An Innovative Approach to Control Steel Reinforcement Corrosion by Self-Healing


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Steel Passivity, Passivity Breakdown and Matrix Carbonation—Background
2.1. Steel Passivity in Reinforced Concrete
2.2. Passivity Breakdown
2.3. Matrix Carbonation—Steel-Corrosion-Related Aspects
3. Experimental Program—Introduction to Sequence and Approach
4. The Concept of Nanoparticle Application in Reinforced Concrete
4.1. The Approach to Corrosion Control via Nanoparticles
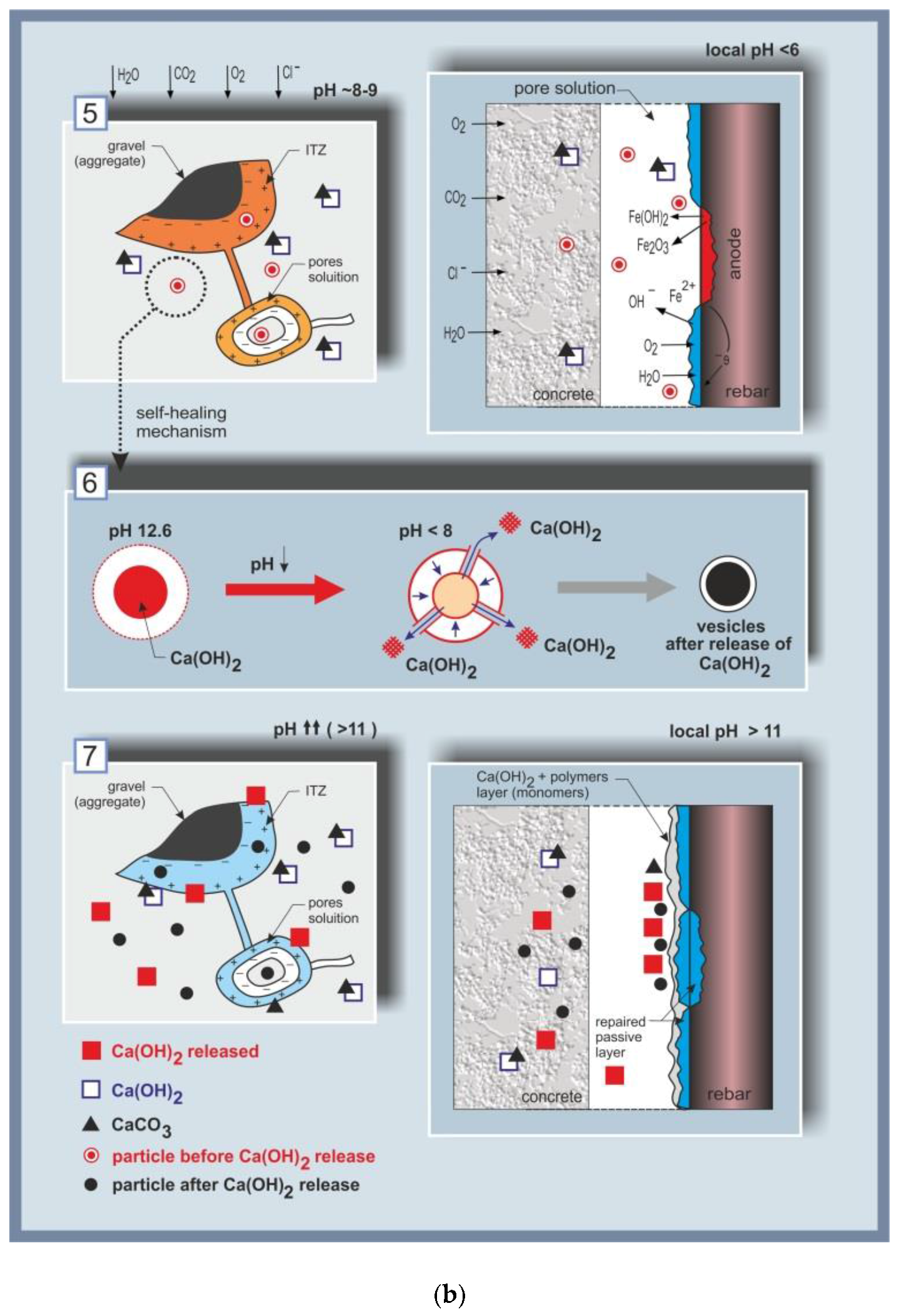
4.2. The Concept with the Added Value of the Self-Healing of Corrosion Damage
5. Credibility of the Concept and Approach
5.1. PEO-b-PS Performance in Contact with Cementitious Materials—Preliminary Studies
5.2. Micelles and Vesicles in (Reinforced) Cementitious Materials—Brief Review of the Main Outcomes
5.3. The Effect of Vesicles on the Corrosion Performance of Steel and on Bulk Matrix Properties
- (i)
- After 1 h and 3 h of treatment, the corroding CEn specimens (vesicle-free medium) presented OCP values in the range of those for the control CE (ca. −180 mV). For CEn specimens, corrosion initiation occurred between 3 h and 24 h and was sustained (and propagated) towards 96 h and 168 h. This is proven by the recorded cathodic OCP shift after 24 h, reaching ca. −400 mV towards the end of the test. For CEn at the initial time intervals (1 h and 3 h), corrosion initiation and propagation compete with passive layer formation in the alkaline medium, while with treatment, the reaction mechanisms as previously described in Section 2.2., were determined by the rate of chloride ion adherence, surface layer dissolution and passivity breakdown;
- (ii)
- For the time intervals of 1 h and 3 h, the specimens in both the corroding and control conditions, where vesicles were present (i.e., CEV, CEVn and CEVC and CEVCn), initially exhibited more cathodic OCP values (between −200 and −230 mV). This is due to the competitive mechanisms of passive layer formation in the alkaline medium and the effect of vesicles and/or chloride ions. In other words, when vesicles were present, these acted as a barrier towards both passive layer formation and chloride-induced corrosion. The vesicles induced a resistance polarization for the oxidation and reduction reactions on the steel surface;
- (iii)
- For the case of steel treated in chloride-free, empty-vesicle-containing solution (CEV specimens), surface stabilization was gradually achieved towards 168h of treatment. However, the final OCP values were not as noble as those for the control (vesicle-free) CE case (ca. −30 mV for CE and ca. −70 mV for CEV). This result is due to the abovementioned limitations, which are not relevant for CE specimens. There was only supportive evidence for barrier effects and resistance polarization for the CEV specimens; the CEV specimens also had the highest global Rp value, recorded at the end of the test (Figure 6b). The empty vesicles in the corroding CEVn specimens initially induced the same barrier effect regarding passive layer formation, but also exerted a delay in corrosion initiation. This is evident from the ennoblement of OCP from 1 h to 24 h (ca. −200 mV to ca. −180 mV). After 24 h, however, the OCP for CEVn specimens shifted in the cathodic direction, reaching approx. −240 mV after 96 h;
- (iv)
- In contrast to all above cases, the steel electrodes in the medium with Ca-bearing vesicles (CEVC and CEVCn specimens) present similar values at 1 h and 3 h, irrespective of the presence of chloride ions in the medium. These values are more cathodic, accounting for limitations regarding passive layer formation. For the control case (CEVC) an anodic shift was observed around 168 days, similar to the CE and CEV groups. Contrary to the corroding cases (CEn and CEVn) discussed above, the corroding specimens (CEVCn) show ennoblement only at all time intervals, with the most noble OCP at the end of the test. This accounts for a restructuring of the passive film on the steel surface for CEVCn, most likely the formation of a Ca-substituted product layer with higher corrosion resistance (phenomena previously discussed in Section 2). The superior corrosion resistance for CEVCn specimens was obviously triggered by the Ca-bearing vesicles in this case.
6. Conclusions and Outlook
Supporting Information: Materials and Methods
Model Medium and (Reinforced) Cement-Based Systems
Steel Electrodes and Steel Reinforcement
Nanoparticles (Micelles and Vesicles)
Electrochemical Methods and Microscopy
Acknowledgments
Conflicts of Interest
References
- Bertolini, L.; Elsener, B.; Pedeferri, P.; Polder, R. Corrosion of Steel in Concrete: Prevention, Diagnosis, Repair, 1st ed.; Wiley: Weinheim, Germany, 2004. [Google Scholar]
- Elsener, B. Macrocell corrosion of steel in concrete—Implications for corrosion monitoring. Cem. Concr. Compos. 2002, 24, 65–72. [Google Scholar] [CrossRef]
- Garces, A.; Andrade, M.C.; Saez, A.; Alonso, M.C. Corrosion of reinforcing steel in neutral and acid solutions simulating the electrolytic environments in the micropores of concrete in the propagation period. Corros. Sci. 2005, 47, 289–306. [Google Scholar] [CrossRef]
- Petterson, K. Corrosion and Corrosion Protection of Steel in Concrete; Swamy, R.N., Ed.; Academic Press: Sheffield, UK, 1994; p. 461. [Google Scholar]
- Pedeferri, P. Cathodic protection and cathodic prevention. Constr. Build. Mater. 1996, 10, 391–402. [Google Scholar] [CrossRef]
- Castle, J.E. Cathodic disbondment. AIP Conf. Proc. 1996, 354, 165–175. [Google Scholar]
- Andrade, C.; Keddam, M.; Nóvoa, X.R.; Pérez, M.C. Electrochemical behaviour of steel rebars in concrete: Influence of environmental factors and cement chemistry. Electrochim. Acta 2001, 46, 3905–3912. [Google Scholar] [CrossRef]
- Gaidis, J.M. Chemistry of corrosion inhibitors. Cem. Concr. Comp. 2004, 26, 181–189. [Google Scholar] [CrossRef]
- Wombacher, F.; Maeder, U.; Marazzani, B. Aminoalcohol based mixed corrosion inhibitors. Cem. Concr. Compos. 2004, 26, 209–216. [Google Scholar] [CrossRef]
- Volpi, E.; Foiadelli, C.; Trasatti, S.; Koleva, D.A. Development of smart corrosion inhibitors for reinforced concrete structures exposed to a microbial environment. Ind. Eng. Chem. Res. 2017, 20, 5778–5794. [Google Scholar] [CrossRef]
- Bassi, R.; Davies, H. Testing Anti-Carbonation Coatings for Concrete. BRE Information paper 7/96, 1996, ISBN 9781860810930. Available online: http://www.ihsti.com/CIS/document/87656.
- Buenfeld, N.; Zhang, J.Z. Chloride diffusion through surface-treated mortar specimens. Cem. Concr. Res. 1998, 28, 665–674. [Google Scholar] [CrossRef]
- Obada, K.; Yeomans, S.R. Bond of ribbed galvanized reinforcing steel in concrete. Cem. Concr. Compos. 2000, 22, 459–467. [Google Scholar]
- The Concrete Society. Diagnosis of Deterioration in Concrete; Concrete Society TR No56; The Concrete Society: Camberley, UK, 2000. [Google Scholar]
- Andrade, C.; Holst, J.D. Coating Protection for Reinforcement; State of the Art Report; Comite Euro Intemational Du Beton, Ed.; Thomas Telford Publication: London, UK, 1995; p. 51. [Google Scholar]
- Mietz, J. Electrochemical Rehabilitation Methods for Reinforced Concrete Structures: A State of the Art Report; Institute of Materials: London, UK, 1998. [Google Scholar]
- Koleva, D.A.; Hu, J.; Fraaij, A.L.A.; Stroeven, P.; Boshkov, N.; van Breugel, K. Cathodic protection revisited: Impact on structural morphology sheds new light on its efficiency. Cem. Concr. Compos. 2006, 28, 696–706. [Google Scholar] [CrossRef]
- Koleva, D.A.; Copuroglu, O.; van Breugel, K.; Ye, G.; de Wit, J.H.W. Electrical resistivity and microstructural properties of concrete materials in conditions of current flow. Cem. Concr. Compos. 2008, 30, 731–744. [Google Scholar] [CrossRef]
- Corradi, M.; Khurana, R.; Magarotto, R. Controlling performance in ready mixed concrete. Concr. Int. 2004, 26, 123–126. [Google Scholar]
- Li, G. Properties of high-volume fly ash concrete incorporating nano-SiO2. Cem. Concr. Res. 2004, 34, 1043–1049. [Google Scholar]
- Dalton, A.B.; Collins, S.; Muñoz, E.; Razal, J.M.; von Howard, E.; Ferraris, J.P.; Coleman, J.N.; Kim, G.B.; Baughman, R.H. Super-tough carbon nanotube fibres. Nature 2003, 423, 703. [Google Scholar] [CrossRef] [PubMed]
- Silva, D.; Monteiro, P. Hydration evolution of C3S-EVA composite analyzed by soft X-ray microscopy. Cem. Concr. Res. 2005, 35, 351–357. [Google Scholar] [CrossRef]
- Li, C.Z.; Feng, N.Q.; Li, Y.D.; Chen, R.J. Effects of polyethlene oxide chains on the performance of poly-carboxylate-type water-reducers. Cem. Concr. Res. 2005, 35, 867–873. [Google Scholar] [CrossRef]
- Nicoleau, L. New Calcium Silicate Hydrate Network. Transp. Res. Rec. J. Transp. Res. Board 2010, 2142, 42–51. [Google Scholar] [CrossRef]
- Dennis, R.V.; Viyannalage, L.T.; Gaikwad, A.V.; Rout, T.K.; Banerjee, S. Graphene nanocomposite coatings for protecting low-alloy steels from corrosion. ACS Bull. 2013, 92, 18–24. [Google Scholar]
- Kirkland, N.T.; Schiller, T.; Medhekar, N.; Birbilis, N. Exploring graphene as a corrosion protection barrier. Corros. Sci. 2012, 56, 1–4. [Google Scholar] [CrossRef]
- Lv, S.; Ma, Y.; Qiu, C.; Zhou, Q. Regulation of GO on cement hydration crystals and its toughening effect. Mag. Concr. Res. 2013, 65, 1246–1254. [Google Scholar] [CrossRef]
- Collepardi, S.; Borsoi, A.; Ogoumah Olagot, J.J.; Troli, R.; Collepardi, M.; Cursio, A.Q. Influence of nano-sized mineral additions on performance of SCC. In Proceedings of the 6th International Congress Global Construction, Ultimate Concrete Opportunities, Dundee, UK, 5–7 July 2005. [Google Scholar]
- Zhang, M.H.; Li, H. Pore structure and chloride permeability of concrete containing nano-particles for pavement. Constr Build.. Mater. 2011, 25, 608–616. [Google Scholar] [CrossRef]
- Hui, L.; Xiao, H.; Yuan, J.; Ou, J. Microstructure of cement mortar with nano-particles. Compos. B 2004, 35, 185–189. [Google Scholar]
- Sanchez, F.; Sobolev, K. Nanotechnology in concrete—A review. Constr. Build. Mater. 2010, 24, 2060–2071. [Google Scholar] [CrossRef]
- Plank, J.; Sachsenhauser, B. Experimental determination of the effective anionic charge density of polycarboxylate superplasticizers in cement pore solution. Cem. Concr. Res. 2009, 39, 1–5. [Google Scholar] [CrossRef]
- Bassioni, G. A study towards greener construction. Appl. Energy 2012, 93, 132–137. [Google Scholar] [CrossRef]
- Albani, O.A.; Zerbino, J.O.; Vilche, J.R.; Arvia, A.J. A comparative electrochemical and ellipsometric study of the iron electrodes in different alkaline electrolytes. Electrochim. Acta 1986, 31, 1403–1411. [Google Scholar] [CrossRef]
- Misawa, T. The thermodynamic consideration for Fe-H2O system at 25 °C. Corros. Sci. 1973, 13, 659–676. [Google Scholar] [CrossRef]
- Sudakar, C.; Subbanna, G.N.; Kutty, T.R.N. Effect of anions on the phase stability of γ-FeOOH nanoparticles and the magnetic properties of gamma-ferric oxide derived from lepidocrocite. J. Phys. Chem. Solids 2004, 64, 2337–2349. [Google Scholar] [CrossRef]
- Bentur, A.; Diamond, S.; Berke, N.S. Steel Corrosion in Concrete; E & FN: London, UK, 1997; p. 27. [Google Scholar]
- MacDougall, B.; Graham, M.J. Corrosion Mechanisms in Theory and Practice, 2nd ed.; Marcus, P., Oudar, J., Eds.; Marcel Dekker Inc.: New York, NY, USA, 2002; p. 143. [Google Scholar]
- Diéz-Perez, I.; Vericat, C.; Gorostiza, P.; Sanz, F. The iron passive film breakdown in chloride media may be mediated by transient chloride-induced surface states located within the band gap. Electrochem. Commun. 2006, 8, 627–632. [Google Scholar] [CrossRef]
- Sato, N. An overview on the passivity of metals. Corros. Sci. 1990, 31, 1–19. [Google Scholar] [CrossRef]
- Angst, U.M.; Boschmann, C.; Wagner, M.; Elsener, B. Experimental protocol to determine the chloride threshold value for corrosion in samples taken from reinforced concrete structures. J. Vis. Exp. 2017, 126, 56229. [Google Scholar] [CrossRef] [PubMed]
- Page, C.L. Advances in understanding and techniques for controlling reinforcement corrosion. In Proceedings of the 15th International Corrosion Congress, Granada, Spain, 22–27 September 2002. [Google Scholar]
- Vassie, P. Reinforcement corrosion and the durability of concrete bridges. PRW Proc. Inst. Civ. Eng. 1984, 76, 713–723. [Google Scholar] [CrossRef]
- Glass, G.K.; Buenfeld, N.R. The presentation of the chloride threshold level for corrosion of steel in concrete. Corros. Sci. 1997, 39, 1001–1013. [Google Scholar] [CrossRef]
- Gaal, G.C.M. Prediction of Deterioration of Concrete Bridges. Ph.D. Thesis, Delft University of Technology, Delft, The Netherlands, 2004. [Google Scholar]
- Poursaee, A. Corrosion of Steel in Concrete; Tuutti, K., Ed.; Swedish Cement and Concrete Research Institute: Stockholm, Sweden, 1982. [Google Scholar]
- Reddy, B.; Glass, G.K.; Lim, P.J.; Buenfeld, N.R. On the corrosion risk presented by chloride bound in concrete. Cem. Concr. Compos. 2002, 24, 1–5. [Google Scholar] [CrossRef]
- Hussain, S.E.; Rasheeduzzafar, S.; Al-Gahtani, A.S. Influence of sulfates on chloride binding in cements. Cem. Concr. Res. 1994, 24, 8–24. [Google Scholar] [CrossRef]
- Leek, D.S.; Poole, A.B. The breakdown of the passive film on high yield mild steel by chloride ions. In Proceedings of the Third International Symposium on Corrosion of Reinforcement in Concrete, Wishaw, UK, 21–24 May 1990; Elsevier Applied Science: London, UK, 1990; pp. 65–73. [Google Scholar]
- Oranowska, H.; Szklarska-Smialowska, Z. An electrochemical and ellipsometric investigation of surface films grown on iron in saturated calcium hydroxide solutions with or without chloride ions. Corros. Sci. 1981, 21, 735–747. [Google Scholar] [CrossRef]
- Koleva, D.A.; Boshkov, N.; van Breugel, K.; De Wit, J.H.W. Steel corrosion resistance in model solutions, containing waste materials. Electrochim. Acta 2011, 58, 628–646. [Google Scholar] [CrossRef]
- Koleva, D.A. Electrochemical behavior of corroded and protected construction steel in cement extract. Mater. Corros. 2011, 62, 240–251. [Google Scholar] [CrossRef]
- Koleva, D.A.; Hu, J.; Fraaij, A.L.A.; Stroeven, P.; Boshkov, N.; de Wit, J.H.W. Quantitative characterisation of steel/cement paste interface microstructure and corrosion phenomena in mortars suffering from chloride attack. Corros. Sci. 2006, 48, 4001–4019. [Google Scholar] [CrossRef]
- Koleva, D.A.; van Breugel, K.; De Wit, J.H.W.; van Westing, E.; Boshkov, N.; Fraaij, A.L.A. Electrochemical behavior, microstructural analysis, and morphological observations in reinforced mortar subjected to chloride ingress. J. Electrochem. Soc. 2007, 154, E45–E56. [Google Scholar] [CrossRef]
- Koleva, D.A.; Hu, J.; Fraaij, A.L.A.; van Breugel, K.; de Wit, J.H.W. Microstructural analysis of plain and reinforced mortars under chloride-induced deterioration. Cem. Concr. Res. 2007, 37, 604–617. [Google Scholar] [CrossRef]
- Koleva, D.A.; Denkova, A.G.; Boshkov, N.; van Breugel, K. Electrochemical performance of steel in cement extract and bulk matrix properties of cement paste in the presence of Pluronic 123 micelles. J. Mater. Sci. 2013, 48, 2490–2503. [Google Scholar] [CrossRef]
- Koleva, D.A.; Hu, J.; Milkova, V.; van Breugel, K. Hybrid nano/micro-particles for increased steel corrosion resistance: Particles’ alterations with pH change and steel behavior in cement extract and mortar. J. Mech. 2013, 1612, 3. [Google Scholar] [CrossRef]
- Koleva, D.A.; van Breugel, K.; Ye, G.; Zhou, J.; Chamululu, G.; Koenders, E.A.B. Porosity and permeability of mortar specimens incorporating PEO113-b-PS2l8 micelles. Am. Concr. Inst. 2009, 267, 101–110. [Google Scholar]
- Gawin, D.; Pesavento, F.; Schrefler, B. Modeling deterioration of cementitious materials exposed to calcium leaching in non-isothermal conditions. Comput. Methods Appl. Mech. Eng. 2009, 198, 3051–3083. [Google Scholar] [CrossRef]
- Kamali, S.; Gerard, B.; Moranville, M. Modelling the leaching kinetics of cement-based materials—Influence of materials and environment. Cem. Concr. Compos. 2003, 25, 451–458. [Google Scholar] [CrossRef]
- Marchand, J.; Bentz, D.P.; Samson, E.; Maltais, Y. Influence of calcium hydroxide dissolution on the transport properties of hydrated cement systems. In Workshop on the Role of Calcium Hydroxide in Concrete; Anna Maria Island: Holmes Beach, FL, USA, 2000; pp. 113–129. [Google Scholar]
- Carde, C.; Francois, R.; Torrenti, J.M. Leaching of both calcium hydroxide and C-S-H from cement paste: Modeling the mechanical behaviour. Cem. Concr. Res. 1996, 26, 1257–1268. [Google Scholar] [CrossRef]
- Koleva, D.A. Corrosion and Protection in Reinforced Concrete. Ph.D. Thesis, TU Delft, Delft, The Netherlands, 2007. [Google Scholar]
- Wang, G.; de Kruijff, R.; Stuart, M.C.A.; Mendes, E.; Wolterbeek, H.T.; Denkova, A.G. Polymersomes as radionuclide carriers loaded via active ion transport through the hydrophobic bilayer. Soft Matter. 2013, 9, 727–734. [Google Scholar] [CrossRef]
- Koleva, D.; Boshkov, N.; Raichevski, G.; Veleva, L. Electrochemical corrosion behaviour and surface morphology of electrodeposited zinc, zinc-cobalt and their composite coatings. Trans. Inst. Metal Finish. 2005, 83, 188–193. [Google Scholar] [CrossRef]
- Koleva, D.A.; Boshkov, N.; Bachvarov, V.; Zhan, H.; de Wit, J.H.W.; van Breugel, K. Application of PEO113-b-PS218 nano-aggregates for improved protective characteristics of composite zinc coatings in chloride-containing environment. Surf. Coat. Technol. 2010, 204, 3760–3772. [Google Scholar] [CrossRef]
- Lee, J.; Mahendra, S.; Alvarez, P.J.J. Nanomaterials in the construction industry: A review of their applications and environmental health and safety considerations. ACS Nano 2010, 4, 3580–3590. [Google Scholar] [CrossRef] [PubMed]
- Chiappetta, D.A.; Sosnik, A. Poly(ethylene oxide)-poly(propylene oxide) block copolymer micelles as drug delivery agents: Improved hydrosolubility, stability and bioavailability of drugs. Eur. J. Pharmac. Biopharmac. 2007, 66, 303–317. [Google Scholar] [CrossRef] [PubMed]
- Pitto-Barry, A.; Barry, N.P.E. Pluronic® block-copolymers in medicine: From chemical and biological versatility to rationalisation and clinical advances. Polym. Chem. 2014, 5, 3291–3297. [Google Scholar] [CrossRef]
- Zhu, Y.Q.; Wang, F.Y.K.; Zhang, C.; Du, J.Z. Preparation and mechanism insight of nuclear envelope-like polymer vesicles for facile loading of biomacromolecules and enhanced biocatalytic activity. ACS Nano 2014, 8, 6644–6654. [Google Scholar] [CrossRef] [PubMed]
- Zhua, Y.; Bo, Y.; Shuai, C.; Dua, J. Polymer vesicles: Mechanism, preparation, application, and responsive behaviour. Prog. Polym. Sci. 2017, 64, 1–22. [Google Scholar] [CrossRef]
- Meeuwissen, S.A.; Rioz-Martinez, A.; de Gonzalo, G.; Fraaije, M.W.; Gotor, V.; van Hest, J.C.M. Cofactor regeneration in polymersome nano-reactors: Enzymatically catalysed Baeyer–Villiger reactions. J. Mater. Chem. 2011, 21, 18923–18926. [Google Scholar] [CrossRef]
- Berthier, D.L.; Schmidt, I.; Fieber, W.; Schatz, C.; Furrer, A.; Wong, K.; Lecom-mandoux, S. Controlled release of volatile fragrance molecules from PEO-b-PPO-b-PEO block copolymer micelles in ethanol–water mixtures. Langmuir 2010, 26, 7953–7961. [Google Scholar] [CrossRef] [PubMed]
- Geng, Q.R.; Du, J.Z. Reduction of 4-nitrophenol catalyzed by silver nanoparticles supported on polymer micelles and vesicles. RSC Adv. 2014, 4, 16425–16428. [Google Scholar] [CrossRef]
- Zhu, Y.Q.; Fan, L.; Yang, B.; Du, J.Z. Multifunctional homopolymer vesicles for facile immobilization of gold nanoparticles and effective water remediation. ACS Nano 2014, 8, 5022–5031. [Google Scholar] [CrossRef] [PubMed]
- Du, J.Z.; Fan, L.; Liu, Q.M. pH-sensitive block copolymer vesicles with variable trigger points for drug delivery. Macromolecules 2012, 45, 8275–8283. [Google Scholar] [CrossRef]
- Qin, S.H.; Geng, Y.; Discher, D.E.; Yang, S. Temperature-controlled assembly and release from polymer vesicles of poly(ethylene oxide)-block-poly(N-isopropylacrylamide). Adv. Mater. 2006, 18, 2905–2909. [Google Scholar] [CrossRef]
- Cerritelli, S.; Velluto, D.; Hubbell, J.A. PEG-SS-PPS: Reduction-sensitive disulfide block copolymer vesicles for intracellular drug delivery. Biomacromolecules 2007, 8, 1966–1972. [Google Scholar] [CrossRef] [PubMed]
- Babin, J.; Pelletier, M.; Lepage, M.; Allard, J.F.; Morris, D.; Zhao, Y. A new two-photon-sensitive block copolymer nanocarrier. Angew. Chem. Int. Ed. 2009, 48, 3329–3332. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Koleva, D.A.; Ma, Y.; Schlangen, E.; Petrov, P.; van Breugel, K. The influence of admixed micelles on the microstructural properties and global performance of cement-based materials. Cem. Concr. Res. 2012, 42, 1122–1133. [Google Scholar] [CrossRef]
- Hu, J.; Koleva, D.A.; van Breugel, K. Corrosion performance of reinforced mortar in the presence of polymeric nano-aggregates: Electrochemical behavior, surface analysis, and properties of the steel/cement paste interface. J. Mater. Sci. 2012, 47, 4981–4995. [Google Scholar] [CrossRef]
- Hu, J.; Koleva, D.A.; Petrov, P.; van Breugel, K. Polymeric vesicles for corrosion control in reinforced mortar: Electrochemical behavior, steel surface analysis and bulk matrix properties. Corros. Sci. 2012, 65, 414–430. [Google Scholar] [CrossRef]
- Hu, J.; Koleva, D.A.; De Wit, J.H.W.; Kolev, H.; van Breugel, K. Corrosion performance of carbon steel in simulated pore solution in the presence of micelles. J. Electrochem. Soc. 2011, 158, C76–C87. [Google Scholar] [CrossRef]
- Hu, J.; Koleva, D.A.; Ma, Y.; Schlangen, E.; van Breugel, K. Early age hydration, microstructure and micromechanical properties of cement paste modified with polymeric vesicles. J. Adv. Concr. Technol. 2013, 11, 291–300. [Google Scholar] [CrossRef]
- Jansen, D.; Neubauer, J.; Goetz-Neunhoeffer, F.; Haerzschel, R.; Hergeth, W.D. Change in reaction kinetics of Portland cement caused by a superplasticizer—Calculation of heat flow curves from XRD data. Cem. Concr. Res. 2012, 42, 327–332. [Google Scholar] [CrossRef]
- Mollah, M.; Adams, W.; Schennach, R.; Cocke, D. A review of cement-superplasticizer interactions and their models. Adv. Cem. Res. 2000, 12, 153–161. [Google Scholar] [CrossRef]
- Matyjaszewski, K.; Xia, J. Atom transfer radical polymerization. Chem. Rev. 2001, 101, 2921–2990. [Google Scholar] [CrossRef] [PubMed]
- Stern, M.; Geary, A.L. Electrochemical Polarization I. A Theoretical Analysis of the Shape of Polarization Curves. J. Electrochem. Soc. 1957, 104, 56–63. [Google Scholar] [CrossRef]
- Andrade, C.; Alonso, C. Corrosion rate monitoring in the laboratory and on-site. Constr. Build. Mater. 1996, 10, 315–328. [Google Scholar] [CrossRef]
- Zornoza, E.; Paya, J.; Garces, P. Chloride-Induced corrosion of steel embedded in mortars containing fly ash and spent cracking catalyst. Corros. Sci. 2008, 50, 1567–1575. [Google Scholar] [CrossRef]









© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koleva, D.A. An Innovative Approach to Control Steel Reinforcement Corrosion by Self-Healing. Materials 2018, 11, 309. https://doi.org/10.3390/ma11020309
Koleva DA. An Innovative Approach to Control Steel Reinforcement Corrosion by Self-Healing. Materials. 2018; 11(2):309. https://doi.org/10.3390/ma11020309
Chicago/Turabian StyleKoleva, Dessi A. 2018. "An Innovative Approach to Control Steel Reinforcement Corrosion by Self-Healing" Materials 11, no. 2: 309. https://doi.org/10.3390/ma11020309
APA StyleKoleva, D. A. (2018). An Innovative Approach to Control Steel Reinforcement Corrosion by Self-Healing. Materials, 11(2), 309. https://doi.org/10.3390/ma11020309