Abstract
The structural, mechanical, electronic and optical properties of SrTMO3 (TM = Rh, Zr) compounds are investigated by using first principle calculations based on density functional theory (DFT). The exchange-correlation potential was treated with the generalized gradient approximation (GGA) for the structural properties. Moreover, the modified Becke-Johnson (mBJ) approximation was also employed for the electronic properties. The calculated lattice constants are in good agreement with the available experimental and theoretical results. The elastic constants and their derived moduli reveal that SrRhO3 is ductile and SrZrO3 is brittle in nature. The band structure and the density of states calculations with mBJ-GGA predict a metallic nature for SrRhO3 and an insulating behavior for SrZrO3. The optical properties reveal that both SrRhO3 and SrZrO3 are suitable as wave reflectance compounds in the whole spectrum for SrRhO3 and in the far ultraviolet region (FUV) for SrZrO3.
1. Introduction
Perovskite structure solids are of great interest in materials science due to their simple crystal structure and their different unique properties such as ferromagnetism, ferroelectricity, superconductivity, thermoelectricity and colossal magneto resistance [1]. Recently, many experimental and theoretical works have been devoted to perovskite oxides RE–TM–O3 (RE represents rare earth and TM represents transition metal elements) [2,3,4].
SrTMO3 (TM = Rh, Zr) compounds crystallize in the cubic perovskite structure with space group of space Pm-3m (# 221). SrTMO3 can be described as Sr2+ and O−2 ions forming a cubic close packed lattice with TM (Rh, Ti) ions occupying the octahedral holes created by the oxygen. The perovskite structure has a three-dimensional net of corner sharing [TMO6] octahedral with Sr2+ ions in the 12-fold cavities in between the polyhedral [5]. The Sr atom (alkaline earth or rare earth element) is located at (0, 0, 0), the TM atom (transition metal) at (1/2, 1/2, 1/2) and the oxygen atoms sit at face centered positions (1/2, 1/2, 0), (0, 1/2, 1/2) and (1/2, 0, 1/2). A large number of metallic elements are stable in the perovskite structure, if the tolerance factor t is in the range of 0.75–1.0 [6].
The elastic, electronic and optical properties of some perovskite compounds were examined by many researchers [7,8,9,10]. In 1992, Roosmalen et al. studied the structure of SrZrO3 [9]. In 2009, Baudali et al. [10] computed the structural, optical, electronic and thermal properties of SrTiO3 pervoskite cubic structure by using the full potential linearized augmented plane wave (FP-LAPW) method integrated in Wien2k code [11]. In 2011, Daga et al. [12] used the first principle study to calculate the lattice constant of the cubic SrMO3 (M = Ti, Zr, Mo, Rh, Ru). They found the lattice constants of SrRhO3 and SrZrO3 to be 3.932 Å and 4.076 Å, respectively.
In 2016 and 2017, Ali and Rahaman [13,14] used the pseudo potential method integrated in CASTEP code [15] to study the structural, elastic, electronic and optical properties of SrTMO3 (TM = Rh, Mo, Ti, Zr, V) compounds of cubic perovskite. They predicted that SrTiO3, SrZrO3 and SrVO3 have a brittle nature, while SrMoO3 and SrRhO3 have a ductile nature. Moreover, they concluded that SrRhO3 has the highest dielectric constant compared to the other compounds.
SrZrO3 compound (with 4d-electrons) is of interest because of its high temperature electronic properties applications such as hydrogen gas sensors and fuel cells. SrZrO3 with high perfection can be grown and used as laser-host materials. Shende et al. [16] suggested that these materials can be used in high-voltage capacitor applications because of their high breakdown strengths and high dielectric constant. SrRhO3 satisfies the conditions for having significant quantum critical fluctuations [17]. A non-Fermi-liquid behavior in transport properties should be observed in SrRhO3 in the range where quantum magnetic fluctuations are active [17]. Considering the heaviness of the band structure, these should be observed in transport [17]. Pseudo gap formation, metal-insulator transitions and high-voltage applications are the other significant properties which draw a considerable attention to 4d TMO. To the best of our knowledge, no ab initio investigations of the optical properties of these compounds have been reported and no experimental work on the electronic, elastic and optical properties have been studied too.
In the present work, we have studied the structural, electronic, elastic and optical properties of SrTMO3 (TM = Rh, Zr) by using FP-LAPW (all electron method) method. The motivation of this work is to improve the calculations and to provide some additional information to the features of SrZrO3 and SrRhO3 compounds using the all electron method (FP-LAPW).
2. Computational Details
The computation has been performed by using FP-LAPW method integrated in Wien2k code [11]. In this method, the unit cell is partitioned into two types of regions: (i) atomic-centered muffin-tin (MT) spheres with radius Rα; and (ii) the remaining interstitial region [18]. The expansion of spherical harmonics is defined within a muffin-tin sphere of radius RMT around each nucleus. The Muffin-Tin radius RMT values used are 2.5, 1.95, 1.67 atomic units (a.u) for Sr, Rh and O atoms for the SrRhO3 compound and 2.5, 1.8 and 1.5 a.u for Sr, Zr and O atoms for the SrZrO3 compound. The plane wave cut-off parameter RMT × Kmax is equal to 8. The cut-off energy to separate the core states from valence states is set to be −9 Ry. Inside the sphere, Fourier expanded up to Gmax = 14 with a cut-off lmax = 12 and 35 k-points in the irreducible Brillion zone with a grid of 10 × 10 × 10 Monkhorst-Pack meshes [19] (equivalent to 1000 k points in full Brillion zone) which are used to obtain self-consistency for SrRhO3 and SrZrO3 compounds to be better than 0.1 mRy. The Perdew-Burke-Ernzerhof–generalized gradient approximation (PBE-GGA) is used for the exchange correlation potential [20], while the modified Becke-Johnson potential (mBJ-GGA) [21] is also used to improve the energy band gap of the herein studied compounds.
3. Results and Discussions
3.1. Structural Properties
Strontium Transition metal oxides (SrTMO3) crystallize in the ABO3 cubic perovskite structure with the space group Pm-3m (221). The crystal structure of SrTMO3 (TM = Rh, Zr) is shown in Figure 1. We have optimized the lattice parameters (a) of these compounds by minimizing the total energy. The total energy at different unit cell volumes for SrTMO3 (TM = Rh, Zr) is shown in Figure 2, the volume versus energy is fitted to the Murnaghan equation of state [22] to estimate the ground-state properties of these compounds such as the lattice constant (a), the bulk modulus (B) and the pressure derivatives of the bulk modulus (B’). These obtained values are listed in Table 1 together with the available experimental and theoretical data for comparison. In general, the computed structural parameters are in good agreement with experimental and theoretical data available in the literature. More precisely, the lattice parameters of SrRhO3 and SrZrO3 are found to be 3.976 Å and 4.176 Å, respectively. The present calculations show that the lattice parameter of SrRhO3 (SrZrO3) is 1.43% (1.63%) larger than the experimental value [23,24]. This guarantees the reliability of the present first-principles computations for the lattice parameters of these compounds using the PBE-GGA method. The calculated bulk modulus for SrZrO3 is 2.11% larger than the experimental value [25].
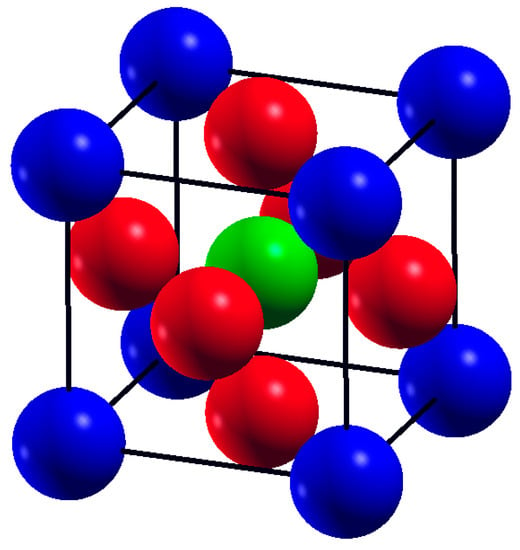
Figure 1.
The crystal structure of SrTMO3 (TM = Rh, Zr), (blue spheres are Sr atoms, red spheres are Oxygen O atoms and green spheres are TM atoms).
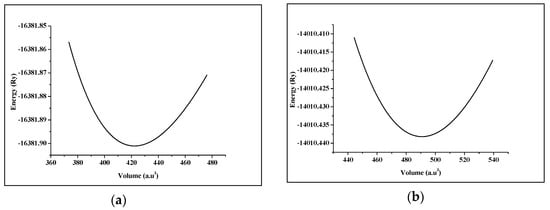
Figure 2.
Total energy as a function of the volume of (a) SrRhO3 compound and (b) SrZrO3 compound.

Table 1.
Calculated lattice parameter (a), bulk modulus (B), first pressure derivative (B’) of SrTMO3 (TM-Rh, Zr).
3.2. Electronic Properties
In this section, we study the electronic properties of SrRhO3 and SrZrO3 via calculating the energy band structure and density of states. The calculated band structures along the high symmetry lines in Brillion-zone of SrRhO3 and SrZrO3 at zero pressure using PBE-GGA approximation [20] are depicted in Figure 3, where the Fermi level is set at zero eV. In SrZrO3, the valence band maximum (VBM) occurs along the M-point symmetry line, while the conduction band minimum occurs along the Γ-point symmetry line with energy band gap 3.69 eV, resulting in an indirect energy band gap (M-Γ) semiconductor.
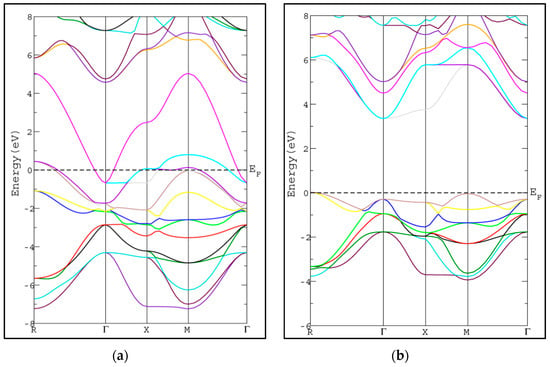
Figure 3.
Band structure of cubic pervoskite using PBE-GGA method for (a) SrRhO3 and (b) SrZrO3 compounds.
The calculated energy band gap is tabulated in Table 2 along with the available previous theoretical value [26]. The calculated energy gap is larger than the theoretical value by 0.32 eV [26]. To the best of our knowledge, there is no experimental value available to compare with. The usual trend of the modified Becke-Johnson potential (mBJ-GGA) method is enlarging the energy band gap values (Eg) of the semiconductor and insulator materials which makes Eg comparable to the experimental results [21]. The band structures of SrRhO3 and SrZrO3 compounds using mBJ-GGA are displayed in Figure 4. For the SrZrO3 compound, the minimum energy gap within mBJ-GGA is still indirect with the same direction as the PBE-GGA approach, but its value increases by about 0.85 eV to become 4.54 eV. SrZrO3 is classified as an insulator within the mBJ-GGA method. SrRhO3 has a metallic nature with no energy gap within the two approaches PBE-GGA and mBJ-GGA.
Table 2.
Calculated energy band gap Eg (eV) of SrZrO3 compound.
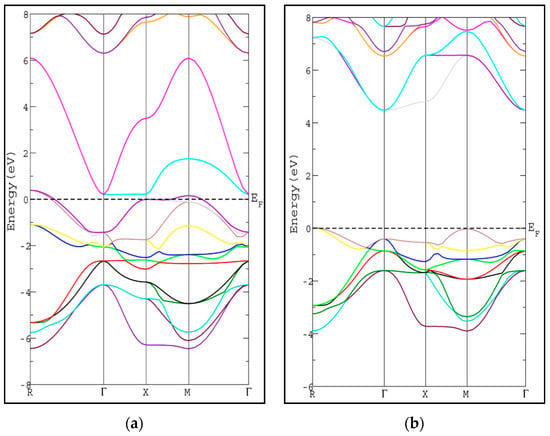
Figure 4.
Band structure of cubic pervoskite (a) SrRhO3 and (b) SrZrO3 by using (mBJ-GGA) method.
The total and partial density of states for SrRhO3 and SrZrO3 are shown in Figure 5 and Figure 6 for an energy range from −15 to 14 eV and −12.5 to 14 eV, respectively. In SrRhO3, the valence band originates from O-p and Rh-d states. The maximum contribution of the Rh-d state is near the Fermi level. In the conduction band, the bands are due to the Sr-d with a small contribution from Rh-d, O-p and O-s states. In SrZrO3, the valence band originates from O-p with few contribution from Zr-d,p and Sr-d states. In the conduction band, the bands are due to Zr-d and Sr-d states with a small contribution from O-p states.
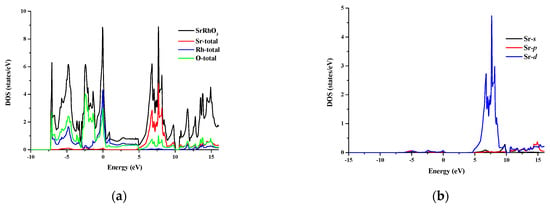
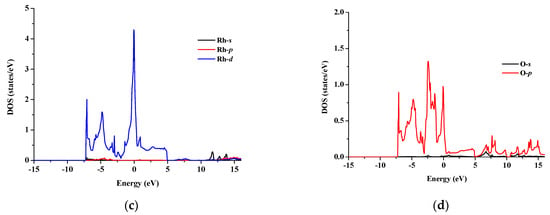
Figure 5.
(a) Total density of states of SrRhO3 compound and partial density of states for (b) Sr atom, (c) Rh atom and (d) O atom in SrRhO3 compound.
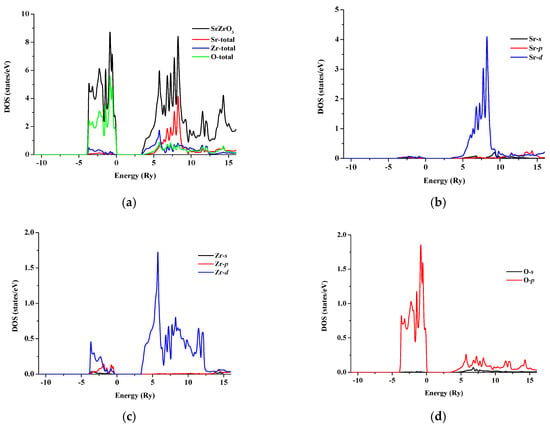
Figure 6.
(a) Total density of states of SrZrO3; Partial density of state for (b) Sr atom, (c) Zr atom and (d) O atom in SrZrO3 compound.
To make a deeper analysis of the bonding nature of our titled compounds, the wave function obtained from a final Wien2k calculation at the optimal geometry has been analyzed in the critic program (version 1.0). Critic is a full-edged program for the topological analysis of solid-state electron densities.
This program uses the quantum theory of atom in molecule (QTAIM) to make a topological analysis of the electronic density (ρ) of a crystal. By means of QTAIM, we can automatically locate all critical points (CP’s) of the electronic density rising from a nil flux of the electron density gradient condition [27]. The procedure implemented in critic is to divide ρ into disjoint regions Ω (basins) through the real space approaches. Here, we can generate topological schemes in two or tridimensional plots. It is clearly shown from the topological distribution of the charge density in Figure 7A, that the charge distribution in the SrZrO3 compound is spherically symmetrical. This suggests that all electronic charges are localized around anions—a typical ionic bonding. The same trend is also displayed for the second compound with some small difference in the distribution of ρ due to the charge transfer, which differs in the two titled compounds (see Figure 7C). We also present in Figure 7B,D, the molecular graph of our two perovskites.
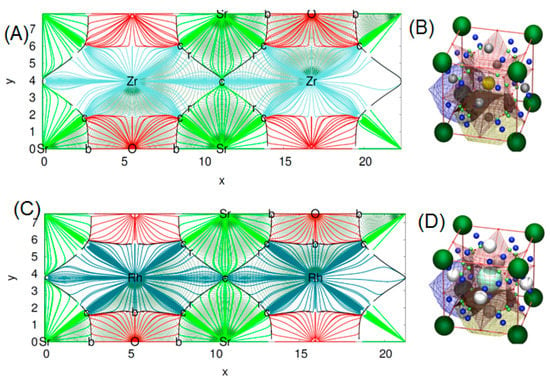
Figure 7.
2D map of topological analysis of electron density of (A) SrZrO3 and (C) SrRhO3, molecular graph representation of (B) SrZrO3 and (D) SrRhO3. In colored trajectories traced out by the electron density gradient, vector field (in electron/bohr3) of upper panel: SrZrO3 and lower panel: SrRhO3 are at their denser plane. Here, the red, dark blue, light blue and green lines refer respectively to O, Rh, Zr and Sr atomic basins. The set of trajectories that terminates at each bond’s critical point (b) defines an interatomic surface. The set of trajectories that originate at the ring critical (r) point defines the perimeter of the interatomic surface. The gradient paths associated with the negative eigenvalues at the (n) point terminate at this CP and define the zero-flux surfaces that partition the crystal into unique fragments (the atomic basins). On the right of the figures, molecular graph representation: the small red balls placed at the Wyckoff’s 3c are the r CPs. The light green balls the b CPs and the dark blue the c cage CPs.
These plots were done following the character of Wyckoff’s, bond CP and ring CP family [28]. The form of the atomic basin generated by the CCP point suggests that both SrRhO3 and SrZrO3 are belonging to the R11 family (see Figure 7B,D) [28]. Here, we can define a topological charge of each atomic basin. As results, Q(Sr) = 1.59 electron, Q(Rh) = 1.47 electron and Q(O) = −1.05 electron for SrRhO3, and Q(Sr) = 1.61 electron, Q(Zr) = 2.49 electron and Q(O) = −1.37 electron for the SrZrO3 compound. When we compare these charges with the oxidation number corresponding to each of the atoms, we find that the charge transfer is mainly due to the strontium one, with an equal percentage of 80% in both perovskites. However, we note that replacing the cation Rh with electro-negativity of 2.2 by the Zr one with low electro-negativity of 1.4 has a negligible effect on the nature of the bonding. The SrZrO3 has a more ionic trend. However, even the constituent cations show different charge transfer (Zr and Rh are transferred by 62% and 37%) and each of them varies as strongly as the oxide ions. The latter adapt their charges according to electro-neutrality requirements. There is a second topological index that has been proposed as a global measurement of the degree of metallicity, the electron density flatness defined as where is the minimal electron density found on the unit cell (it necessarily corresponds to a critical point) and is the maximal electron density among bond critical points. This flatness has values close to one on common metallic compounds and close to zero on localized bonding compounds [29]. The delocalization of electrons (flatness) in the SrRhO3 and SrZrO3 compounds are respectively equal to 80.61% and 5.62%. This suggests that the global chemical behavior of SrRhO3 is metallic, whereas SrZrO3 is nonmetallic. To go further in the characterization of the bonds nature of the titled compounds we have employed another tool named the electron localization function (ELF) [30], which is based on a measure of the likelihood of finding an electron in the neighborhood space of a reference electron located at a given point and with the same spin. Taking into account that the homogeneous electron gas has an ELF value of 0.5, valence electrons of metals should deviate very slightly from this quantity. The ELF isosurfaces, as well as the one-dimensional projections of Sr–O–Rh and Sr–O–Zr bond paths of the investigated perovskites are depicted in Figure 8. It is strikingly clear from the plot that O–Zr and O–Rh are different. The integration of population on the ELF attractor of the SrZrO3 compound shows two types of Zr–O bonds, the first one has ELF maxima equal to 0.78 and the second to 0.83; the former has the negligible population, but the latter has 0.95 electrons. The integration gives a number of the delocalized attractor as lone pair bonds around the Sr and Zr cations; the population in this attractor varies from 1.4 to 1.1 electrons. Regarding the second compound (SrRhO3), no O–Rh bonds have been found, only delocalized lone pair ones with ELF maxima near to 0.5 are reported. Here, we should emphasize that the delocalization of these attractors in the SrRhO3 compound provides a reliable measure of the delocalization of wave function and its localization in the SrZrO3 compound.
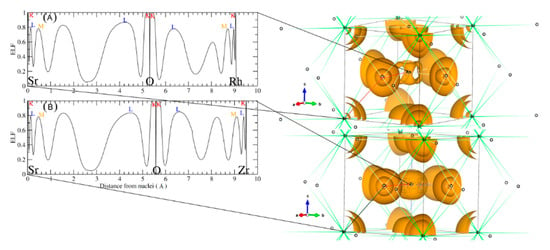
Figure 8.
Left panel shows a one dimensional electron localization profile and the right panel shows the 3D isosurface of ELF basins of (A) SrRhO3 and (B) SrZrO3 compounds.
3.3. Elastic Properties
In this subsection, we turn our attention to study the mechanical properties of SrRhO3 and SrZrO3 via calculating their elastic constants. These constants define the properties of material that undergo stress, deform and then recover, returning to its original shape after stress ceases. They have a significant role in finding information about the brittleness, ductility, stiffness and the mechanical stability of the material [31]. The elastic constants require knowledge of the derivative of the energy as a function of the lattice strain. In the case of the cubic system, this strain is chosen in such a way that the volume of the unit cell is preserved. Thus, for the calculation of elastic constants C11, C12 and C44, for these compounds we have used the method developed by Morteza Jamal [32] and integrated in Wien2k code as the IRelast package (Cubic-elastic_13.2). The calculated Cij constants are listed in Table 3. Our computed Cij data are in reasonable agreement with previous theoretical results. In view of Table 3, it can be noticed that the calculated values of the elastic modulus C11, which are related to the unidirectional compression along the principal crystallographic directions, are much higher than that of C44, which represent the resistance to the shear deformation, indicating the weak resistance to the shear deformation compared to the resistance to the unidirectional compression. The mechanical stability of a cubic material requires that its independent elastic constants should satisfy the following Born’s stability criteria [33,34]:
C11 > 0; C44 > 0; C11 + 2C12 > 0; C11 > B > C12
Table 3.
Calculated elastic constants of SrTMO3 (TM = Rh, Zr).
From Table 3, we can see that all required conditions given in the above Equation (1) are simultaneously satisfied, which clearly indicates that the SrRhO3 and SrZrO3 are mechanically stable.
The three elastic constants C11, C12 and C44 are estimated from first-principles calculations for SrRhO3 and SrZrO3 single-crystals. However, the prepared materials are in general polycrystalline, and therefore it is important to evaluate the corresponding moduli for the polycrystalline species using the Hill’s approach [35]. In this approach, the effective modulus for polycrystals could be approximated by the arithmetic mean of the two well-known bounds for monocrystals according to Voigt [36] and Reuss [37]. Then, for the cubic system, the shear modulus S in the mentioned approximations: Voigt (V), Reuss (R) and Hill (H) are calculated from the elastic constants of the single crystal, in the following form:
To compute the Young’s modulus (Y), Poisson’s ratio (ν), and the anisotropic factor (A), the following equations are used, respectively:
The computed bulk modulus, shear modulus, Young’s modulus, Poisson’s ratio and anisotropic factor are listed in Table 3 along with the theoretical results [14,38]. The bulk modulus and shear modulus can be used to measure the material hardness [39]. In general, when values increase, the materials become stiffer. From the obtained values of shear modulus one can remark that SrZrO3 is stiffer than SrRhO3.
To categorize the brittle and ductility behaviors of a material, the ratio of the bulk modulus to the shear modulus, B/S, an empirical relationship related to the plastic and elastic properties of the material, is used. According to Pugh [40], a high B/S value is associated with ductility, while a low value is consistent with brittleness. The critical value that separates the two behaviors has been determined to be 1.75. The results listed in Table 3 clearly indicate that the SrRhO3 (SrZrO3) compound has B/SH ratio higher (smaller) than the critical value of 1.75, which classifies SrRhO3 (SrZrO3) compound as ductile (brittle) material. In addition, to identify the materials as ductile or brittle, we also applied the Cauchy’s pressure rule defined as the difference between the elastic constants C12–C44 [41]. According to this rule, if the Cauchy’s pressure is positive (negative), the material will be ductile (brittle) in nature. As shown in Table 3, It is seen that the Cauchy’s pressure is positive for SrRhO3 and negative for SrZrO3, confirming the ductile nature for SrRhO3 and the brittle behavior for the SrZrO3 compound. We may also refer to Frantsevich et al. [42] who distinguishes the ductility and brittleness of materials in terms of Poisson’s ratio (ν). According to this rule, if the Poisson’s ratio is less than 0.26, the material will be brittle in nature; otherwise, the material will be ductile. As shown, the computed Poisson’s ratiosare 0.30 for SrRhO3 and 0.23 for SrZrO3, categorizing SrZrO3 as brittle compounds and SrRhO3 as ductile compounds. These results exactly agree with the results of the B/S ratio and Cauchy’s pressure.
The anisotropy factor is an important parameter to measure the degree of materials anisotropy; also, it has a significant usage in engineering science to inspect the potential of micro-cracks in the material [43,44]. For completely isotropic materials, the anisotropy factor A takes the value of the unity and the deviation from unity measures the degree of elastic anisotropy. The calculated values of the anisotropic factor A are found to be equal to 1.85 for SrRhO3 and 0.71 for SrZrO3, suggesting that both compounds are anisotropic in nature and SrRhO3 is characterized by a profound anisotropy.
3.4. Optical Properties
Since the investigated compounds have cubic symmetry, we need to calculate only one dielectric tensor component to completely characterize their linear optical properties. The frequency-dependent complex dielectric function ε(ω) = ε1(ω) + iε2(ω); where ε1(ω) and ε2(ω) are the real and imaginary components of the dielectric function, respectively; is known to describe the optical response of the medium at all photon energies , using the formalism of Ehrenreich and Cohen [45].
Complex dielectric function can be derived from the definition given by Hedin [46]
where P is the polarization propagator and is the Coulomb interaction; P can be given by the following form:
where V is the unit cell volume, f0 is the Fermi distribution function and εk is the single particle energy. The matrix element can be given by: Mn,n’(k,q) = ⟨unk|e−iq,r|un’k⟩, q is the wave vector of light and it is much smaller than the wave vector of electrons in the system; the matrix elements Mn,n’(k,q) with small q can be given by:
The sum over n’ and n must be split into two terms, one with n’ = n corresponding to intra-band electronic transitions, and the second with n’ ≠ n, corresponding to inter-band transitions, the intra-band part of the dielectric function can be given by:
while the inter band part can be written as
where n’ ≠ n in Equation (10).
The imaginary part of the ε(ω) in the long wavelength limit has been obtained directly from the electronic structure calculation, using the joint density of states (JDOS) and the transition moments elements Mn,n’(k,q):
The integral is over the first Brillouin zone. The real part of ε(ω) can be derived from the imaginary part using the Kramers–Kronig relations.
where P implies the principal value of the integral. The knowledge of the real and imaginary parts of the dielectric function allows the calculation of other important optical functions such as the refractive index n(ω), reflectivity R(), extinction coefficient k(ω), energy loss function L(ω) and absorption coefficient α(ω) by using the following expressions [47,48,49]:
Real and imaginary parts of the dielectric constant are displayed in Figure 9a–d for the SrRhO3 compound. The value of the static real part of the dielectric function ε1(0) for the SrRhO3 compound within intra and inter band transition in Figure 9a is negative, while the imaginary part of the static dielectric function ε2(0) within intra and inter band transition Figure 9c is positive; this implies two important facts: Firstly, the SrRhO3 compound has considerable metallic behavior, which agrees with the energy band structure calculations. Secondly, the negative value of (ε1) especially in the energy range 0–1.15 eV in Figure 9a and the highly positive value of ε2(ω) at the early beginning of Figure 9c, reveal the loss of light transit. Real dielectric constant within intra band transition for SrRhO3 in Figure 9b has small peaks; the first two of them are centered at 1.42 eV and 3.73 eV. We see that (ε1) for the SrRhO3 compound in Figure 9a, it has roots in the 1.15 eV, 1.55 eV, 5.0 eV, 8.45 eV and 13.5 eV. When these roots occur, (ε1) = 0, the compound does not respond to incident light, this fact is mainly due to the Plasmon oscillation. The sharp increase in ε1(ω) with intra and inter band transition, in Figure 9a, at an energy range 0–1.15 eV, indicates that the compound does not interact with the incident photons at this energy range. The static dielectric constant ε1(0) of SrRhO3 with inter + intra in Figure 9a and intra band transition Figure 9b are −180 and 32, respectively. Negative value ε1(0) of with inter + intra band transition ensures the metallic behavior of SrRhO3 compound. By taking only the intra band transition into account as seen in Figure 9b,d, both the real and imaginary parts of dielectric constant have positive values, which indicates a mix of metallic and semiconducting behavior for the SrRhO3 compound. This implies that for metallic compounds inter and intra band transitions must be taken into account.
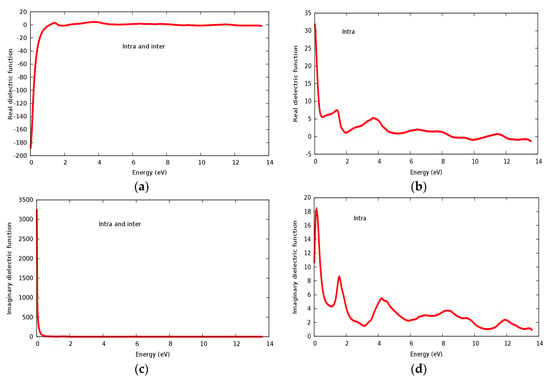
Figure 9.
The dielectric function of SrRhO3 compound: (a,b) Real part; (c,d) Imaginary part.
Optical conductivity is a quantity depending on the inter band and intra band transitions. In Figure 10a,b, the real and imaginary parts of conductivity are illustrated for the SrRhO3 compound, by taking the inter and intra band transition into account; the static real conductivity is high, while it is zero when only intra band transition is taken into account. As the incident light energy increases, the real and imaginary parts of conductivity with the two approaches; intra + inter and intra band transition; both almost have the same behavior.
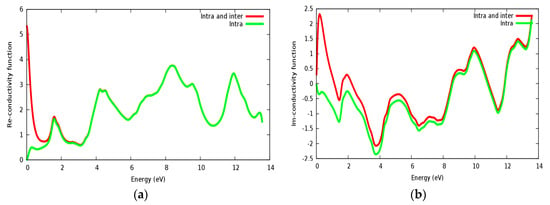
Figure 10.
The conductivity function of SrRhO3 compound: (a) Real part; (b) Imaginary part.
The refractive index n(ω) and extinct factor k(ω) for the SrRhO3 compound are illustrated in Figure 11a,b and Figure 12a,b, respectively. The SrRhO3 has a high n(0) and k(0) with intra and inter band transition, which indicates metallic behavior for the real and imaginary parts of the dielectric. As the incident light energy increases the n(ω); and k(ω) goes to a lower values. Many peaks are shown in n(ω) and k(ω) spectra, these peaks originate from intra-band transition. The extinction coefficient depends on the amount of absorption of the photon when it propagates in the material, while the refractive index indicates the phase velocity of the electromagnetic wave.
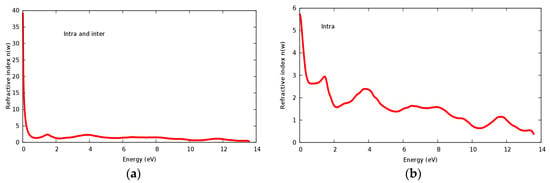
Figure 11.
(a) Refractive index n(ω) within inter and intra; (b) Refractive index n(ω) within intra for SrRhO3 compound.
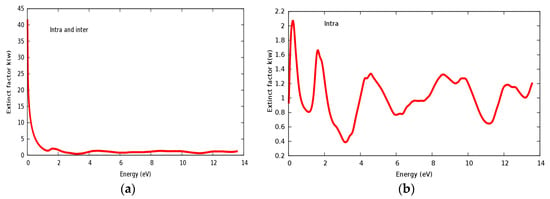
Figure 12.
(a) Extinct factor k(ω) within inter and intra; (b) Extinct factor k(ω) within intra for SrRhO3 compound.
The reflectivity spectra of SrRhO3 as a function of energy are shown in Figure 13a. The static reflectivity of SrRhO3 within intra and inter transition is 0.95, while it is 0.5 within intra transition. The reflectivity of SrRhO3 in the low energy region goes down as the incident light energy increases, while it increases in the high energy region—the far ultraviolet region (FUV). The absorption coefficient spectra of SrRhO3 is plotted in Figure 13b, absorption spectra, as shown, begins at the early beginning and increases as the incident photon energy increases with some peaks along the spectrum. The observed peaks in the spectra related to electron transitions from conduction to valence bands, sharp peaks in the absorption spectrum may be accordance with transitions between valance and conduction band (inter band transitions) that can be considerably far from each other. The SrRhO3 compound is a good absorbent compound because it is a metal compound and it is identical with intra + inter and intra band transition. Peaks in the spectrum of the absorption coefficient are proportional to the peaks in the (ε2) spectrum. The energy loss function L(ω) is describing the energy loss of the fast electrons that propagate inside the material. The energy loss spectrum of SrRhO3 is depicted in Figure 13c. Energy loss for SrRhO3 is high along the whole spectrum and it is identical with intra + inter and intra band transition in the high energy region. We observe some peaks, the highest peaks are related to the plasma frequency [50]. From these Figures, the plasma frequency of SrRhO3 occurs at 10.9 eV and 13.2 eV.
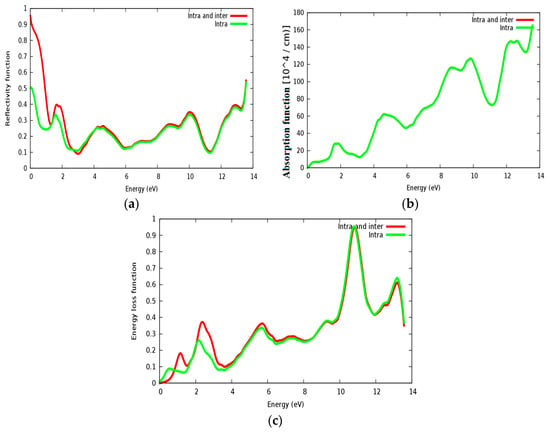
Figure 13.
(a) Reflectivity function R(ω); (b) absorption function; and (c) energy loss function L(ω) for SrRhO3 compound.
Figure 14a,b displays the calculated real and imaginary parts of the dielectric function for the SrZrO3 compound for a radiation up to 14 eV. It is seen that the calculated linear optical components ε1(ω) and ε2(ω) spectra for SrZrO3 are different from SrRhO3. The calculated ε2(ω) spectra show that the first critical point (threshold energy) of the dielectric function occurs at about 4.52 eV within the mBJ approach (GGA: 3.6 eV) for SrZrO3; the first critical points are comparable with the energy band gap computed from the energy band structure. We can see from Figure 14b, that ε2(ω) displaced to the high energy region within the mBJ approach, but the first peak in ε2(ω) within the GGA approach has a higher value. The threshold’s energy is followed by some peaks that originate from direct optical transition between the valence band and conduction bands. The main peak in the absorptive spectra is positioned at about 7.2 eV within mBJ (GGA: 6.2 eV). The real part ε1(ω) gives us information about the material’s polarizability; the static dielectric constant ε1(0) of SrZrO3 is 3 within the mBJ approach (GGA: 3.92), as we can see that the mBJ approach has a lower value of ε1(0). The value of ε1(ω) within mBJ displaced to the higher energy region along the spectrum. The behavior with the high dielectric constant makes SrZrO3 a possible useful candidate for manufacturing high capacitors [51]. The negative value in the spectra of ε1(ω) for the SrZrO3 compound is located only in the ultraviolet spectrum; and shows the metallic behavior of this compound in the mentioned region.
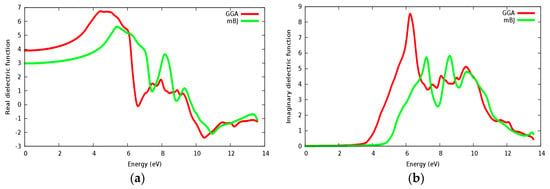
Figure 14.
Dielectric function for SrZrO3: (a) Real part; (b) Imaginary part using both PBE-GGA and mBJ-GGA methods.
In Figure 15a,b, the real and imaginary parts of conductivity are illustrated for SrZrO3, respectively. It can be seen from Figure 15a, that the real part of conductivity starts to have considerable values at about 4.75 eV within the mBJ approach (GGA: 3.6 eV). From the conductivity and imaginary part of the dielectric function spectrums (Figure 14b), it is shown that both of them start to have a considerable value from approximately the same value. We have also observed some peaks in the conductivity spectra as also observed in the ε2(ω) spectra. The conductivity increases as the material becomes more photon (energy) absorbent.
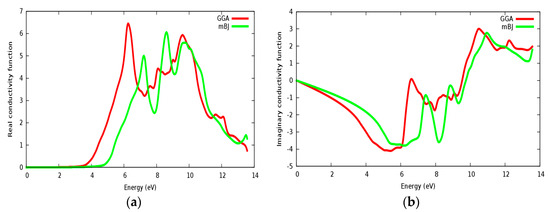
Figure 15.
Conductivity function for SrZrO3: (a) Real part; (b) Imaginary part using both PBE-GGA and mBJ-GGA methods.
Figure 16a,b displaying the extinction coefficient and refractive index n(ω). One can remark that the extinction coefficient of SrZrO3 starts to have considerable value at 4.75 eV within mBJ (GGA: 3.6 eV). We can clearly see that k(ω), conductivity and ε2(ω) start to have considerable values at the same point. Some peaks are presented along the spectrum, these peaks are related to the electrons transitions from valence to conduction bands. It is clearly seen that the extinction coefficient and the imaginary part of the epsilon vary in the same way. The static value of the refractive index n(0) in Figure 16b for the SrZrO3 compound is 1.75 within the mBJ approach (GGA: 1.9), which is a small value compared to n(0) for the SrRhO3 compound.
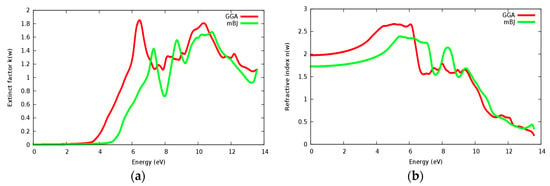
Figure 16.
(a) Extinct factor k(ω); (b) Refractive index n(ω) for SrZrO3 compound using both PBE-GGA and mBJ-GGA methods.
Reflectivity, energy loss and absorption functions are illustrated in Figure 17a–c, respectively. The static reflectivity of SrZrO3 is about 0.07 within the mBJ approach (GGA: 0.11). The static reflectivity of SrRhO3 is more than nine times greater than the reflectivity of SrZrO3. The reflectivity value increases rapidly in the high energy region; far ultraviolet region (FUV); ab = nd for the SrZrO3 compound, indicating that SrZrO3 is suitable as a wave reflectance compound in the far ultraviolet region (FUV). Energy loss and absorption functions are seen in Figure 17b,c; both of them quietly behave in the same way. Both absorption and energy loss for SrZrO3 begin at about 4.8 eV within the mBJ approach (GGA: 3.8 eV). The SrZrO3 is a good absorbent compound, but not in the low energy region; it is good absorbent in the far ultraviolet region, as it shows metallic behavior in the high energy region.
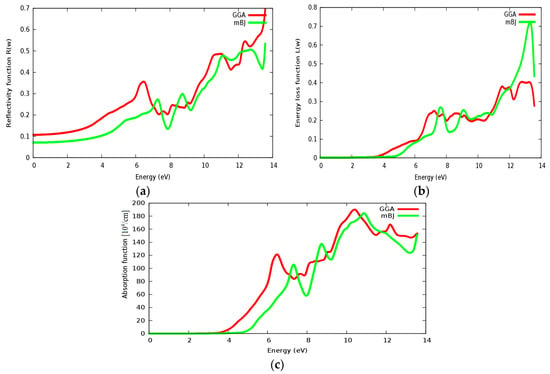
Figure 17.
(a) Reflectivity function R(ω); (b) Energy loss function L(ω) and (c) absorption function for SrZrO3 compound using both PBE-GGA and mBJ-GGA methods.
4. Conclusions
In summary, we have studied the structural, electronic, elastic and optical properties of SrTMO3 (TM = Rh, Zr) using the FP-LAPW method within the PBE-GGA and mBJ-GGA approaches in the framework of density functional theory. The lattice parameter is found to be in good agreement with experimental and theoretical results. From the band structure, we found that the SrRhO3 compound has a metallic nature within both BPE-GGA and mBJ-GGA approaches, while SrZrO3 is found to be an insulator within mBJ-GGA, while it is a wide energy band gap semiconductor within PBE-GGA. The elastic constants (C11, C12, C44), bulk modulus, shear modulus and Young’s modulus are also calculated and discussed. By analyzing the B/S ratio and Poisson’s ratio, we found that SrRhO3 has a ductile nature, while SrZrO3 has brittle nature. SrZrO3 has a more ionic trend compared to the SrRhO3. The optical properties such as dielectric constant, absorption coefficient, reflectivity coefficient, refractive index, optical conductivity and energy loss function were investigated in the energy range (0–14) eV. According to the dielectric constant, SrZrO3 has a large static dielectric constant which may make it promising as a good dielectric material; while SrRhO3 has a negative real dielectric constant which indicates a metallic behavior. It would be interesting to make detailed measurements of the transport and thermodynamic properties of SrRhO3 under applied fields looking for metamagnetic behavior and non-Fermi-liquid scalings. In conclusion, our theoretical calculations on SrTMO3 (TM = Rh, Zr) will help to use these materials for practical applications.
Author Contributions
Conceptualization, M.A.-J.; Data Curation, A.S., T.O. and A.M.; Formal Analysis, A.S., R.T.J. and T.O.; Investigation, M.A.-J. and R.K.; Methodology, M.A.-J.; Project Administration, M.A.-J.; Supervision, M.A.-J.; Visualization, M.A.-J.; Writing–Original Draft, A.S., T.O. and A.M.; Writing–Review & Editing, R.K. and K.F.I.
Funding
This research received no external funding.
Acknowledgments
This work has been carried out in the Computational Physics Laboratory, Physics Department, An-Najah N. University.
Conflicts of Interest
The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, and in the decision to publish the results.
References
- Bouadjemi, B.; Bentata, S.; Abbad, A.; Benstaali, W.; Bouhafs, B. Half-metallic ferromagnetism in PrMnO3 perovskite from first principles calculations. Solid State Commun. 2013, 168, 6–10. [Google Scholar] [CrossRef]
- Rahaman, M.Z.; Rahman, M.A.; Sarker, M.A.R. Prediction of a new transition metal oxide MgRhO3 with SrTiO3-type structure: Stability, structure and physical characteristics Chin. J. Phys. 2017, 55, 1489–1494. [Google Scholar]
- Yamaura, K.; Huang, Q.; Young, D.P.; Arai, M.; Takayama-Muromachi, E. Electronic properties of the novel 4d metallic oxide SrRhO3. Phys. B Phys. Condens. Matter 2003, 329–333, 820–821. [Google Scholar] [CrossRef]
- Saha, S.; Sinha, T.P.; Mookerjee, A. Structural and optical properties of paraelectric SrTiO3. J. Phys. Condens. Matter 2000, 12, 3325. [Google Scholar] [CrossRef]
- Johnsson, M.; Lemmens, P. Crystallography and chemistry of perovskites. In Handbook of Magnetism and Advanced Magnetic Materials; Kronmüller, H., Parkin, S., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2007. [Google Scholar]
- Pena, M.A.; Fierro, J.L. Chemical structures and performance of perovskite oxides. Chem. Rev. 2001, 101, 1981–2018. [Google Scholar] [CrossRef] [PubMed]
- Ghebouli, B.; Ghebouli, M.A.; Chihi, T.; Fatmi, M.; Boucetta, S.; Reffas, M. First-principles study of structural, elastic, electronic and optical properties of SrMO3 (M = Ti and Sn). Solid State Commun. 2009, 149, 2244–2249. [Google Scholar] [CrossRef]
- Sakhya, A.; Maibam, J.; Saha, S.; Chanda, S.; Dutta, A.; Sharama, B.; Thapa, R.K.; Sinha, T.B. Electronic structure and elastic properties of ATiO3 (A = Ba, Sr, Ca) perovskites: A first principles study. Indian J. Pure Appl. Phys. 2015, 53, 102–109. [Google Scholar]
- Van Roosmalen, J.A.M.; van Vlaanderen, P.; Cordfunke, E.H.P. On the structure of SrZrO3. J. Solid State Chem. 1992, 101, 59–65. [Google Scholar] [CrossRef]
- Boudali, A.; Khodja, M.D.; Amrani, B.; Bourbie, D.; Amara, K.; Abada, A. First-principles study of structural, elastic, electronic, and thermal properties of SrTiO3 perovskite cubic. Phys. Lett. A 2009, 373, 879–884. [Google Scholar] [CrossRef]
- Blaha, P.; Schwarz, K.; Madsen, G.K.H.; Kvasnicka, D.; Luitz, J. WIEN2k: An Augmented Plane Wave plus Local Orbitals Program for Calculating Crystal Properties; Vienne University of Technology: Vienna, Austria, 2001. [Google Scholar]
- Daga, A.; Sharma, S.; Sharma, K.S. First principle study of cubic SrMO3 perovskites (M = Ti, Zr, Mo, Rh, Ru). J. Mod. Phys. 2011, 2, 812–816. [Google Scholar] [CrossRef]
- Ali, M.L.; Rahaman, M.Z. The structural, elastic, electronic and optical properties of cubic perovskite SRVO3 compound: An ab initio study. Int. J. Mater. Sci. Appl. 2016, 5, 202–206. [Google Scholar]
- Ali, M.L.; Rahaman, M.Z. Variation of the physical proprties of four transition metal oxides SrTMO3 (TM = Rh, Ti, Mo, Zr) under pressure: An ab initio study. J. Adv. Phys. 2017, 6, 197–205. [Google Scholar] [CrossRef]
- CASTEP Guide. Materials Studio 8.0; BIOVIA: San Diego, CA, USA, 2010; pp. 261–262.
- Shende, R.V.; Krueger, D.S.; Rosetti, G.A.; Lombardo, S.J. Strontium zirconate and strontium titanate ceramics for high-voltage applications: Synthesis, processing, and dielectric properties. J. Am. Ceram. Soc. 2001, 84, 1648–1650. [Google Scholar] [CrossRef]
- Singh, D.J. Prospects for quantum criticality in perovskite SrRhO3. Phys. Rev. B 2003, 67, 054507. [Google Scholar] [CrossRef]
- Blaha, P.; Schwarz, K.; Sorantin, P.; Trickey, S.B. Full-potential, linearized augmented plane wave programs for crystalline systems. Comput. Phys. Commun. 1990, 59, 399–415. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.; Johnson, E.R. A simple effective potential for exchange. J. Chem. Phys. 2006, 124, 221101. [Google Scholar] [CrossRef] [PubMed]
- Murnaghan, F.D. The compressibility of media under extreme pressures. Proc. Natl. Acad. Sci. USA 1944, 30, 244–247. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Lee, J.S.; Noh, T.W.; Byun, D.Y.; Yoo, K.S.; Yamaura, K.; Takayama-Muromachi, E. Systematic trends in the electronic structure parameters of the 4d transition-metal oxides SrMO3 (M = Zr, Mo, Ru, and Rh). Phys. Rev. B 2003, 67, 113101. [Google Scholar] [CrossRef]
- Smith, A.J.; Welch, A.J.E. Some mixed metal oxides of perovskite structure. J. Acta Crystallogr. 1960, 13, 653–656. [Google Scholar] [CrossRef]
- Ligny, D.D.; Richet, P. High-temperature heat capacity and thermal expansion of SrTiO3 and SrZrO3 perovskites. Phys. Rev. B 1996, 53, 3013. [Google Scholar] [CrossRef]
- Mete, A.; Shaltaf, R.; Ellialtioglu, S. Electronic and structural properties of a 4d perovskite: Cubic phase of SrZrO3. Phys. Rev. B 2003, 68, 035119. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Luana, V.; Costales, A.; Pendas, A.M. Ions in crystals: The topology of the electron density in ionic materials. II. The cubic alkali halide perovskites. Phys. Rev. B 1997, 55, 4285. [Google Scholar] [CrossRef]
- Mori-Sánchez, P.; Pendás, A.M.; Luaña, V. A classification of covalent, ionic, and metallic solids based on the electron density. J. Am. Chem. Soc. 2002, 124, 14721–14723. [Google Scholar] [CrossRef]
- Becke, A.D.; Edgecombe, K.E. A simple measure of electron localization in atomic and molecular systems. J. Chem. Phys. 1990, 92, 5397–5403. [Google Scholar] [CrossRef]
- Sun, Z.; Music, D.; Ahuja, R.; Schneider, J.M. Electronic origin of shearing in M2AC (M = Ti, V, Cr, A = Al, Ga). J. Phys. Condens. Matter 2005, 17, 7169–7176. [Google Scholar] [CrossRef]
- IRelast Package. Cubic-elastic_13.2; Part of the Commercial Code WIEN2k. Available online: http://susi.theochem.tuwien.ac.at/ (accessed on 1 September 2018).
- Born, M. On the stability of crystal lattices. I. Math. Proc. Camb. Philos. Soc. 1940, 36, 160–172. [Google Scholar] [CrossRef]
- Born, M.; Huang, K. Dynamical Theory of Crystal Lattices; Oxford University Press: Oxford, UK, 1956. [Google Scholar]
- Hill, R. The elastic behaviour of a crystalline aggregate. Proc. Phys. Soc. Sect. A 1952, 65, 349. [Google Scholar] [CrossRef]
- Voigt, W. Lehrbuch der Kristallphysik (mit Ausschluss der Kristalloptik); B.G. Teubner: Berlin, Germany, 1910; pp. 1850–1919. [Google Scholar]
- Reuss, A. Berechnung der fließgrenze von mischkristallen auf grund der plastizitätsbedingung für einkristalle. Z. Angew. Math. Mech. 1929, 9, 49–58. [Google Scholar] [CrossRef]
- Terki, R.; Feraoun, H.; Bertrand, G.; Aourag, H. Full potential calculation of structural, elastic and electronic properties of BaZrO3 and SrZrO3. Basic Solid State Phys. 2005, 242, 1054–1062. [Google Scholar] [CrossRef]
- Teter, D.M. Computational alchemy: The search for new superhard materials. MRS Bull. 2013, 23, 22–27. [Google Scholar] [CrossRef]
- Pugh, S.F. XCII. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals. Philos. Mag. 1954, 45, 823–843. [Google Scholar] [CrossRef]
- Pettifor, D.G. Theoretical predictions of structure and related properties of intermetallics. Mater. Sci. Technol. 1992, 8, 345. [Google Scholar] [CrossRef]
- Frantsevich, I.N.; Voronov, F.F.; Bokuta, S.A. Elastic Constants and Elastic Moduli of Metals and Insulators Handbook; Frantsevich, I.N., Ed.; Naukova Dumka: Kiev, Ukraine, 1983; p. 60. [Google Scholar]
- Zener, C. Elasticity and Anelasticity of Metals; University of Chicago Press: Chicago, IL, USA, 1948. [Google Scholar]
- Tvergaard, V.; Hutshinson, J.W. Microcracking in ceramics induced by thermal expansion or elastic anisotropy. J. Am. Ceram. Soc. 1988, 71, 157–166. [Google Scholar] [CrossRef]
- Ehrenreich, H.; Cohen, M.H. Self-consistent field approach to the many-electron problem. Phys. Rev. 1959, 115, 786. [Google Scholar] [CrossRef]
- Hedin, L. New Method for Calculating the One-Particle Green’s function with application to the electron-gas problem. Phys. Rev. A 1965, 139, A796. [Google Scholar] [CrossRef]
- Fox, M. Optical Properties of Solids; Oxford University Press: Oxford, UK, 2001. [Google Scholar]
- Abeles, F. Optical Properties of Solids; American Elsevier: Atlanta, GA, USA, 1972. [Google Scholar]
- Wang, H.; Wang, Y.; Cao, X.; Zhang, L.; Feng, M.; Lan, G. Simulation of electronic density of states and optical properties of PbB4O7 by first-principles DFT method. Phys. Status Solidi B 2009, 246, 437–443. [Google Scholar] [CrossRef]
- He, Y.; Zeng, T. First-principles study and model of dielectric functions of silver nanoparticles. J. Phys. Chem. C 2010, 114, 18023–18030. [Google Scholar] [CrossRef]
- Koontz, R.; Blokhina, G.; Gold, S.; Krasnykh, A. High dielectric constant materials for pulsed energy storage capacitors. In Proceedings of the 1998 Annual Report Conference on Electrical Insulation and Dielectric Phenomena (Cat. No. 98CH36257), Atlanta, GA, USA, 25–28 October 1998. [Google Scholar]


















© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).