1. Introduction
In 2001, I surveyed aromaticity in transition metal oxide structures in a publication based on a presentation at a mathematical chemistry conference [
1]. At that time, some particularly interesting chemistry of vanadoborates was beginning to emerge. A key species is the ion [V
6B
20O
50H
8]
8− shown by X-ray crystallography to have a macrohexagon of d
1 vanadium(IV) ions as V–O–V units imbedded into an electronically inactive borate matrix [
2,
3]. In this V(IV) macrohexagon, the interaction between the single d electrons of each vanadium atom is mathematically similar to the interaction between the single π electrons provided by each CH vertex in benzene. However, because of the oxygen spacers between the vanadium atoms in [V
6B
20O
50H
8]
8−, these vanadium-vanadium interactions are too weak to completely overcome electron-electron repulsion. This prevents complete spin pairing in [V
6B
20O
50H
8]
8−, thereby leading to a paramagnetic species in contrast to the diamagnetic benzene. The implied the possibility of interactions between transition metals separated by oxygen bridges in polyoxometalate structures relates to the 1988 experimental observation by Baker and co-workers of ring currents in wholly inorganic heteropolyoxometalate blue structures using a modification of Evans’ susceptibility method [
4].
The field of vanadoborates has expanded significantly since the publication of my 2001 article in view of their potential applications as building blocks for the construction of mesoporous frameworks, including materials with unusual magnetic properties. Typically, hydrothermal reactions have been used for their synthesis. Of particular interest as a new high-spin aromatic system is the [V
10B
28O
74H
8]
16− anion, in which a V
10 macrodecagon is imbedded into a borate matrix [
5,
6,
7,
8], making it a high-spin analogue of the the hydrocarbon cyclodecapentaene, also known as [10]annulene [
9]. In this paper, I review the vanadium-vanadium interactions in the V
6 macrohexagon in [V
6B
20O
50H
8]
8− mentioned briefly in my earlier article [
1]. I then show how related concepts suggest an analogue of aromaticity in the V
10 unit of [V
10B
28O
74H
8]
16−, thereby making it a high-spin analogue of the experimentally known hydrocarbon [10]annulene (cyclodecapentaene).
2. Aromaticity in Polyoxometalates
The concept of aromaticity was initially applied to a specific class of organic substances approximately two centuries ago, when chemical bonding in general was very poorly understood [
10]. The initial aromatic substances, notably benzene, first obtained by pyrolysis of whale oil [
11], were initially characterized by their odors, which were considered to be more pleasant than other organic substances, considered as aliphatic species. Kekulé recognized that the common structural feature of these early aromatic compounds is a hexagonal ring of six carbon atoms [
12]. The C
6H
6 formula of benzene, combined with the tetravalence of carbon, suggested a cyclohexatriene structure with six π-electrons in the three C=C double bonds. Thus, one of the four valence electrons from the carbon atom of each CH unit in benzene remains as a π electron after using two of the carbon valence electrons for bonding to adjacent carbon atoms and a third electron for bonding to the external hydrogen atom.
A noteworthy feature of benzene is the unusually low chemical reactivity of its C=C double bonds as compared, for example, with the cyclic tetraolefin cyclooctatetraene. Hückel [
13] used molecular orbital theory to show that planar hydrocarbons with six π-electrons are delocalized systems exhibiting special stability, as indicated by a large HOMO-LUMO gap [
14,
15,
16,
17]. More generally, Hückel predicted special stability for planar cyclic hydrocarbons having 4
k + 2 π-electrons, where
k is an integer. The six π-electrons in benzene, as well as in the unusually stable ionic species tropylium (C
7H
7+) and cyclopentadienide (C
5H
5−), correspond to 4
k + 2 where
k = 1. The strength of the interaction between the π-orbitals on adjacent vertices linked by direct C–C bonds in such aromatic systems is characterized by a parameter
β.
During the past several decades the concept of aromaticity has extended far beyond the original examples of planar cyclic hydrocarbons and their derivatives. Deltahedral boranes [
18,
19] and carboranes [
20] of the type B
nH
n2−, CB
n–1H
n−, and C
2B
n−2H
n, and their substitution products provide examples of three-dimensional aromatic systems [
21]. A multicenter two-electron bond in the center of the deltahedra of such species corresponds to the π-bonding in planar cyclic hydrocarbons. Thus, the deltahedral boranes and carboranes fit into the 4
k + 2 delocalized electron scheme where
k = 0. The
β values for the interaction between adjacent vertices in aromatic deltahedral boranes, which are linked by direct B–B, B–C, or C–C bonds, are similar to those in planar cyclic hydrocarbons.
Metal-metal interactions involving metal d orbitals that are related to aromaticity can occur in certain groups of polyoxometalates [
22]. However, these interactions are much weaker since the metals involved are not directly bonded to each other in polyoxometalate structures. Thus, the metal polyhedra in polyoxometalates can be considered as macropolyhedra with relatively long edges consisting of M–O–M units rather than direct M–M bonds. This leads to considerably lower
β values as compared with planar cyclic hydrocarbons or deltahedral boranes. As a result of the low
β value, complete spin pairing of the delocalized electrons does not occur in polyoxometalates thereby leading to higher spin state analogues of aromatic systems. Thus the distinction between the aromaticity in diamagnetic planar cyclic hydrocarbons and deltahedral boranes and related delocalization in paramagnetic polyoxometalates is analogous to the distinction between low-spin coordination complexes with high ligand field splitting, such as Fe(CN)
63− with one unpaired electron, and high-spin coordination complexes with low ligand field splitting, such as FeF
63− with five unpaired electrons [
23].
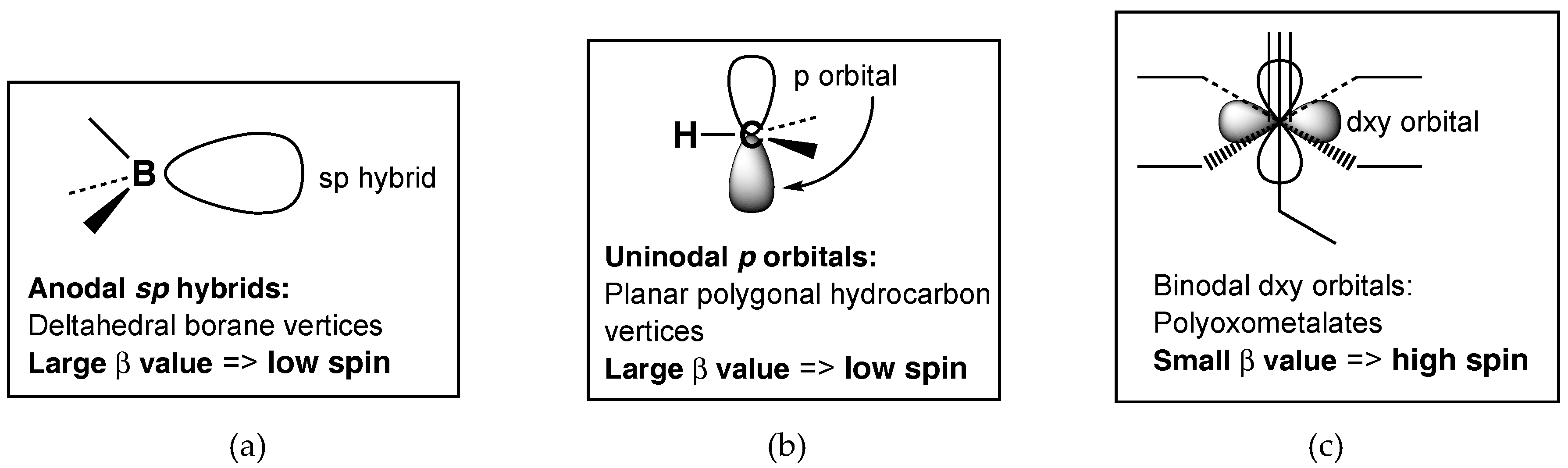
Systems displaying aromaticity and related delocalization can be classified by the nodality of the interacting orbitals. The two-dimensional aromaticity in planar polygonal hydrocarbons involves interaction between carbon
p orbitals with a single node, and thus can be considered as uninodal aromaticity (
Figure 1). Similarly, the three-dimensional aromaticity in deltahedral boranes involves interaction between boron and carbon
sp hybrid orbitals in the center of the deltahedron. Since these
sp hybrid orbitals have no nodes, such aromaticity can be considered as anodal aromaticity. In both uninodal and anodal aromaticity, the interacting orbitals are located on adjacent directly bonded atoms leading to relatively large values of the interaction parameter
β. This interaction parameter is larger than the electron repulsion energy, thereby leading to low-spin systems.
In the vanadoborates as in other polyoxometalates the interacting orbitals on each transition metal are
d orbitals that have two nodes (
Figure 1). However, the transition metals in such systems are not directly bonded to each other, but are separated by oxygen bridges. Therefore, the binodal interactions between transition metal atoms in polyoxometalates, including vanadium (IV) atoms in vanadoborates, are relatively weak interactions corresponding to low values of the interaction parameter
β. Such low values of
β are comparable to electron-electron repulsion energies, leading to high spin systems.
3. The High-Spin Vanadoborate Analogue of Benzene
Since the publication of my previous paper in 2001 [
1] additional examples of vanadoborates have been synthesized. The known vanadoborates have now been classified into the following four categories [
24]:
- (1)
V
6B
20 species containing a V
6 macrohexagon imbedded in a B
20 borate matrix, typically corresponding to a [V
6B
20O
50H
8]
8− anion building block [
2,
3,
25].
- (2)
V
10B
28 species containing a V
10 macrodecagon imbedded in a B
28 borate matrix, typically corresponding to a [V
10B
28O
74H
8]
16− building block [
5,
6].
- (3)
V
12B
18 species containing a planar V
12 unit imbedded in a B
18 borate matrix, typically corresponding to a [V
12B
18O
60]
6− building block [
6,
26].
- (4)
V
12B
16 species containing a three-dimensional V
12 polyhedron imbedded in a B
16 borate matrix, typically corresponding to a [V
12B
16O
50(OH)
8]
12− building block [
27].
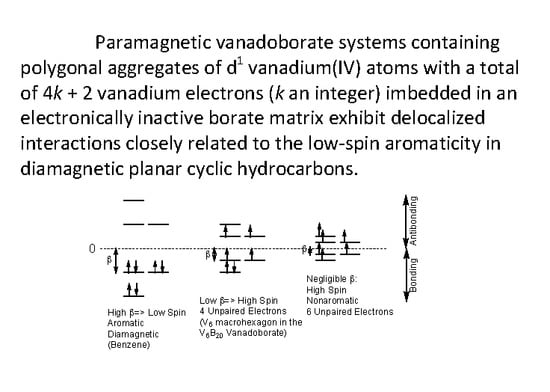
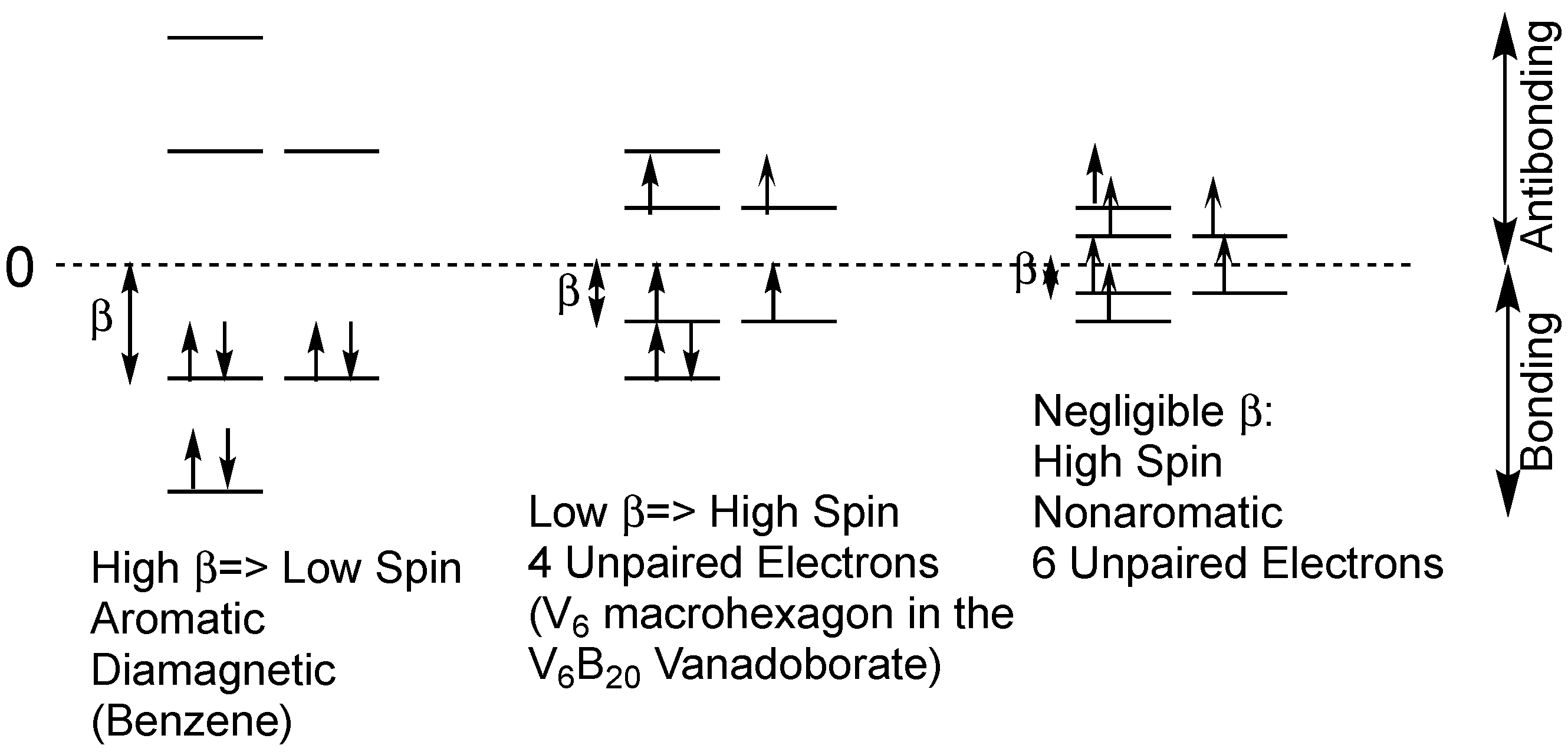
In the V6B20, V10B28, and V12B16 species all vanadium atoms are in the d1 +4 oxidation state and thus can provide an electron for delocalization throughout the vanadium cluster similar to the single electron provided CH vertices in benzene. However, in the V12B18 species, all of the vanadium atoms are in the d0 +5 oxidation state and thus do not have electrons available for delocalization. These +5 oxidation state vanadium atoms are analogous to the carbon atoms in the CH2 groups in a saturated cyclic hydrocarbon such as cyclohexane. Furthermore, the first two vanadoborate types, namely V6B20 and V10B28, can be regarded as high-spin analogues of planar cyclic hydrocarbons.
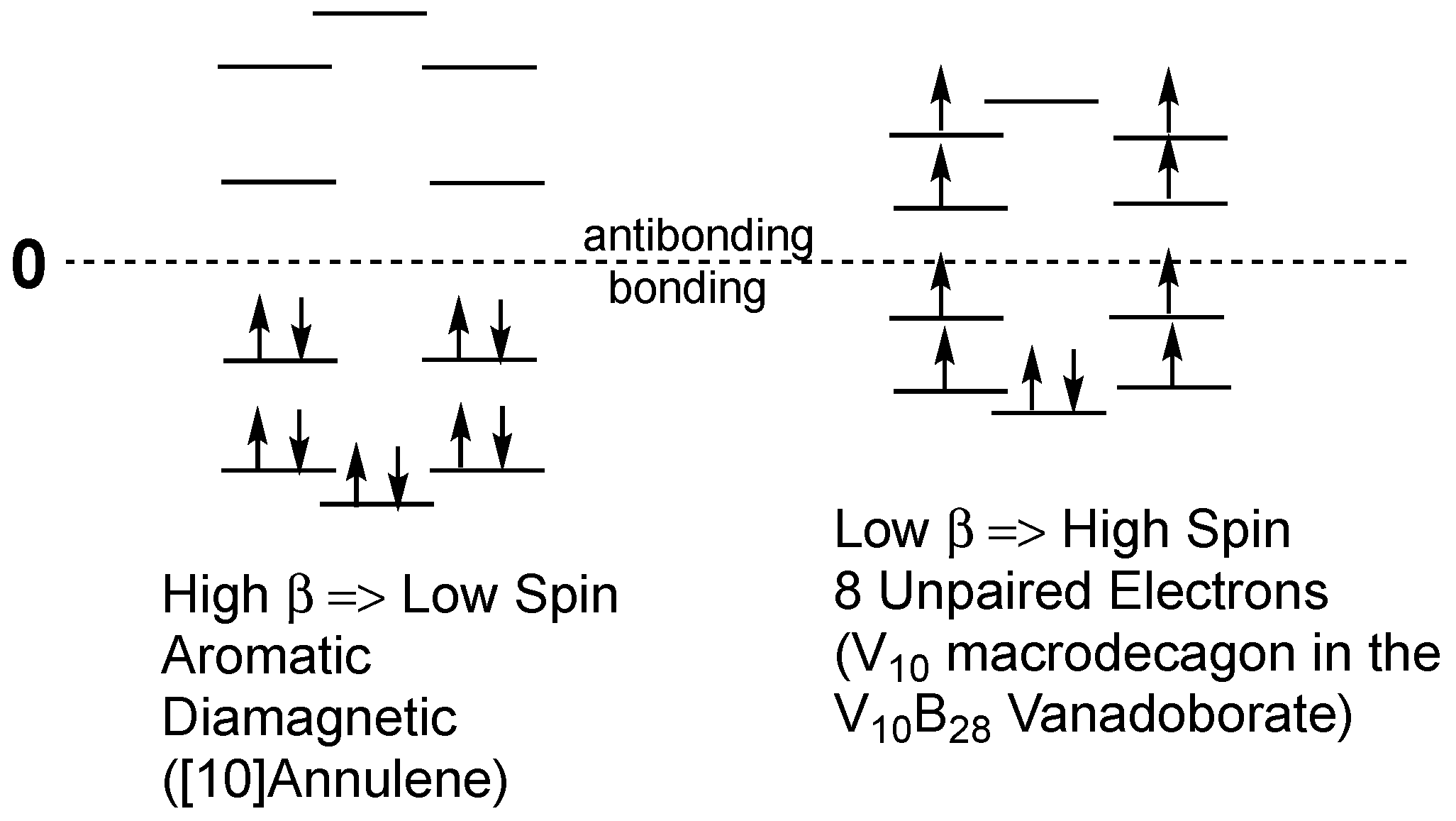
The relationship between the energy levels in benzene and those in the V
6B
20 vanadoborates is illustrated in
Figure 2. In benzene the
β value is large when compared with the spin pairing repulsion analogous to a strong ligand field in a low-spin coordination complex. All six π-electrons in benzene are paired leading to a diamagnetic species. However, in the V
6B
20 vanadoborates, the low
β value is comparable to the spin pairing repulsion so that only partial spin pairing occurs, leading to a paramagnetic species. The experimental magnetic moment of [V
6B
20O
50H
8]
8− of 4.1 µ
B is close to the spin-only value of 4.9 µ
B for four unpaired electrons. Note that for a low
β value that is small as compared with electron-electron repulsion energy can theoretically lead to a non-aromatic hexagonal system with six unpaired electrons (
Figure 2).
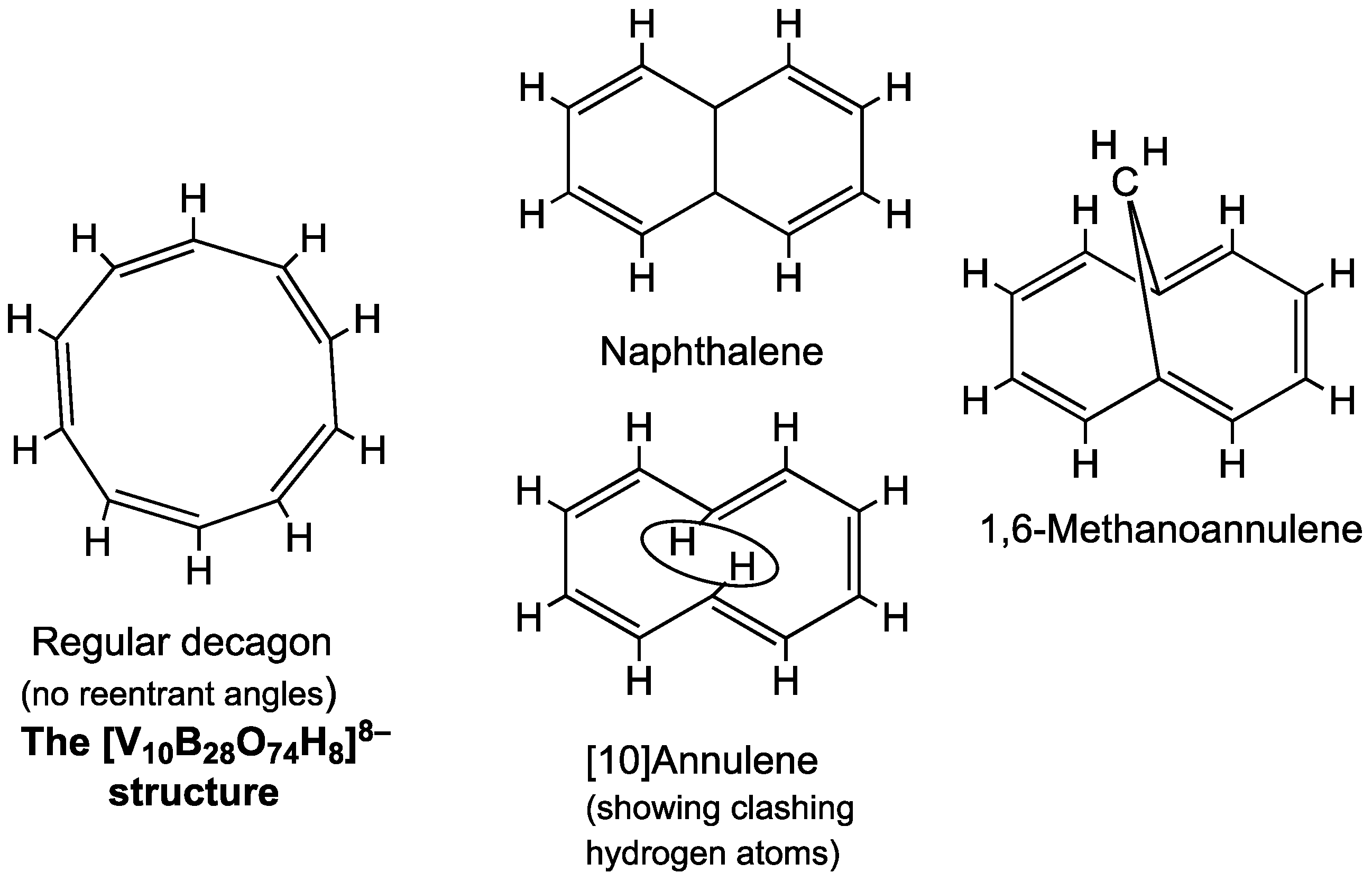
4. The High-Spin Vanadoborate Analogue of [10]Annulene
The next member of the 4
k + 2 π-electron series after benzene is cyclodecapentaene, also known more compactly as [10]annulene [
9]. [10]Annulene has 10 π-electrons corresponding to 4
k + 2 for
k = 2. However, a regular C
10H
10 decagon with alternating C=C double bonds in a Kekulé-like structure for [10]annulene has unfavorable C–C–C angles, leading to considerable strain (
Figure 3). Instead an arrangement of the ten carbons in the 10-membered C
10 ring that is similar to that in naphthalene (C
10H
8) with some reentrant C–C–C angles has less angular strain. However, even that arrangement has a problem with transannular steric interference between the hydrogen atoms located at the pair of carbon atoms at the center of the two reentrant angles of the C
10 ring. This difficulty can be overcome by replacing the offending hydrogen atoms with a transannular –CH
2– bridge leading to the stable hydrocarbon 1,6-methano[10]annulene [
9].
The V10B28 anion [V10B28O74H8]16−, found in species such as [Zn(OH2)(en)]4[Zn4(B2O4H2)(BO2H)2(V10B28O74H8)]·10H2O, is the vanadoborate analogue of [10]annulene. However, the central V10 system in this structure is essentially a regular decagon rather than a 10-membered naphthalene-like ring with reentrant angles similar to 1,6-methano[10]annulene. Thus the borate matrix combined with the flexibility of the V–O–V edges in the V10 ring keeps the V10 ring in a macrodecagon configuration without any reentrant angles.
The electronic properties of the V
10 ring in the [V
10B
28O
74H
8]
16– anion appear to be analogous to those of the V
6 ring in the [V
6B
20O
50H
8]
8− anion discussed above, but with an additional layer of doubly degenerate orbitals in the molecular orbital pattern of the delocalization in the V
10 system (
Figure 4). The experimental magnetic moment of 0.961 µ
B per vanadium atom corresponds to ~9.6 µ
B for the entire V
10 system. This suggests a low
β value corresponding approximately to eight unpaired electrons with an electron pair in only the lowest energy non-degenerate orbital. However, the decrease of the χ
MT value to nearly zero obtained upon cooling to 2 K suggests that this system becomes diamagnetic with complete electron pairing at low temperatures.
Comparison of
Figure 2 and
Figure 4 show a common feature of the patterns of the macropolygon molecular orbitals electron distributions in the V
6 macrohexagons in [V
6B
20O
50H
8]
8− and the V
10 macrodecagons in [V
10B
28O
74H
8]
16−. These patterns relate to the experimentally observed magnetic properties of these systems. Thus, for both V
6B
20 and V
10B
28, the non-degenerate lowest energy molecular orbital still retains an electron pair whereas all of the higher lying doubly degenerate orbitals are half-filled with single electrons.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



