Theoretical Study of Hydrogen on LaFeO3 (010) Surface Adsorption and Subsurface Diffusion


Abstract
:1. Introduction
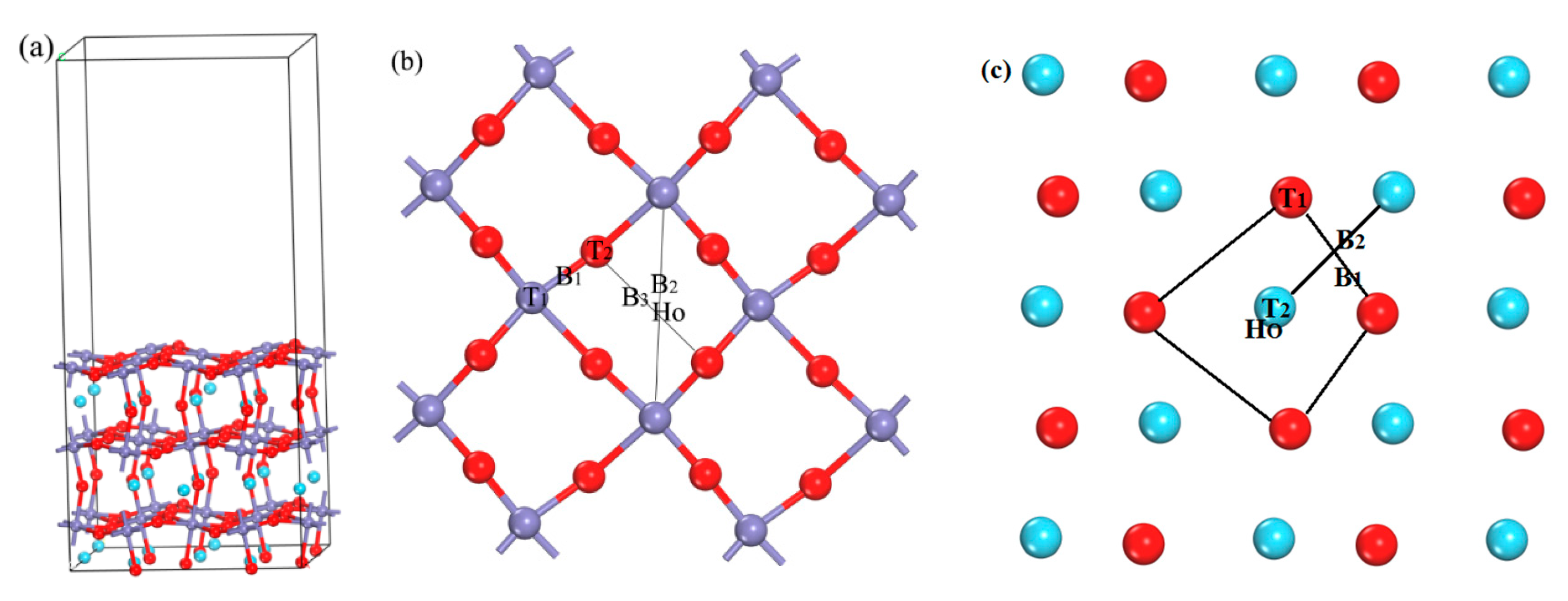
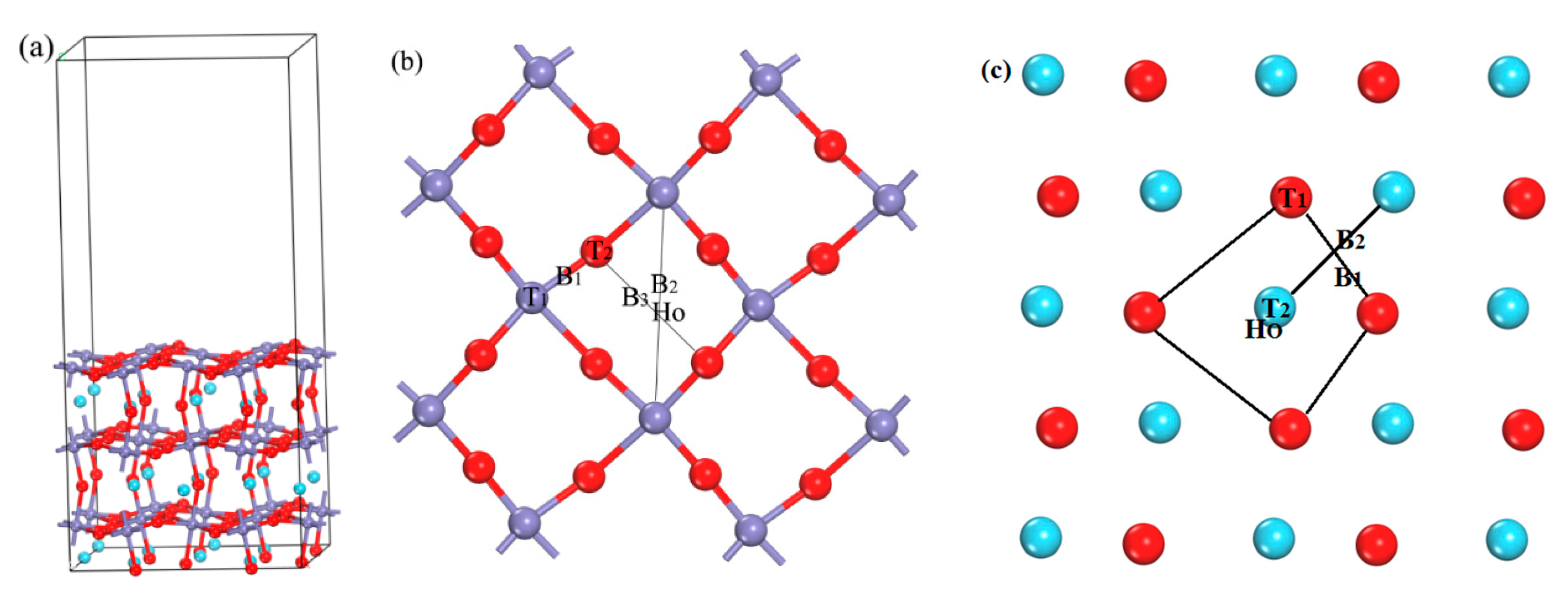
2. Calculation Method and Model
3. Results and Discussion
3.1. Adsorption of H Atom on the Surface and Occupation in the Subsurface
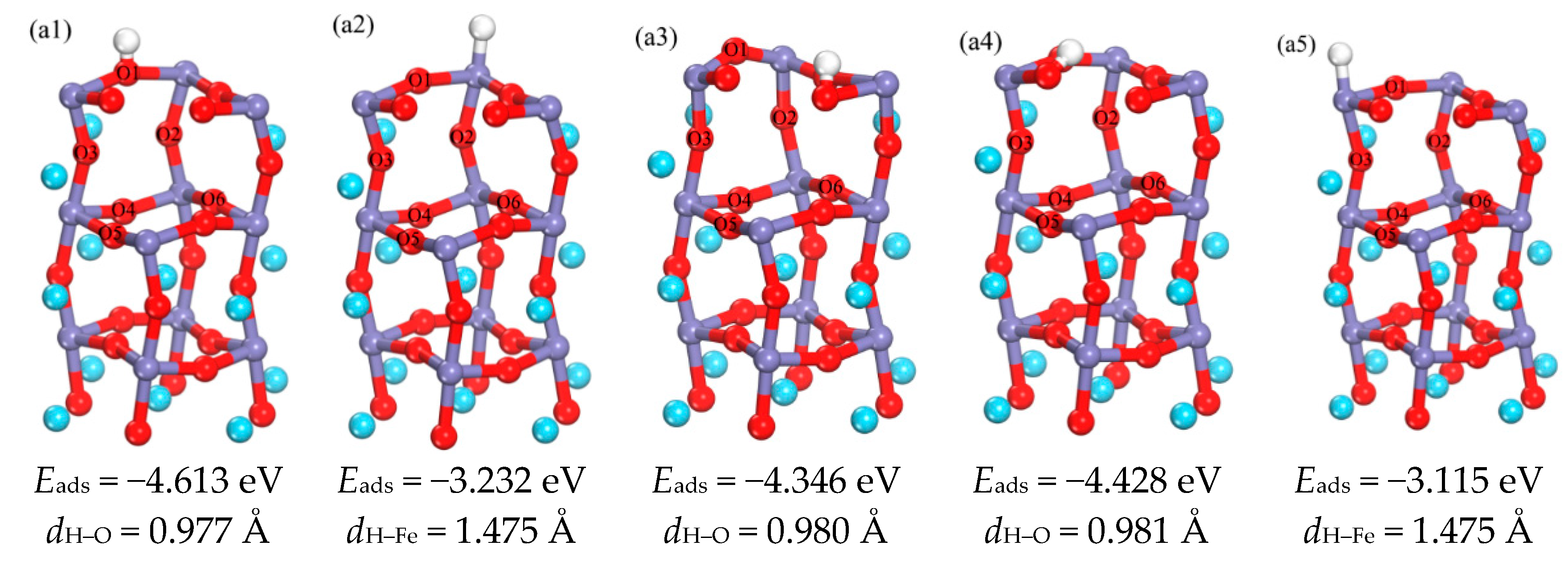
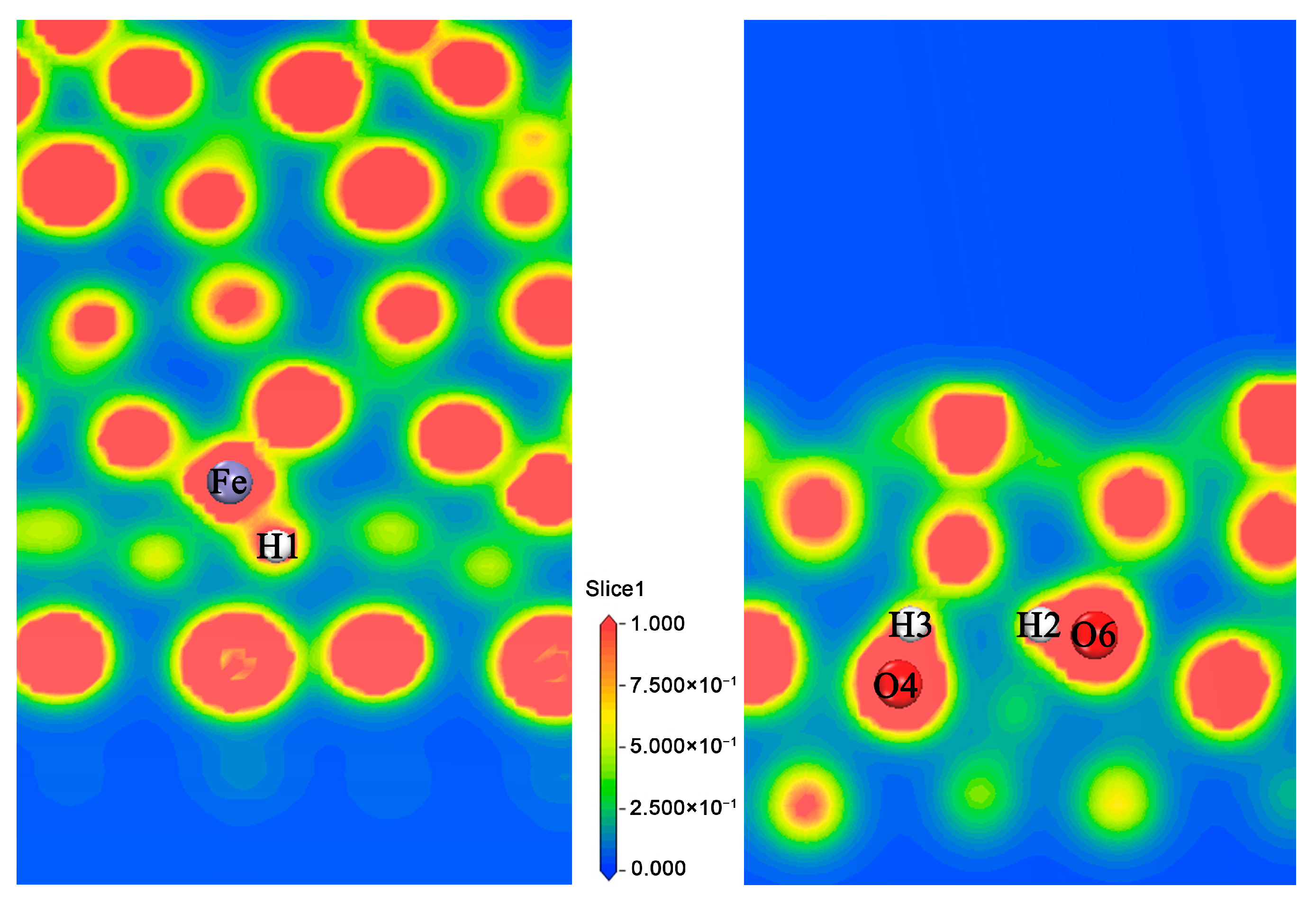
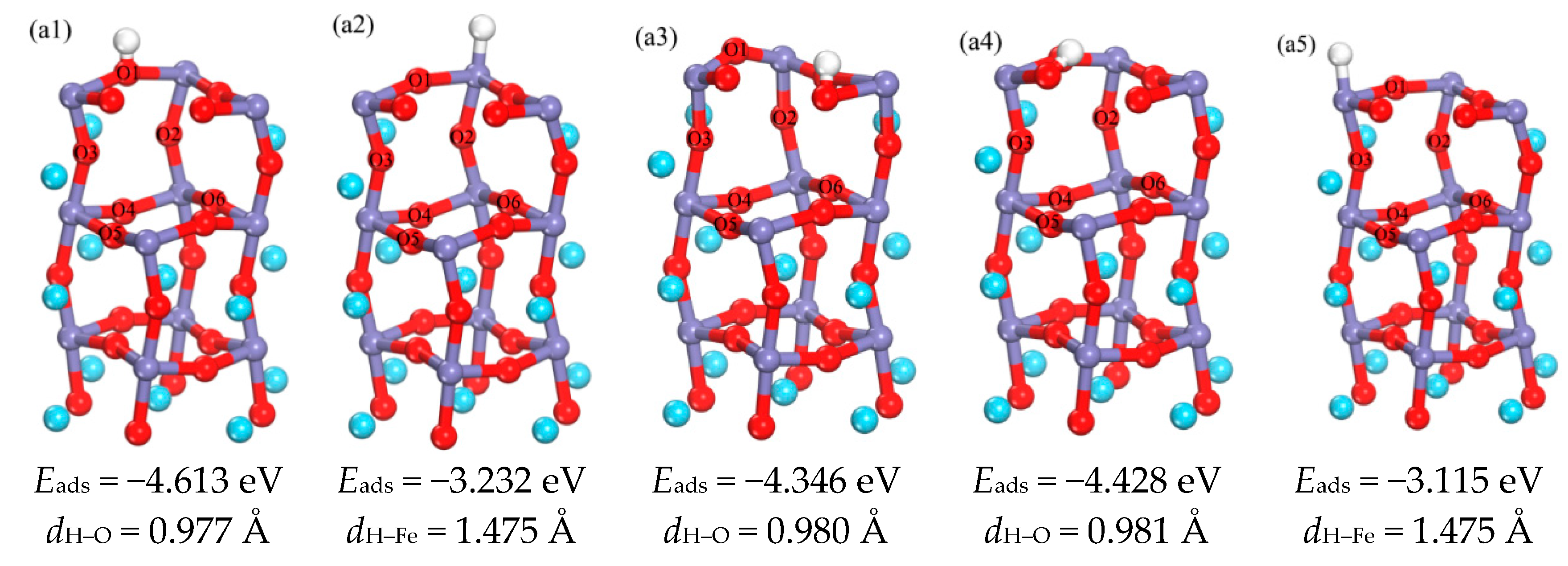
3.1.1. Adsorption of H Atom on Surface
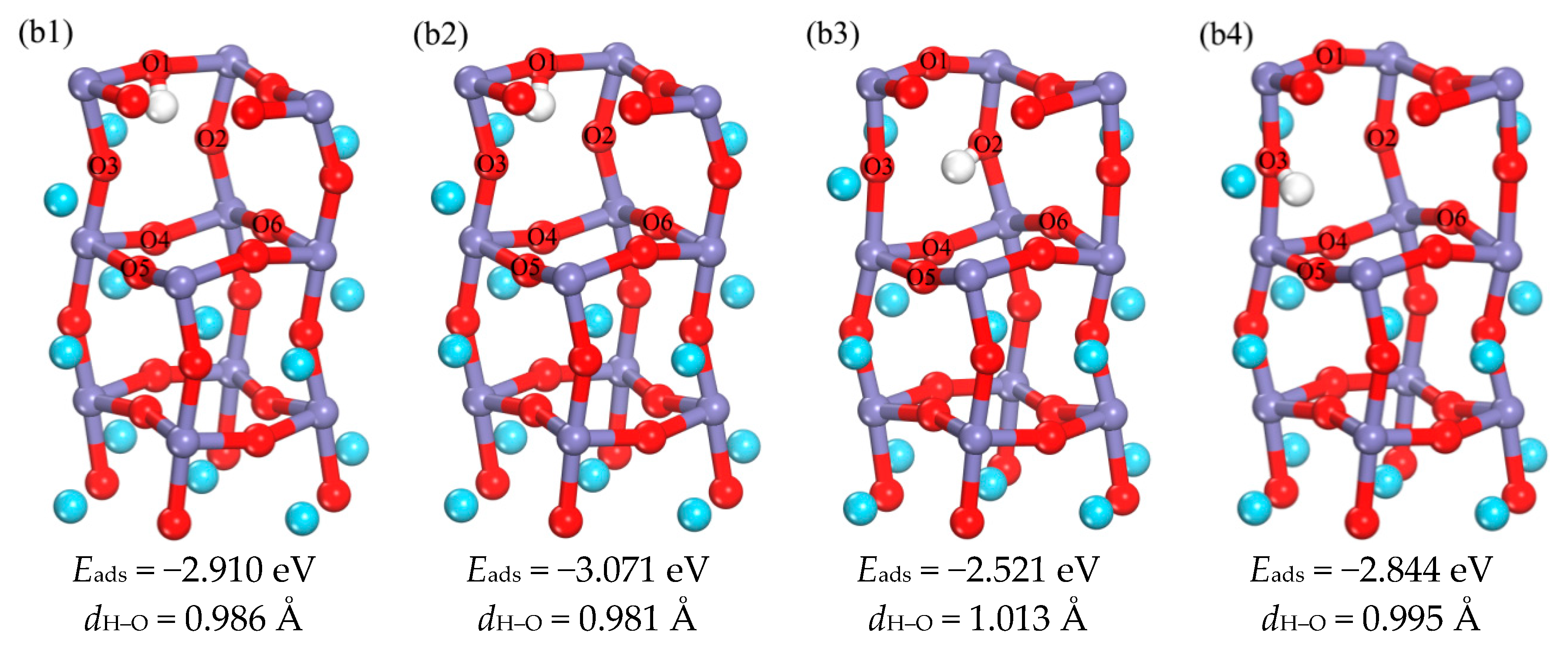
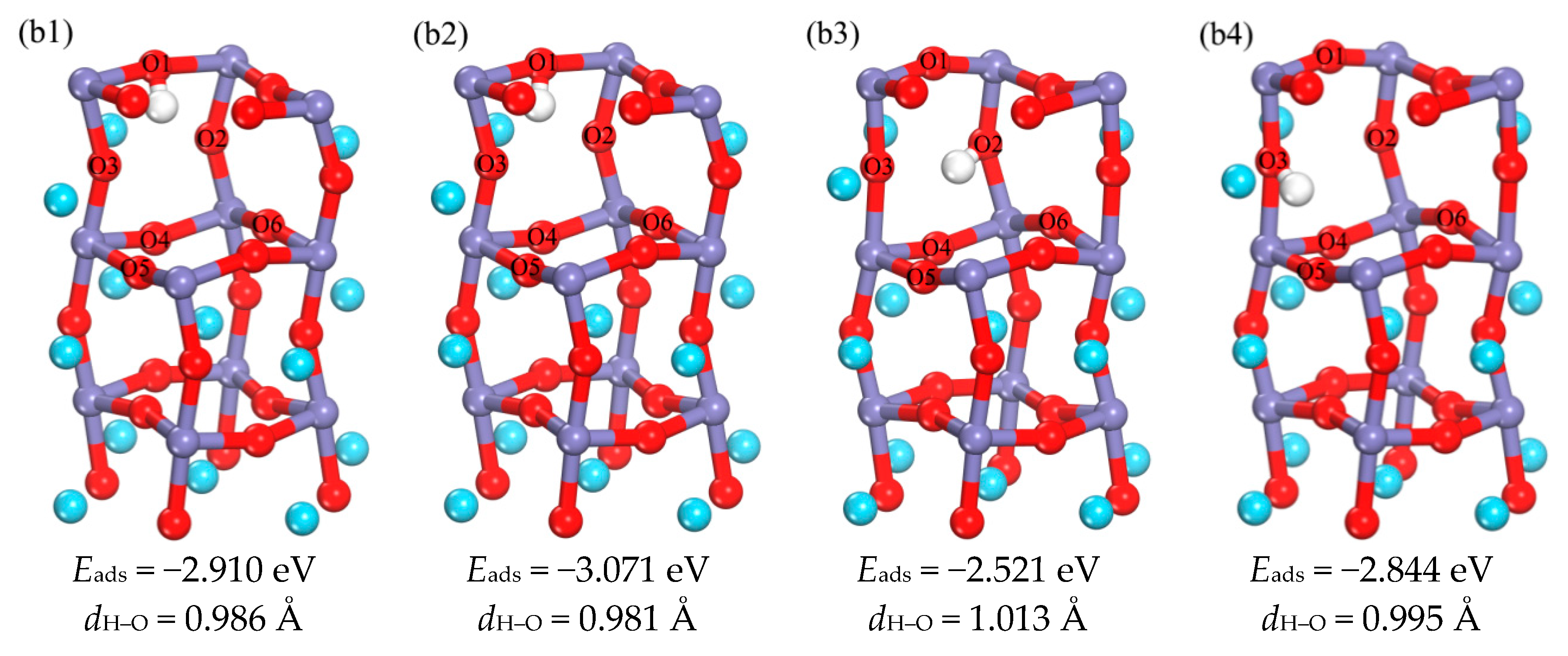
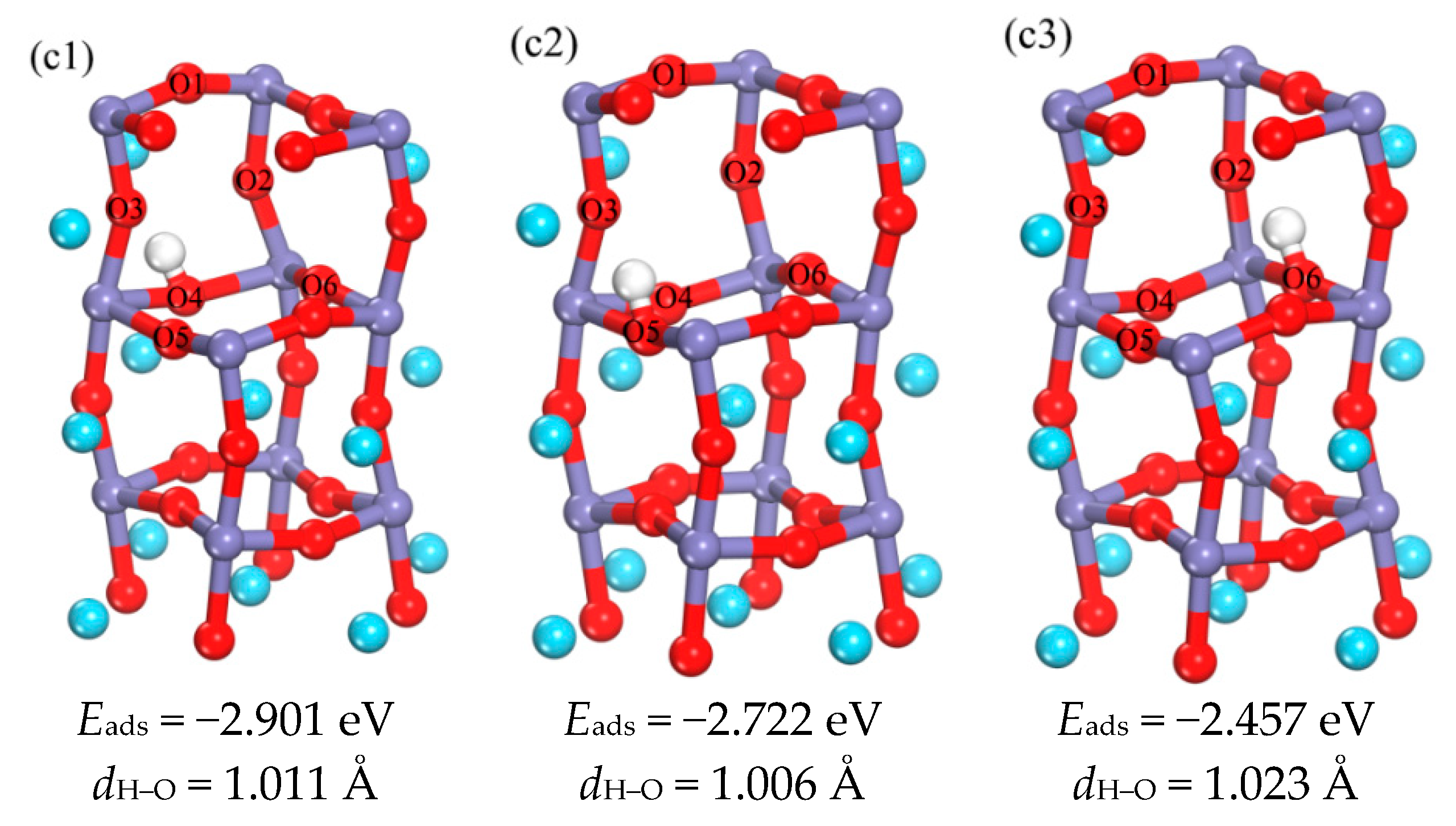
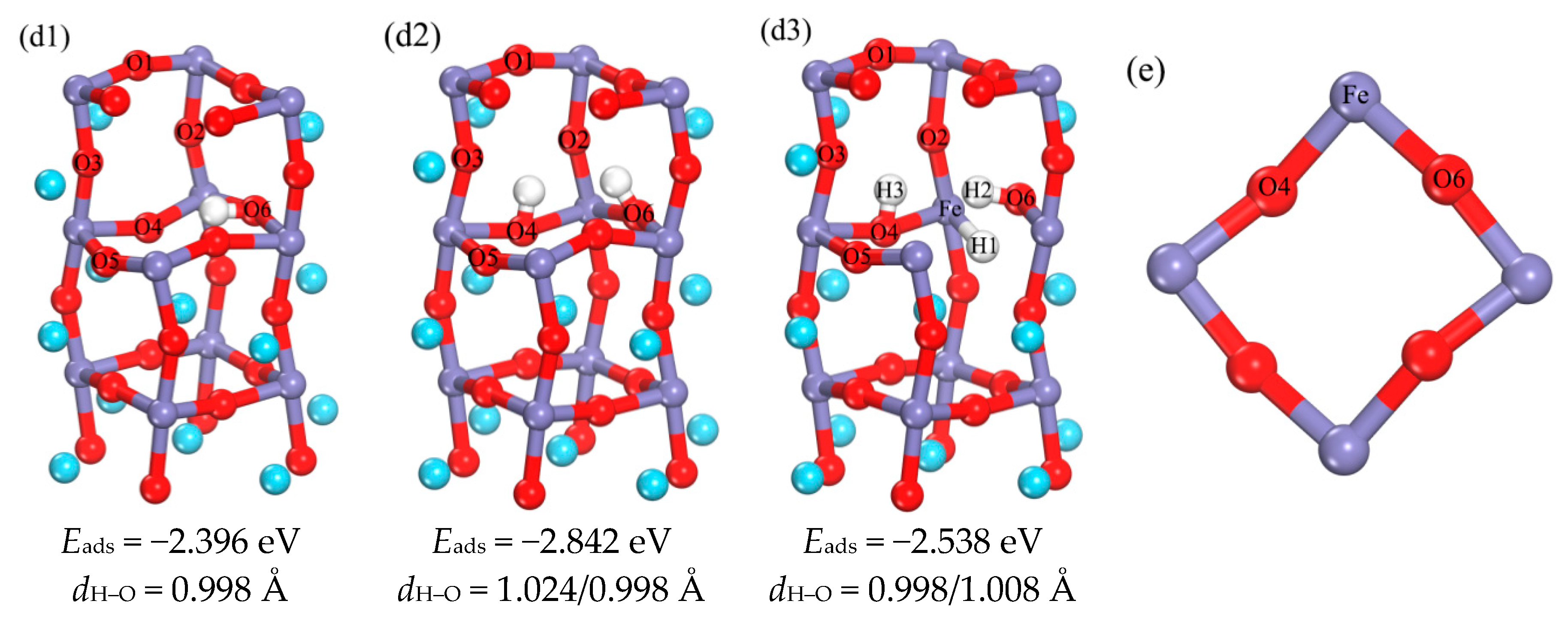
3.1.2. The Occupation of H in the Subsurface
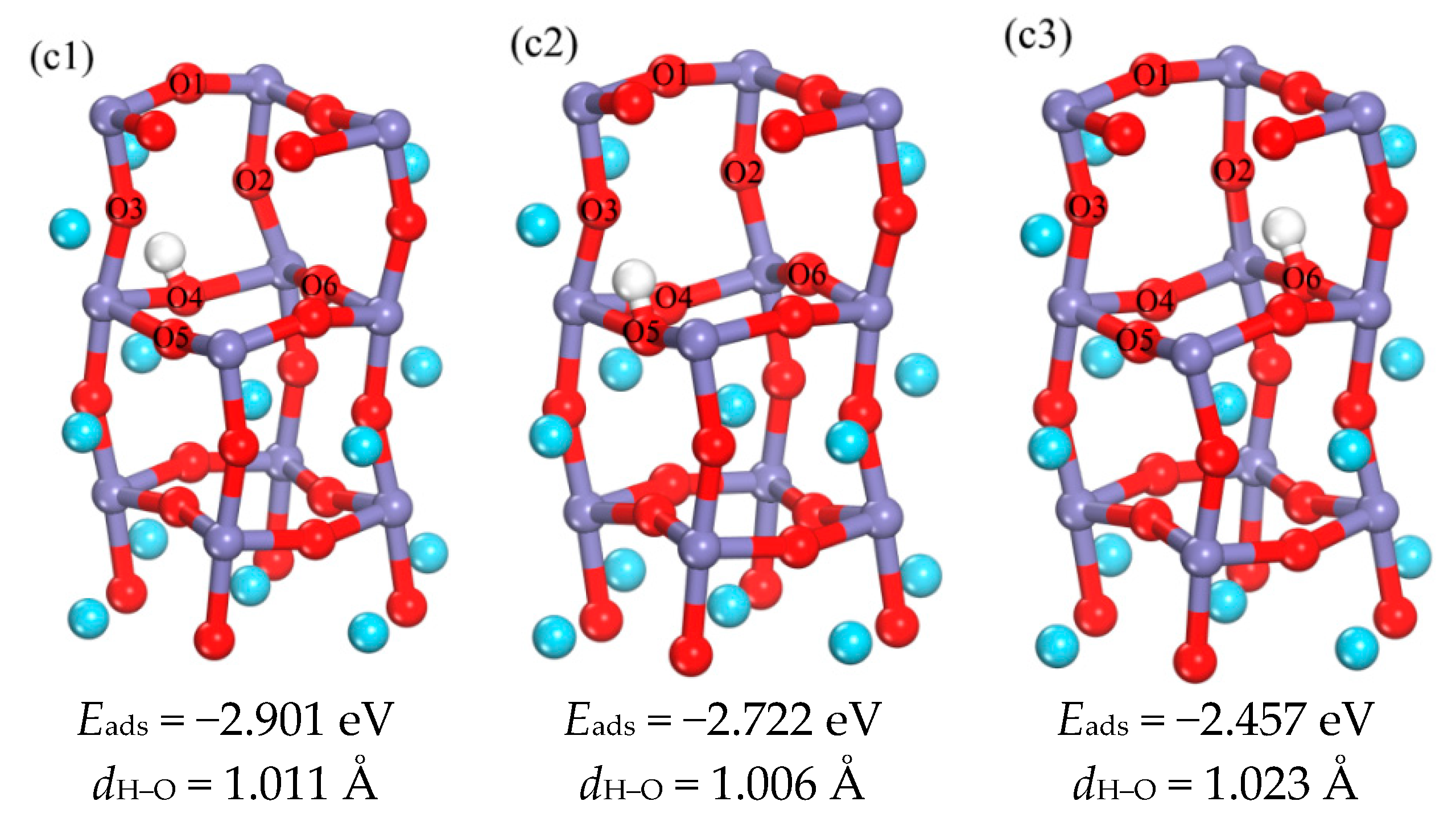
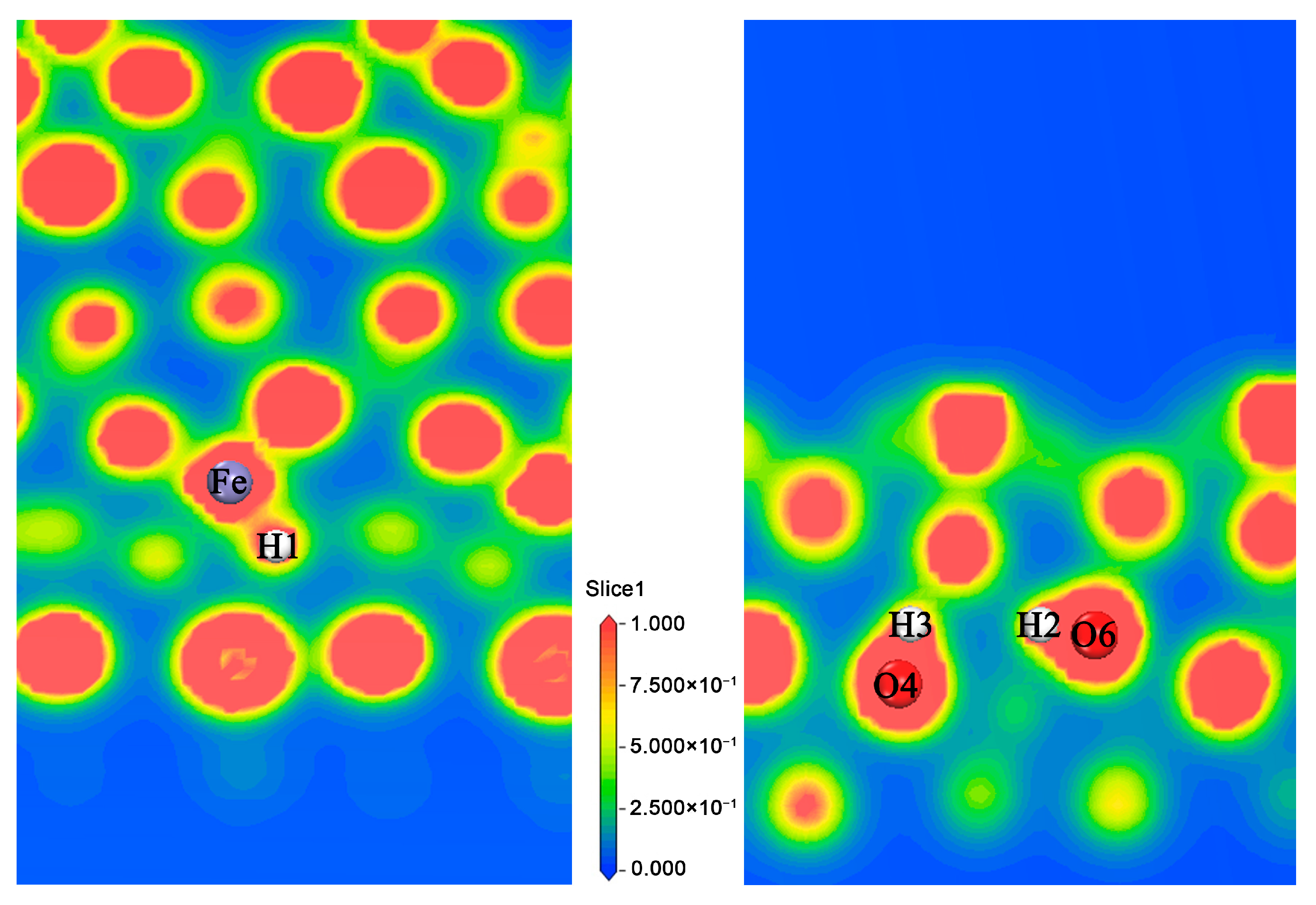
3.1.3. Analysis of the Properties of d3 Structure
3.2. Diffusion of H on the Surface and in the Subsurface
3.2.1. Diffusion of H Atom on the Surface
3.2.2. Diffusion of H from Surface to Sub-Surface Layer
3.2.3. The Diffusion of H from the Sub-Surface Layer to the Third Layer
3.2.4. The Main Diffusion Path of H Diffusion from the Surface to the Subsurface
4. Summary
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Lim, D.K.; Im, H.N.; Singh, B.; Park, C.-J.; Song, S.-J. Electrochemical hydrogen charge and discharge properties of La0.1Sr0.9Co1−yFeyO3−δ (y = 0, 0.2, 1) electrodes in alkaline electrolyte solution. Electrochim. Acta 2013, 102, 393–399. [Google Scholar] [CrossRef]
- Deng, G.; Chen, Y.G.; Tao, M.D.; Wu, C.; Shen, X.; Yang, H.; Liu, M. Preparation and electrochemical properties of La0.4Sr0.6FeO3 as negative electrode of Ni/MH batteries. Int. J. Hydrog. Energy 2009, 34, 5568–5573. [Google Scholar] [CrossRef]
- Hibino, T.; Mizutani, K.; Yajima, T.; Iwahara, H. Evaluation of proton conductivity in SrCeO3, BaCeO3, CaZrO3 and SrZrO3 by temperature programmed desorption method. Solid State Ionics 1992, 57, 303–306. [Google Scholar] [CrossRef]
- Deng, G.; Chen, Y.G.; Tao, M.D.; Wu, C.; Shen, X.; Yang, H.; Liu, M. Study of the electrochemical hydrogen storage properties of the proton-conductive perovskite-type oxide LaCrO3 as negative electrode for Ni/MH batteries. Electrochim. Acta 2010, 55, 884–886. [Google Scholar] [CrossRef]
- Wang, Q.; Chen, Z.Q.; Chen, Y.G.; Cheng, N.; Hui, Q. Hydrogen storage in perovskite-type oxides ABO3 for Ni/MH battery applications: A density functional investigation. Ind. Eng. Chem. Res. 2012, 51, 11821–11827. [Google Scholar] [CrossRef]
- Yugami, H.; Chiba, Y.; Ishigame, M. Local structures in Y3+-doped SrCeO3 crystals studied by site-selective spectroscopy. Solid State Ionics 1995, 77, 201–206. [Google Scholar] [CrossRef]
- Mandal, T.K.; Sebastian, L.; Gopalakrishnan, J.; Abrams, L.; Goodenough, J.B. Hydrogen uptake by barium manganite at atmospheric pressure. Mater. Res. Bull. 2004, 39, 2257–2264. [Google Scholar] [CrossRef]
- Esaka, T.; Sakaguchi, H.; Kobayashi, S. Hydrogen storage in proton-conductive perovskite-type oxides and their application to nickel-hydrogen batteries. Solid State Ionics 2004, 166, 351–357. [Google Scholar] [CrossRef]
- Deng, G.; Chen, Y.G.; Tao, M.D.; Wu, C.; Shen, X.; Yang, H.; Liu, M. Electrochemical properties and hydrogen storage mechanism of perovskite-type oxide LaFeO3 as a negative electrode for Ni/MH batteries. Electrochim. Acta 2010, 55, 1120–1124. [Google Scholar] [CrossRef]
- Wang, Q.; Deng, G.; Chen, Z.; Chen, Y.; Cheng, N. Electrochemical hydrogen property improved in nano-structured perovskite oxide LaFeO3 for Ni/MH battery. J. Appl. Phys. 2013, 113, 053305. [Google Scholar] [CrossRef]
- Deng, G.; Chen, Y.G.; Tao, M.D.; Wu, C.; Shen, X.; Yang, H. Electrochemical properties of La1-xSrxFeO3 (x = 0.2, 0.4) as negative electrode of Ni-MH batteries. Electrochim. Acta 2009, 54, 3910–3914. [Google Scholar] [CrossRef]
- Deng, G.; Chen, Y.G.; Tao, M.D.; Wu, C.; Shen, X.; Yang, H.; Liu, M. Preparation and electrochemical property of LaFeO3 electrode material. Acta Chim. Sin. 2009, 67, 2001–2004. (In Chinese) [Google Scholar]
- Nalbandian, L.; Evdou, A.; Zaspalis, V. La1−xSrxMyFe1−yO3−δ perovskites as oxygen-carrier materials for chemical-looping reforming. Int. J. Hydrog. Energy 2011, 36, 6657–6670. [Google Scholar] [CrossRef]
- Sun, Y.; Hla, S.S.; Duffy, G.J.; Cousins, A.J.; French, D.; Morpeth, L.D.; Edwards, J.H.; Roberts, D.G. Effect of Ce on the structural features and catalytic properties of La0.9−xCexFeO3 perovskite-like catalysts for the high temperature water-gas shift reaction. Int. J. Hydrog. Energy 2011, 36, 79–86. [Google Scholar] [CrossRef]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.I.; Refson, K.; Payne, M.C. First principles methods using CASTEP. Zeitschrift Kristallographie 2005, 220, 567–570. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burke, K.; Wang, Y. Generalized gradient approximation for the exchange-correlation hole of a many-electron system. Phys. Rev. B Condens Matter. 1996, 54, 16533–16539. [Google Scholar] [CrossRef] [PubMed]
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B Condens. Matter. 1990, 41, 7892–7895. [Google Scholar] [CrossRef] [PubMed]
- Aarons, J.; Sarwar, M.; Thompsett, D.; Skylaris, C.K. Perspective: Methods for large-scale density functional calculations on metallic systems. J. Chem. Phys. 2016, 145, 220901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.L.; Kleis, J.; Rossmeisl, J.; Morgan, D. Ab initio energetics of LaBO3 (001) (B = Mn, Fe, Co, and Ni) for solid oxide fuel cell cathodes. Phys. Rev. B 2009, 80, 224101. [Google Scholar] [CrossRef]
- Lee, Y.L.; Morgan, D.; Kleis, J.; Rossmeisl, J. Ab initio defect energetics in LaBO3 perovskite solid oxide fuel cell materials. ECS Trans. 2009, 25, 2761–2767. [Google Scholar]
- Chen, Y.H.; Pan, C.C.; Zhang, M.L.; Lihua, Y.; Zhang, C.-R.; Kang, L.; Luo, Y.C. First principles study of the adsorption of H2 on LaFeO3 surface. Chin. J. Inorg. Chem. 2016, 32, 945–953. (In Chinese) [Google Scholar]
- Sun, J.; Wang, H.T.; He, J.L.; Tian, Y. Ab initio investigations of optical properties of the high-pressure phases of ZnO. Phys. Rev. B 2005, 71, 125132. [Google Scholar] [CrossRef]
- Sundell, P.G.; Björketun, M.E.; Wahnström, G. Density-functional calculations of prefactors and activation energies for H diffusion in BaZrO3. Phys. Rev. B 2007, 76, 094301. [Google Scholar] [CrossRef]
- Taguchi, H.; Masunaga, Y.; Hirota, K.; Yamaguchi, O. Synthesis of perovskite-type (La1−xCax) FeO3 (0 ≤ x ≤ 0.2) at low temperature. Mater. Res. Bull. 2005, 40, 773–780. [Google Scholar] [CrossRef]
- Zhong, W.; Liu, Y.; Zhang, D. Theoretical study of methanol oxidation on the PtAu (111) bimetallic surface: CO pathway vs. non-CO pathway. J. Phys. Chem. C 2012, 116, 2994–3000. [Google Scholar] [CrossRef]
- Wang, D.Y.; Gong, M.; Chou, H.L.; Pan, C.J.; Chen, H.A.; Wu, Y.; Lin, M.C.; Guan, M.; Yang, J.; Chen, C.W.; et al. Highly active and stable hybrid catalyst of cobalt-doped FeS2 nanosheets-carbon nanotubes for hydrogen evolution reaction. J. Am. Chem. Soc. 2015, 137, 1587–1592. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Li, L.; Xu, J.; Fang, T. Density functional periodic study of the dehydrogenation of methaneon Pd (111) surface. Appl. Surf. Sci. 2013, 286, 115–120. [Google Scholar] [CrossRef]
- Govind, N.; Petersen, M.; Fitzgerald, G.; King-Smith, D.; Andzelm, J. A generalized synchronous transit method for transition state location. Comput. Mater. Sci. 2003, 28, 250–258. [Google Scholar] [CrossRef]
- Jiang, Z.; Fang, T. DFT study on the synergistic effect of Pd-Cu bimetal on the adsorption and dissociation of H2O. J. Phys. Chem. C 2016, 120, 25289–25295. [Google Scholar] [CrossRef]
- Li, J.; Xie, Y.P.; Chen, Y.X.; Wang, B.W.; Zhao, S.J. First-principles study of the hydrogen adsorption and diffusion onordered Ni3Fe (111) surface and in the bulk. Intermetallics 2014, 44, 64–72. [Google Scholar] [CrossRef]
- Sun, L.H.; Hu, J.F.; Gao, F.; Zhang, Y.; Qin, H. First-principle study of NO adsorption on the LaFeO3 (010) surface. Phys. B Condens. Matter. 2011, 406, 4105–4108. [Google Scholar] [CrossRef]
- Lie, G.C.; Clementi, E. Molecular-dynamics simulation of liquid water with an ab initio flexible water-water interaction potential. Phys. Rev. A 1986, 33, 2679–2693. [Google Scholar] [CrossRef]
- Sorescu, D.C. First principles calculations of the adsorption and diffusion of hydrogen on Fe (100) surface and in the bulk. Catal. Today 2005, 105, 44–65. [Google Scholar] [CrossRef]
- Sanchez-Portal, D.; Artacho, E.; Soler, J.M. Projection of plane-wave calculations into atomic orbitals. Solid State Commun. 1995, 95, 685–690. [Google Scholar] [Green Version]
- Mulliken, R.S. Electronic population analysis on LCAO–MO molecular wave functions. I. J. Chem. Phys. 1955, 23, 1833–1840. [Google Scholar] [CrossRef]
- Segall, M.D.; Pickard, C.J.; Shah, R.; Payne, M.C. Population analysis in plane wave electronic structure calculations. Mol. Phys. 1996, 89, 571–577. [Google Scholar] [CrossRef]
- Segall, M.D.; Shah, R.; Pickard, C.J.; Payne, M.C. Population analysis of plane-wave electronic structure calculations of bulk materials. Phys. Rev. B Condens. Matter. 1996, 54, 16317–16320. [Google Scholar] [PubMed]
- Becke, A.D.; Edgecombe, K.E. A simple measure of electron localization in atomic and molecular systems. J. Chem. Phys. 1990, 92, 5397–5403. [Google Scholar] [CrossRef]
- Burdett, J.K.; McCormick, T.A. Electron localization in molecules and solids: The meaning of ELF. J. Phys. Chem. A 1998, 102, 6366–6372. [Google Scholar] [CrossRef]
- Lee, S.B.; Chen, R.; Balents, L. Metal-insulator transition in a two-band model for the perovskite nickelates. Phys. Rev. B 2011, 84, 165119. [Google Scholar]
- Islam, M.M.; Calatayud, M.; Pacchioni, G. Hydrogen adsorption and diffusion on the anatase TiO2 (101) surface: A first-principles investigation. J. Phys. Chem. C 2011, 115, 6809–6814. [Google Scholar] [CrossRef]
- Kuzubov, A.A.; Eliseeva, N.S.; Krasnov, P.O.; Kuklin, A.V.; Kovaleva, E.A.; Kholtobina, A.S. Theoretical investigation of the adsorption and diffusion of hydrogen on the surface and in the bulk of the intermetallic compound Mg2Ni. Phys. Solid State 2014, 56, 2035–2042. [Google Scholar] [CrossRef]
- Zhang, G.K.; Wang, X.L.; Xiong, Y.F.; Shi, Y.; Song, J.; Luo, D. Mechanism for adsorption, dissociation and diffusion of hydrogen in hydrogen permeation barrier of a-Al2O3: A density functional theory study. Int. J. Hydrog. Energy 2013, 38, 1157–1165. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Atom | Pure Host (e) | Adsorption 2 H (e) | Adsorption 3 H (e) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| s | p | d | Charge | s | p | d | Charge | s | p | d | Charge | |
| Fe | 0.33 | 0.58 | 6.51 | 0.59 | 0.32 | 0.45 | 6.60 | 0.63 | 0.33 | 0.57 | 6.65 | 0.45 |
| O4 | 1.86 | 4.86 | - | −0.72 | 1.83 | 4.91 | - | −0.74 | 1.83 | 4.88 | - | −0.71 |
| O6 | 1.85 | 4.86 | - | −0.71 | 1.82 | 4.91 | - | −0.73 | 1.81 | 4.87 | - | −0.69 |
| H1 | 1.00 | - | - | - | 1.00 | - | - | - | 1.33 | - | - | −0.33 |
| H2 | 1.00 | - | - | - | 0.80 | - | - | 0.20 | 0.77 | - | - | 0.23 |
| H3 | 1.00 | - | - | - | 0.79 | - | - | 0.21 | 0.77 | - | - | 0.23 |
| Bond | Population (e) | Length (Å) | ||||
|---|---|---|---|---|---|---|
| Pure Host | Adsorption 2 H | Adsorption 3 H | Pure Host | Adsorption 2 H | Adsorption 3 H | |
| Fe–H1 | - | - | 0.50 | - | - | 1.534 |
| O4–H3 | - | 0.70 | 0.70 | - | 0.998 | 1.005 |
| O6–H2 | - | 0.68 | 0.72 | - | 1.024 | 0.995 |
| Fe–O4 | 0.33 | 0.12 | 0.15 | 1.984 | 2.126 | 2.125 |
| Fe–O6 | 0.30 | 0.11 | −0.23 | 1.974 | 2.087 | 2.807 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pan, C.; Chen, Y.; Zhang, M.; Yuan, L.; Zhang, C. Theoretical Study of Hydrogen on LaFeO3 (010) Surface Adsorption and Subsurface Diffusion. Materials 2018, 11, 2347. https://doi.org/10.3390/ma11122347
Pan C, Chen Y, Zhang M, Yuan L, Zhang C. Theoretical Study of Hydrogen on LaFeO3 (010) Surface Adsorption and Subsurface Diffusion. Materials. 2018; 11(12):2347. https://doi.org/10.3390/ma11122347
Chicago/Turabian StylePan, Changchang, Yuhong Chen, Meiling Zhang, Lihua Yuan, and Cairong Zhang. 2018. "Theoretical Study of Hydrogen on LaFeO3 (010) Surface Adsorption and Subsurface Diffusion" Materials 11, no. 12: 2347. https://doi.org/10.3390/ma11122347
APA StylePan, C., Chen, Y., Zhang, M., Yuan, L., & Zhang, C. (2018). Theoretical Study of Hydrogen on LaFeO3 (010) Surface Adsorption and Subsurface Diffusion. Materials, 11(12), 2347. https://doi.org/10.3390/ma11122347