2.1. Molecular Design
In the introduction, it was briefly mentioned that in the series of diazinium ethenes developed by Hünig et al., an effect related to benzocondensation emerges, resulting in a shift of the reduction potentials to less negative values [
4,
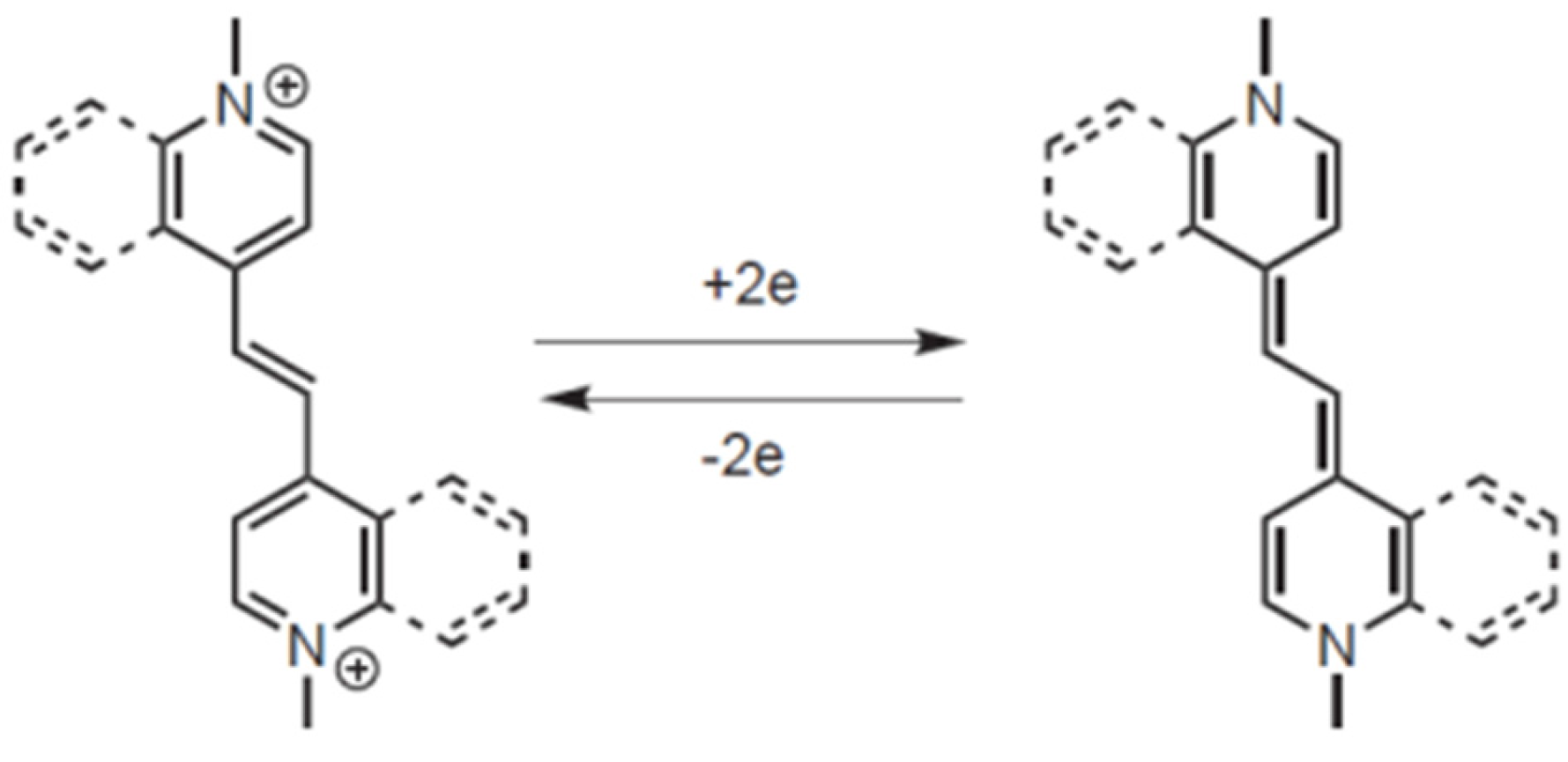
5]. This can be attributed to the strongly π-accepting capabilities of the quinoline ring with respect to pyridine (lower LUMO and lower HOMO), and to the aromatic stabilization of the reduced form. It has to be considered, in fact, that the aromatic-to-quinoid transition takes place upon reduction to the fully reduced form. Benzocondensation allows us to retain a certain grade of aromatic stabilization, as illustrated in
Figure 2, according to the fact that one of the two rings in the terminal group gains full benzenoid aromaticity upon reduction.
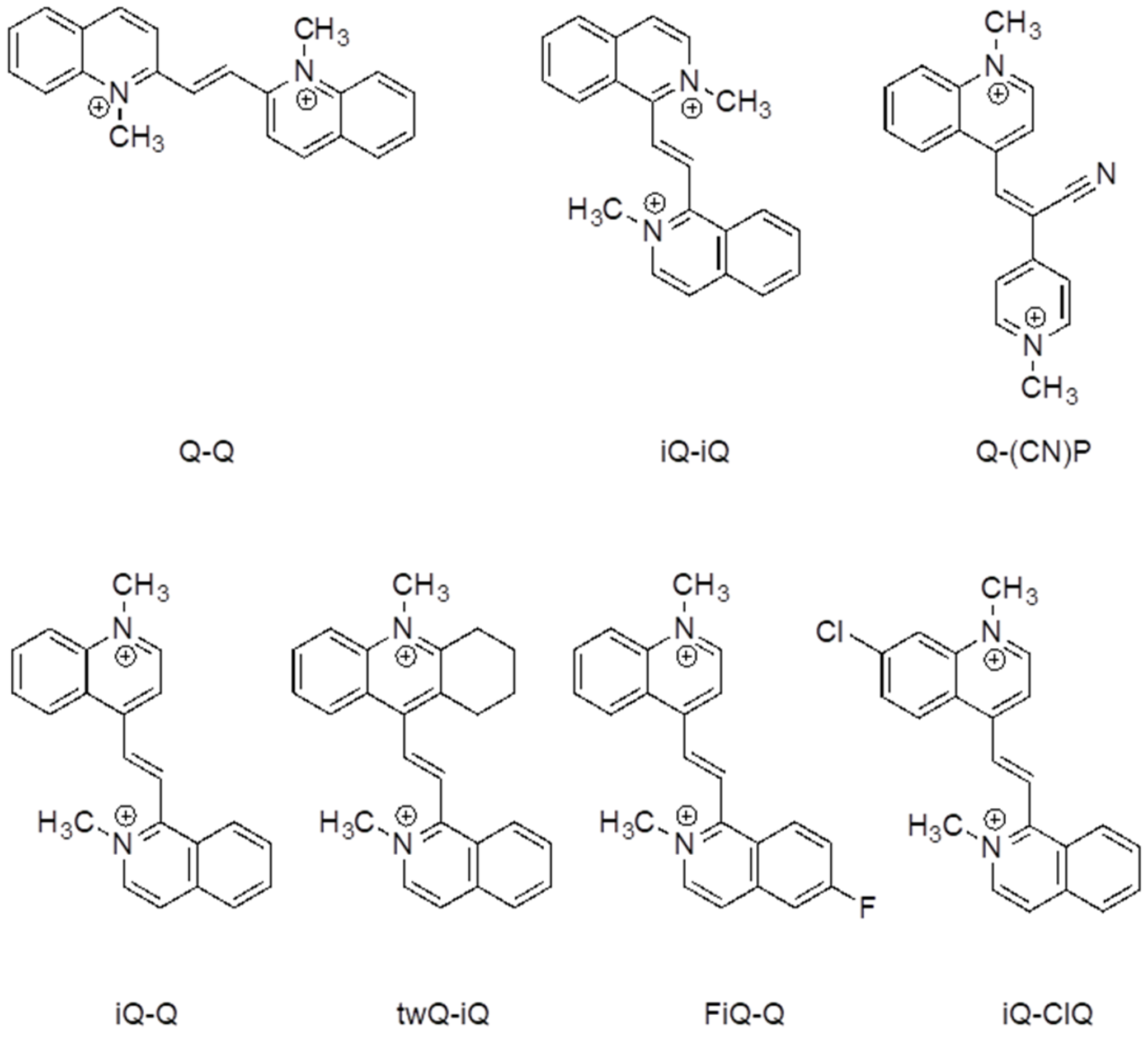
Despite the huge number of Weitz-type violene systems studied by Hünig et al., for some reason, they always limited their investigation to symmetric systems, even in those cases where asymmetric derivatives were synthetically accessible. We decided to take full advantage of the benzocondensation effect, also exploring the design of asymmetric structures in order to further increase the tailoring of the properties by mixing different heterocycles with similar redox potentials. The effect of the presence of different substituents on the electrochemical and spectroelectrochemical properties was also studied, with the aim of developing the optimal strategy to tune the color of its reduced state. For this purpose, derivates twQ-iQ, FiQ-Q, and iQ-ClQ (
Figure 1) were also prepared. In detail, to maximize the effect of substitution, the halogen groups in the derivatives FiQ-Q and iQ-ClQ were placed in the p-position with respect to the position of the ethylene bridge. In the compound twQ-iQ, the choice of the tetrahydroacridine end group used to explore the effect of the alkyl donating groups was dictated by synthetic convenience. The introduction of electron withdrawing groups stimulates a shift of the redox wave to higher potential values, as expected. A relevant feature of azinic derivatives, particularly those obtained through the stepwise approach we employed, is the introduction of different functional solubilizing chains in the nitrogen atoms. In fact, along with solubility, those chains enabled conjugation with other, possibly electro-active, components, as some of us have recently showed.
2.2. Electrochemical and Spectroelectrochemical Properties
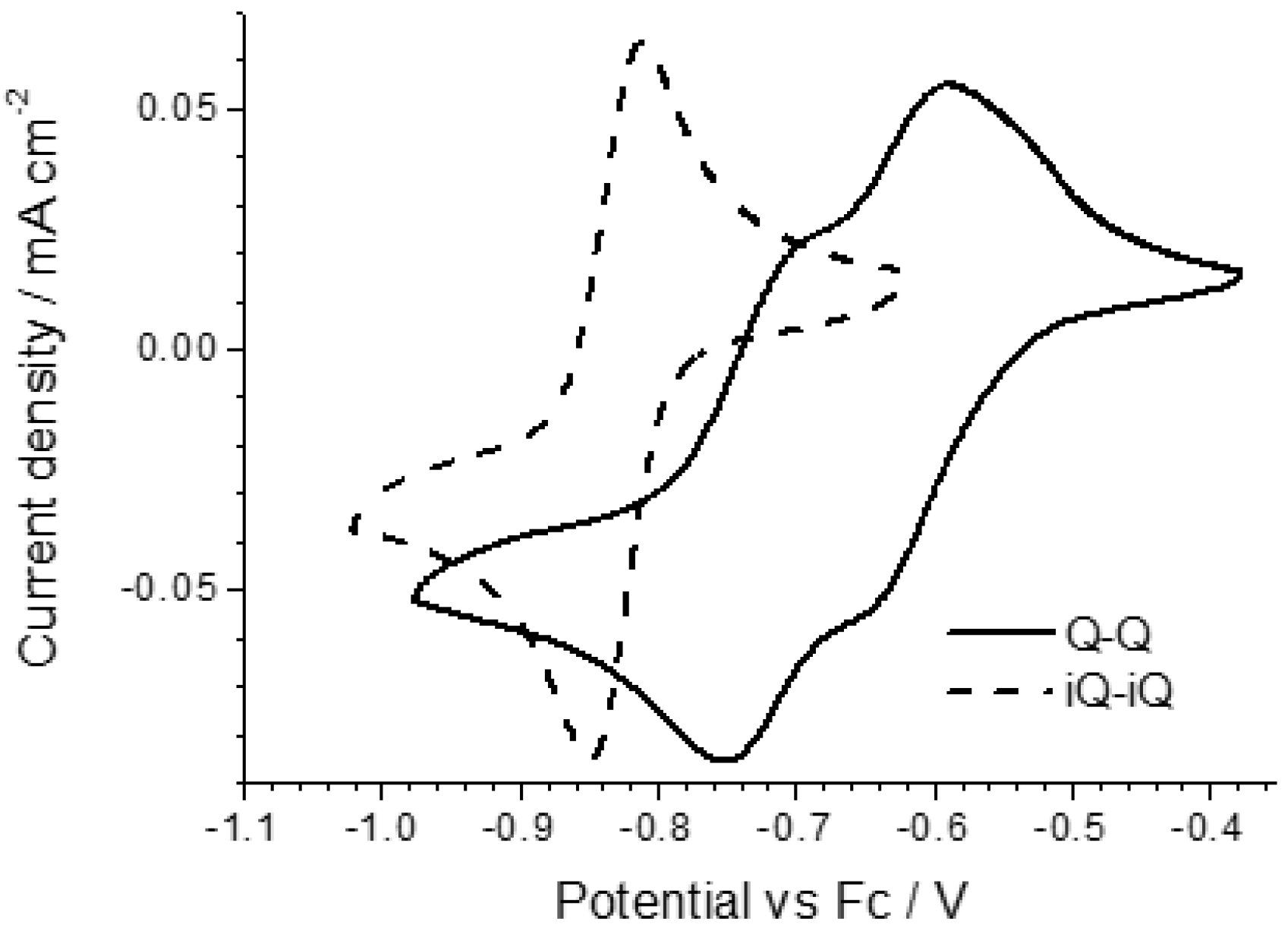
The electrochemical properties of the diazinium ethene compounds were obtained by cyclic voltammetry measurements. The voltammograms of the symmetric Q-Q and iQ-iQ molecules are depicted in
Figure 3. Both systems exhibit reversible redox behavior. In particular, the presence of two distinct redox waves, corresponding to the E
1 and E
2 processes (see Introduction), is evident in the Q-Q compound. The process E
1 is characterized by a oxidation shoulder and a reduction peak, while E
2 by a reduction shoulder and an oxidation peak. To separate the two processes, we also performed a differential pulsed voltammetry (DPV) analysis (
Figure SI_1). The peak to peak separation, consistent for the both reductions and oxidations, is 100 ± 5 mV, which can be used to estimate the K
IRF as 50 ± 10. In contrast, a highly reversible two-electron process is involved in iQ-iQ, as testified by the low peak separation observed (ΔE = 38 mV) in the sharply-peaked redox waves. Similar behavior is observed in the asymmetric compound iQ-Q (
Figure 4). In this case, the measured peak separation is 40 mV, thus confirming the involvement of a two-electron process. Q-(CN)P, conversely, has a less negative reduction potential, as expected by the presence of the strong electron withdrawing –CN group. However, the electrochemical process shows two partially irreversible mono-electronic waves. Indeed, in each process, the separation between the reduction and oxidation peaks is about 80 mV, larger than the Nernstian theoretical value. However, the voltage difference between the oxidation peaks of E
1 and E
2 is consistent with the difference between the reduction peaks, and was calculated as 230 ± 5 mV, which gives a K
IRF value of 7.8 × 10
3 ± 1.7 × 10
3, two orders of magnitude larger than for the Q-Q compound. From the reduction potential waves, it is also possible to calculate the corresponding LUMO energy levels of the dication forms, assuming the vacuum level to coincide with the ferrocene potential in the electrolyte is (−5.2 eV).
The electrochemical data are summarized in
Table 1, wherein E
1 and E
2 correspond to the first and second redox processes, according to the above reported equation (see Introduction). Absolute redox potentials, and thus the corresponding LUMO levels, can be relatively straightforwardly rationalized in terms of molecular structure. In the case of the symmetric structures, indeed, the larger electron withdrawing character of the quinolinic rings leads to a higher reduction potential and a lower LUMO level. Dealing with the asymmetric structures, the LUMO level of the iQ-Q compound is intermediate between those of the corresponding symmetric systems (iQ-iQ and Q-Q). As for Q-(CN)P, the strong electron withdrawing character of the cyanopyridinic substituent results in the highest reduction potential and thus the lowest LUMO level.
It is more difficult to rationalize in such simple terms why some derivatives feature mono- instead of two-electron redox processes. The computational investigation reported below was specifically targeted to investigate this issue.
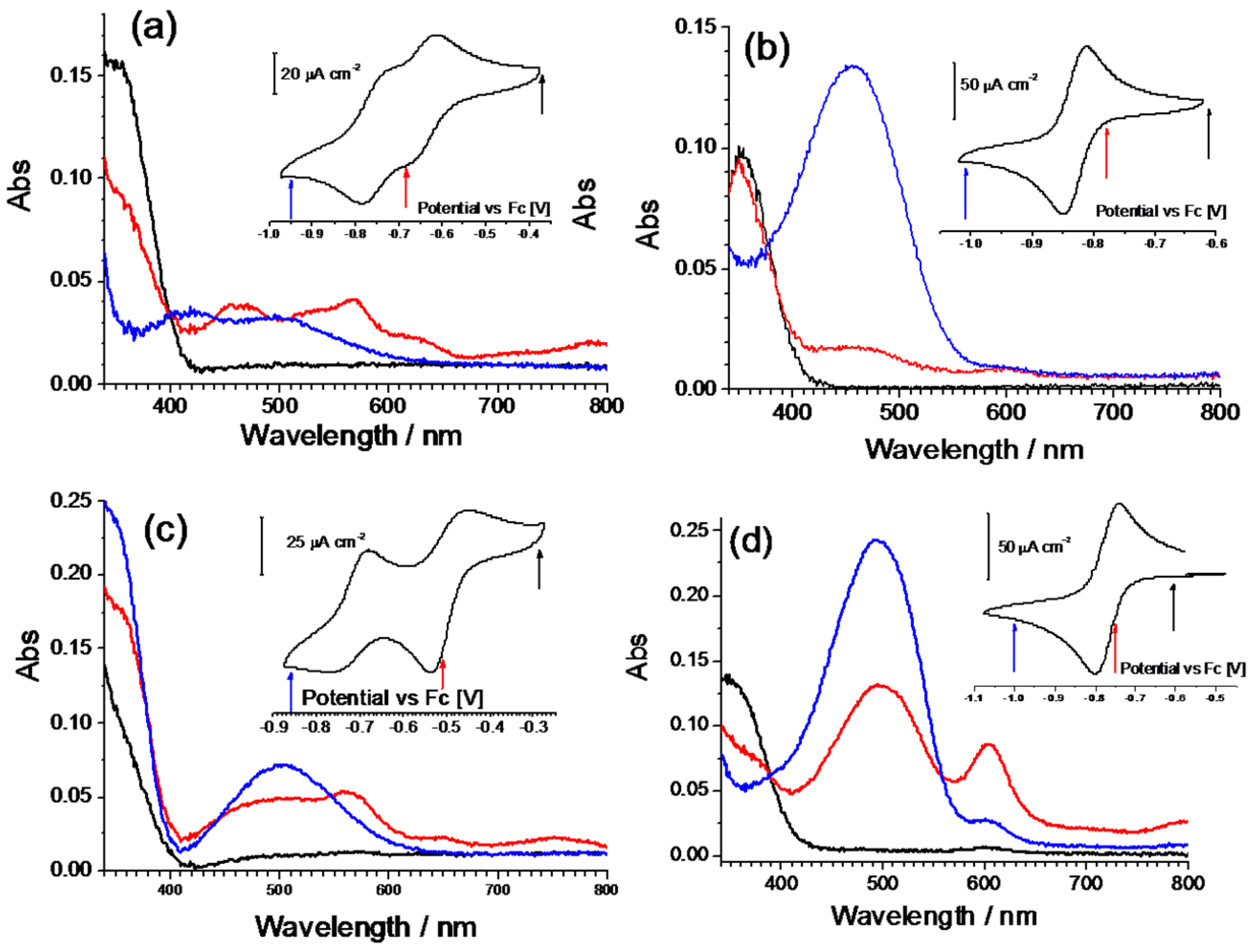
These compounds were further characterized by an in situ spectroelectrochemical measurement using a 10
–4 M solution in a thin layer cell. All the dication forms give a strong UV absorption centered at about 350 nm (black lines in
Figure 5), only the Q-(CN)P molecule shows a higher energy shift (
Figure 5c). Since no other absorption is present in the spectra, the solutions are colorless. The electrochemical reduction provokes, generally, a dimming of the main UV peak along with the formation of a low energy broad band, right in the middle of the visible region around 500 nm. In particular, in the case of the derivate iQ-iQ, a strong absorption band centered at 456 nm is present, together with a noticeable decrease in the intensity of the 350 nm UV band. A weak absorption at 594 nm is also present at intermediate potentials, but is subsequently bleached while the reaction proceeds. A similar behavior is observed with the asymmetric compound iQ-Q. Nevertheless, in this case, the absorption band of the reduced form is shifted to higher wavelengths (493 nm), and an incomplete bleaching of the strong band localized at 603 nm takes place. The spectra of the oxidized forms are recovered upon reoxidation, by switching back to the initial potentials. As can be noted from the spectroelectrogram of the iQ-Q compound, a broad weak absorption from 400 to 700 nm can be detected in the visible region. Although the reason for this kind of behavior cannot be clearly identified, a possible explanation might be the formation of stable intermolecular aggregates or dimers. A similar phenomenon has been observed for viologens-based electrochromic devices during ageing tests. Possible causes have been attributed to the radical cation salt deposition on the electrode surface. These deposits might contain spin-paired radical cation dimers with a sandwiched structure, reorientating into ordered phase during open-circuit periods [
7,
8]. Further investigation on deposited heptyl violognes radicals revealed the presence of a contribution from dimerization of radical cations in solution [
9]. A second mechanism for dimer formation can involve a comproportionation reaction between the neutral completely reduced specie (DE
0) and the oxidized specie (DE
2+) [
10,
11]:
The dimer has lower electroactivity as a consequence of its very slow oxidation rate [
12], and a decreasing of the write-erase efficiency of the system is thus observed [
13]. It has been demonstrated that the use of redox mediators can have a beneficial effect [
14] on the functionalization of the nitrogen atoms of the bipyridine core [
15,
16,
17]. In our case, we suppose that a similar aggregation or dimerization process can take place in the solution during spectroelectrochemical measurements, resulting in a loss of electroactivity and a deviation of the observed spectrum from the one expected from the reduced form. The symmetric structure and the higher stability of the intermediate radical cation in Q-Q can have a contribution in making this effect even more evident in this derivative.
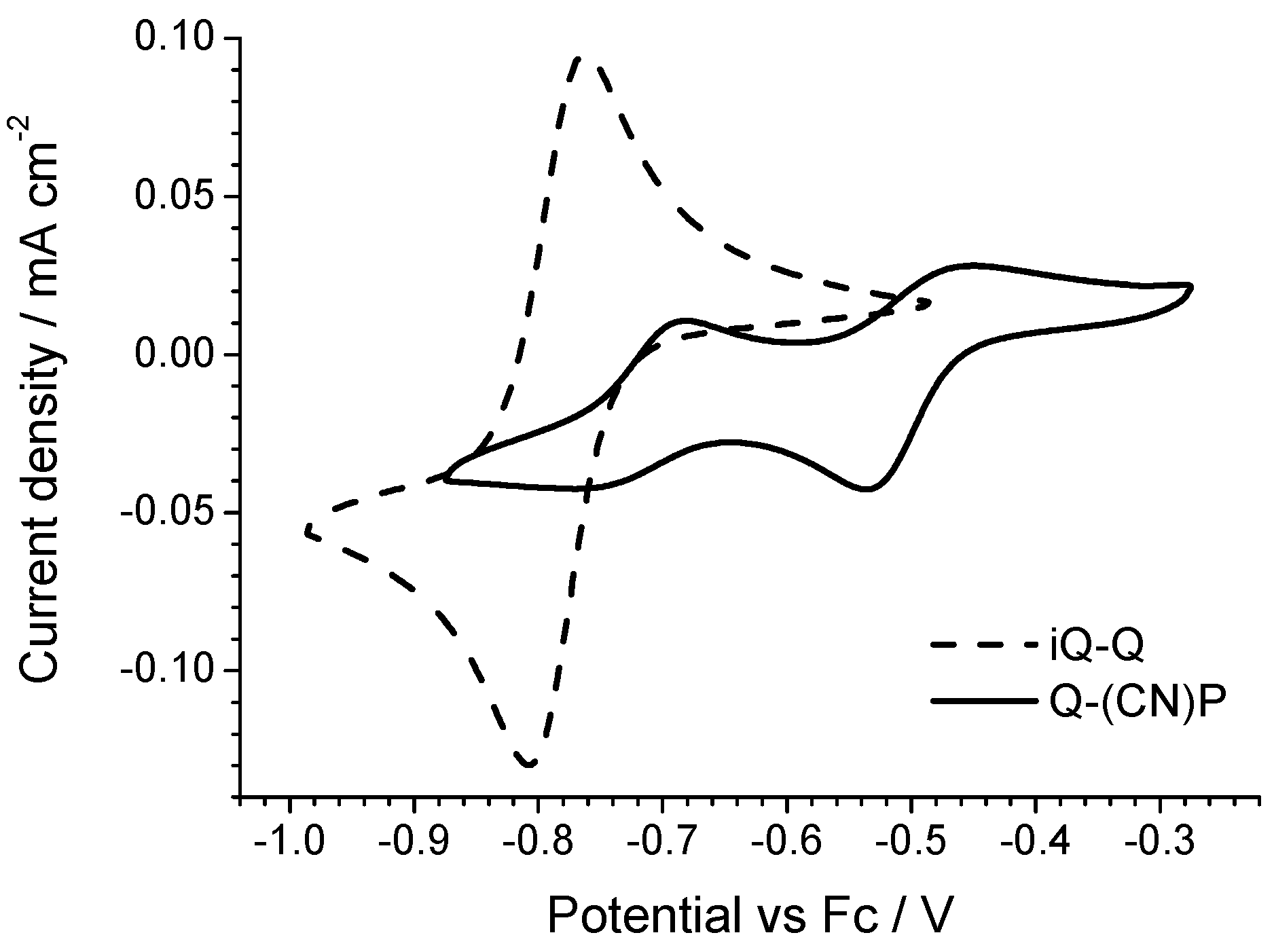
In view of its good electrochemical and spectroelectrochemical behavior, iQ-Q was selected for further derivatization with different substituents. The synthetic details are reported in the supporting information, together with the compounds’ coding (
Table SI_1). Compound iQ-iQ was not further developed, as the functionalization of its nitrogen atoms proved to be impractical from the synthetic point of view. Derivatives of iQ-Q carrying halogen substituents on the 6-position of the isoquinoline ring (fluorine for derivate FiQ-Q) or on the 7-position of the quinoline ring (Chlorine in iQ-ClQ), and one with a tetramethylene tether connecting the 2- and 3-position of quinoline ring (twQ-iQ), were prepared (
Figure 1).
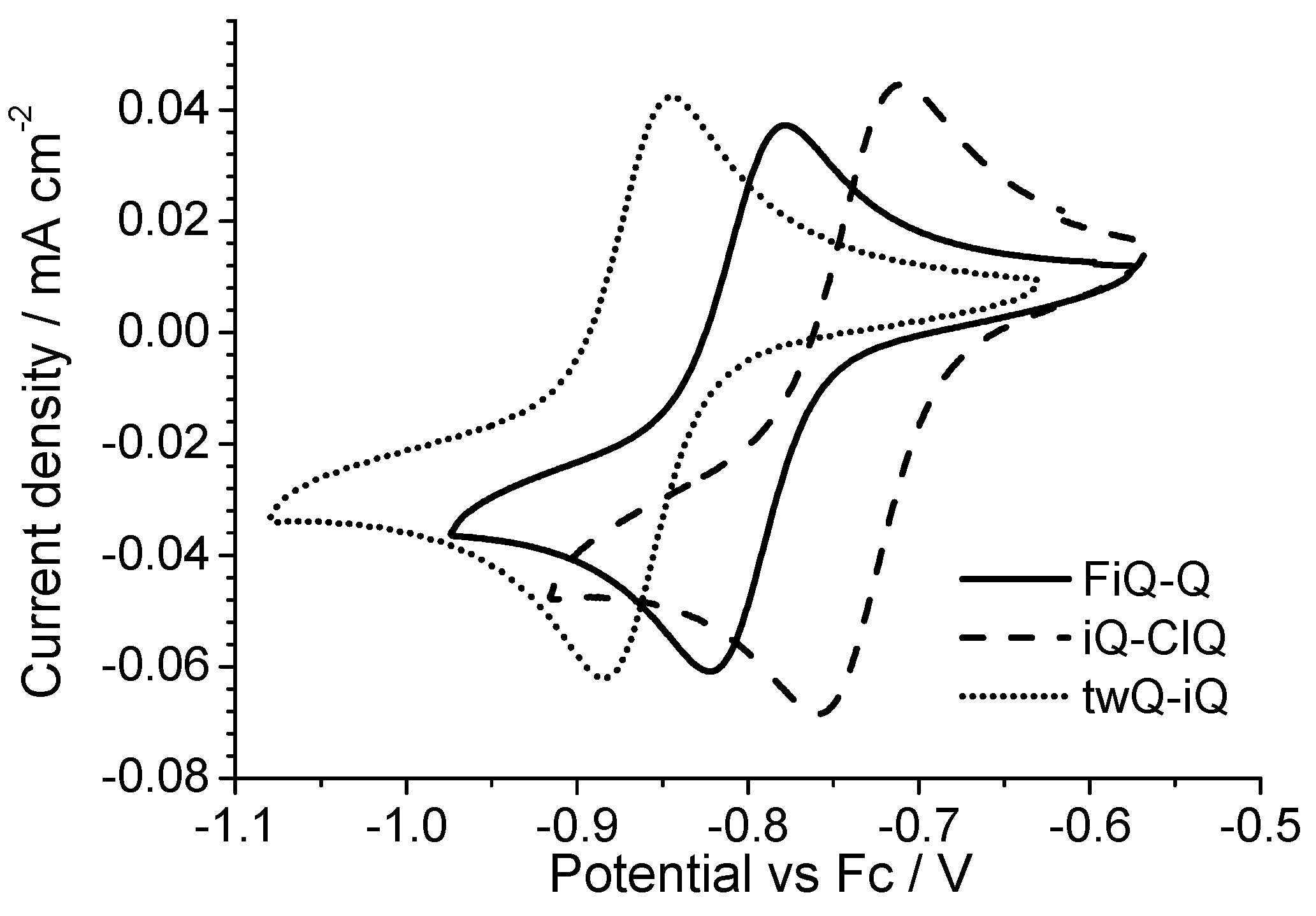
All of the derivatives show a two-electron reversible electron transfer (
Figure 6) with a low peak separation (<40 mV). The Cl- and F-substituted derivatives show a small but significant deviation from the reduction potential measured on the parent compound iQ-Q. The Cl-derivate is slightly easier to reduce, while the F-substituted is not. In the case of the chlorine presence this result is not surprising, even if often the mesomeric effect mitigates the potential shift due to the strong chlorine electron affinity. However, in the case of the small, polarizing fluorine atom, the behavior cannot be rationalized taking in account just the nature of the substituent. Finally, the twQ-iQ (with the tetramethylene moiety connecting 2- and 3-positions on the quinolinic end) is the hardest compound to be reduced, and shows the highest LUMO energy in
Table 1. This can be explained as the consequence of the electron-donating effect of the alkyl groups on LUMO energy. From this analysis, it is evident that the chemical features of the substituent groups play an important but not crucial role in driving the electronic (and thus the electrochemical) molecular properties. A more complete scheme can be drawn only considering the effect of the substituent on the molecular structure.
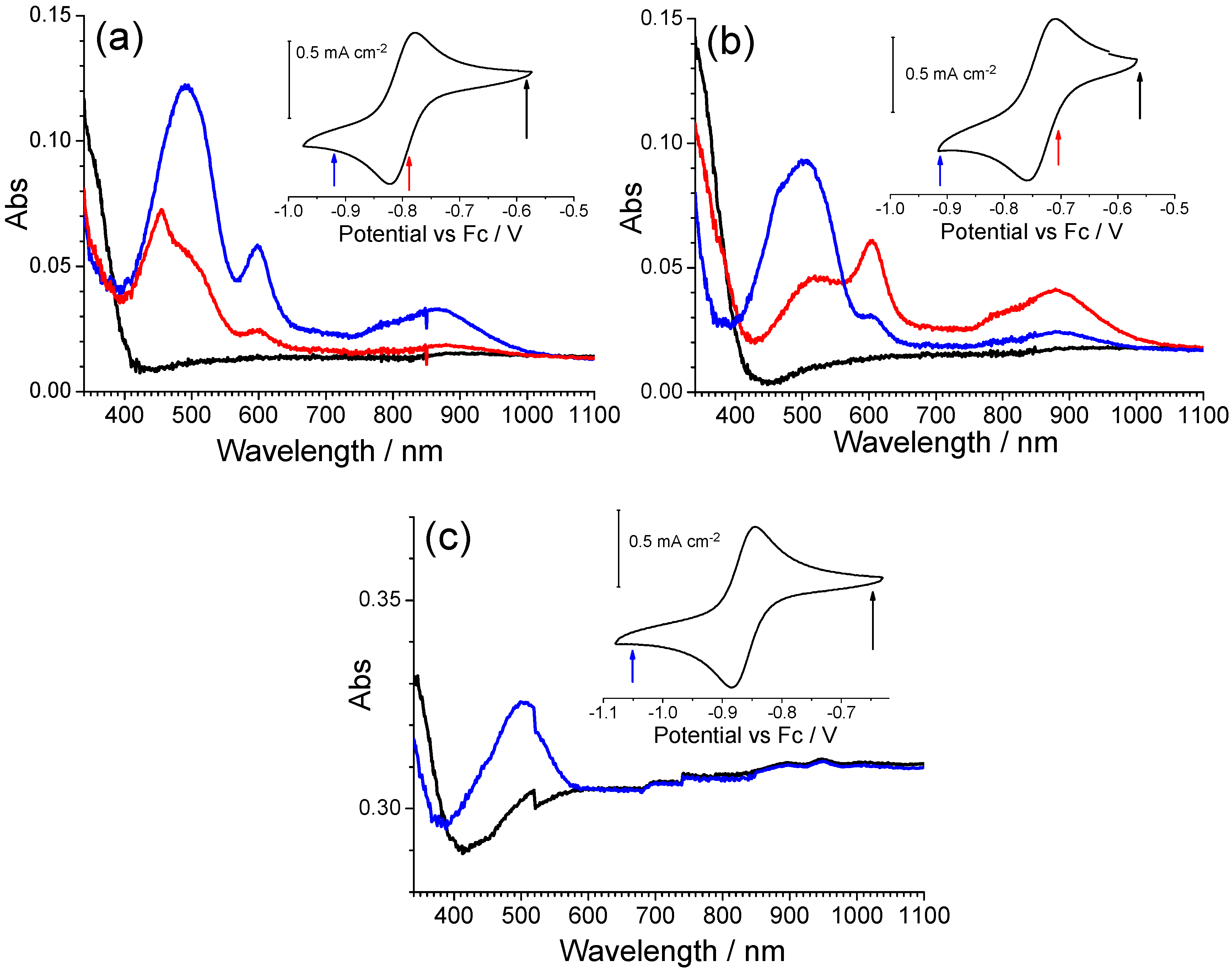
Derivatives of the iQ-Q compound were also characterized by spectroelectrochemistry (
Figure 7).
In iQ-ClQ (chlorine in the 7-position on the quinolinic end), the absorption bands of the reduced form are centered at 503 and 506 nm, respectively, thus being a little red shifted in comparison with the parent compound. An opposite behavior is observed for compounds FiQ-Q (490 nm) and iQ-twQ (467 nm). In this case, in fact, a blue shift of the band is observed. The effect is more intense in derivative twQ-iQ, demonstrating that the substituent on the 2-3-position of the quinoline ring can strongly alter the electrochemical and optical properties of the compound. A closer look at the spectroelectrochemistry of compounds FiQ-Q and iQ-ClQ reveals that the reduction process is associated with the formation of a third species, absorbing around 455–460 nm. The extra absorption is not bleached upon reoxidation, a behavior similar to the one previously observed for compound Q-Q. As we already discussed before, this phenomenon can be ascribed to the formation of radical-cation dimers with low electroactivity.
2.3. Ab Initio Calculations
Figure 2 shows that the DE
2+ and DE
0 species are characterized by a completely different electronic structure. Indeed, the parent dicationic species features two rigid and planar azinium endcapping units connected to the central ethenylic bridge by C-C carbon bonds. This arrangement is connected with a certain rotational freedom, thus leading to a severely distorted ground state. In contrast, the neutral reduced species features a quinoid electronic structure showing only one rotational degree of freedom in the bridge connecting the two heterocycles. In principle, such a structure allows for smaller deviations from planarity. Thus, the variation of geometrical structural factors at the oxidation state is expected to play a major role.
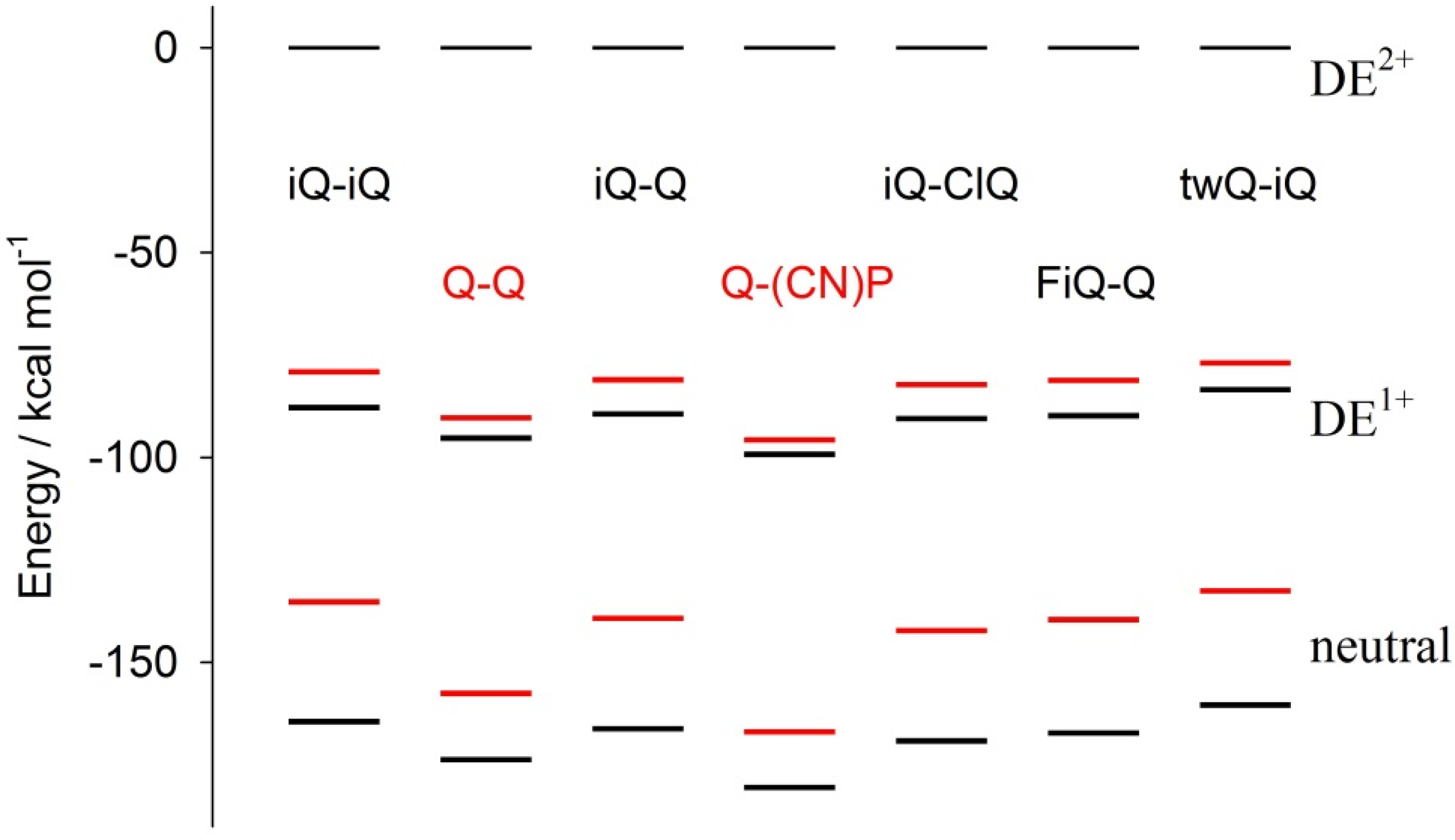
Figure 8 shows the molecular total electronic energy of all of the compounds as a function of the oxidation state and calculated following a full optimization procedure, which implies a relaxation of the molecular geometry (black bar) or the estimated maintaining of the geometry fixed (red bar) to the geometry of the original parent DE
2+ species (the energy of the DE
2+ parent species is set as the conventional zero energy). This investigation gives a direct insight into the incidence of redox-induced geometrical distortion as a function of the connectivity and substitution pattern of the specific investigated compound. Moreover, as the investigation was carried out as a function of the oxidation state, the study gives insight on the role of geometrical factors on the relative stabilization of monocation over dication.
Note that the energy differences between the vertical and relaxed structures of the DE
1+ states of the Q-Q and Q-(CN)P compounds, i.e. the distance between the red and black bars, is about 50% of those found for all of the other compounds; the same holds for the neutral species. Thus, the diazinium ethenes here investigated can be grouped into two families: (i) the Q-Q and Q-(CN)P compounds featuring a virtually planar geometry; and (ii) the remaining compounds, where the intramolecular reciprocal ring-to-ring disposition is significantly “distorted”, showing a ring-to-ring dihedral angle in the 60° to 90° range (
Table 2). It is noteworthy that the value of the aforementioned dihedral angle decreases with the oxidation state. In this respect, Q-Q and Q-(CN)P again show a peculiar behavior, indeed, the Q-Q compound’s calculated dihedral angles are 0°, 25.3°, and 0° for oxidation states of 2
+, 1
+, and 0, respectively, while Q-(CN)P features a flat geometrical disposition invariant with the oxidation state. Remarkably, the structure of the final product neutral species is the most planar of all of the compounds. On the whole, a substantial difference in the electrochemical results can be expected due to the interplay between the benzenoid/quinoid resonance stabilization, as well as to steric effects inducing large differences in the geometry relaxation energies of the parent compound.
From the theoretical point of view, the experimental electrochemical behaviour of the diazinium ethenes (DE) derivatives here investigated is assumed to be represented as two successive single-step reversible reduction processes [
18,
19].
The relevant elementary step is the first reduction process:
characterized by a redox potential E
2 (experimental values in
Table 1) and the corresponding electron affinity EA(I), and the second reduction process:
characterized by a redox potential E
1 (experimental value in
Table 1) and the corresponding electron affinity EA (II).
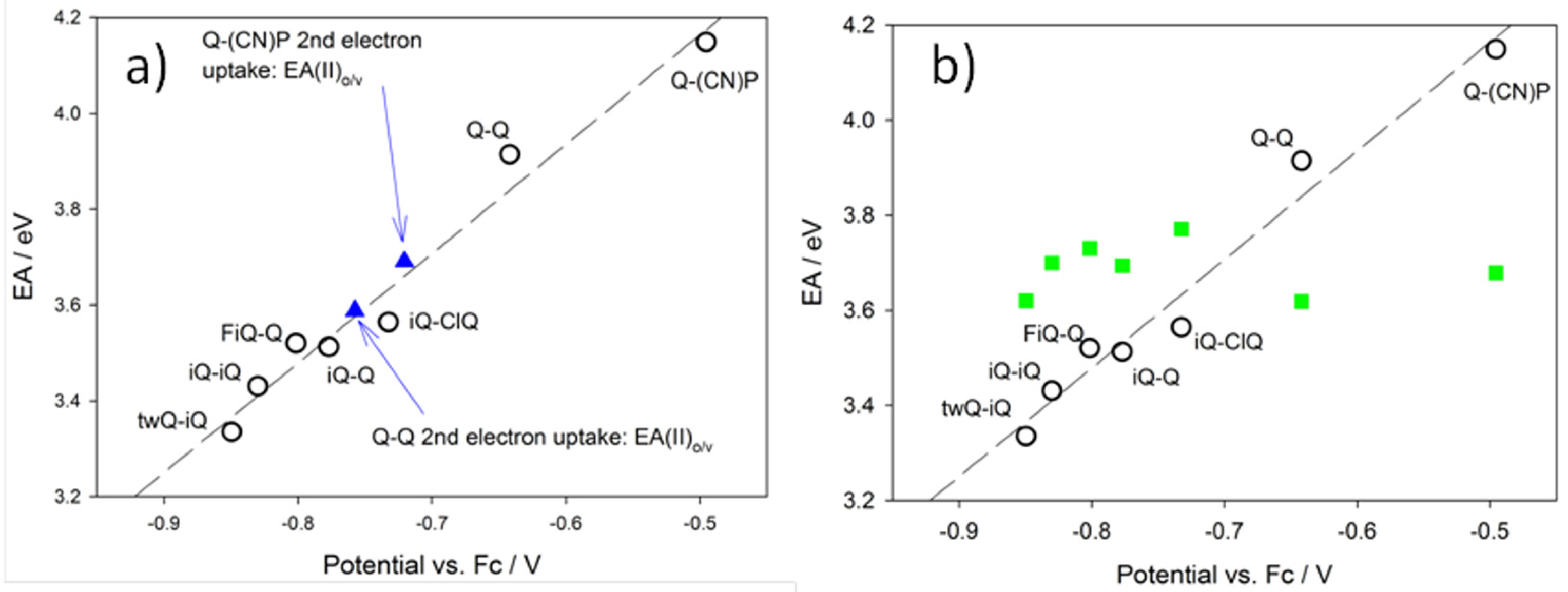
Figure 9 shows theoretical Electron Affinity (“EA”) values as a function of the experimental potential. Theoretical EAs were calculated using a number of different strategies, aiming at the rationalization of the most peculiar feature of the redox processes investigated: a single two-electron redox wave versus two distinct waves, each of one with a single-electron character [
18,
20].
Figure 9a reports the EA data calculated, allowing, or not allowing, for structural relaxation (geometry rearrangement as a function of the various oxidation states). The black circles in
Figure 9a are representative of the so-called “vertical electron affinity” vs. first process experimental reduction potential (compare E
2 in
Table 1). In this case, EA(I)
vert is calculated as:
where the subscript “opt” refers to the DE
2+ species calculated at the fully optimized geometry, whilst the subscript “vert” indicates that the energy, in this case of the DE
1+ species, is calculated at the same geometry of the parent DE
2+ species (i.e., with the assumption that the charge transfer process is much faster than the nuclear rearrangement time scale). Thus, the first electron uptake is considered as an instantaneous process, so fast that the relaxation process involving the DE
1+ species does not affect the reduction potential (this is the opposite of an adiabatic reduction process, where the geometry relaxation of the species involved in the redox process must be taken into account [
18]). Note that a correct physical slope is obtained in
Figure 9; that is, larger EA values imply less negative reduction potentials. The application of the same principle to the second reduction process (obviously, for the case of Q-Q and Q-(CN)P alone) leads to:
the values obtained for Q-(CN)P and Q-Q are 3.09 and 2.92 eV, respectively, which clearly do not fit the EA(I)
vert vs. E
2 correlation, shown as the black dashed line in
Figure 9 (in fact, the E
1 values for Q-(CN)P and Q-Q, compare
Table 1, are −0.70 and −0.74 V, respectively). Conversely, if we take into account the structural relaxation process coupled with the second charge transfer process, shown as the blue solid triangles in
Figure 9a, we obtain again a quite satisfactory correlation with all of the other data. In this case, the relevant equation is:
where: Energy (DE°)
0 is the energy relevant to the neutral relaxed (optimized geometry) species.
As a result, by a comparison of the experimental and theoretical results, we can infer that the first electron uptake can be considered as a vertical/non-adiabatic process where the species involved in the reduction process does not relax to the relevant cation (DE
1+)-relaxed state. Remarkably, a unique linear pattern is fitted by EA(I)
vert vs. E
2 for all of the compounds, regardless of the presence or not of a second distinct redox wave. The EA(II) values for Q-Q and Q-(CN)P fall on the same line, only if the relaxation of the reduced species to the equilibrium geometries are taken into account (thus, only if EA(II)
o/v instead of EA(II)
vert values are used).
Figure 9b shows the EA(II)
o/v values (green squares) plotted against the E
2 data for all compounds displaying only one two-electron redox wave. The EA(II)
o⁄v values are consistently larger than the EA(I)
vert ones, and are not linearly correlated with the E
2 data. This would imply that at the potential of the first reduction, also the uptake of the second electron occurs. Conversely, the EA(II)
o⁄v values of Q-Q and Q-(CN)P are too low to allow for the second simultaneous electron uptake to occur (compare the green squares in
Figure 9b). This is likely the rationale for the observation of two single electron steps for such compounds. Dynamical effects seem to play a major role, as already found in reduction processes involving complex systems [
21,
22]. Thus, the initial substantial differences (found in the parent DE
2+ species) in the reciprocal orientation of quinolinic rings of flat (Q-Q and Q-(CN)P) to orthogonal orientation (all remaining compounds save Q-Q and Q-(CN)P), play a major role in determining a qualitative difference in the electrochemical reduction process. Note also that the regular variation in the carbon−carbon bond distance going from the DE
2+ to the DE
0 species, (
Table SI_1). The C=C distance in the central moiety changes from 1.35 Å to 1.438 Å, and, on average, the opposite is found for the C-Benzene ring distance. This variation in the C-C distances accounts for a change from aromatic to quinoid character in the cyclic π-systems, going from the oxidized to the reduced neutral species (
Figure 2).

 ,
, 











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}