
Vibrational Spectroscopy in Studies of Atmospheric Corrosion


Abstract
:1. Introduction
2. Results and Discussion
2.1. Overwiew
2.1.1. Infrared Spectroscopy in General
2.1.2. IR Transmission Spectroscopy
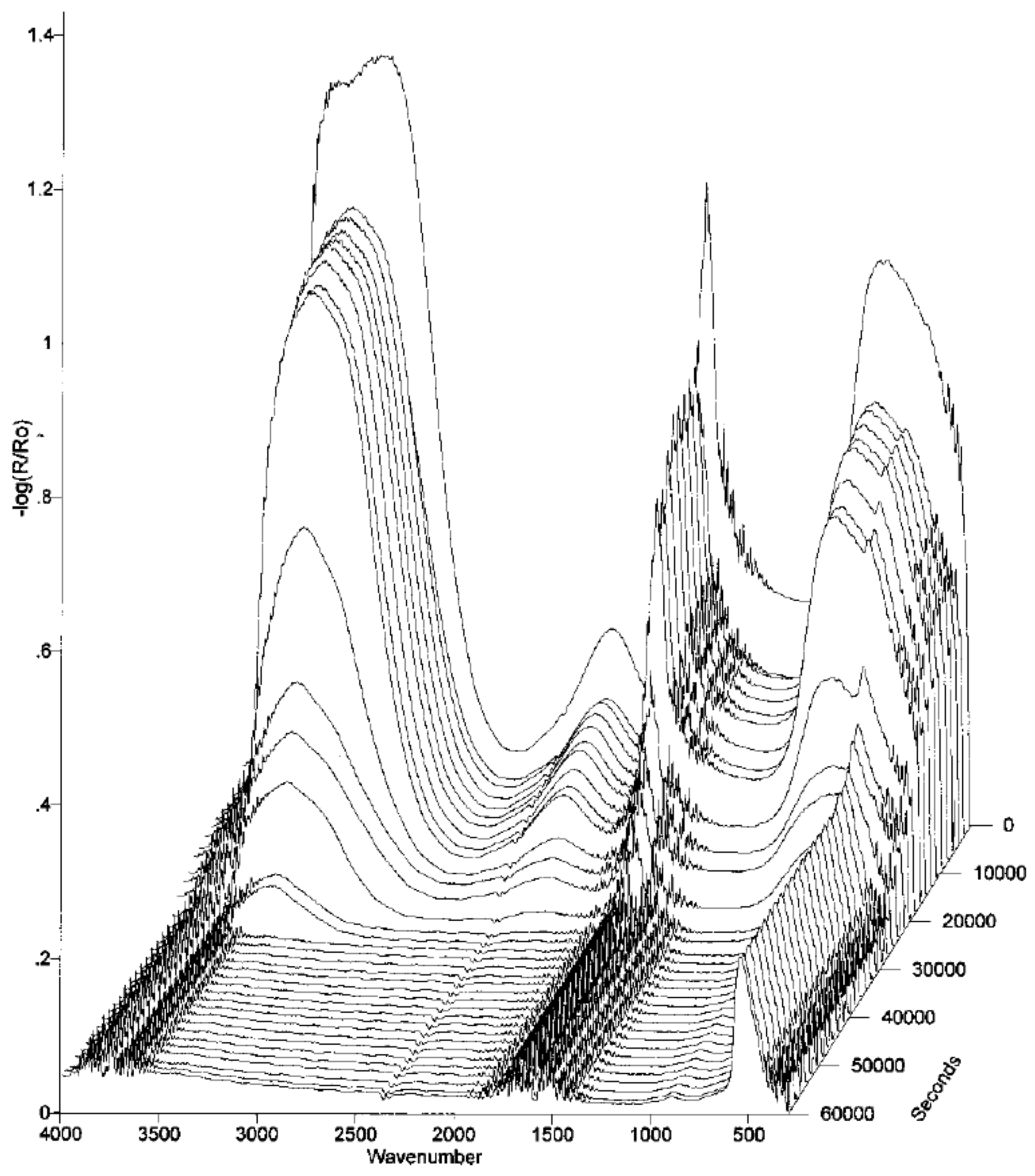
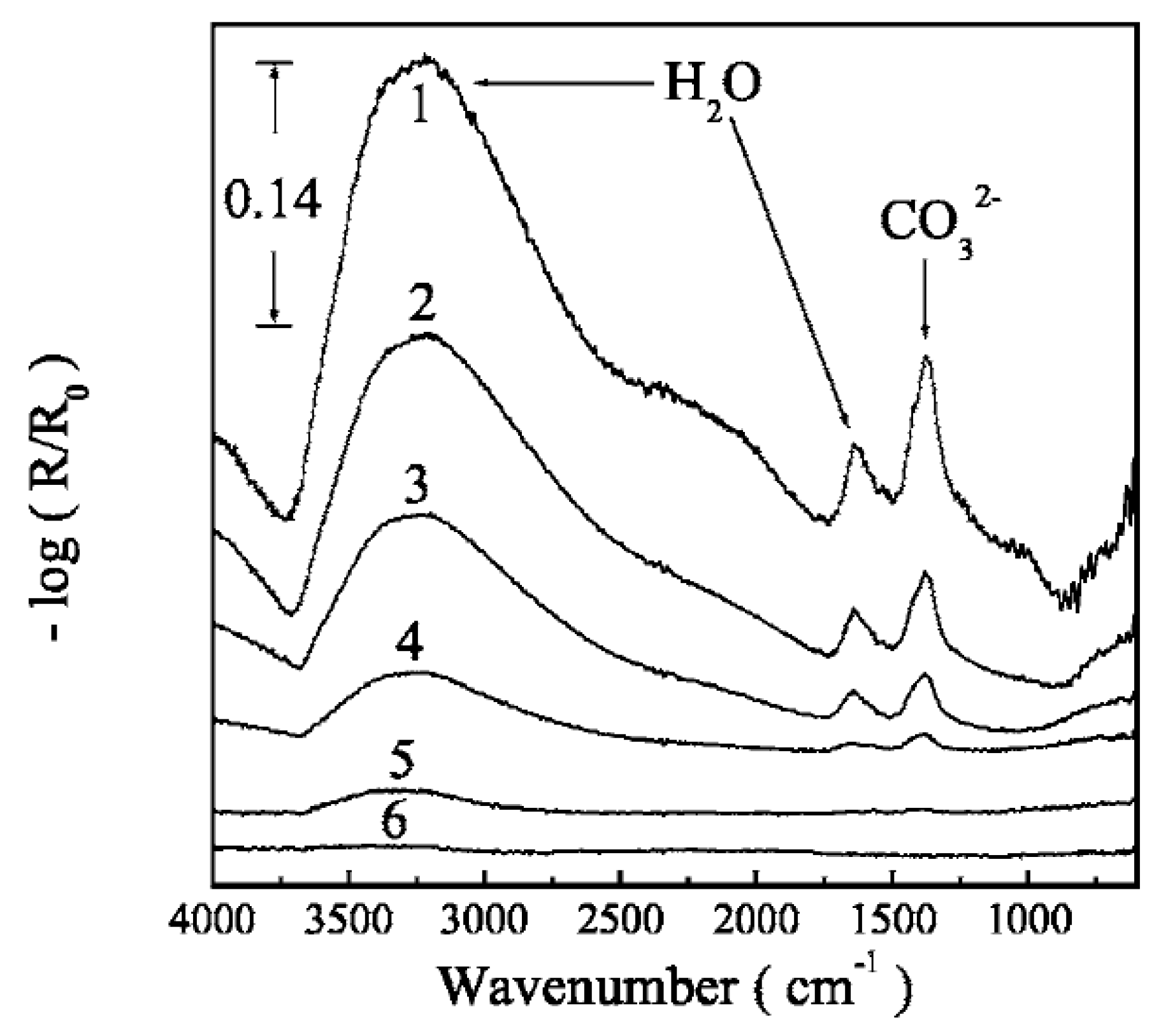
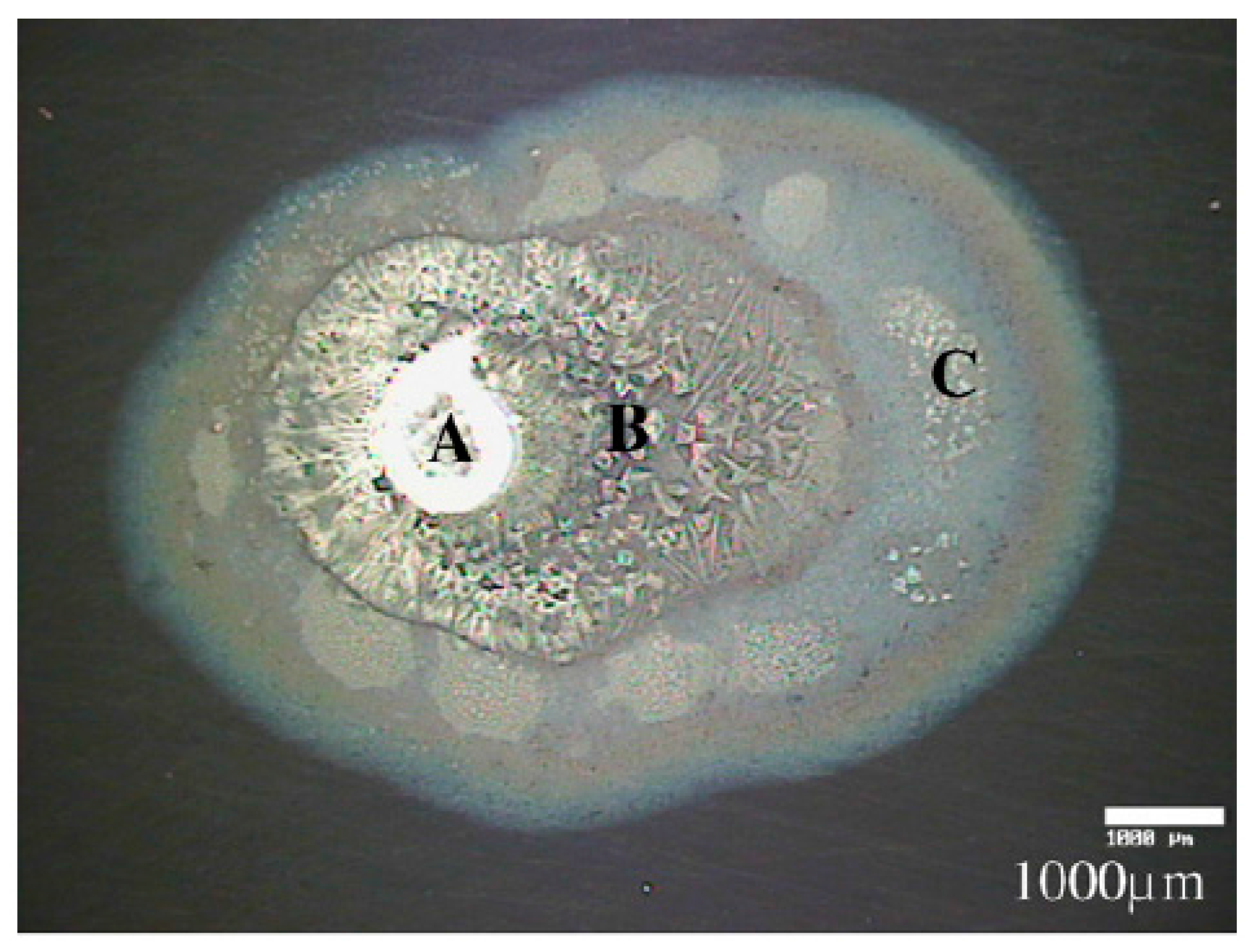
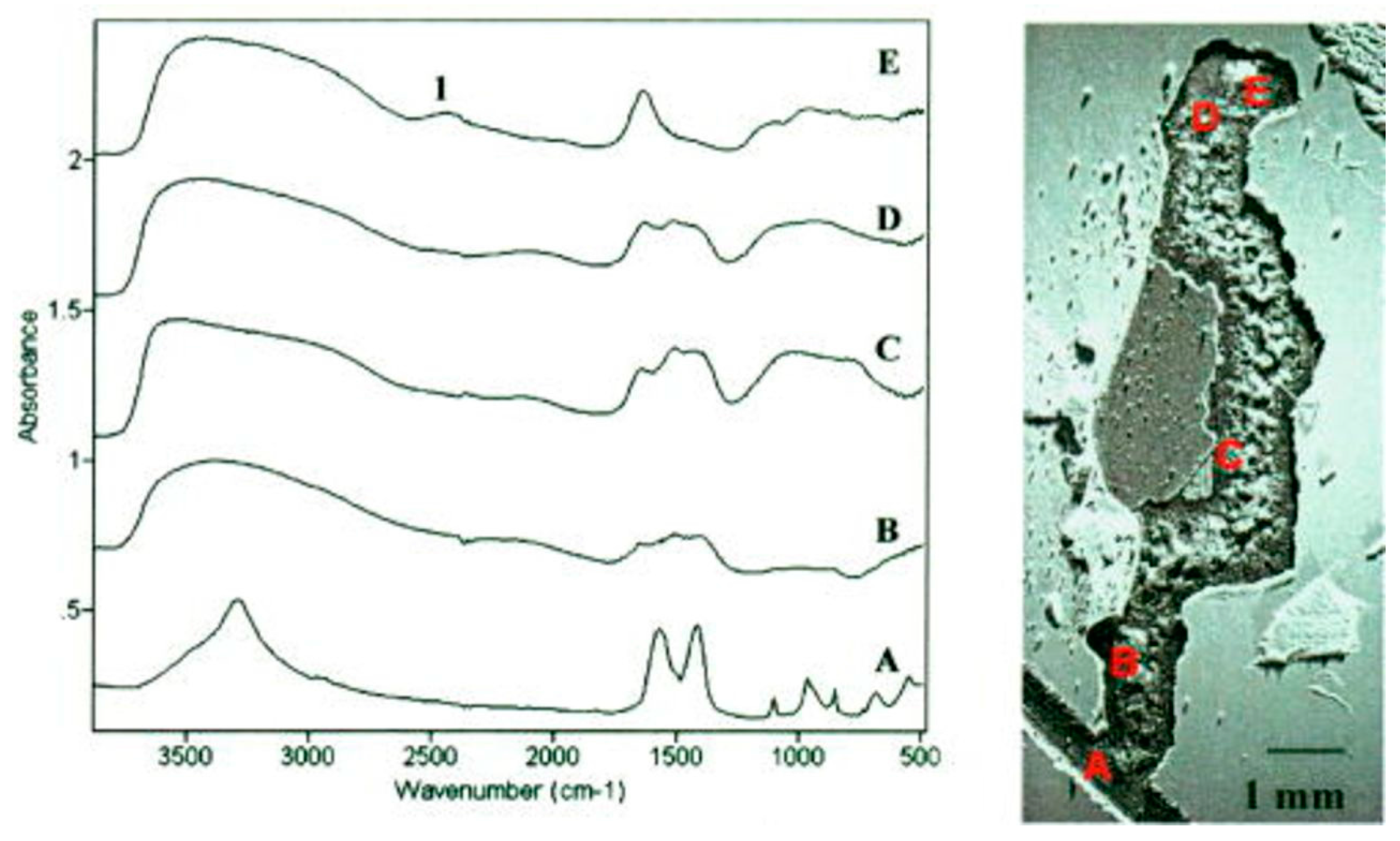
2.1.2.1. Steel
2.1.2.2. Copper
2.1.2.3. Zinc
2.1.2.4. Aluminum and Magnesium Alloys
2.1.2.5. Corrosion Inhibitors
2.1.3. Diffuse Reflectance (DRIFTS)
Nickel
2.1.4. Infrared Reflection/Absorption Spectroscopy (IRRAS)
2.1.4.1. Iron
2.1.4.2. Steel
2.1.4.3. Copper
2.1.4.4. Silver
2.1.4.5. Zinc
2.1.4.6. Brass
2.1.4.7. Tin
2.1.4.8. Bronze
2.1.4.9. Aluminum
2.1.5. Attenuated Total Reflection (ATR)
2.1.6. IR Microscopy
2.1.6.1. Conventional IR Microscopy
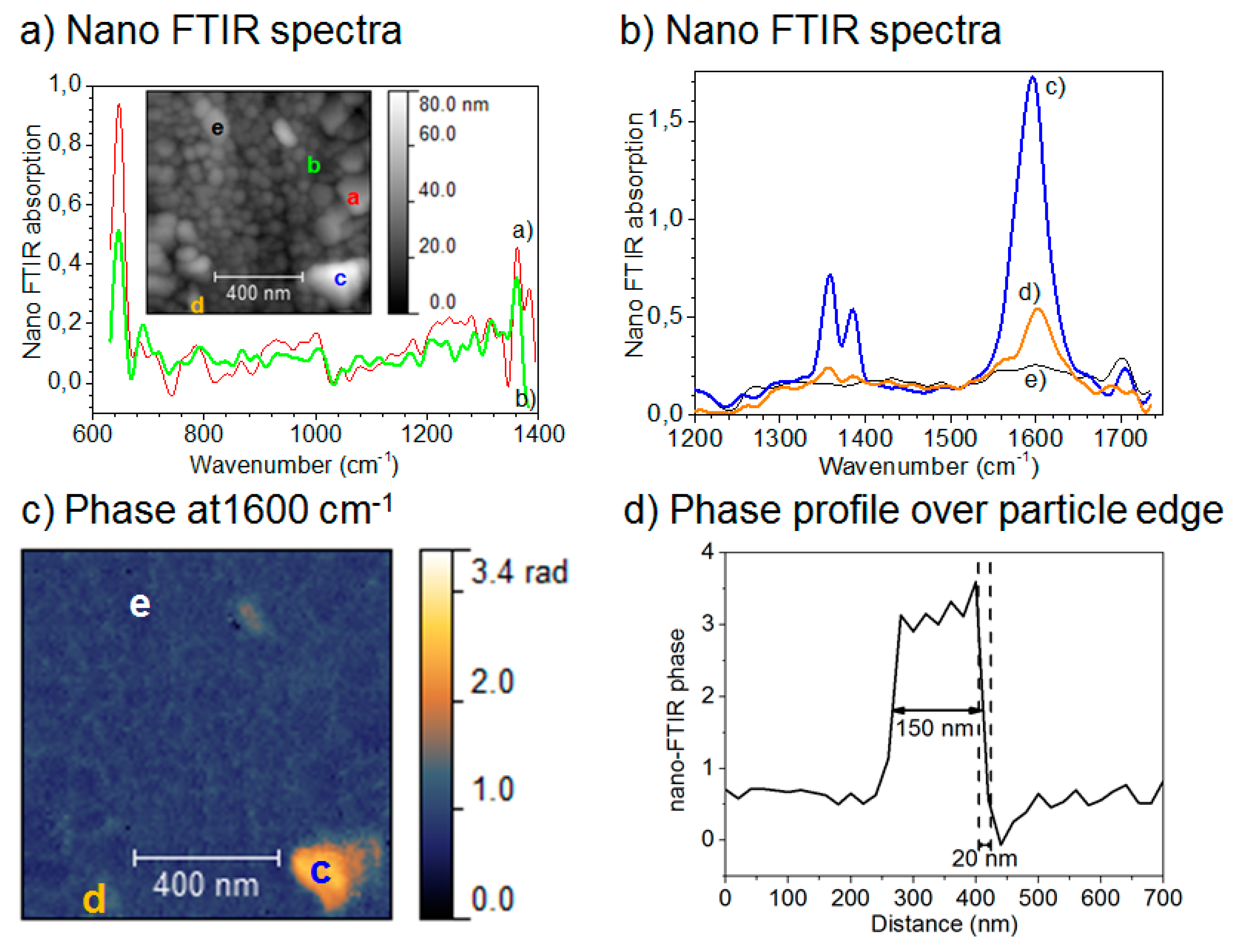
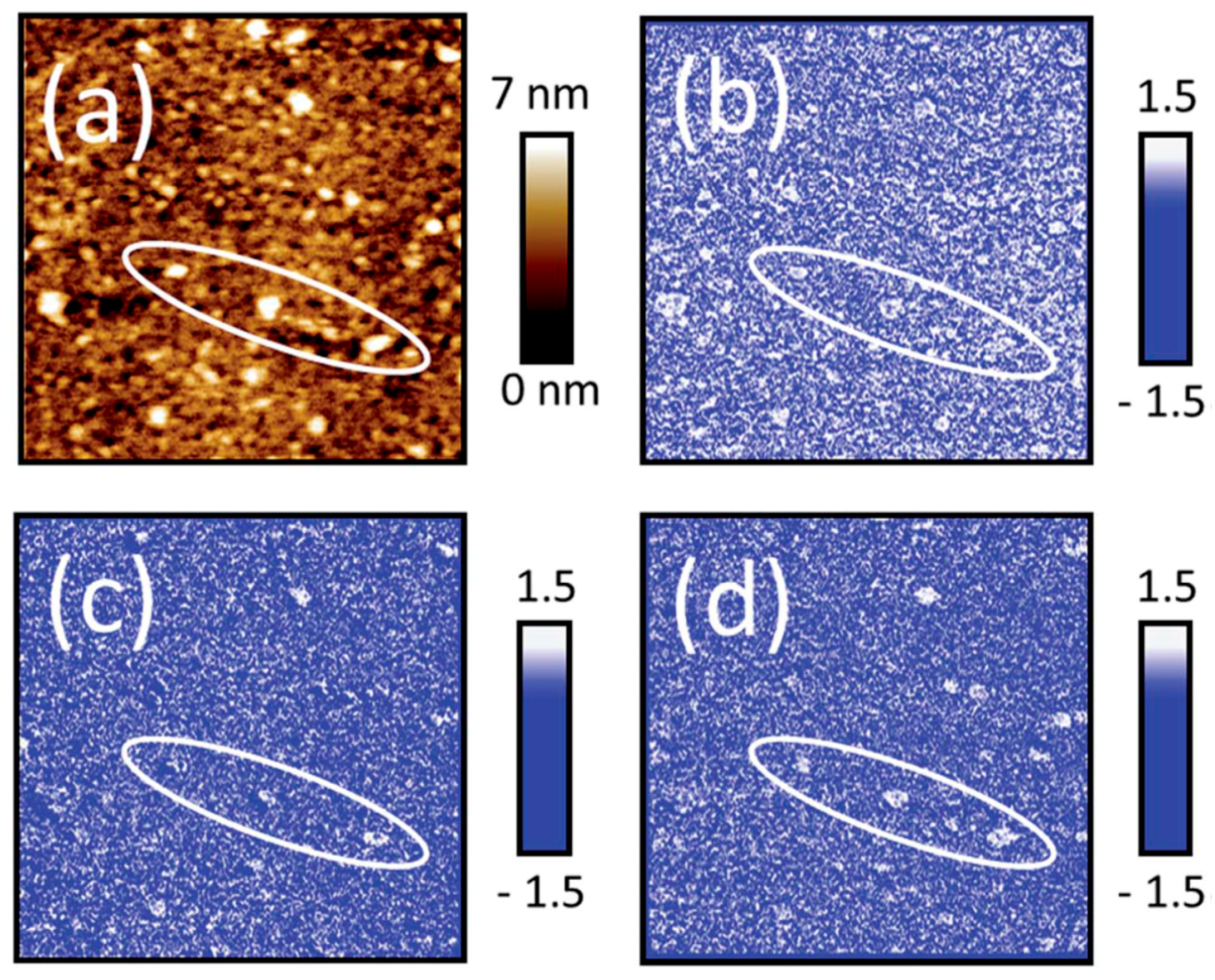
2.1.6.2. Nano IR Microscopy
2.1.7. Photoacoustic Infrared Spectroscopy
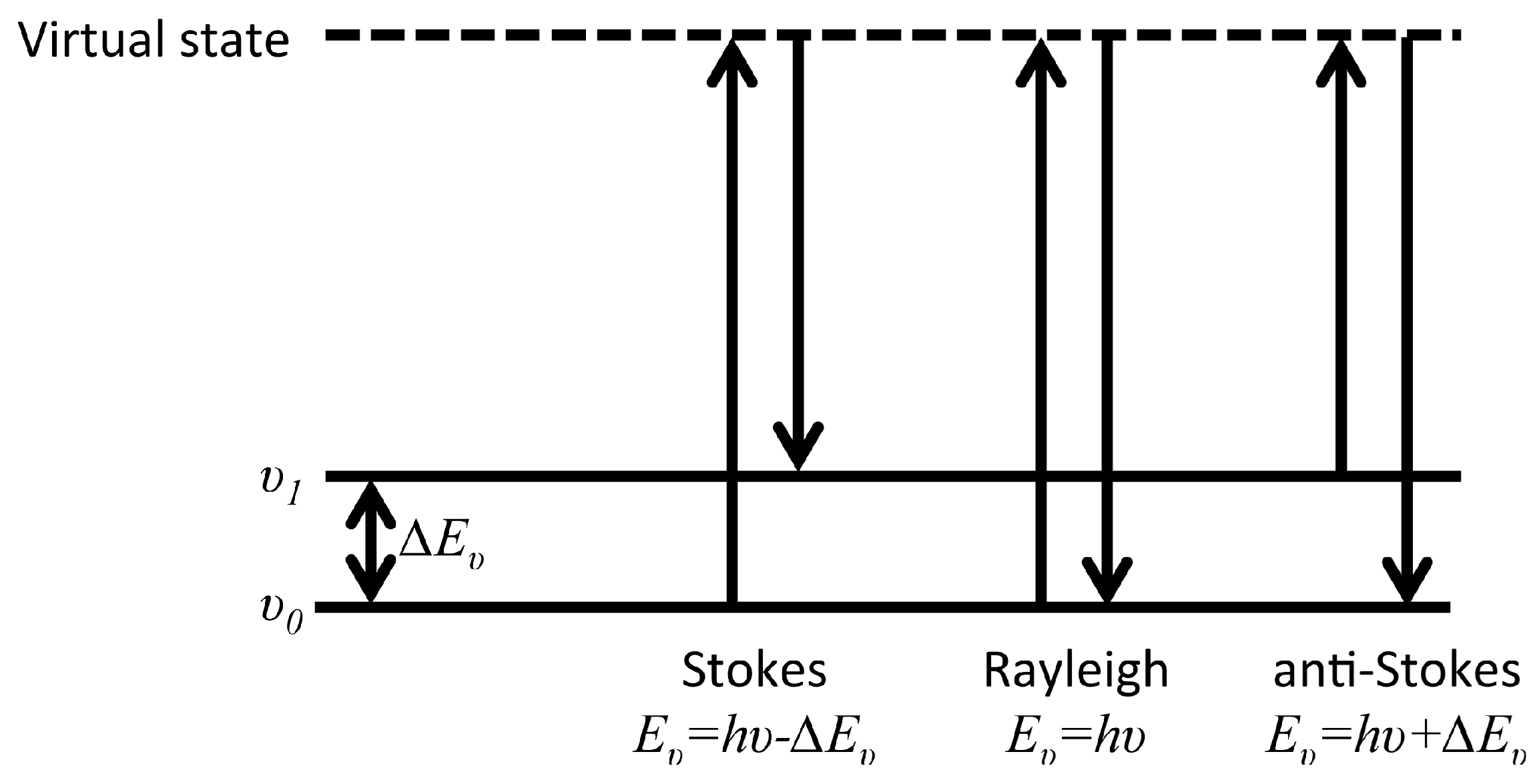
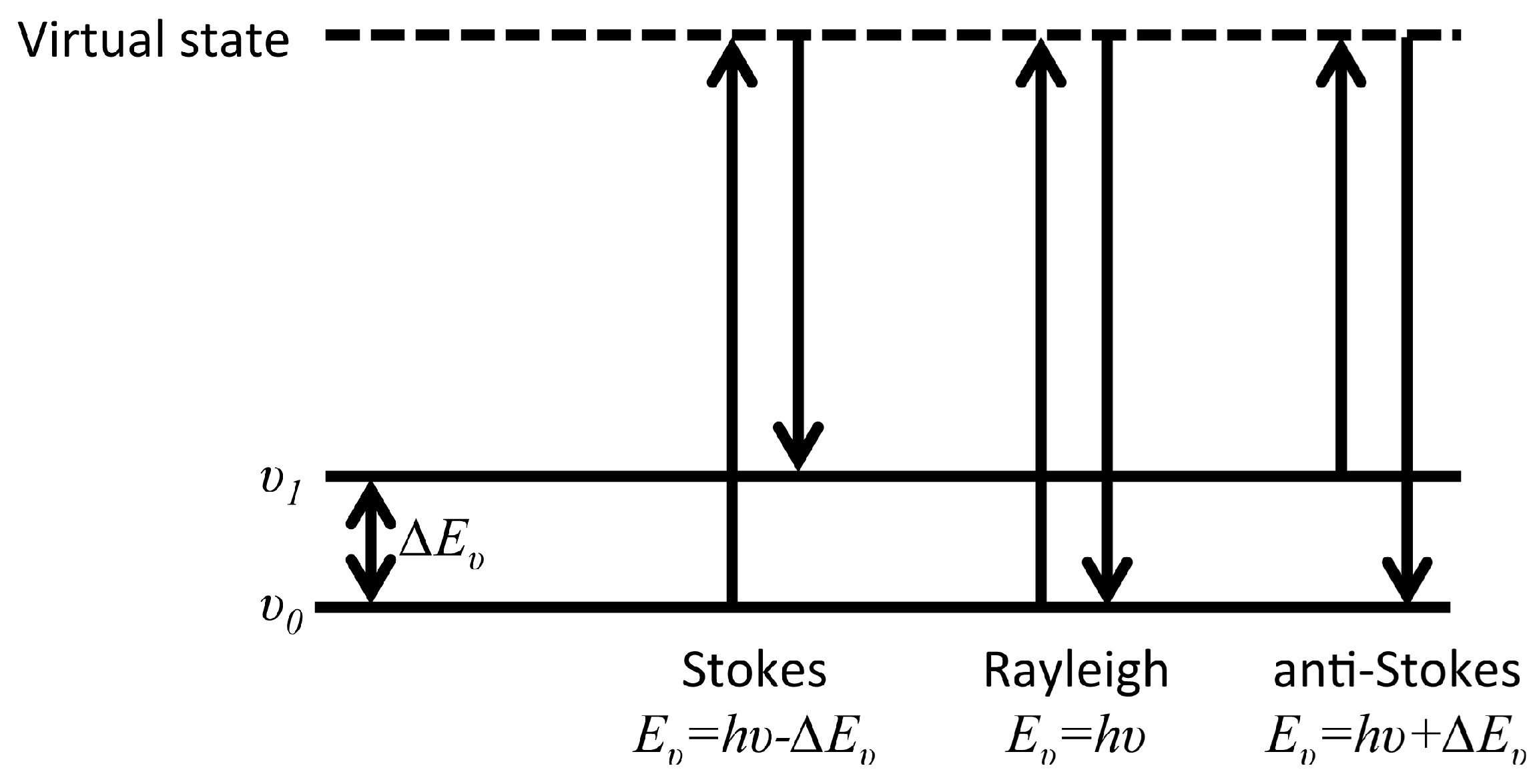
2.2. Raman Spectroscopy
2.2.1. Conventional Raman Spectroscopy
2.2.1.1. Steel
2.2.1.2. Copper
2.2.1.3. Silver
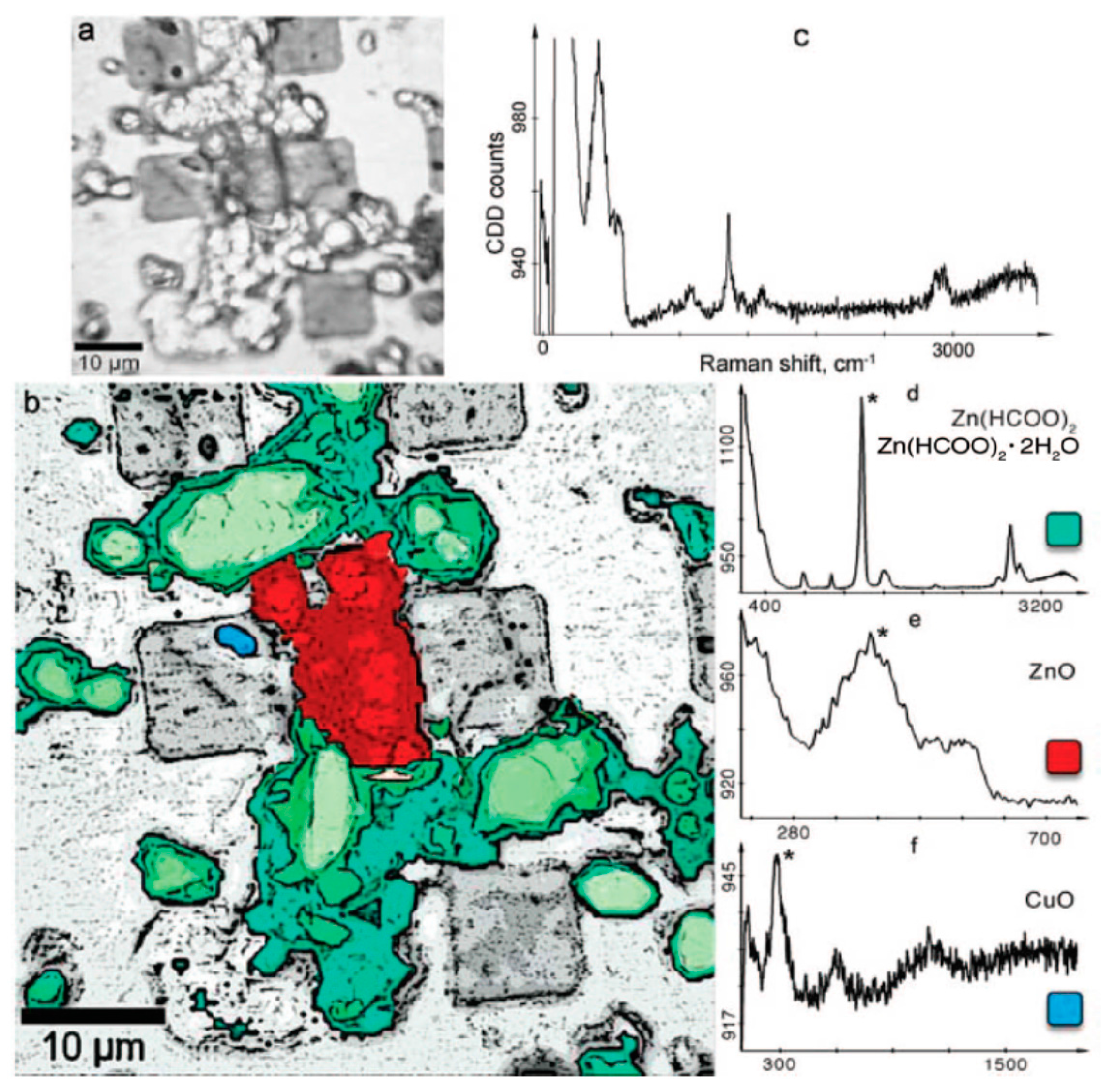
2.2.1.4. Zinc
2.2.1.5. Zinc Alloys
2.2.1.6. Other Elements and Alloys (Mo, Ni, Pb, SiC Fibers, U, Mg, Al, CdS, Graphene, As2S3)
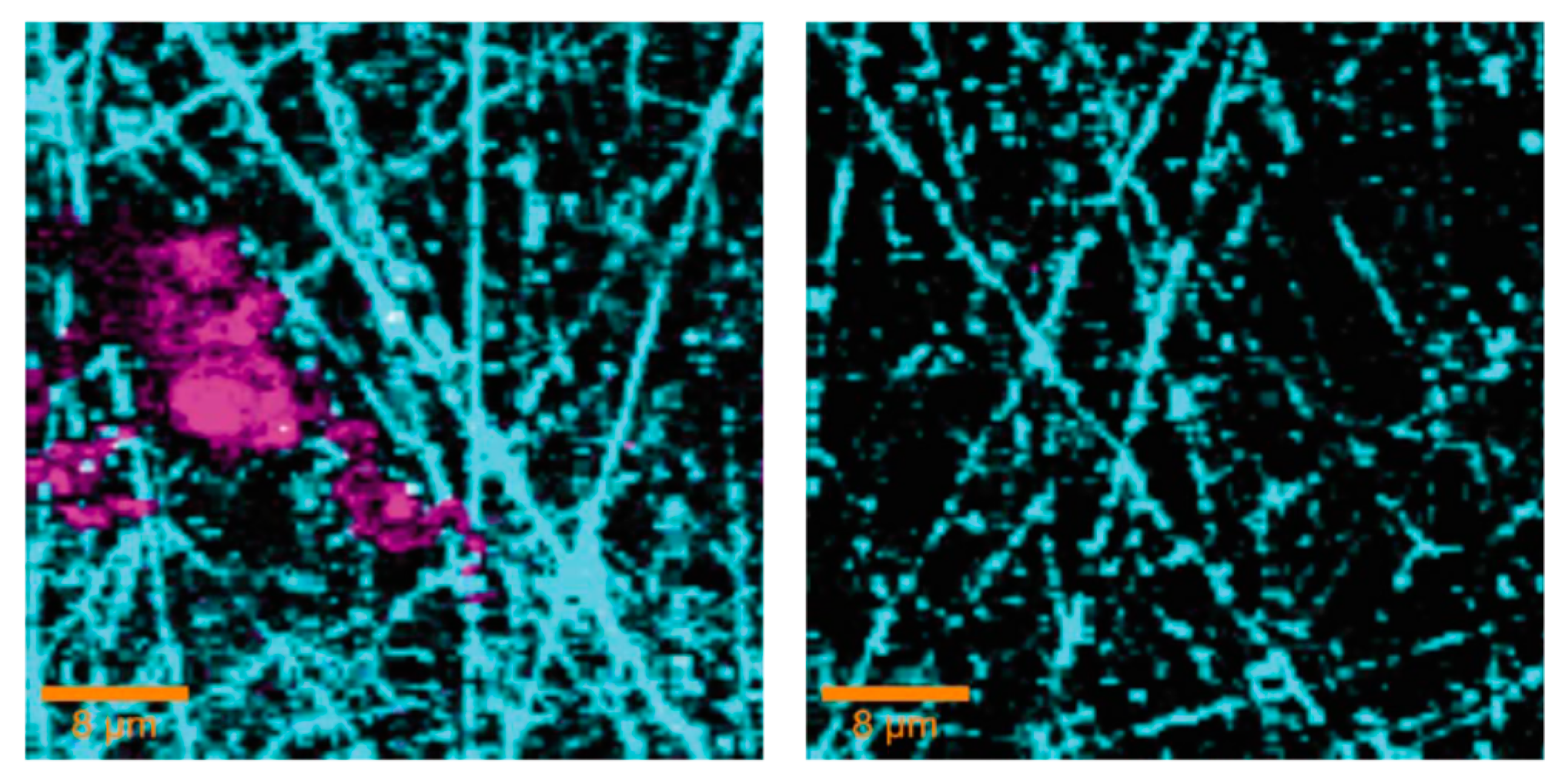
2.2.2. Surface Enhanced Raman Spectroscopy (SERS) and Tip Enhanced Raman Spectroscopy (TERS)
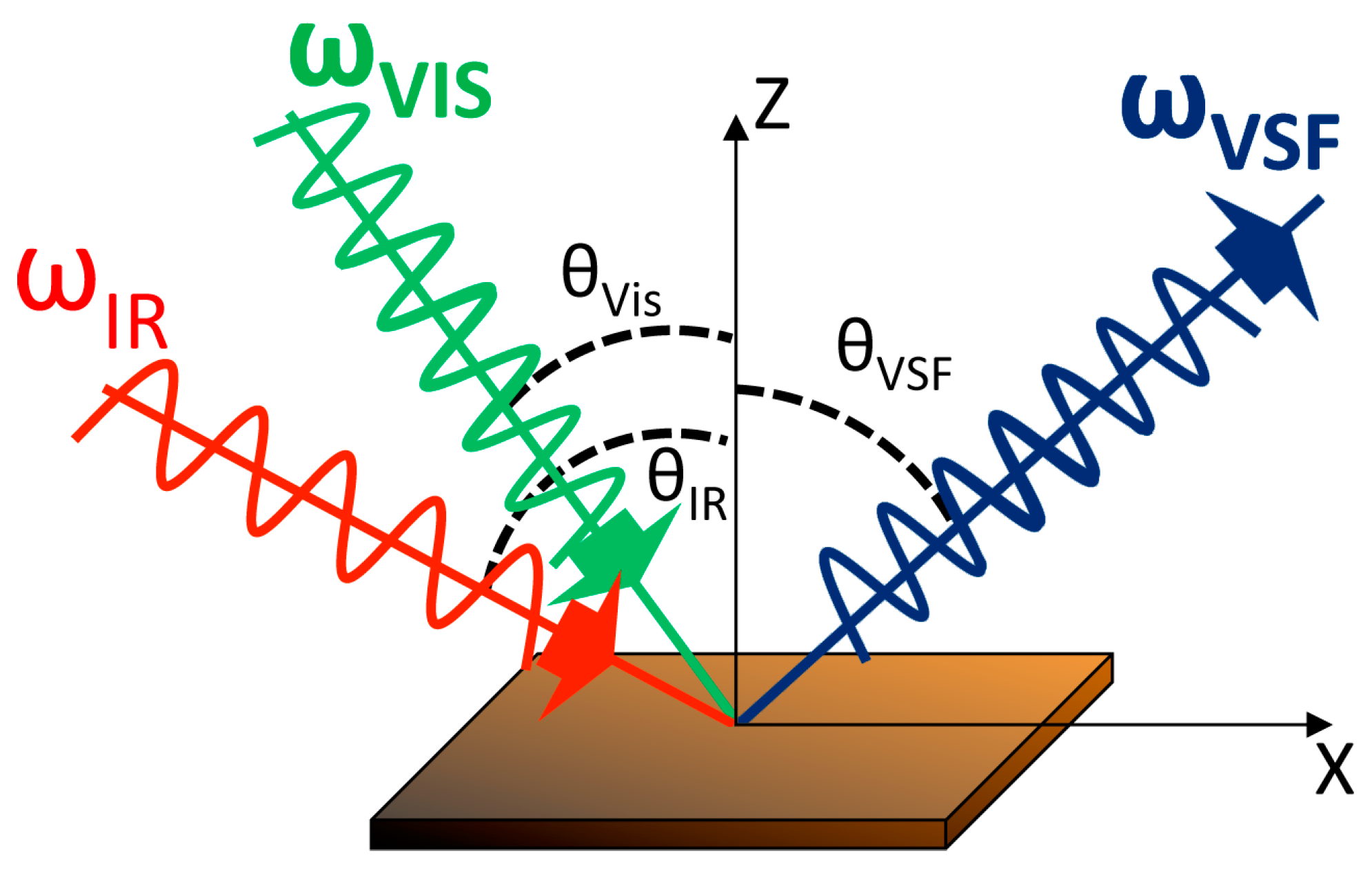
2.3. Vibrational Sum Frequency Spectroscopy (VSFS)
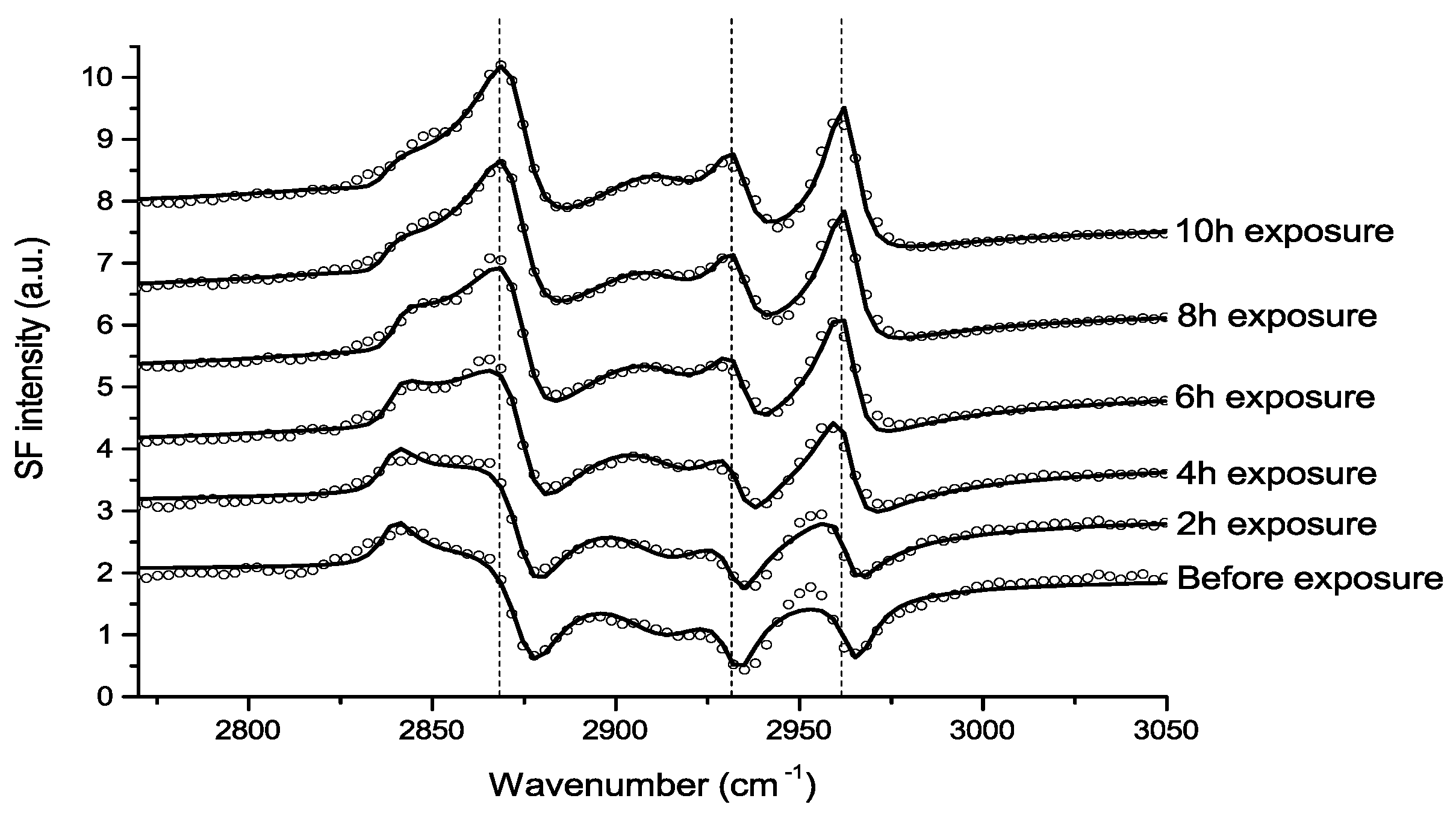
Atmospheric Corrosion Studies by VSFS
3. Conclusions
Author Contributions
Conflicts of Interest
References
- Graedel, T.E. Corrosion mechanisms for silver exposed to the atmosphere. J. Electrochem. Soc. 1992, 139, 1963–1970. [Google Scholar] [CrossRef]
- Persson, D.; Leygraf, C. Metal carboxylate formation during indoor atmospheric corrosion of Cu, Zn, and Ni. J. Electrochem. Soc. 1995, 142, 1468–1477. [Google Scholar] [CrossRef]
- Daly, L.H.; Colthup, N.B.; Wiberley, S.E. Introduction to Infrared and Raman Spectroscopy, 3rd ed.; Academic Press Ltd.: London, UK, 1990. [Google Scholar]
- Nakamoto, K. Infrared and Raman Spectra of Inorganic and Coordination Compounds, 4th ed.; John Wiley & Sons: New York, NY, USA, 1986. [Google Scholar]
- Marcus, P.; Mansfeld, F. Analytical Methods in Corrosion Science and Technology; CRC Press, Taylor & Francis: Boca Raton, FL, USA, 2005. [Google Scholar]
- Greenler, R.G.; Rahn, R.R.; Schwartz, J.P. The effect of index of refraction on the position, shape, and intensity of infrared bands in reflection-absorption spectra. J. Catal. 1971, 23, 42–48. [Google Scholar] [CrossRef]
- Li, Q.X.; Wang, Z.Y.; Han, W.; Han, E.H. Characterization of the corrosion products formed on carbon steel in qinghai salt lake atmosphere. Corrosion 2007, 63, 640–647. [Google Scholar] [CrossRef]
- Wang, J.; Wang, Z.Y.; Ke, W. Characterisation of rust formed on carbon steel after exposure to open atmosphere in qinghai salt lake region. Corros. Eng. Sci. Technol. 2012, 47, 125–130. [Google Scholar] [CrossRef]
- Allam, I.M.; Arlow, J.S.; Saricimen, H. Initial stages of atmospheric corrosion of steel in the arabian gulf. Corros. Sci. 1991, 32, 417–432. [Google Scholar] [CrossRef]
- Raman, A.; Kuban, B.; Razvan, A. The application of infrared spectroscopy to the study of atmospheric rust systems—I. Standard spectra and illustrative applications to identify rust phases in natural atmospheric corrosion products. Corros. Sci. 1991, 32, 1295–1306. [Google Scholar] [CrossRef]
- Pacheco, A.M.G.; Teixeira, M.G.I.B.; Ferreira, M.G.S. Initial stages of chloride induced atmospheric corrosion of iron: An infrared spectroscopic study. Br. Corros. J. 1990, 25, 57–59. [Google Scholar] [CrossRef]
- Morcillo, M.; Almeida, E.; Marrocos, M.; Rosales, B. Atmospheric corrosion of copper in ibero-america. Corrosion 2001, 57, 967–980. [Google Scholar] [CrossRef]
- Almeida, E.; Morcillo, M.; Rosales, B. Atmospheric corrosion of zinc part 1: Rural and urban atmospheres. Br. Corros. J. 2000, 35, 284–288. [Google Scholar] [CrossRef]
- Saxena, A.; Balasubramaniam, R.; Raman, S.; Raman, K. The forge-welded iron cannon at tanjore. In Proceedings of the Corrosion & Prevention and NDT, Melbourne, Australia, 23–26 November 2003; pp. 75/1–75/12. [Google Scholar]
- Zhu, F.; Zhang, X.; Persson, D.; Thierry, D. In situ infrared reflection absorption spectroscopy studies of confined zinc surfaces exposed under periodic wet-dry conditions. Electrochem. Solid State Lett. 2001, 4, B19–B22. [Google Scholar] [CrossRef]
- Wang, B.-B.; Wang, Z.-Y.; Han, W.; Wang, C.; Ke, W. Effects of magnesium chloride-based multicomponent salts on atmospheric corrosion of aluminum alloy 2024. Trans. Nonferr. Met. Soc. China 2013, 23, 1199–1208. [Google Scholar] [CrossRef]
- Yang, L.-J.; Li, Y.-F.; Wei, Y.-H.; Hou, L.-F.; Li, Y.-G.; Tian, Y. Atmospheric corrosion of field-exposed az91d mg alloys in a polluted environment. Corros. Sci. 2010, 52, 2188–2196. [Google Scholar] [CrossRef]
- Focke, W.W.; Nhlapo, N.S.; Vuorinen, E. Thermal analysis and ftir studies of volatile corrosion inhibitor model systems. Corros. Sci. 2013, 77, 88–96. [Google Scholar] [CrossRef]
- Persson, D.; Leygraf, C. Analysis of atmospheric corrosion products of field exposed nickel. J. Electrochem. Soc. 1992, 139, 2243–2249. [Google Scholar] [CrossRef]
- Fuller, M.P.; Griffiths, P.R. Diffuse reflectance measurements by infrared fourier transform spectrometry. Anal. Chem. 1978, 50, 1906–1910. [Google Scholar] [CrossRef]
- Weissenrieder, J.; Kleber, C.; Schreiner, M.; Leygraf, C. In situ studies of sulfate nest formation on iron. J. Electrochem. Soc. 2004, 151, B497–B504. [Google Scholar] [CrossRef]
- Kotenev, V.A.; Petrunin, M.A.; Maksaeva, L.B.; Sokolova, N.P.; Gorbunov, A.M.; Kablov, E.N.; Tsivadze, A.Y. Gravimetry, resistometry, and fourier-transform infrared spectroscopy for monitoring the corrosivity of the atmosphere with the use of an iron-oxide nanocomposite sensor layer. Prot. Met. Phys. Chem. Surf. 2013, 49, 597–603. [Google Scholar] [CrossRef]
- Aramaki, K. Protection of iron corrosion by ultrathin two-dimensional polymer films of an alkanethiol monolayer modified with alkylethoxysilanes. Corros. Sci. 1999, 41, 1715–1730. [Google Scholar] [CrossRef]
- Grundmeier, G.; Matheisen, E.; Stratmann, M. Formation and stability of ultrathin organosilane polymers on iron. J. Adhes. Sci. Technol. 1996, 10, 573–588. [Google Scholar] [CrossRef]
- Persson, D.; Thierry, D.; LeBozec, N.; Prosek, T. In situ infrared reflection spectroscopy studies of the initial atmospheric corrosion of Zn–Al–Mg coated steel. Corros. Sci. 2013, 72, 54–63. [Google Scholar] [CrossRef]
- Jang, J.; Kim, E.K. Corrosion protection of epoxy-coated steel using different silane coupling agents. J. Appl. Polym. Sci. 1999, 71, 585–593. [Google Scholar] [CrossRef]
- Persson, D.; Leygraf, C. Vibrational spectroscopy and xps for atmospheric corrosion studies on copper. J. Electrochem. Soc. 1990, 137, 3163–3169. [Google Scholar] [CrossRef]
- Persson, D.; Leygraf, C. In situ infrared reflection absorption spectroscopy for studies of atmospheric corrosion. J. Electrochem. Soc. 1993, 140, 1256–1260. [Google Scholar] [CrossRef]
- Kleber, C.; Kattner, J.; Frank, J.; Hoffmann, H.; Kraft, M.; Schreiner, M. Design and application of a new cell for in situ infrared reflection–absorption spectroscopy investigations of metal–atmosphere interfaces. Appl. Spectrosc. 2003, 57, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Persson, D.; Leygraf, C. Initial interaction of sulfur dioxide with water covered metal surfaces: An in situ iras study. J. Electrochem. Soc. 1995, 142, 1459–1468. [Google Scholar] [CrossRef]
- Aastrup, T.; Leygraf, C. Simultaneous infrared reflection absorption spectroscopy and quartz crystal microbalance measurements for in situ studies of the metal/atmosphere interface. J. Electrochem. Soc. 1997, 144, 2986–2990. [Google Scholar] [CrossRef]
- Itoh, J.; Sasaki, T.; Seo, M.; Ishikawa, T. In situ simultaneous measurement with ir-ras and qcm for investigation of corrosion of copper in a gaseous environment. Corros. Sci. 1997, 39, 193–197. [Google Scholar] [CrossRef]
- Wadsak, M.; Schreiner, M.; Aastrup, T.; Leygraf, C. Combined in-situ investigations of atmospheric corrosion of copper with sfm and iras coupled with qcm. Surf. Sci. 2000, 454–456, 246–250. [Google Scholar] [CrossRef]
- Gil, H.; Leygraf, C. Quantitative in situ analysis of initial atmospheric corrosion of copper induced by acetic acid. J. Electrochem. Soc. 2007, 154, C272–C278. [Google Scholar] [CrossRef]
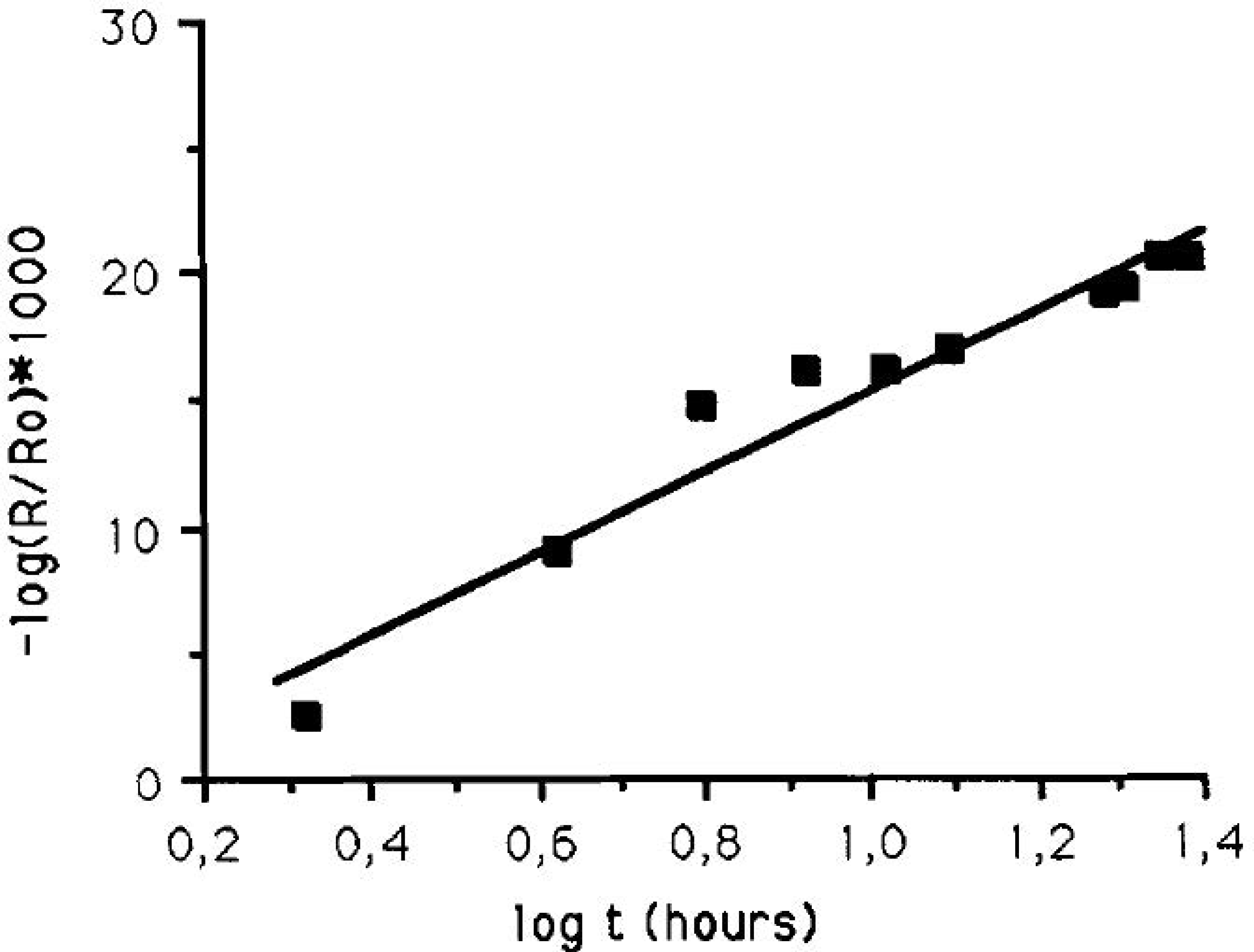
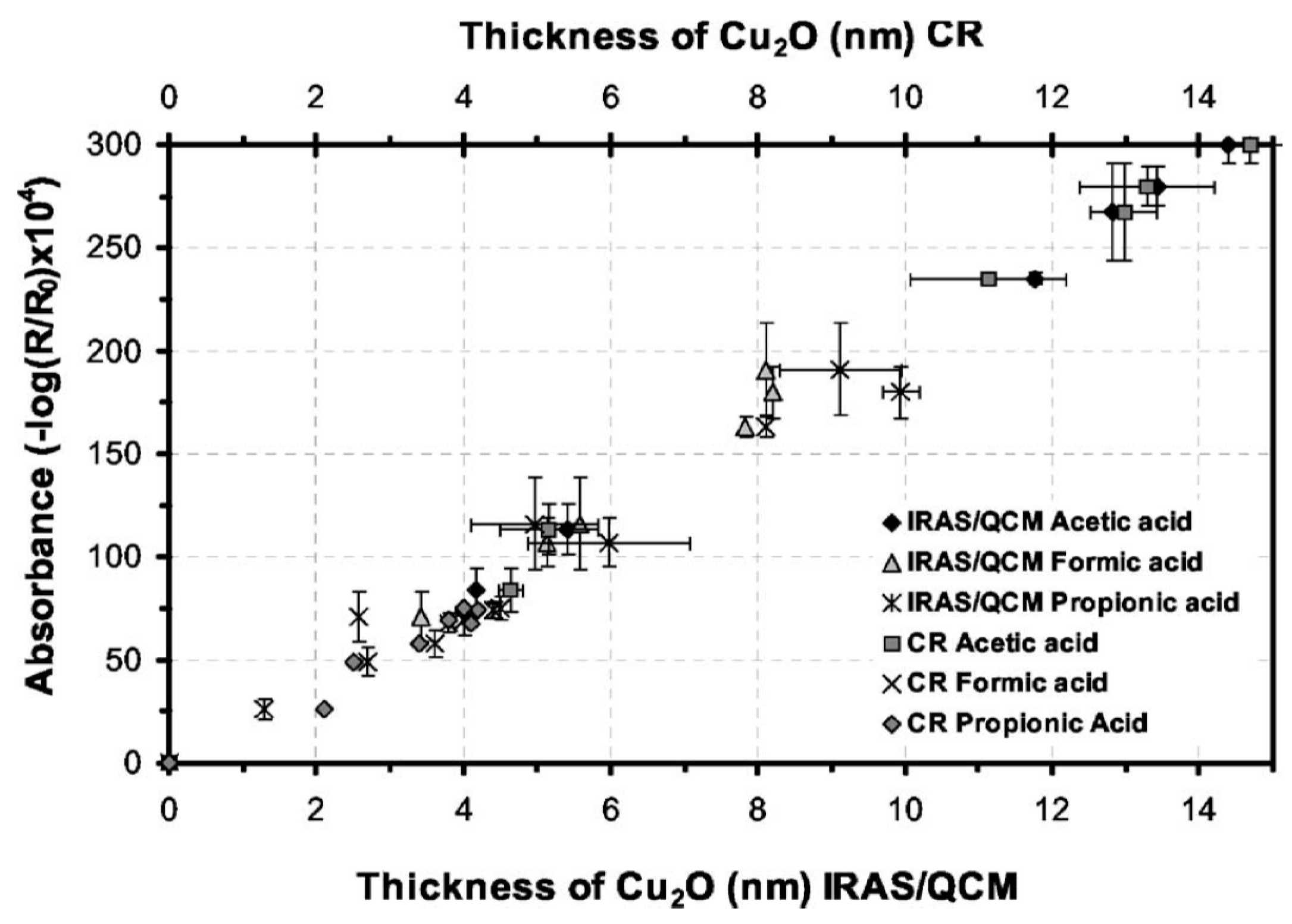
- Gil, H.; Leygraf, C. Initial atmospheric corrosion of copper induced by carboxylic acids: A comparative in situ study. J. Electrochem. Soc. 2007, 154, C611–C617. [Google Scholar] [CrossRef]
- Gil, H.; Leygraf, C.; Tidblad, J. Gildes model simulations of the atmospheric corrosion of zinc induced by low concentrations of carboxylic acids. J. Electrochem. Soc. 2012, 159, C123–C128. [Google Scholar] [CrossRef]
- Kleber, C.; Schreiner, M. Multianalytical in-situ investigations of the early stages of corrosion of copper, zinc and binary copper/zinc alloys. Corros. Sci. 2003, 45, 2851–2866. [Google Scholar] [CrossRef]
- Faguy, P.W.; Richmond, W.N.; Jackson, R.S.; Weibel, S.C.; Ball, G.; Payer, J.H. Real-time polarization modulation in situ infrared spectroscopy applied to the study of atmospheric corrosion. Appl. Spectrosc. 1998, 52, 557–564. [Google Scholar] [CrossRef]
- Malvault, J.Y.; Lopitaux, J.; Delahaye, D.; Lenglet, M. Cathodic reduction and infrared reflectance spectroscopy of basic copper(ii) salts on copper substrate. J. Appl. Electrochem. 1995, 25, 841–845. [Google Scholar] [CrossRef]
- Chen, Z.Y.; Persson, D.; Samie, F.; Zakipour, S.; Leygraf, C. Effect of carbon dioxide on sodium chloride-induced atmospheric corrosion of copper. J. Electrochem. Soc. 2005, 152, B502–B511. [Google Scholar] [CrossRef]
- Chen, Z.Y.; Persson, D.; Leygraf, C. In situ studies of the effect of SO2 on the initial nacl-induced atmospheric corrosion of copper. J. Electrochem. Soc. 2005, 152, B526–B533. [Google Scholar] [CrossRef]
- Itoh, J.; Sasaki, T.; Ohtsuka, T.; Osawa, M. Surface layers formed initially on copper in air containing water vapor and so2 as determined by ir-ras and 2d-ir. J. Electroanal. Chem. 1999, 473, 256–264. [Google Scholar] [CrossRef]
- Patois, T.; Et Taouil, A.; Lallemand, F.; Carpentier, L.; Roizard, X.; Hihn, J.-Y.; Bondeau-Patissier, V.; Mekhalif, Z. Microtribological and corrosion behaviors of 1H,1H,2H,2H-perfluorodecanethiol self-assembled films on copper surfaces. Surf. Coat. Technol. 2010, 205, 2511–2517. [Google Scholar] [CrossRef]
- Nilsson, J.O.; Törnkvist, C.; Liedberg, B. Photoelectron and infrared reflection absorption spectroscopy of benzotriazole adsorbed on copper and cuprous oxide surfaces. Appl. Surf. Sci. 1989, 37, 306–326. [Google Scholar] [CrossRef]
- Yoshida, S.; Ishida, H. A study on the orientation of imidazoles on copper as corrosion inhibitor and possible adhesion promoter for electric devices. J. Chem. Phys. 1983, 78, 6960–6969. [Google Scholar] [CrossRef]
- Yoshida, S.; Ishida, H. An investigation of the thermal stability of undecylimidazole on copper by ft-ir reflection-absorption spectroscopy. Appl. Surf. Sci. 1995, 89, 39–47. [Google Scholar] [CrossRef]
- Yoshida, S.; Ishida, H. The effect of chain length on the thermal stability of 2-alkylimidazoles on copper and 2-alkylimidazolato copper(ii) complexes. Appl. Surf. Sci. 1985, 20, 497–511. [Google Scholar] [CrossRef]
- Kim, H.; Jang, J. Effect of copolymer composition in vinyl silane modified polyvinylimidazole on copper corrosion protection at elevated temperature. Polymer 1998, 39, 4065–4074. [Google Scholar] [CrossRef]
- Opila, R.L.; Krautter, H.W.; Zegarski, B.R.; Duboisa, L.H.; Wenger, G. Thermal stability of azole-coated copper surfaces. J. Electrochem. Soc. 1995, 142, 4074–4077. [Google Scholar] [CrossRef]
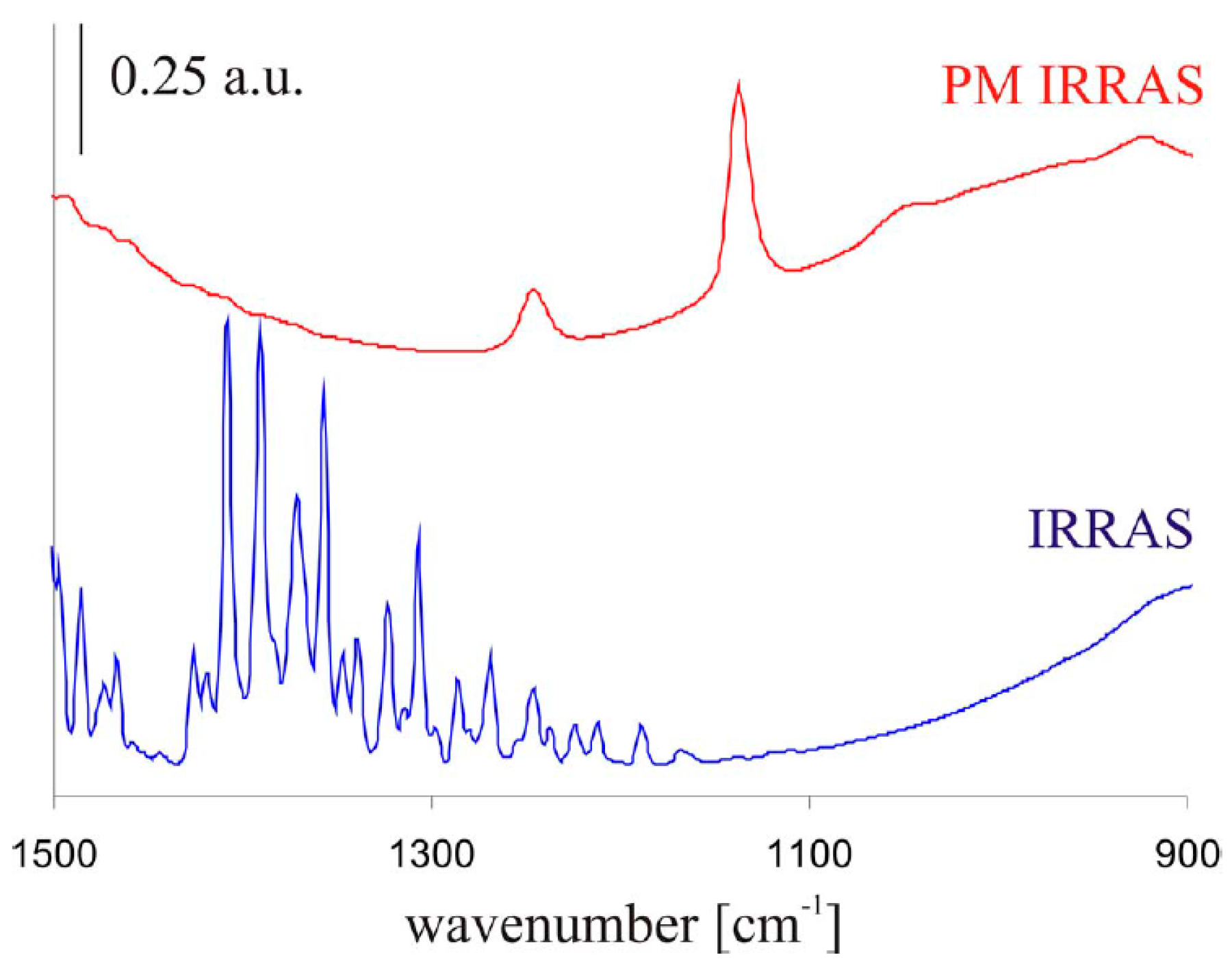
- Wiesinger, R.; Schade, U.; Kleber, C.; Schreiner, M. An experimental set-up to apply polarization modulation to infrared reflection absorption spectroscopy for improved in situ studies of atmospheric corrosion processes. Rev. Sci. Instrum. 2014, 85, 064102. [Google Scholar] [CrossRef] [PubMed]
- Wiesinger, R.; Kleber, C.; Frank, J.; Schreiner, M. A new experimental setup for in situ infrared reflection absorption spectroscopy studies of atmospheric corrosion on metal surfaces considering the influence of ultraviolet light. Appl. Spectrosc. 2009, 63, 465–470. [Google Scholar] [CrossRef] [PubMed]
- Wiesinger, R.S.J.; Hutter, H.; Schreiner, M.; Kleber, C. About the Formation of Basic Silver Carbonate on Silver Surfaces — An In Situ IRRAS Study. Open Corros. J. 2009, 2, 96–104. [Google Scholar] [CrossRef]
- Kleber, C.; Hilfrich, U.; Schreiner, M. In situ investigations of the interaction of small inorganic acidifying molecules in humidified air with polycrystalline metal surfaces by means of tm-afm, irras, and qcm. Surf. Interface Anal. 2007, 39, 702–710. [Google Scholar] [CrossRef]
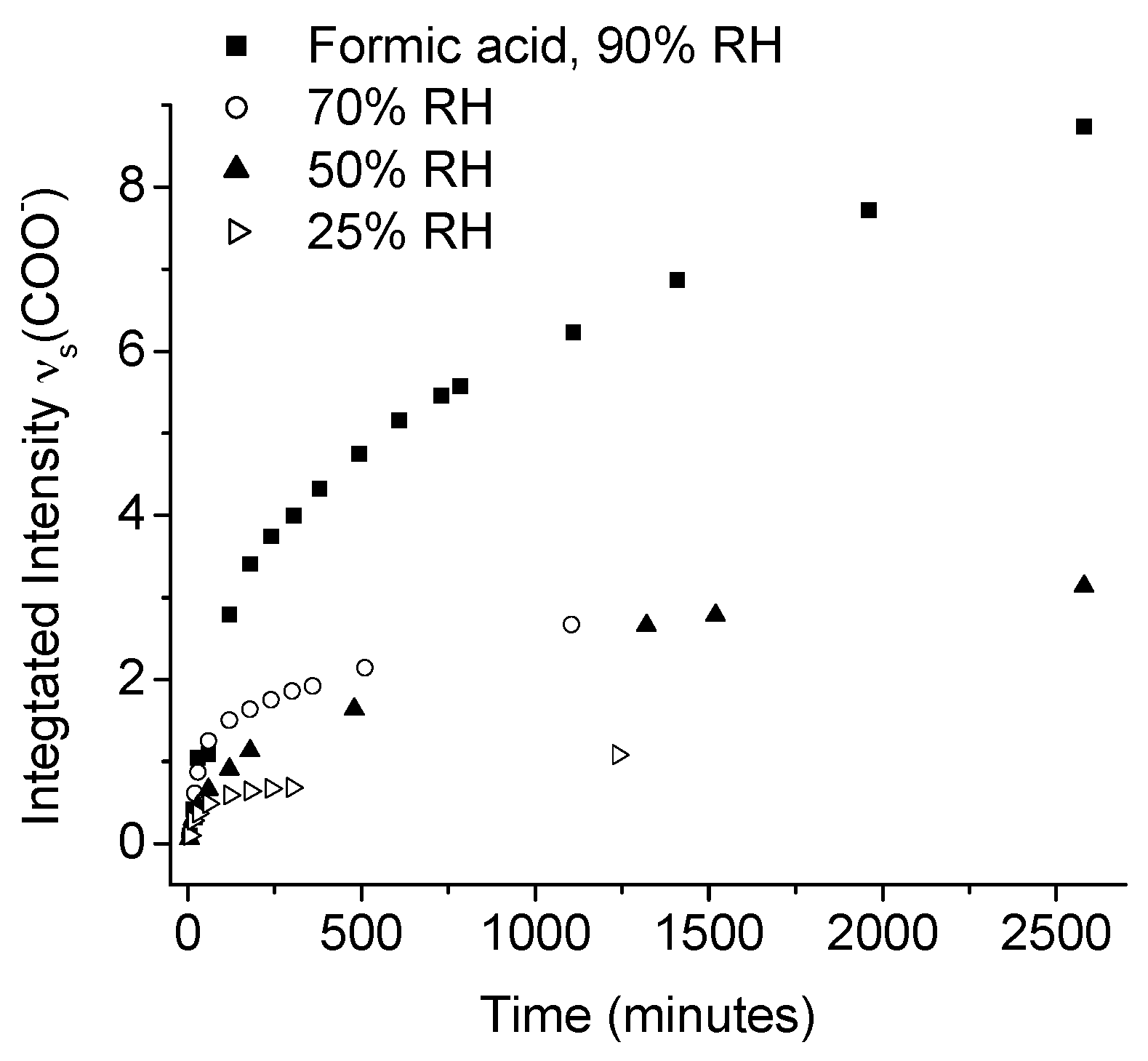
- Johnson, C.M.; Leygraf, C. Atmospheric corrosion of zinc by organic constituents: Iii. An infrared reflection-absorption spectroscopy study of the influence of formic acid. J. Electrochem. Soc. 2006, 153, B547–B550. [Google Scholar] [CrossRef]
- Johnson, C.M.; Leygraf, C. Atmospheric corrosion of zinc by organic constituents: Ii. Reaction routes for zinc-acetate formation. J. Electrochem. Soc. 2006, 153, B542–B546. [Google Scholar] [CrossRef]
- Johnson, C.M.; Tyrode, E.; Leygraf, C. Atmospheric corrosion of zinc by organic constituents: I. The role of the zinc/water and water/air interfaces studied by infrared reflection/absorption spectroscopy and vibrational sum frequency spectroscopy. J. Electrochem. Soc. 2006, 153, B113–B120. [Google Scholar] [CrossRef]
- Qiu, P.; Persson, D.; Leygraf, C. Initial atmospheric corrosion of zinc induced by carboxylic acids: A quantitative in situ study. J. Electrochem. Soc. 2009, 156, C441–C447. [Google Scholar] [CrossRef]
- Petrie, W.T.; Vohs, J.M. An hreels investigation of the adsorption and reaction of formic acid on the (0001)-zinc surface of zinc oxide. Surf. Sci. 1991, 245, 315–323. [Google Scholar] [CrossRef]
- Persson, D.; Axelsen, S.; Zou, F.; Thierry, D. Simultaneous in situ infrared reflection absorption spectroscopy and kelvin probe measurements during atmospheric corrosion. Electrochem. Solid State Lett. 2001, 4, B7–B10. [Google Scholar] [CrossRef]
- Qiu, P.; Leygraf, C. Initial oxidation of brass induced by humidified air. Appl. Surf. Sci. 2011, 258, 1235–1241. [Google Scholar] [CrossRef] [PubMed]
- Qiu, P.; Leygraf, C. Multi-analysis of initial atmospheric corrosion of brass induced by carboxylic acids. J. Electrochem. Soc. 2011, 158, C172–C177. [Google Scholar] [CrossRef]
- Takeshi, S.; Ryoji, K.; Toshiaki, O. In situ ir-ras investigation of corrosion of tin in air containing H2O, NO2 and SO2 at room temperature. J. Univ. Sci. Technol. Beijing 2002, 10, 35–38. [Google Scholar]
- Wadsak, M.; Aastrup, T.; Odnevall Wallinder, I.; Leygraf, C.; Schreiner, M. Multianalytical in situ investigation of the initial atmospheric corrosion of bronze. Corros. Sci. 2002, 44, 791–802. [Google Scholar] [CrossRef]
- Dai, Q.; Freedman, A.; Robinson, G.N. Sulfuric acid-induced corrosion of aluminum surfaces. J. Electrochem. Soc. 1995, 142, 4063–4069. [Google Scholar] [CrossRef]
- Matheisen, E.; Nazarov, A.P.; Stratmann, M. In situ investigation of the adsorption of alkyltrimethoxysilanes on iron surfaces. Fresenius’ J. Anal. Chem. 1993, 346, 294–296. [Google Scholar] [CrossRef]
- Chen, Z.Y.; Persson, D.; Nazarov, A.; Zakipour, S.; Thierry, D.; Leygraf, C. In situ studies of the effect of CO2 on the initial nacl-induced atmospheric corrosion of copper. J. Electrochem. Soc. 2005, 152, B342–B351. [Google Scholar] [CrossRef]
- Chen, Z.Y.; Persson, D.; Leygraf, C. Initial nacl-particle induced atmospheric corrosion of zinc—Effect of CO2 and SO2. Corros. Sci. 2008, 50, 111–123. [Google Scholar] [CrossRef]
- Le Bozec, N.; Persson, D.; Nazarov, A.; Thierry, D. Investigation of filiform corrosion on coated aluminum alloys by ftir microspectroscopy and scanning kelvin probe. J. Electrochem. Soc. 2002, 149, B403–B408. [Google Scholar] [CrossRef]
- LeBozec, N.; Persson, D.; Thierry, D. In situ studies of the initiation and propagation of filiform corrosion on aluminum. J. Electrochem. Soc. 2004, 151, B440–B445. [Google Scholar] [CrossRef]
- LeBozec, N.; Persson, D.; Thierry, D.; Axelsen, S.B. Effect of climatic parameters on filiform corrosion of coated aluminum alloys. Corrosion 2004, 60, 584–593. [Google Scholar] [CrossRef]
- Nazarov, A.; Olivier, M.G.; Thierry, D. Skp and ft-ir microscopy study of the paint corrosion de-adhesion from the surface of galvanized steel. Prog. Org. Coat. 2012, 74, 356–364. [Google Scholar] [CrossRef]
- Nazarov, A.P.; Thierry, D. Probing of atmospheric corrosion of metals: Carbon steel. Prot. Met. 2004, 40, 377–388. [Google Scholar] [CrossRef]
- Lefez, B.; Jouen, S.; Kasperek, J.; Hannoyer, B. Ft-ir microscopic base imaging system: Applications for chemical analysis of zn and ni atmospheric corrosion. Appl. Spectrosc. 2001, 55, 935–938. [Google Scholar] [CrossRef]
- Johnson, C.M.; Böhmler, M. Nano-ftir microscopy and spectroscopy studies of atmospheric corrosion with a spatial resolution of 20 nm. Corros. Sci. 2016, 108, 60–65. [Google Scholar] [CrossRef]
- Morsch, S.; Liu, Y.; Lyon, S.B.; Gibbon, S.R. Insights into epoxy network nanostructural heterogeneity using afm-ir. ACS Appl. Mater. Interfaces 2016, 8, 959–966. [Google Scholar] [CrossRef] [PubMed]
- Morsch, S.; Lyon, S.; Smith, S.D.; Gibbon, S.R. Mapping water uptake in an epoxy-phenolic coating. Prog. Org. Coat. 2015, 86, 173–180. [Google Scholar] [CrossRef]
- Morsch, S.; Lyon, S.; Greensmith, P.; Smith, S.D.; Gibbon, S.R. Mapping water uptake in organic coatings using afm-ir. Faraday Discuss. 2015, 180, 527–542. [Google Scholar] [CrossRef] [PubMed]
- Faubel, W.; Heissler, S.; Palmer, R.A. Quantitative analysis of corroded copper patina by step scan and rapid scan photoacoustic fourier transform infrared spectroscopy. Rev. Sci. Instrum. 2003, 74, 331–333. [Google Scholar] [CrossRef]
- Raman, C.V.; Krishnan, K.S. A new type of secondary radiation. Nature 1928, 121, 501–502. [Google Scholar] [CrossRef]
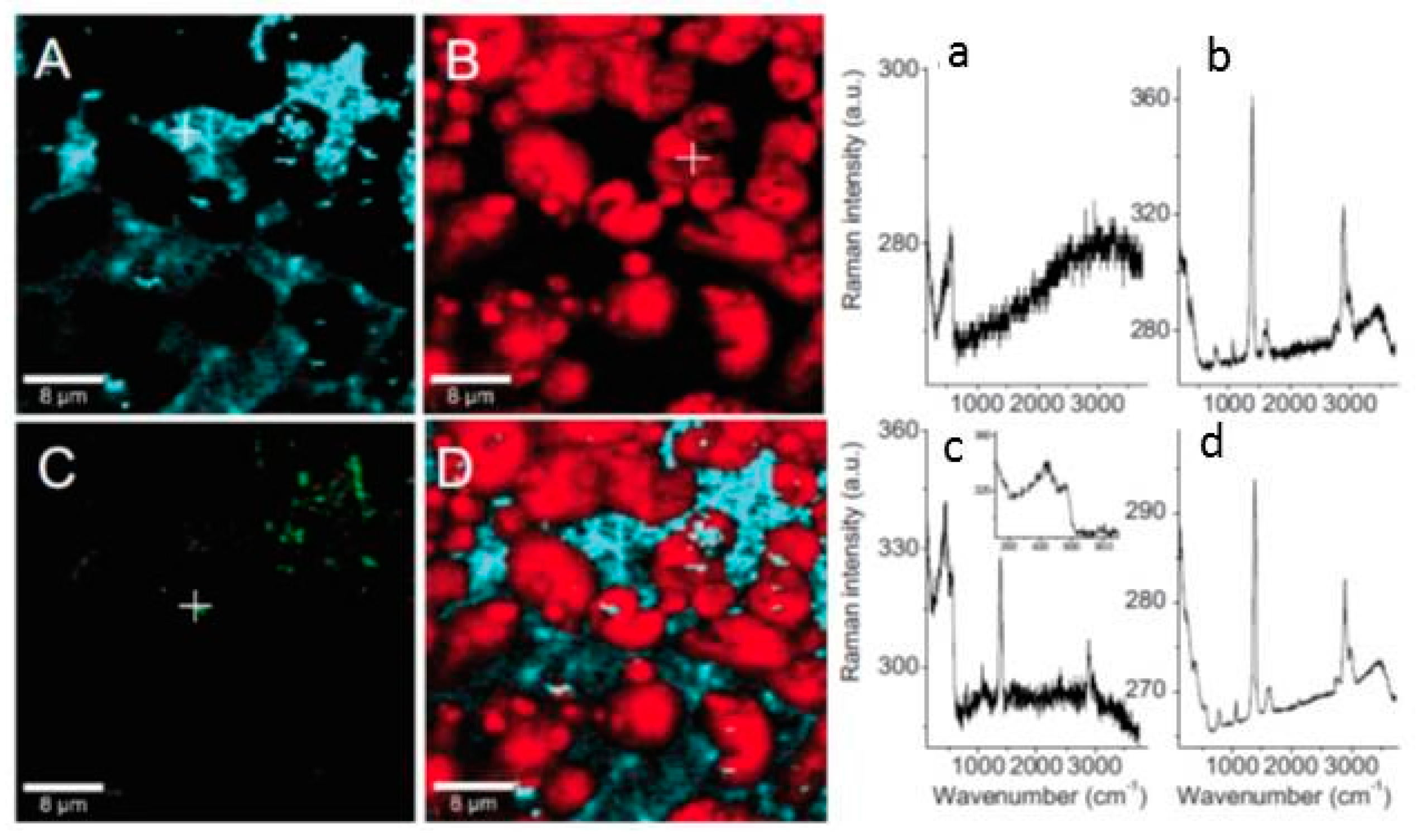
- Li, S.; Hihara, L.H. A micro-raman spectroscopic study of marine atmospheric corrosion of carbon steel: The effect of akaganeite. J. Electrochem. Soc. 2015, 162, C495–C502. [Google Scholar] [CrossRef]
- Dhaiveegan, P.; Elangovan, N.; Nishimura, T.; Rajendran, N. Corrosion behavior of 316l and 304 stainless steels exposed to industrial-marine-urban environment: Field study. RSC Adv. 2016, 6, 47314–47324. [Google Scholar] [CrossRef]
- Morcillo, M.; Chico, B.; Alcantara, J.; Daiaz, I.; Wolthuis, R.; de la Fuente, D. Sem/micro-raman characterization of the morphologies of marine atmospheric corrosion products formed on mild steel. J. Electrochem. Soc. 2016, 163, C426–C439. [Google Scholar] [CrossRef]
- Li, S.; Hihara, L.H. In situ raman spectroscopic study of nacl particle-induced marine atmospheric corrosion of carbon steel. J. Electrochem. Soc. 2012, 159, C147–C154. [Google Scholar] [CrossRef]
- Cano, H.; Neff, D.; Morcillo, M.; Dillmann, P.; Diaz, I.; de la Fuente, D. Characterization of corrosion products formed on ni 2.4 wt %-Cu 0.5 wt %-Cr 0.5 wt % weathering steel exposed in marine atmospheres. Corros. Sci. 2014, 87, 438–451. [Google Scholar] [CrossRef]
- Cook, D.C.; Oh, S.J.; Balasubramanian, R.; Yamashita, M. The role of goethite in the formation of the protective corrosion layer on steels. Hyperfine Interact. 1999, 122, 59–70. [Google Scholar] [CrossRef]
- Zhang, Q.C.; Wu, J.S.; Wang, J.J.; Zheng, W.L.; Chen, J.G.; Li, A.B. Corrosion behavior of weathering steel in marine atmosphere. Mater. Chem. Phys. 2003, 77, 603–608. [Google Scholar] [CrossRef]
- Oh, S.J.; Cook, D.C.; Townsend, H.E. Atmospheric corrosion of different steels in marine, rural and industrial environments. Corros. Sci. 1999, 41, 1687–1702. [Google Scholar] [CrossRef]
- de la Fuente, D.; Alcantara, J.; Chico, B.; Diaz, I.; Jimenez, J.A.; Morcillo, M. Characterization of rust surfaces formed on mild steel exposed to marine atmospheres using xrd and sem/micro-raman techniques. Corros. Sci. 2016, 110, 253–264. [Google Scholar] [CrossRef]
- Diaz, I.; Cano, H.; de la Fuente, D.; Chico, B.; Vega, J.M.; Morcillo, M. Atmospheric corrosion of ni-advanced weathering steels in marine atmospheres of moderate salinity. Corros. Sci. 2013, 76, 348–360. [Google Scholar] [CrossRef]
- Hazan, E.; Sadia, Y.; Gelbstein, Y. Characterization of aisi 4340 corrosion products using raman spectroscopy. Corros. Sci. 2013, 74, 414–418. [Google Scholar] [CrossRef]
- Duennwald, J.; Otto, A. An investigation of phase transitions in rust layers using raman spectroscopy. Corros. Sci. 1989, 29, 1167–1176. [Google Scholar] [CrossRef]
- Evans, U.R. Electrochemical mechanism of atmospheric rusting. Nature 1965, 206, 980–982. [Google Scholar] [CrossRef]
- Aramendia, J.; Gomez-Nubla, L.; Castro, K.; Martinez-Arkarazo, I.; Vega, D.; Sanz Lopez de Heredia, A.; Garcia Ibanez de Opakua, A.; Madariaga, J.M. Portable raman study on the conservation state of four corten steel-based sculptures by eduardo chillida impacted by urban atmospheres. J. Raman Spectrosc. 2012, 43, 1111–1117. [Google Scholar] [CrossRef]
- Veneranda, M.; Aramendia, J.; Gomez, O.; Fdez-Ortiz de Vallejuelo, S.; Garcia, L.; Garcia-Camino, I.; Castro, K.; Azkarate, A.; Madariaga, J.M. Characterization of archaeometallurgical artefacts by means of portable raman systems: Corrosion mechanisms influenced by marine aerosol. J. Raman Spectrosc. 2016, 48, 258–266. [Google Scholar] [CrossRef]
- Yucel, N.; Kalkanli, A.; Caner-Saltik, E.N. Investigation of atmospheric corrosion layers on historic iron nails by micro-raman spectroscopy. J. Raman Spectrosc. 2016, 47, 1486–1493. [Google Scholar] [CrossRef]
- Monnier, J.; Bellot-Gurlet, L.; Baron, D.; Neff, D.; Guillot, I.; Dillmann, P. A methodology for raman structural quantification imaging and its application to iron indoor atmospheric corrosion products. J. Raman Spectrosc. 2011, 42, 773–781. [Google Scholar] [CrossRef]
- Neff, D.; Bellot-Gurlet, L.; Dillmann, P.; Reguer, S.; Legrand, L. Raman imaging of ancient rust scales on archaeological iron artefacts for long-term atmospheric corrosion mechanisms study. J. Raman Spectrosc. 2006, 37, 1228–1237. [Google Scholar] [CrossRef]
- Hayez, V.; Guillaume, J.; Hubin, A.; Terryn, H. Micro-raman spectroscopy for the study of corrosion products on copper alloys: Setting up of a reference database and studying works of art. J. Raman Spectrosc. 2004, 35, 732–738. [Google Scholar] [CrossRef]
- Hayez, V.; Costa, V.; Guillaume, J.; Terryn, H.; Hubin, A. Micro raman spectroscopy used for the study of corrosion products on copper alloys: Study of the chemical composition of artificial patinas used for restoration purposes. Analyst (Camb. UK) 2005, 130, 550–556. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, E.; Chiavari, C.; Martini, C.; Morselli, L. The atmospheric corrosion of quaternary bronzes: An evaluation of the dissolution rate of the alloying elements. Appl. Phys. A Mater. Sci. Process. 2008, 92, 83–89. [Google Scholar] [CrossRef]
- Martina, I.; Wiesinger, R.; Jembrih-Simbuerger, D.; Schreiner, M. Micro-raman characterisation of silver corrosion products: Instrumental set up and reference database. e-Preserv. Sci. 2012, 9, 1–8. [Google Scholar]
- Martina, I.; Wiesinger, R.; Schreiner, M. Micro-raman investigations of early stage silver corrosion products occurring in sulfur containing atmospheres. J. Raman Spectrosc. 2013, 44, 770–775. [Google Scholar] [CrossRef]
- Ohtsuka, T.; Matsuda, M. In situ raman spectroscopy for corrosion products of zinc in humidified atmosphere in the presence of sodium chloride precipitate. Corrosion (Houston, TX, USA) 2003, 59, 407–413. [Google Scholar] [CrossRef]
- Jayasree, R.S.; Mahadevan Pillai, V.P.; Nayar, V.U.; Odnevall, I.; Keresztury, G. Raman and infrared spectral analysis of corrosion products on zinc NaZn4Cl(OH)6SO4·6H2O and Zn4Cl2(OH)4SO4·5H2O. Mater. Chem. Phys. 2006, 99, 474–478. [Google Scholar] [CrossRef]
- Hedberg, J.; Baldelli, S.; Leygraf, C.; Tyrode, E. Molecular structural information of the atmospheric corrosion of zinc studied by vibrational spectroscopy techniques: I. Experimental approach. J. Electrochem. Soc. 2010, 157, C357–C362. [Google Scholar] [CrossRef]
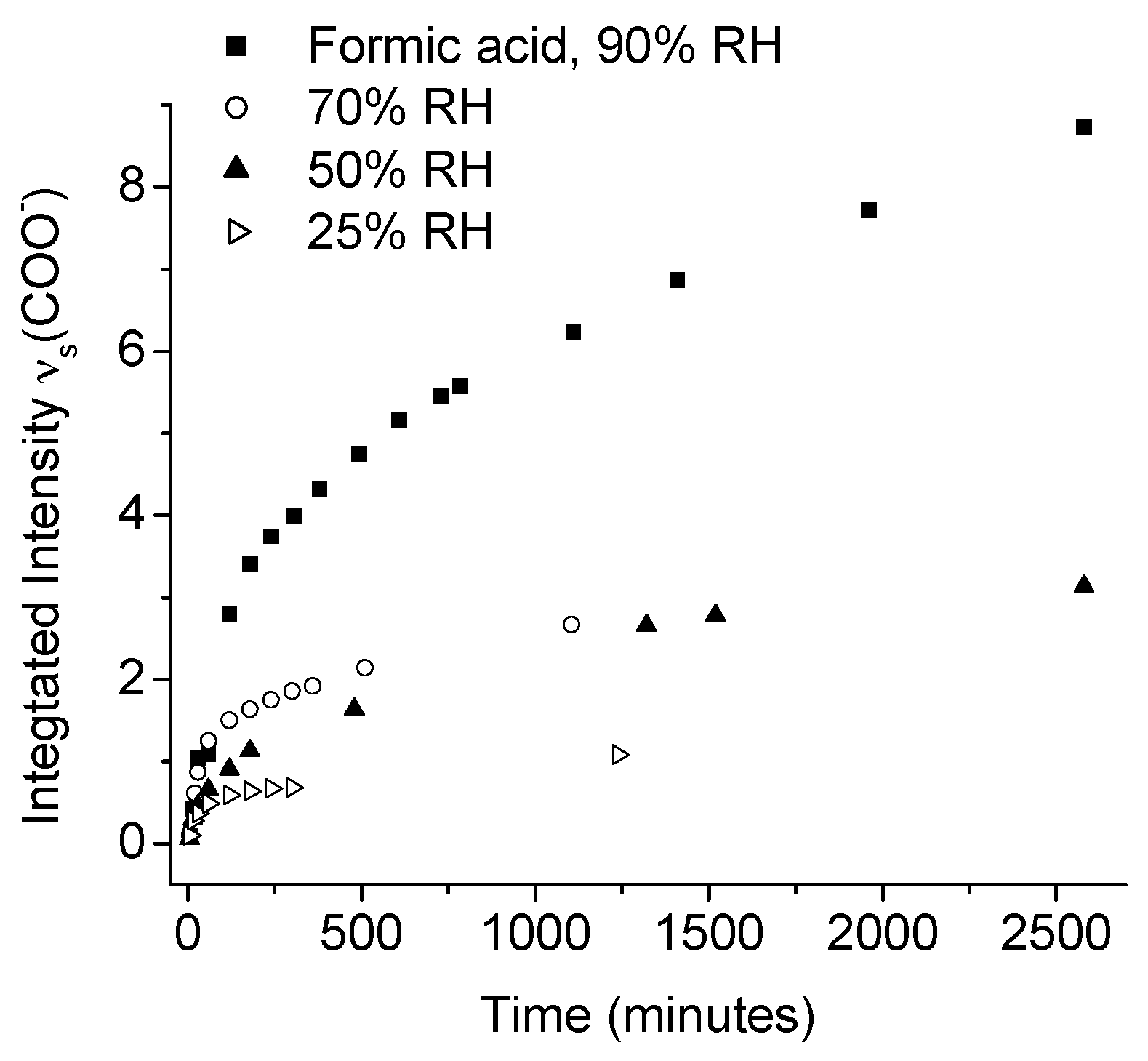
- Hedberg, J.; Baldelli, S.; Leygraf, C. Molecular structural information of the atmospheric corrosion of zinc studied by vibrational spectroscopy techniques. Ii. Two and 3-dimensional growth of reaction products induced by formic and acetic acid. J. Electrochem. Soc. 2010, 157, C363–C373. [Google Scholar] [CrossRef]
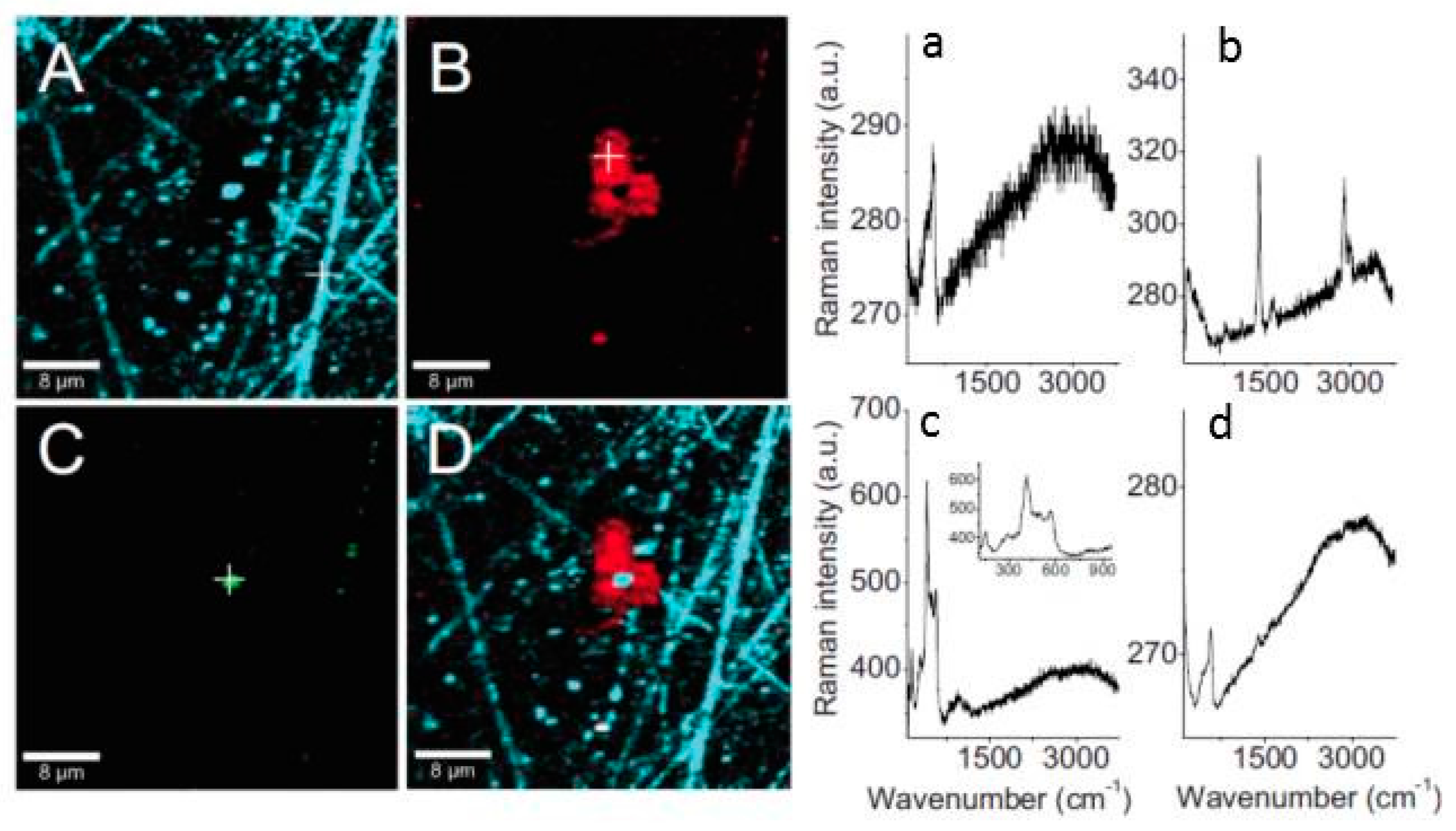
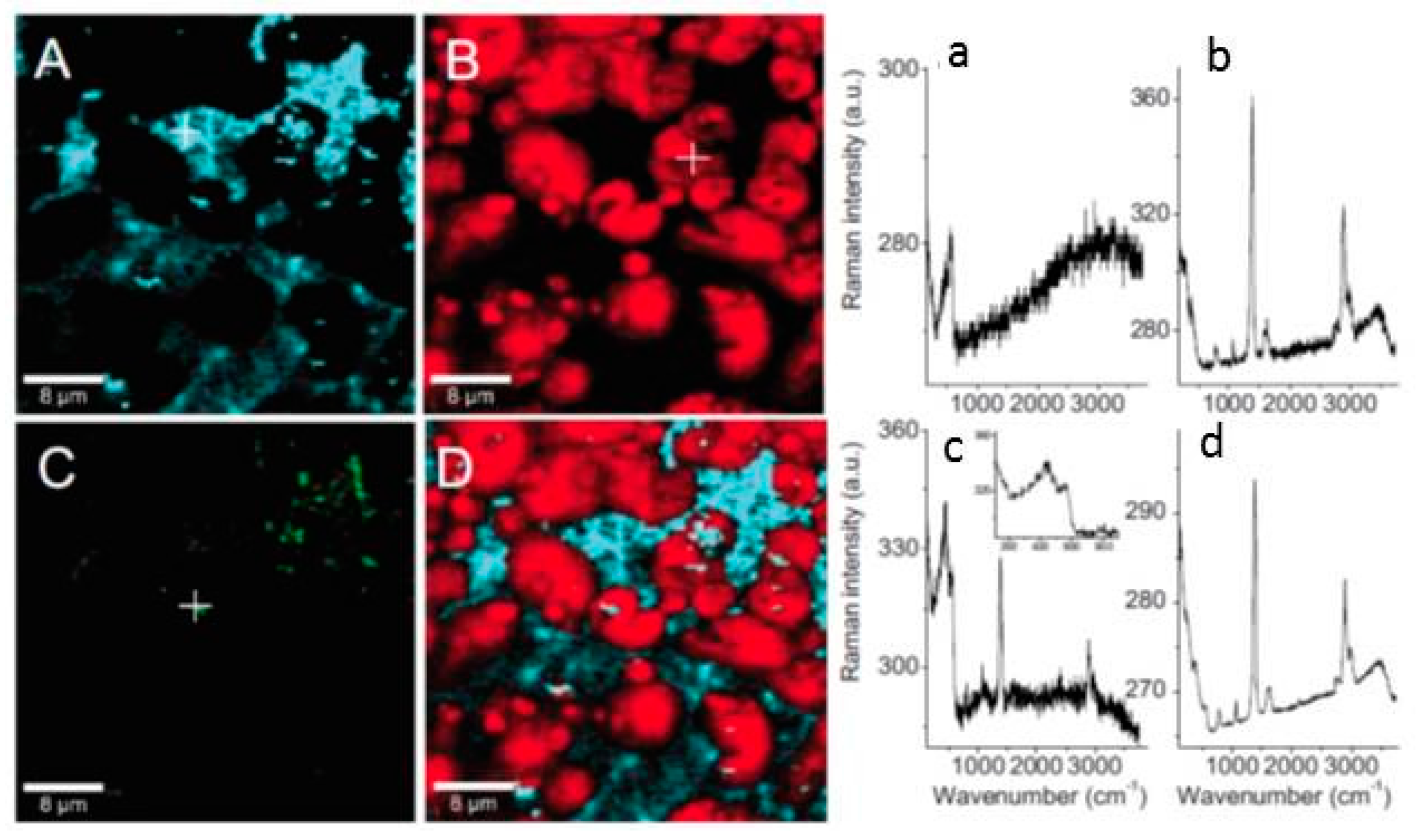
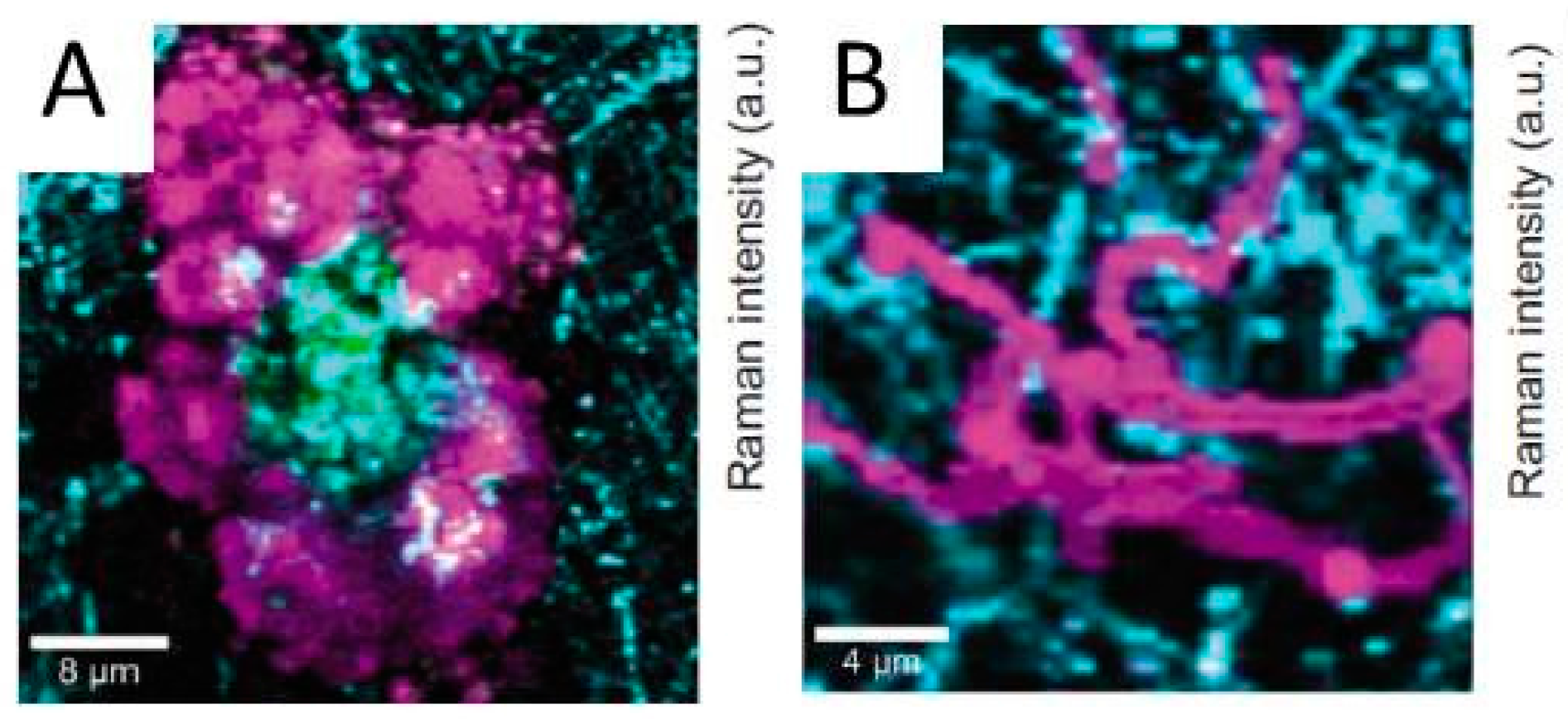
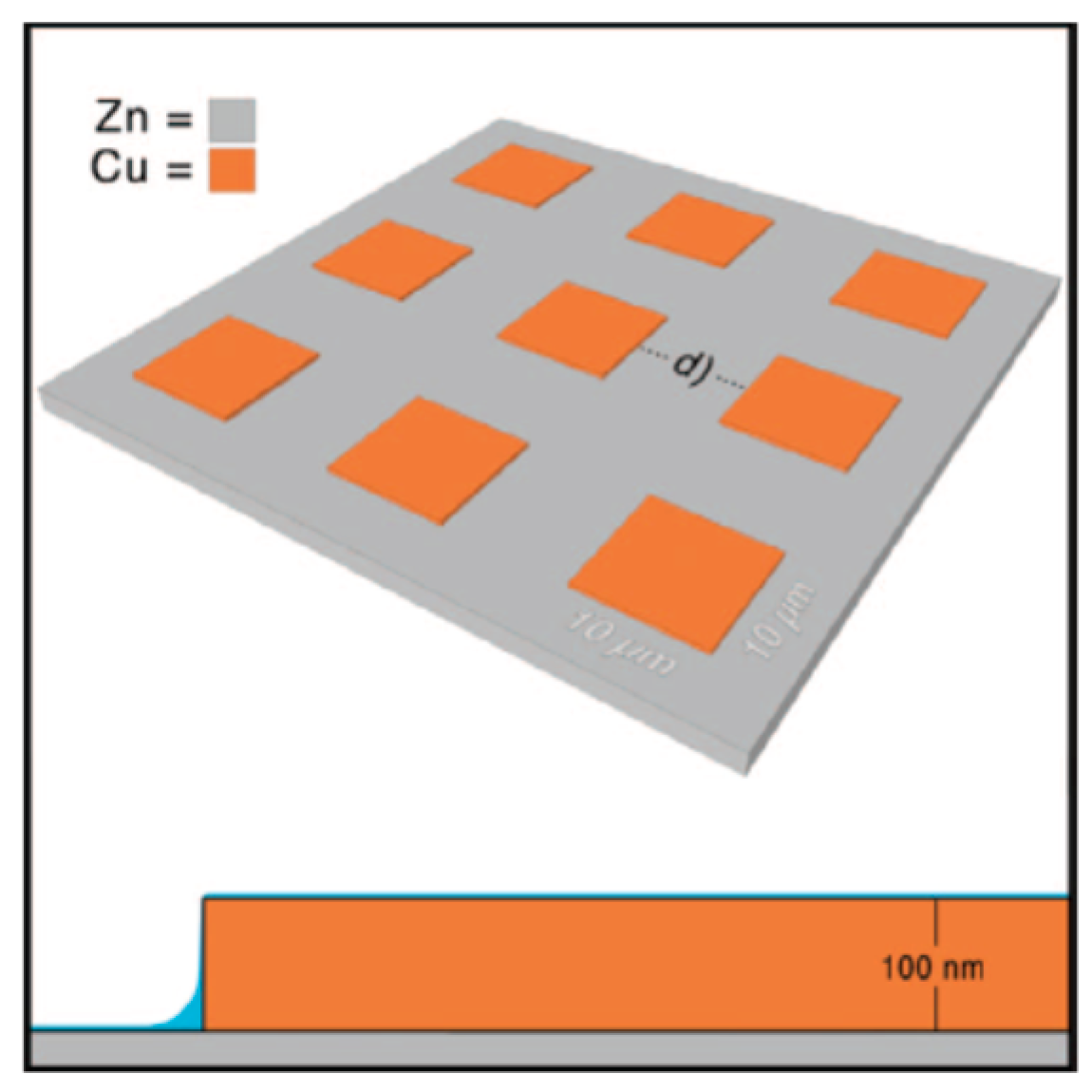
- Forslund, M.; Leygraf, C.; Claesson, P.M.; Lin, C.; Pan, J. Micro-galvanic corrosion effects on patterned copper-zinc samples during exposure in humidified air containing formic acid. J. Electrochem. Soc. 2013, 160, C423–C431. [Google Scholar] [CrossRef]
- Forslund, M.; Leygraf, C.; Claesson, P.M.; Pan, J. Octadecanethiol as corrosion inhibitor for zinc and patterned zinc-copper in humidified air with formic acid. J. Electrochem. Soc. 2014, 161, C330–C338. [Google Scholar] [CrossRef]
- De Faria, D.L.A.; Cavicchioli, A.; Puglieri, T.S. Indoors lead corrosion: Reassessing the role of formaldehyde. Vib. Spectrosc. 2010, 54, 159–163. [Google Scholar] [CrossRef]
- Matikainen, A.; Nuutinen, T.; Itkonen, T.; Heinilehto, S.; Puustinen, J.; Hiltunen, J.; Lappalainen, J.; Karioja, P.; Vahimaa, P. Atmospheric oxidation and carbon contamination of silver and its effect on surface-enhanced raman spectroscopy (sers). Sci. Rep. 2016, 6, 37192. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Spencer, S.J.; Imbraguglio, D.; Rossi, A.M.; Wain, A.J.; Weckhuysen, B.M.; Roy, D. Extending the plasmonic lifetime of tip-enhanced raman spectroscopy probes. PCCP 2016, 18, 13710–13716. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, Y.-S.; He, P. In situ identification of surface species on molybdenum in different media. Electrochim. Acta 1998, 43, 2459–2467. [Google Scholar] [CrossRef]
- Tormoen, G.; Burket, J.; Dante, J.F.; Sridhar, N. Monitoring the adsorption of volatile corrosion inhibitors in real time with surface-enhanced raman spectroscopy. Corrosion 2006, 62, 1082–1091. [Google Scholar] [CrossRef]
- Miranda, P.B.; Shen, Y.R. Liquid interfaces: A study by sum-frequency vibrational spectroscopy. J. Phys. Chem. B 1999, 103, 3292–3307. [Google Scholar] [CrossRef]
- Hosseinpour, S.; Hedberg, J.; Baldelli, S.; Leygraf, C.; Johnson, M. Initial oxidation of alkanethiol-covered copper studied by vibrational sum frequency spectroscopy. J. Phys. Chem. C 2011, 115, 23871–23879. [Google Scholar] [CrossRef]
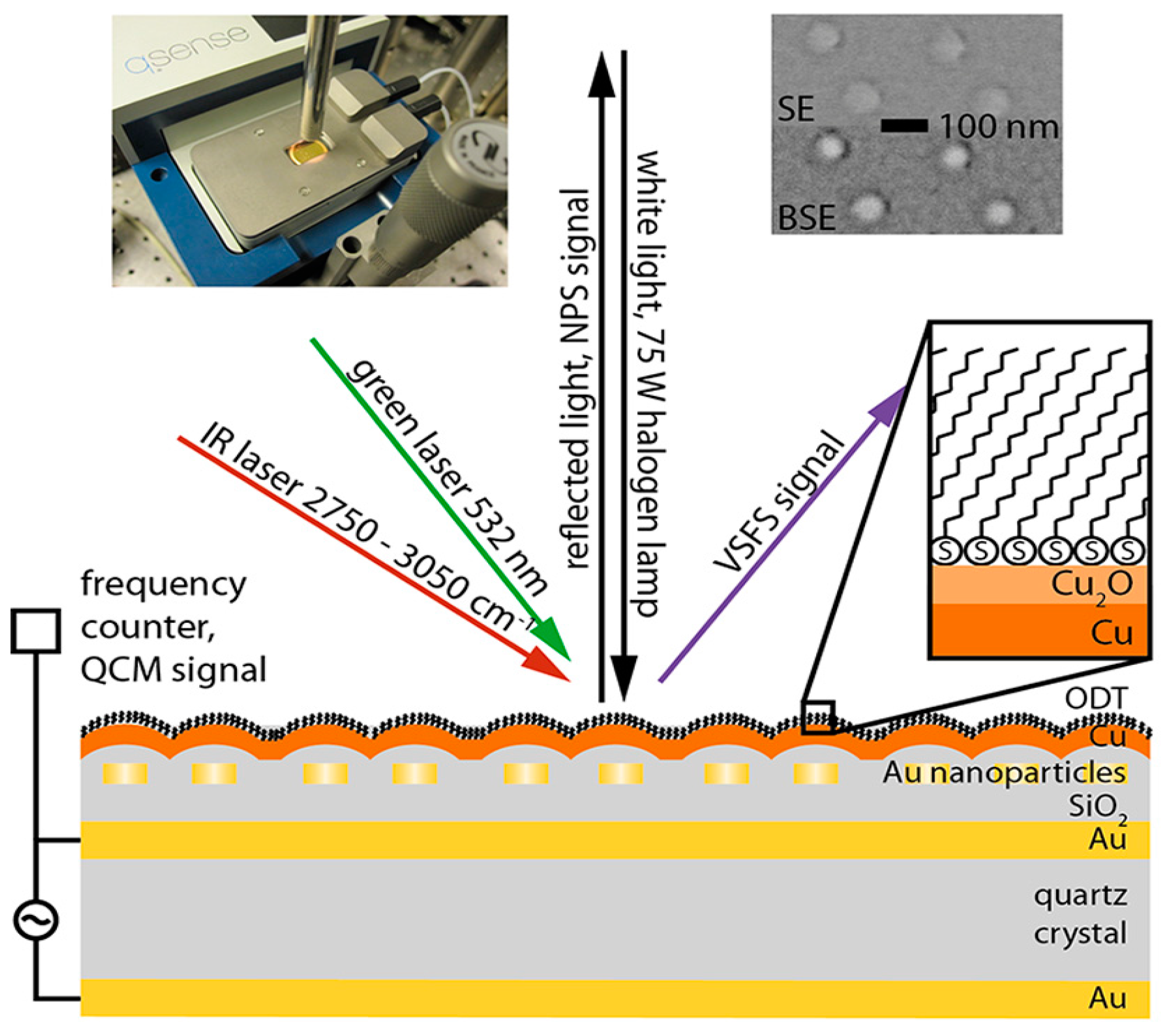
- Hosseinpour, S.; Schwind, M.; Kasemo, B.; Leygraf, C.; Johnson, C.M. Integration of quartz crystal microbalance with vibrational sum frequency spectroscopy–quantification of the initial oxidation of alkanethiol-covered copper. J. Phys. Chem. C 2012, 116, 24549–24557. [Google Scholar] [CrossRef]
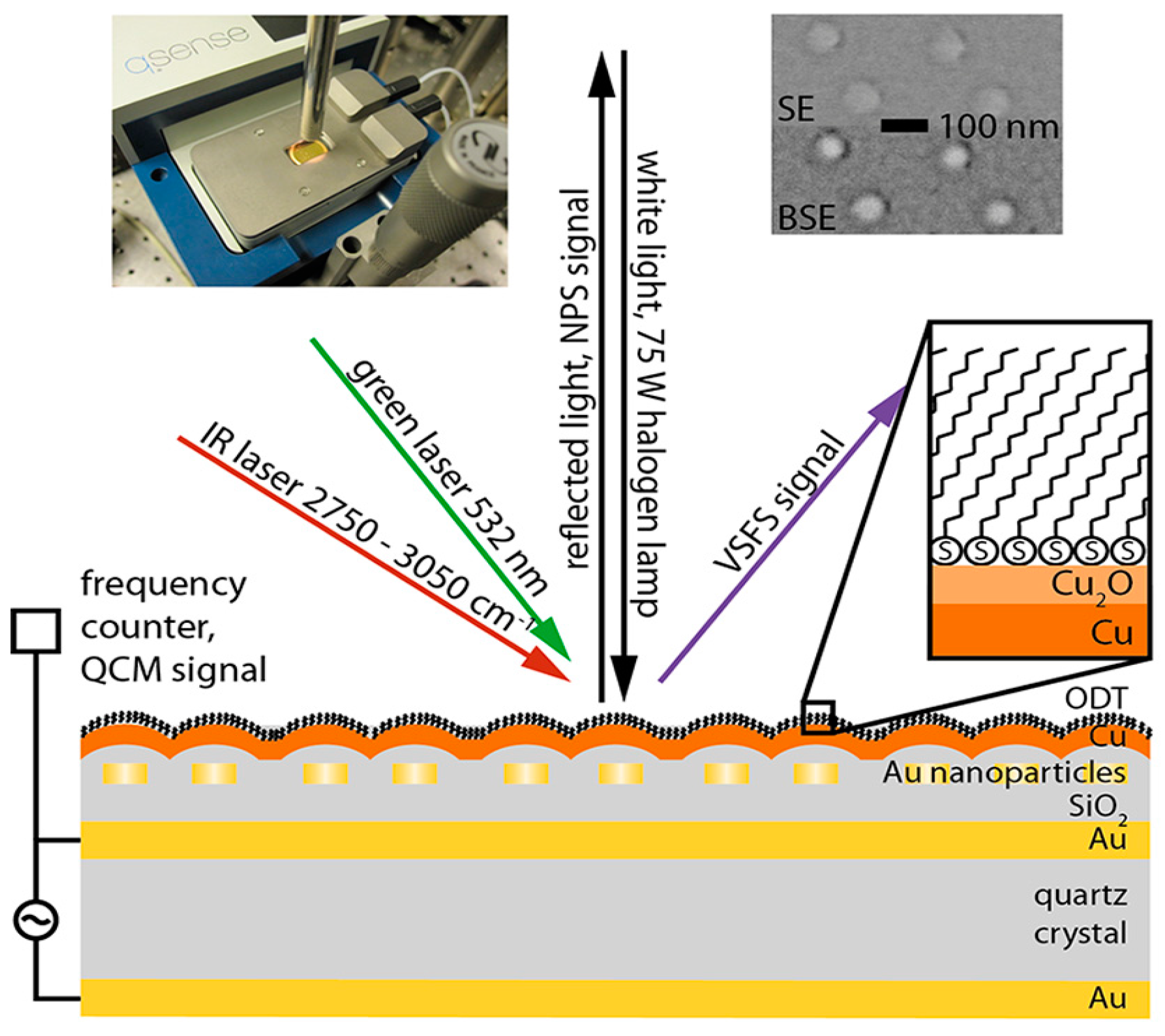
- Schwind, M.; Hosseinpour, S.; Johnson, C.M.; Langhammer, C.; Zorić, I.; Leygraf, C.; Kasemo, B. Combined in situ quartz crystal microbalance with dissipation monitoring, indirect nanoplasmonic sensing, and vibrational sum frequency spectroscopic monitoring of alkanethiol-protected copper corrosion. Langmuir 2013, 29, 7151–7161. [Google Scholar] [CrossRef] [PubMed]
- Hosseinpour, S.; Johnson, C.M.; Leygraf, C. Alkanethiols as inhibitors for the atmospheric corrosion of copper induced by formic acid: Effect of chain length. J. Electrochem. Soc. 2013, 160, C270–C276. [Google Scholar] [CrossRef]
- Hosseinpour, S.; Göthelid, M.; Leygraf, C.; Johnson, C.M. Self-assembled monolayers as inhibitors for the atmospheric corrosion of copper induced by formic acid: A comparison between hexanethiol and hexaneselenol. J. Electrochem. Soc. 2014, 161, C50–C56. [Google Scholar] [CrossRef]
- Forslund, M.; Pan, J.; Hosseinpour, S.; Zhang, F.; Johnson, M.; Claesson, P.; Leygraf, C. Corrosion inhibition of two brass alloys by octadecanethiol in humidified air with formic acid. Corrosion 2015, 71, 908–917. [Google Scholar] [CrossRef]
- Santos, G.M.; Baldelli, S. Monitoring localized initial atmospheric corrosion of alkanethiol-covered copper using sum frequency generation imaging microscopy: Relation between monolayer properties and Cu2O formation. J. Phys. Chem. C 2013, 117, 17591–17602. [Google Scholar] [CrossRef]
- Hedberg, J.; Henriquez, J.; Baldelli, S.; Johnson, C.M.; Leygraf, C. Initial atmospheric corrosion of zinc exposed to formic acid, investigated by in situ vibrational sum frequency spectroscopy and density functional theory calculations. J. Phys. Chem. C 2009, 113, 2088–2095. [Google Scholar] [CrossRef]
- Hedberg, J.; Baldelli, S.; Leygraf, C. Initial atmospheric corrosion of zn: Influence of humidity on the adsorption of formic acid studied by vibrational sum frequency spectroscopy. J. Phys. Chem. C 2009, 113, 6169–6173. [Google Scholar] [CrossRef]
- Hedberg, J.; Baldelli, S.; Leygraf, C. Evidence for the molecular basis of corrosion of zinc induced by formic acid using sum frequency generation spectroscopy. J. Phys. Chem. Lett. 2010, 1, 1679–1682. [Google Scholar] [CrossRef]
- Lagutchev, A.; Hambir, A.; Dlott, D. Nonresonant Background Suppression in Broadband Vibrational Sum-Frequency Generation Spectroscopy. J. Phys. Chem. C 2007, 111, 13645–13647. [Google Scholar] [CrossRef]
- Hajdari Gretić, Z.; Kristan Mioč, E.; Čadež, V.; Šegota, S.; Otmačić Ćurković, H.; Hosseinpour, S. The influence of thickness of stearic acid self-assembled film on its protective properties. J. Electrochem. Soc. 2016, 163, C937–C944. [Google Scholar] [CrossRef]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Typical indoor Concentration (ppb) | H b | Equilibrium Solution Concentration (µM) |
|---|---|---|---|
| O2 | 2.1 (8) a | 1.7 (−3) | 3.6 (2) |
| O3 | 18 | 1.8 (−2) | 3.2 (−4) |
| H2O2 | 5 | 2.4 (5) | 1.2 (3) |
| H2S | 0.3 | 1.5 (−1) | 4.5 (−5) |
| COS | 0.6 | 3.7 (−2) | 2.2 (−5) |
| SO2 | 30 | 1.4 | 4.2 (−2) |
| HCl | 0.4 | 2.0 (1) | 8.0 (−3) |
| Cl2 | 0 | 6.2 (−2) | 0 |
| NH3 | 10 | 1.0 (1) | 1.0 (−1) |
| NO2 | 4 | 7.0 (−3) | 2.8 (−5) |
| HNO3 | 3 | 9.1 (4) | 2.7 (2) |
| CO2 | 6.0 (5) | 3.4 (−2) | 2.0 (1) |
| HCHO | 10 | 1.4 (4) | 1.4 (2) |
| HCOOH | 20 | 3.7 (3) | 7.4 (1) |
| CH3COOH | 20 | 8.8 (3) | 8.8 (1) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hosseinpour, S.; Johnson, M. Vibrational Spectroscopy in Studies of Atmospheric Corrosion. Materials 2017, 10, 413. https://doi.org/10.3390/ma10040413
Hosseinpour S, Johnson M. Vibrational Spectroscopy in Studies of Atmospheric Corrosion. Materials. 2017; 10(4):413. https://doi.org/10.3390/ma10040413
Chicago/Turabian StyleHosseinpour, Saman, and Magnus Johnson. 2017. "Vibrational Spectroscopy in Studies of Atmospheric Corrosion" Materials 10, no. 4: 413. https://doi.org/10.3390/ma10040413
APA StyleHosseinpour, S., & Johnson, M. (2017). Vibrational Spectroscopy in Studies of Atmospheric Corrosion. Materials, 10(4), 413. https://doi.org/10.3390/ma10040413