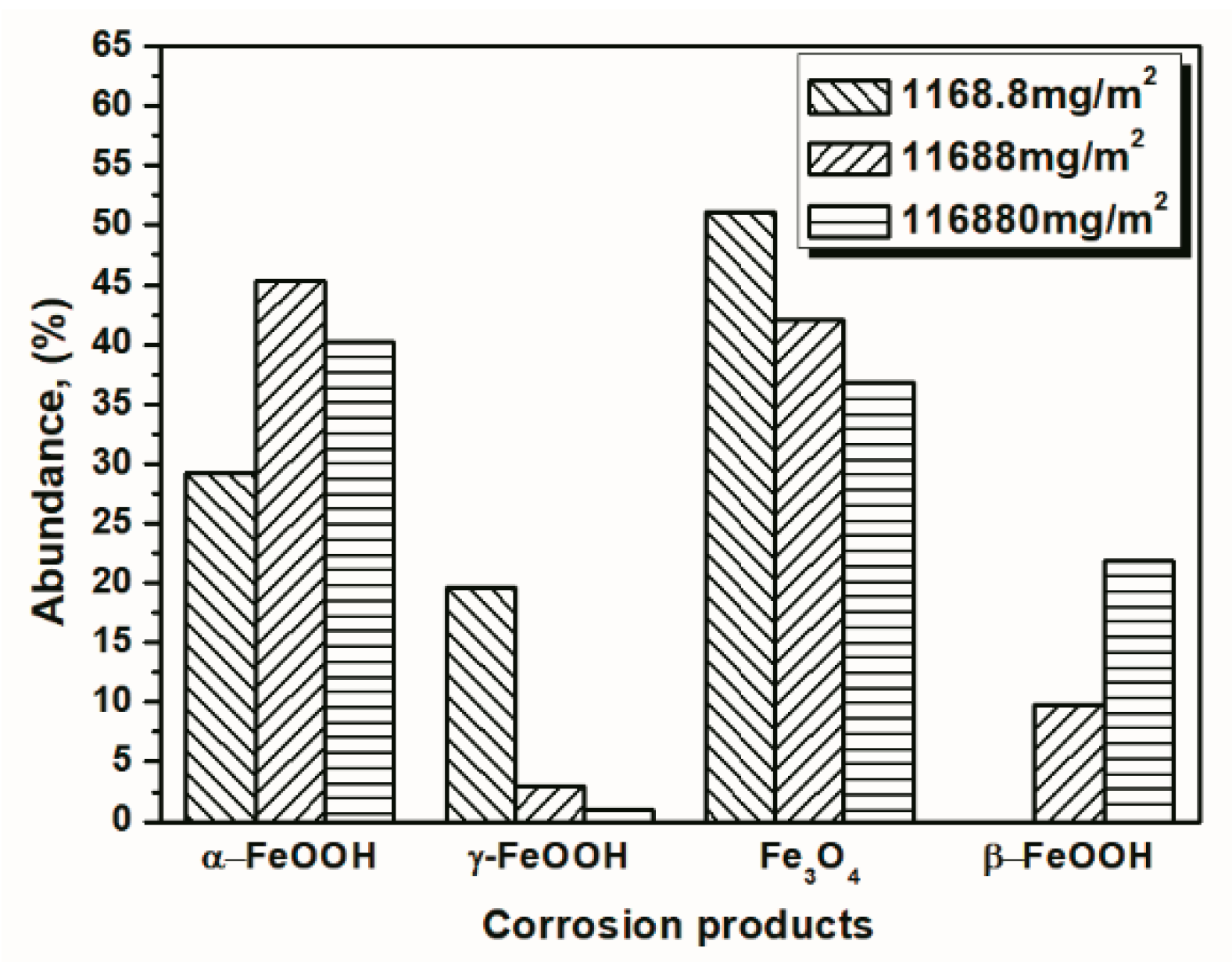
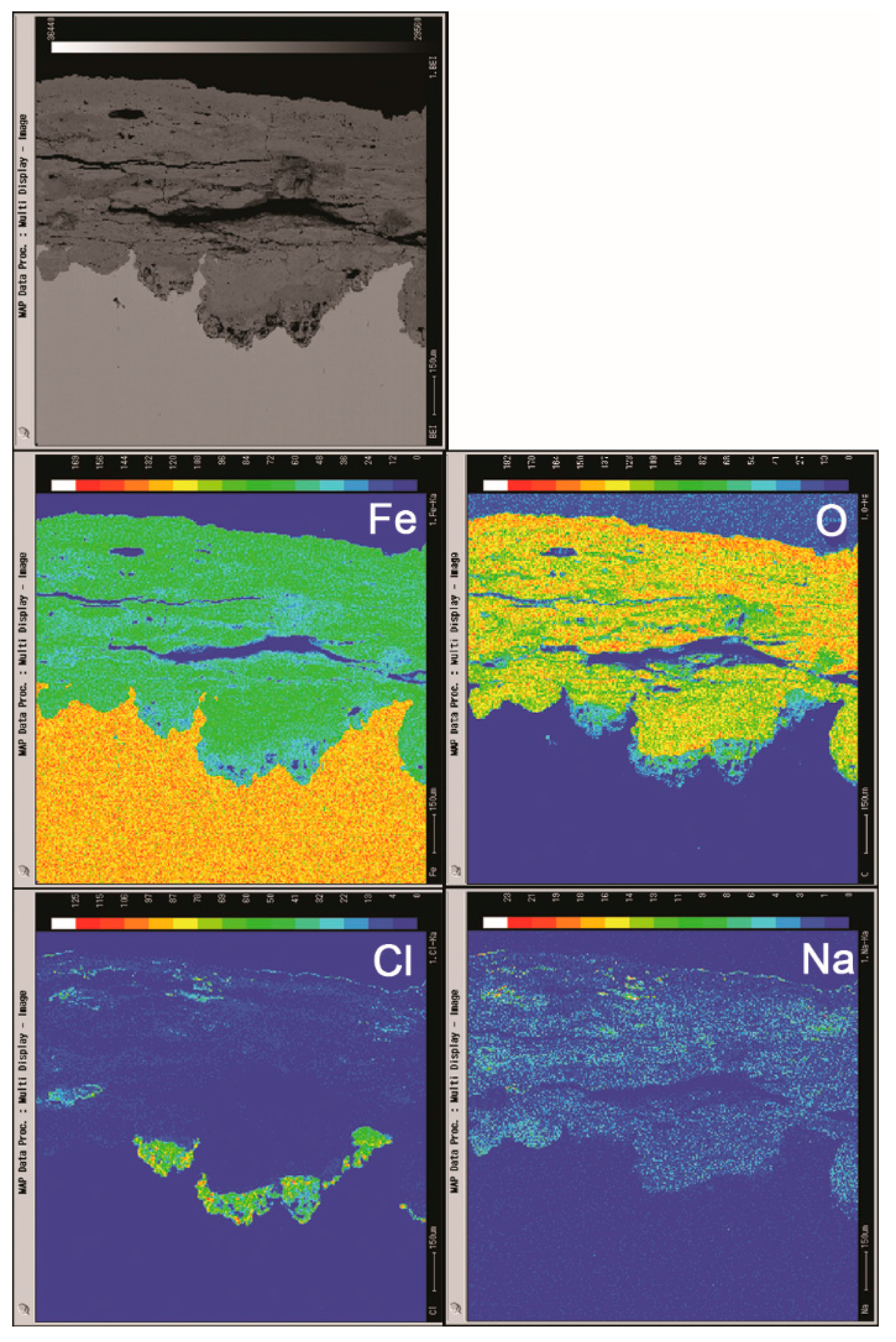
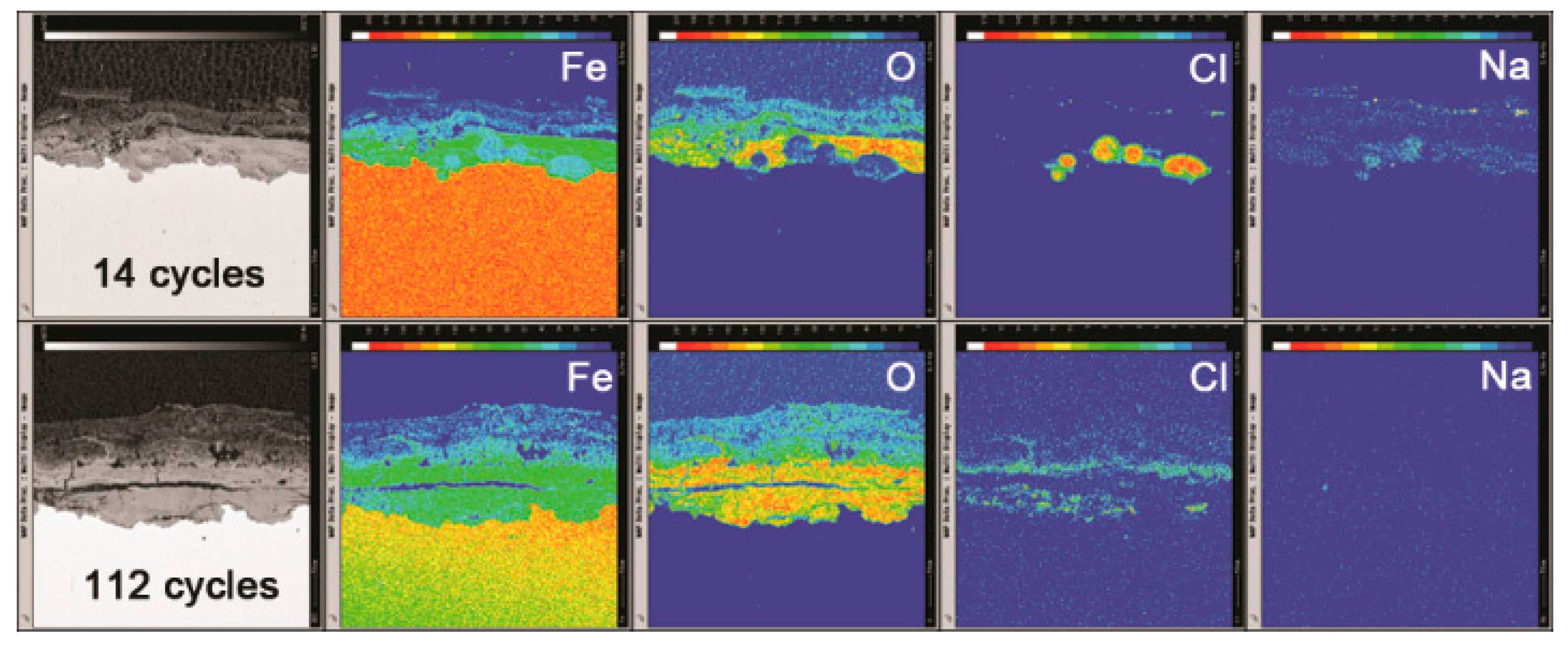
3.2. The Influence of Salt Deposition on the Evolution of Akaganeite
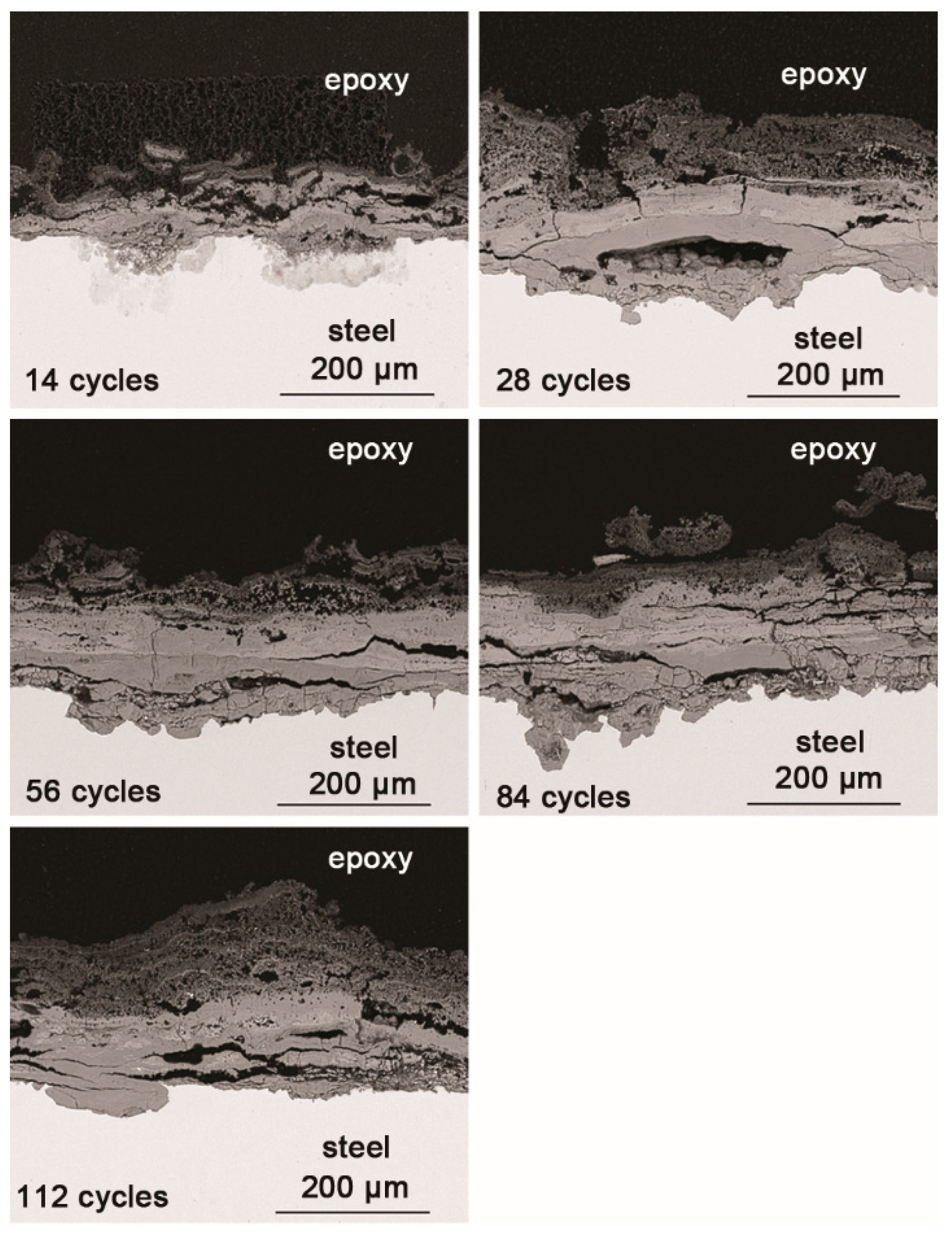
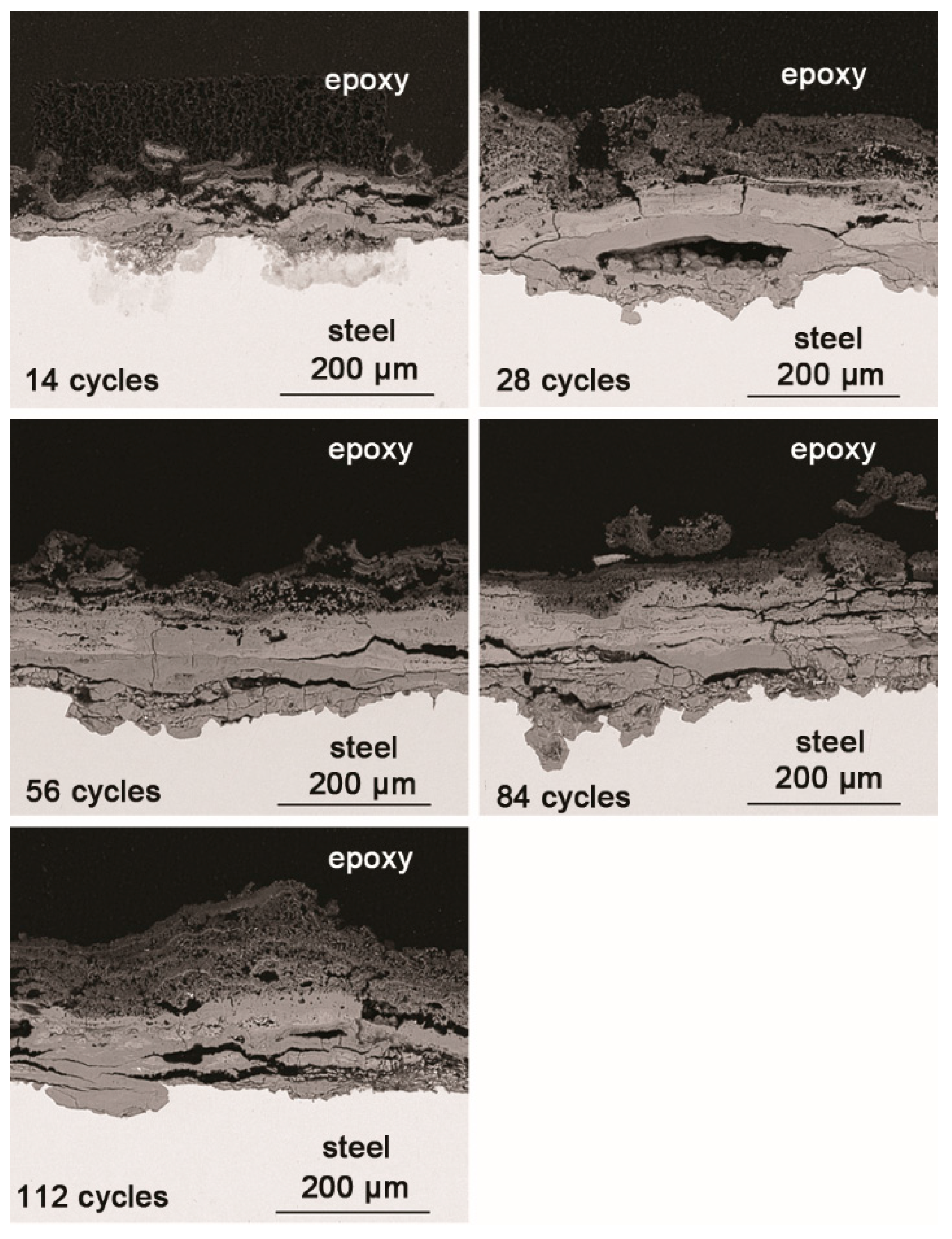
3.3. The Process of the Evolution of Akaganeite in the Rust Layer
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Yamashita, H.M.M.; Matsuda, Y.; Nagano, H.; Misawa, T. The long term growth of the protective rust layer formed on weathering steel by atmospheric corrosion during a quarter of a century. Corros. Sci. 1994, 36, 283–299. [Google Scholar] [CrossRef]
- Misawa, T.; Asami, K.; Hashimoto, K.; Shimodaira, S. The mechanism of atmospheric rusting and the protective amorphous rust on low alloy steel. Corros. Sci. 1974, 14, 279–289. [Google Scholar] [CrossRef]
- Keiser, J.T.; Brown, C.W.; Heidersbach, R.H. Characterization of the passive film formed on weathering steels. Corros. Sci. 1983, 23, 251–259. [Google Scholar] [CrossRef]
- Kamimura, T.; Hara, S.; Miyuki, H.; Yamashita, M.; Uchida, H. Composition and protective ability of rust layer formed on weathering steel exposed to various environments. Corros. Sci. 2006, 48, 2799–2812. [Google Scholar] [CrossRef]
- Yamashita, M.; Maeda, A.; Uchida, H.; Kamimura, T.; Miyuki, H. Crystalline rust compositions and weathering properties of steels exposed in nation-wide atmospheres for 17 years. J. Jpn. Inst. Met. 2001, 65, 967–971. [Google Scholar] [CrossRef]
- Kamimura, T.; Nasu, S.; Tazaki, T.; Kuzushita, K.; Morimoto, S. Mossbauer spectroscopic study of rust formed on a weathering steel and a mild steel exposed for a long term in an industrial environment. Mater. Trans. 2002, 43, 694–703. [Google Scholar] [CrossRef]
- Hara, S.; Kamimura, T.; Miyuki, H.; Yamashita, M. Taxonomy for protective ability of rust layer using its composition formed on weathering steel bridge. Corros. Sci. 2007, 49, 1131–1142. [Google Scholar] [CrossRef]
- Stahl, K.; Nielsen, K.; Jiang, J.Z.; Lebech, B.; Hanson, J.C.; Norby, P.; van Lanschot, J. On the akaganeite crystal structure, phase transformations and possible role in post-excavational corrosion of iron artifacts. Corros. Sci. 2003, 45, 2563–2575. [Google Scholar] [CrossRef]
- Nishimura, T.; Katayama, H.; Noda, K.; Kodama, T. Electrochemical behavior of rust formed on carbon steel in a wet/dry environment containing chloride ions. Corrosion 2000, 56, 935–941. [Google Scholar] [CrossRef]
- Refait, P.; Genin, J.M.R. The mechanisms of oxidation of ferrous hydroxychloride beta-Fe2(OH)3Cl in aqueous solution: The formation of akaganeite vs goethite. Corros. Sci. 1997, 39, 539–553. [Google Scholar] [CrossRef]
- Morcillo, M.; Gonzalez-Calbet, J.M.; Jimenez, J.A.; Diaz, I.; Alcantara, J.; Chico, B.; Mazario-Fernandez, A.; Gomez-Herrero, A.; Llorente, I.; de la Fuente, D. Environmental Conditions for Akaganeite Formation in Marine Atmosphere Mild Steel Corrosion Products and Its Characterization. Corrosion 2015, 71, 872–886. [Google Scholar] [CrossRef]
- Lair, V.; Antony, H.; Legrand, L.; Chaussé, A. Electrochemical reduction of ferric corrosion products and evaluation of galvanic coupling with iron. Corros. Sci. 2006, 48, 2050–2063. [Google Scholar] [CrossRef]
- Tanaka, H.; Mishima, R.; Hatanaka, N.; Ishikawa, T.; Nakayama, T. Formation of magnetite rust particles by reacting iron powder with artificial alpha-, beta- and gamma-FeOOH in aqueous media. Corros. Sci. 2014, 78, 384–387. [Google Scholar] [CrossRef]
- Wang, J.; Wang, Z.Y.; Ke, W. A study of the evolution of rust on weathering steel submitted to the Qinghai salt lake atmospheric corrosion. Mater. Chem. Phys. 2013, 139, 225–232. [Google Scholar] [CrossRef]
- Oh, S.J.; Cook, D.C.; Townsend, H.E. Atmospheric corrosion of different steels in marine, rural and industrial environments. Corros. Sci. 1999, 41, 1687–1702. [Google Scholar] [CrossRef]
- Ma, Y.T.; Li, Y.; Wang, F.H. The effect of beta-FeOOH on the corrosion behavior of low carbon steel exposed in tropic marine environment. Mater. Chem. Phys. 2008, 112, 844–852. [Google Scholar] [CrossRef]
- Morcillo, M.; Diaz, I.; Chico, B.; Cano, H.; de la Fuente, D. Weathering steels: From empirical development to scientific design. A review. Corros. Sci. 2014, 83, 6–31. [Google Scholar] [CrossRef]
- Refait, P.; Remazeilles, C. On the formation of beta-FeOOH (akaganeite) in chloride-containing environments. Corros. Sci. 2007, 49, 844–857. [Google Scholar]
- Ma, Y.; Li, Y.; Wang, F. Corrosion of low carbon steel in atmospheric environments of different chloride content. Corros. Sci. 2009, 51, 997–1006. [Google Scholar] [CrossRef]
- Alcantara, J.; Chico, B.; Diaz, I.; de la Fuente, D.; Morcillo, M. Airborne chloride deposit and its effect on marine atmospheric corrosion of mild steel. Corros. Sci. 2015, 97, 74–88. [Google Scholar] [CrossRef]
- Li, Q.X.; Wang, Z.Y.; Han, W.; Han, E.H. Characterization of the rust formed on weathering steel exposed to Qinghai salt lake atmosphere. Corros. Sci. 2008, 50, 365–371. [Google Scholar] [CrossRef]
- Program, M. Materials Analysis Uring Diffraction by L. Lutterotti. Version: 2.33. 2010. Available online: http://www.ing.unitn.it/luttero/maud (accessed on 15 March 2016).
- Raman, A.; Kuban, B.; Razvan, A. The application of infrared spectroscopy to the study of atmospheric rust systems—I. Standard spectra and illustrative applications to identify rust phases in natural atmospheric corrosion products. Corros. Sci. 1991, 32, 1295–1306. [Google Scholar] [CrossRef]
- Murad, E.; Bishop, J.L. The infrared spectrum of synthetic akaganeite, beta-FeOOH. Am. Mineral. 2000, 85, 716–721. [Google Scholar] [CrossRef]
- Tamura, H. The role of rusts in corrosion and corrosion protection of iron and steel. Corros. Sci. 2008, 50, 1872–1883. [Google Scholar] [CrossRef]
- Singh, A.K.; Ericsson, T.; Häggström, L.; Gullman, J. Mössbauer and X-ray diffraction phase analysis of rusts from atmospheric test sites with different environments in Sweden. Corros. Sci. 1985, 25, 931–945. [Google Scholar] [CrossRef]
- Dillmann, P.; Mazaudier, F.; Hœrlé, S. Advances in understanding atmospheric corrosion of iron. I. Rust characterisation of ancient ferrous artefacts exposed to indoor atmospheric corrosion. Corros. Sci. 2004, 46, 1401–1429. [Google Scholar] [CrossRef]
- Asami, K.; Kikuchi, M. In-depth distribution of rusts on a plain carbon steel and weathering steels exposed to coastal-industrial atmosphere for 17 years. Corros. Sci. 2003, 45, 2671–2688. [Google Scholar] [CrossRef]
- Rémazeilles, C.; Refait, P. Formation, fast oxidation and thermodynamic data of Fe(II) hydroxychlorides. Corros. Sci. 2008, 50, 856–864. [Google Scholar] [CrossRef]
- Neff, D.; Dillmann, P.; Bellot-Gurlet, L.; Beranger, G. Corrosion of iron archaeological artefacts in soil: Characterisation of the corrosion system. Corros. Sci. 2005, 47, 515–535. [Google Scholar] [CrossRef]
- De la Fuente, D.; Alcántara, J.; Chico, B.; Díaz, I.; Jiménez, J.A.; Morcillo, M. Characterisation of rust surfaces formed on mild steel exposed to marine atmospheres using XRD and SEM/Micro-Raman techniques. Corros. Sci. 2016, 110, 253–264. [Google Scholar] [CrossRef]
- Nomura, K.; Tasaka, M.; Ujihira, Y. Conversion Electron Mossbauer Spectrometric Study of Corrosion Products of Iron Immersed in Sodium-Chloride Solution. Corrosion 1988, 44, 131–135. [Google Scholar] [CrossRef]
- Li, S.X.; Hihara, L.H. A Micro-Raman Spectroscopic Study of Marine Atmospheric Corrosion of Carbon Steel: The Effect of Akaganeite. J. Electrochem. Soc. 2015, 162, C495–C502. [Google Scholar] [CrossRef]
- Tewary, N.K.; Kundu, A.; Nandi, R.; Saha, J.K.; Ghosh, S.K. Microstructural characterisation and corrosion performance of old railway girder bridge steel and modern weathering structural steel. Corros. Sci. 2016, 113, 57–63. [Google Scholar] [CrossRef]
- Cano, H.; Neff, D.; Morcillo, M.; Dillmann, P.; Diaz, I.; de la Fuente, D. Characterization of corrosion products formed on Ni 2.4 wt %–Cu 0.5 wt %–Cr 0.5 wt % weathering steel exposed in marine atmospheres. Corros. Sci. 2014, 87, 438–451. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
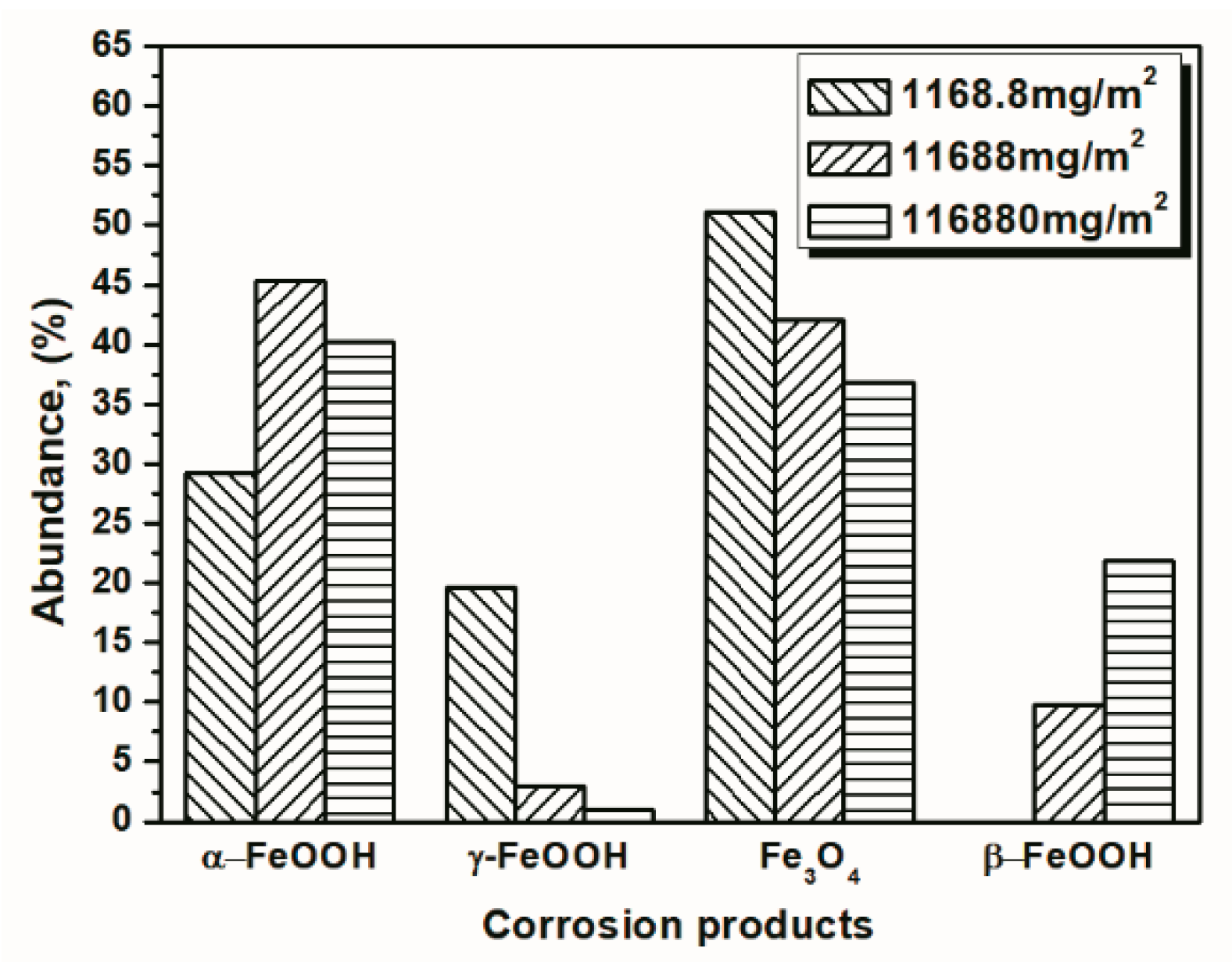
| Numbers of Cycles | α-FeOOH | γ-FeOOH | Fe3O4 | β-FeOOH |
|---|---|---|---|---|
| 14 cycles | 38.25 | 1.22 | 38.82 | 21.70 |
| 28 cycles | 43.77 | 3.34 | 34.50 | 13.28 |
| 56 cycles | 53.14 | 3.58 | 32.18 | 11.10 |
| 84 cycles | 47.11 | 6.60 | 36.23 | 10.06 |
| 112 cycles | 45.29 | 2.90 | 42.02 | 9.78 |
| Numbers of Cycles | α-FeOOH | γ-FeOOH | Fe3O4 | β-FeOOH |
|---|---|---|---|---|
| 14 cycles | 24.49 | 0.94 | 54.77 | 19.80 |
| 28 cycles | 27.69 | 0.73 | 45.02 | 26.56 |
| 42 cycles | 30.28 | 1.43 | 38.76 | 29.52 |
| 112 cycles | 41.66 | 6.73 | 37.85 | 13.75 |
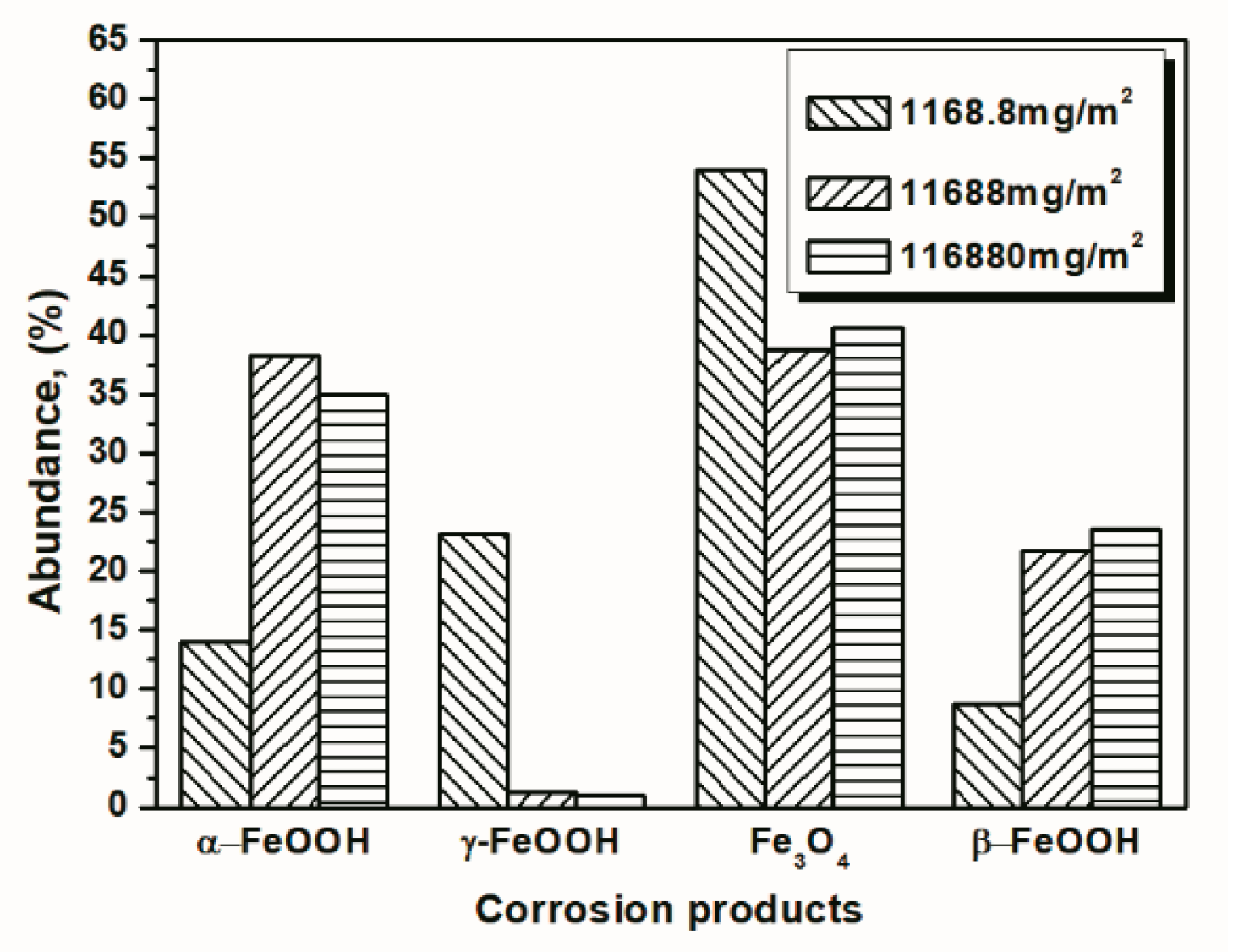
| Numbers of Cycles (Salt Deposition) | α-FeOOH | γ-FeOOH | Fe3O4 | β-FeOOH |
|---|---|---|---|---|
| 14 cycles (1168.8 mg/m2) | 14.04 | 23.17 | 54.06 | 8.73 |
| 112 cycles (1168.8 mg/m2) | 29.20 | 19.69 | 51.11 | 0 |
| 14 cycles (116,880 mg/m2) | 34.96 | 0.94 | 40.59 | 23.51 |
| 112 cycles (116,880 mg/m2) | 40.28 | 1.04 | 36.77 | 21.91 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, H.; Ye, W.; Song, X.; Ma, Y.; Li, Y. Evolution of Akaganeite in Rust Layers Formed on Steel Submitted to Wet/Dry Cyclic Tests. Materials 2017, 10, 1262. https://doi.org/10.3390/ma10111262
Xiao H, Ye W, Song X, Ma Y, Li Y. Evolution of Akaganeite in Rust Layers Formed on Steel Submitted to Wet/Dry Cyclic Tests. Materials. 2017; 10(11):1262. https://doi.org/10.3390/ma10111262
Chicago/Turabian StyleXiao, Haigang, Wei Ye, Xiaoping Song, Yuantai Ma, and Ying Li. 2017. "Evolution of Akaganeite in Rust Layers Formed on Steel Submitted to Wet/Dry Cyclic Tests" Materials 10, no. 11: 1262. https://doi.org/10.3390/ma10111262
APA StyleXiao, H., Ye, W., Song, X., Ma, Y., & Li, Y. (2017). Evolution of Akaganeite in Rust Layers Formed on Steel Submitted to Wet/Dry Cyclic Tests. Materials, 10(11), 1262. https://doi.org/10.3390/ma10111262