Optical Aggregation of Gold Nanoparticles for SERS Detection of Proteins and Toxins in Liquid Environment: Towards Ultrasensitive and Selective Detection

 ,
,  , ,
, ,  ,
,
Abstract
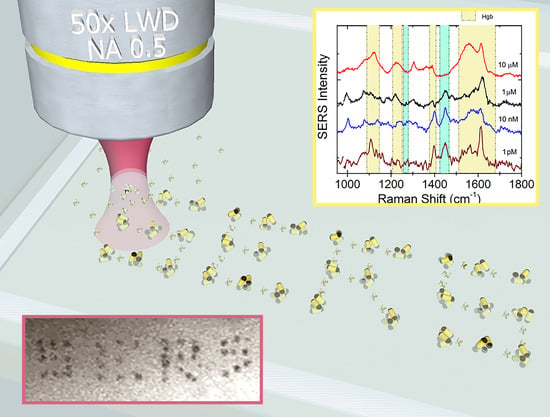
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Gold Nanorods
2.2. Glass Microcell
2.3. Protein Solutions
2.4. Gold Nanorods Functionalization
2.5. Toxin Solutions Preparation
2.6. Dynamic Light Scattering
2.7. Extinction Spectroscopy
2.8. Raman Spectroscopy Setups
3. Results and Discussion
3.1. LIQUISOR Operation
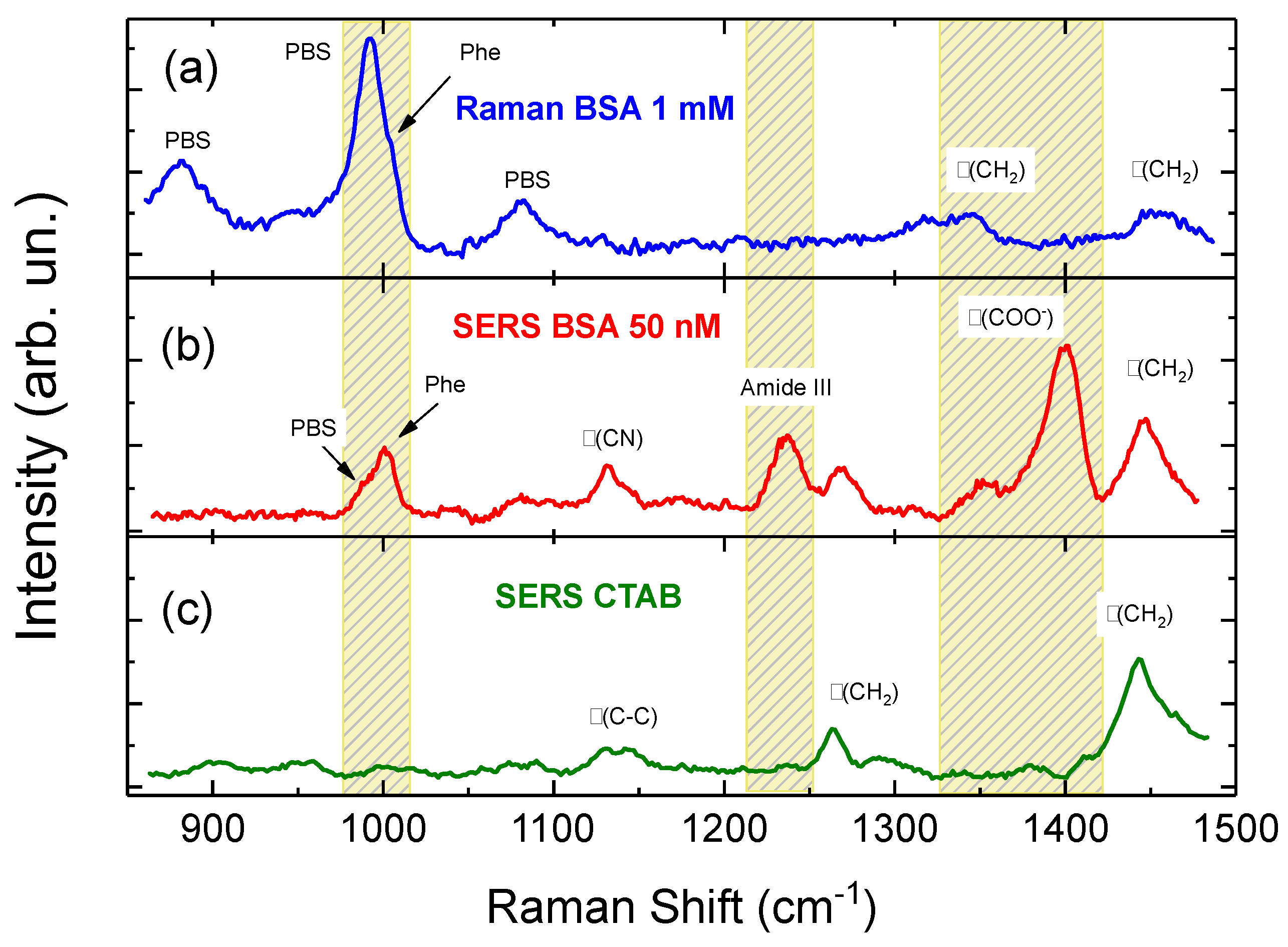
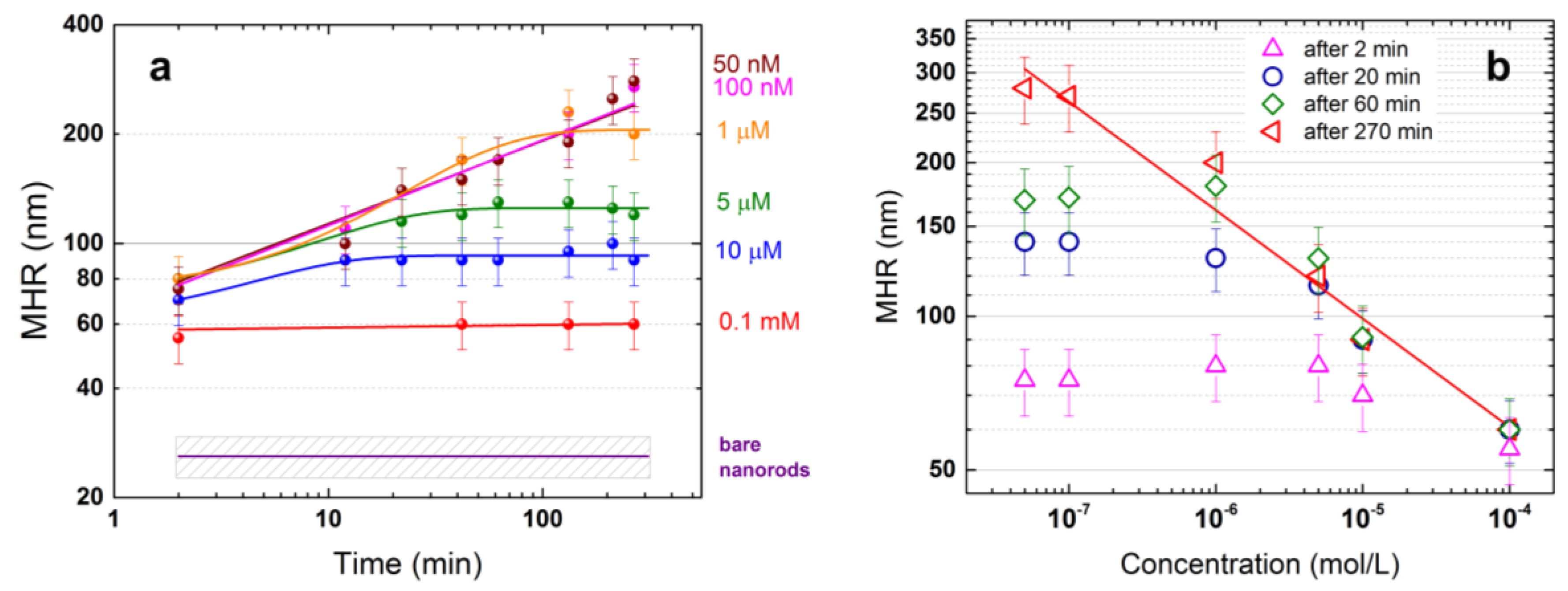
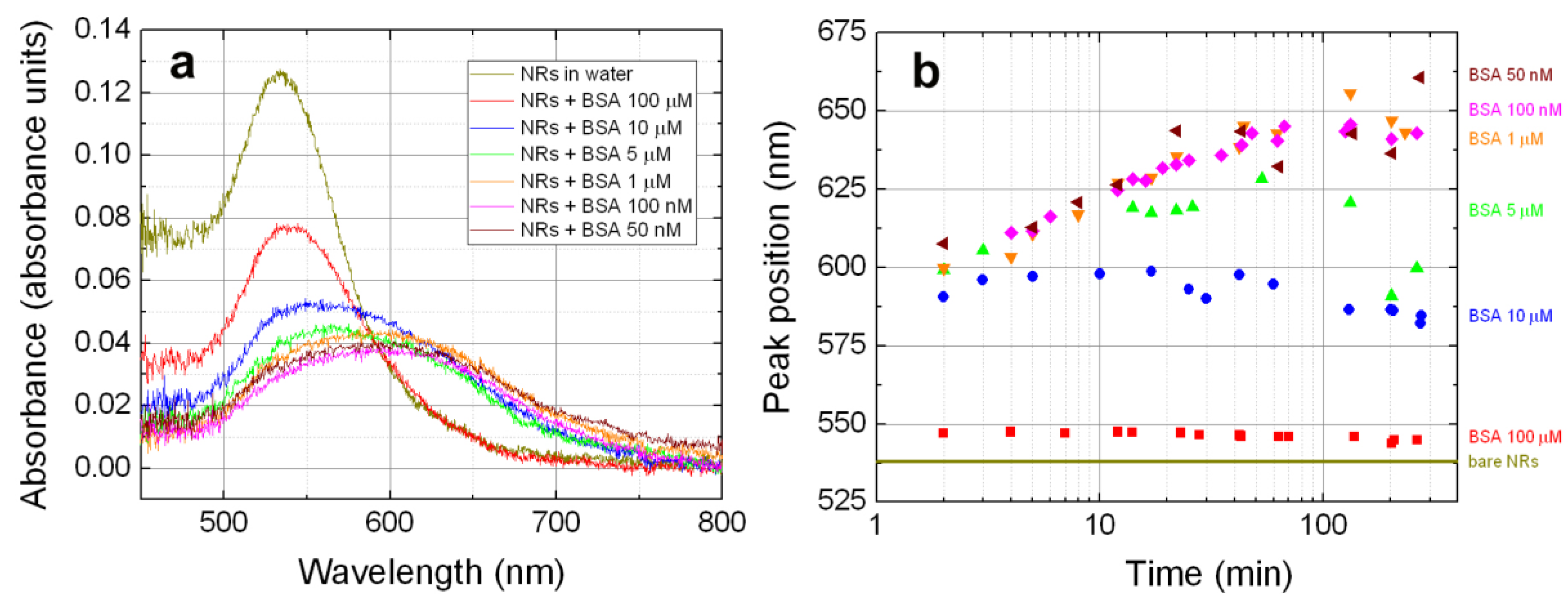
3.2. NRs-Biomolecules Interactions in Buffered Solutions
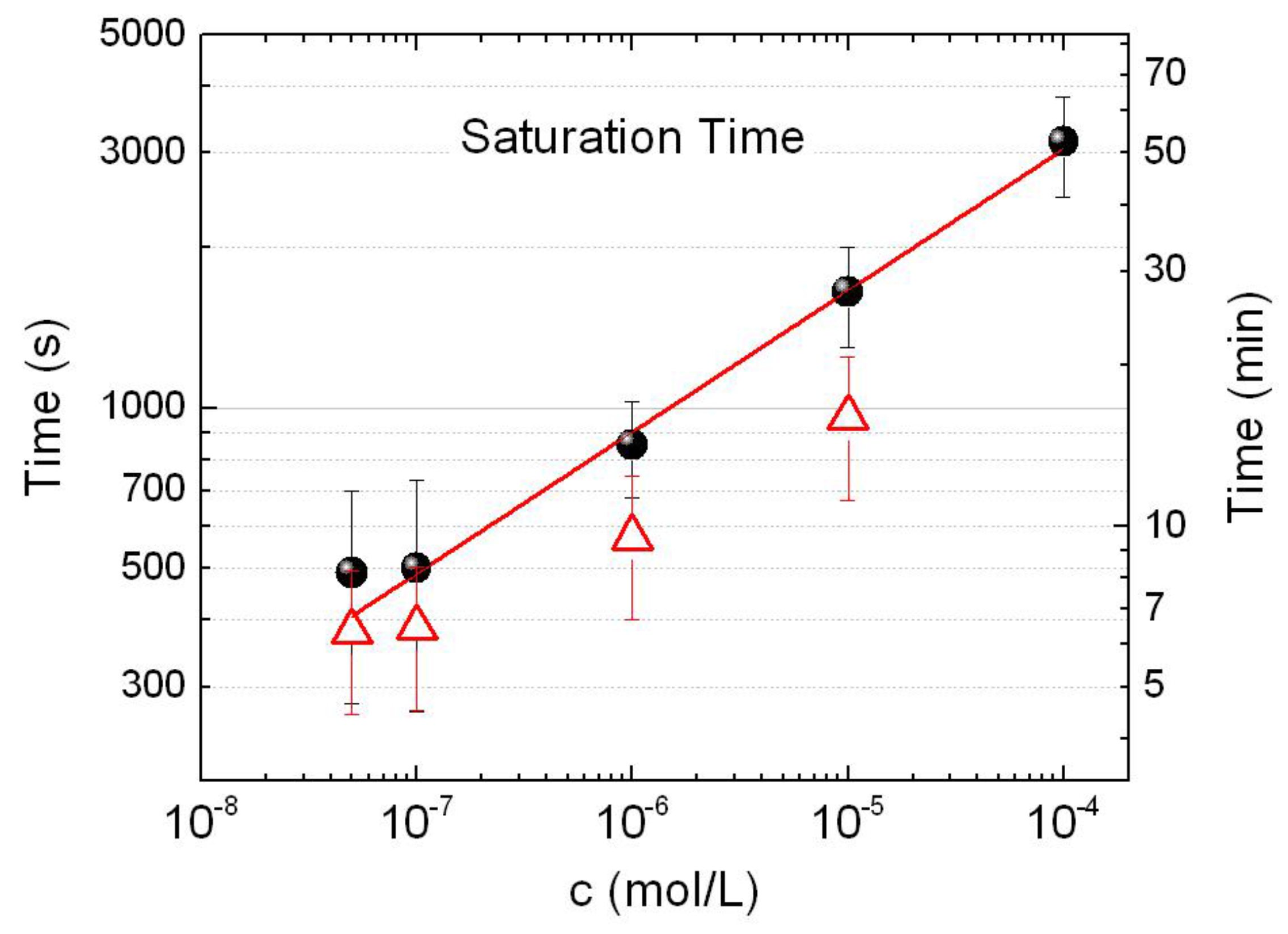
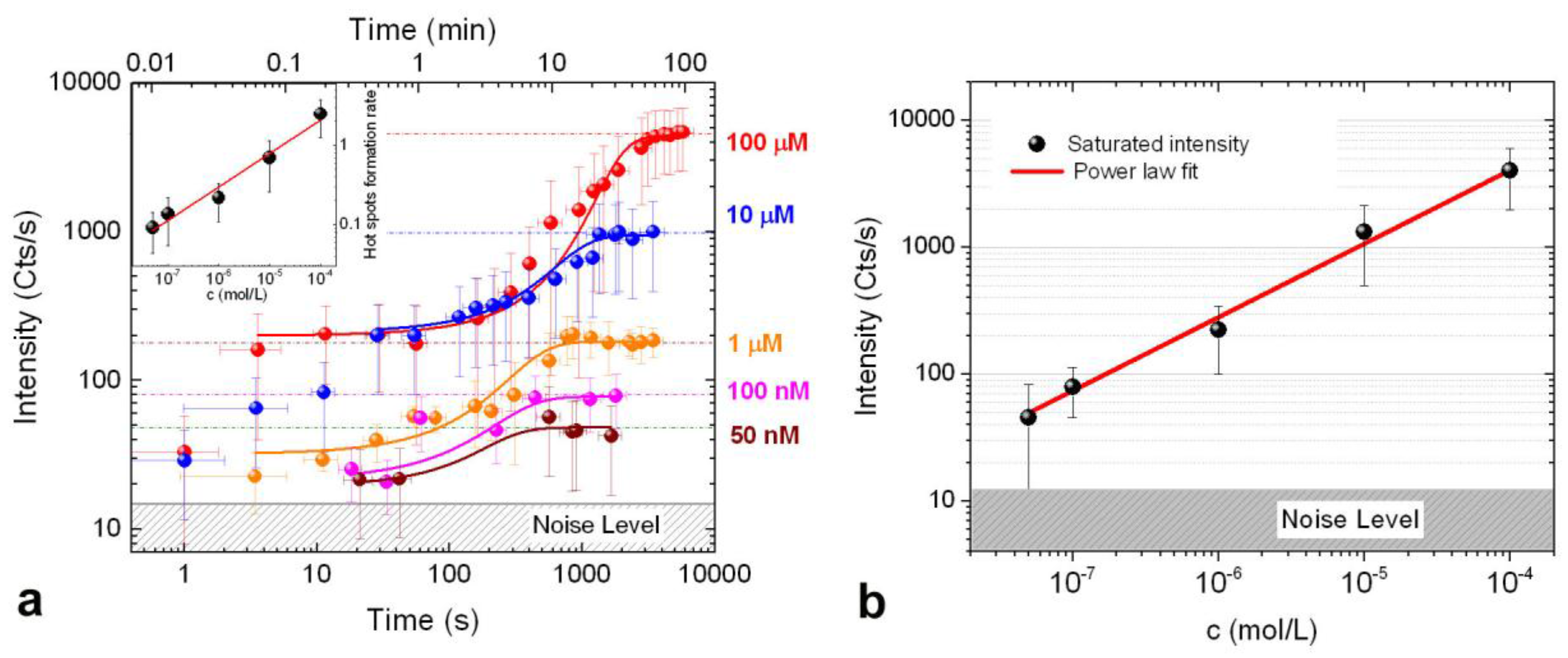
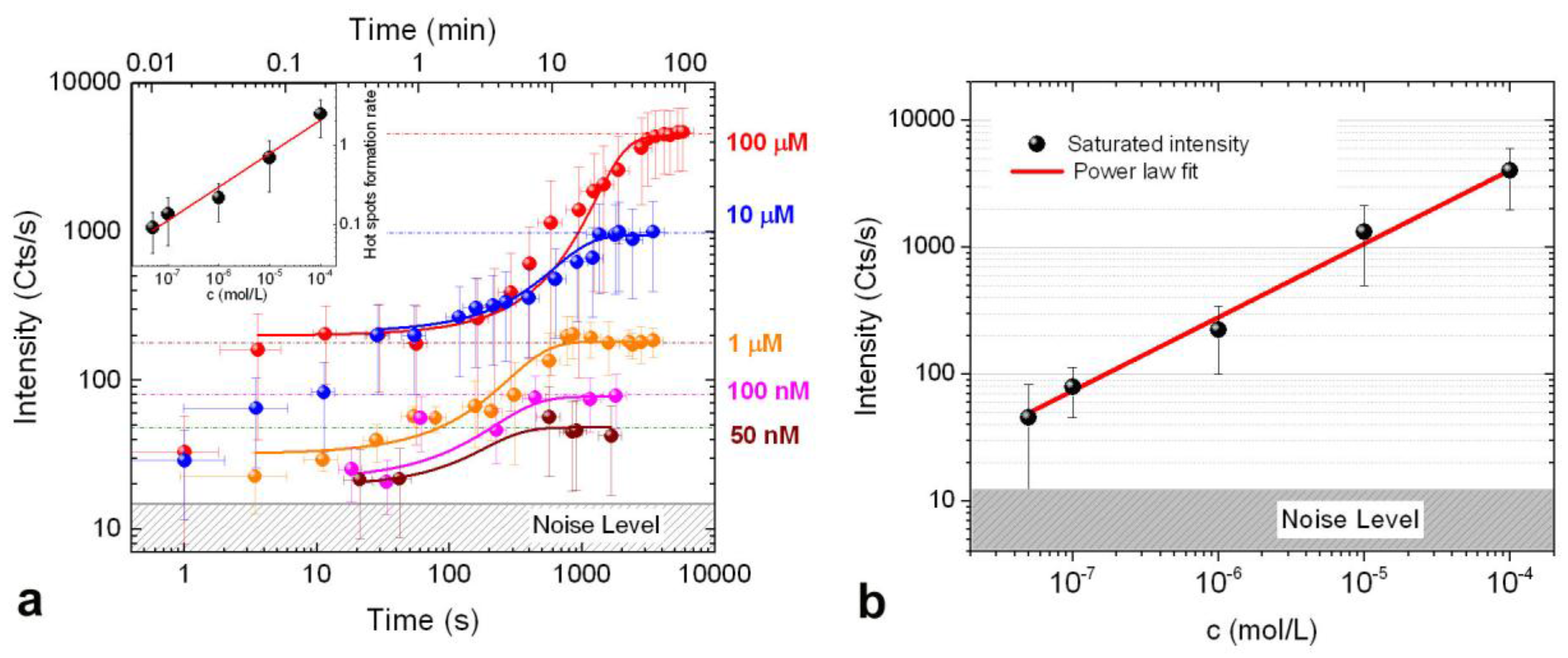
3.3. SERS Intensity vs. Concentration
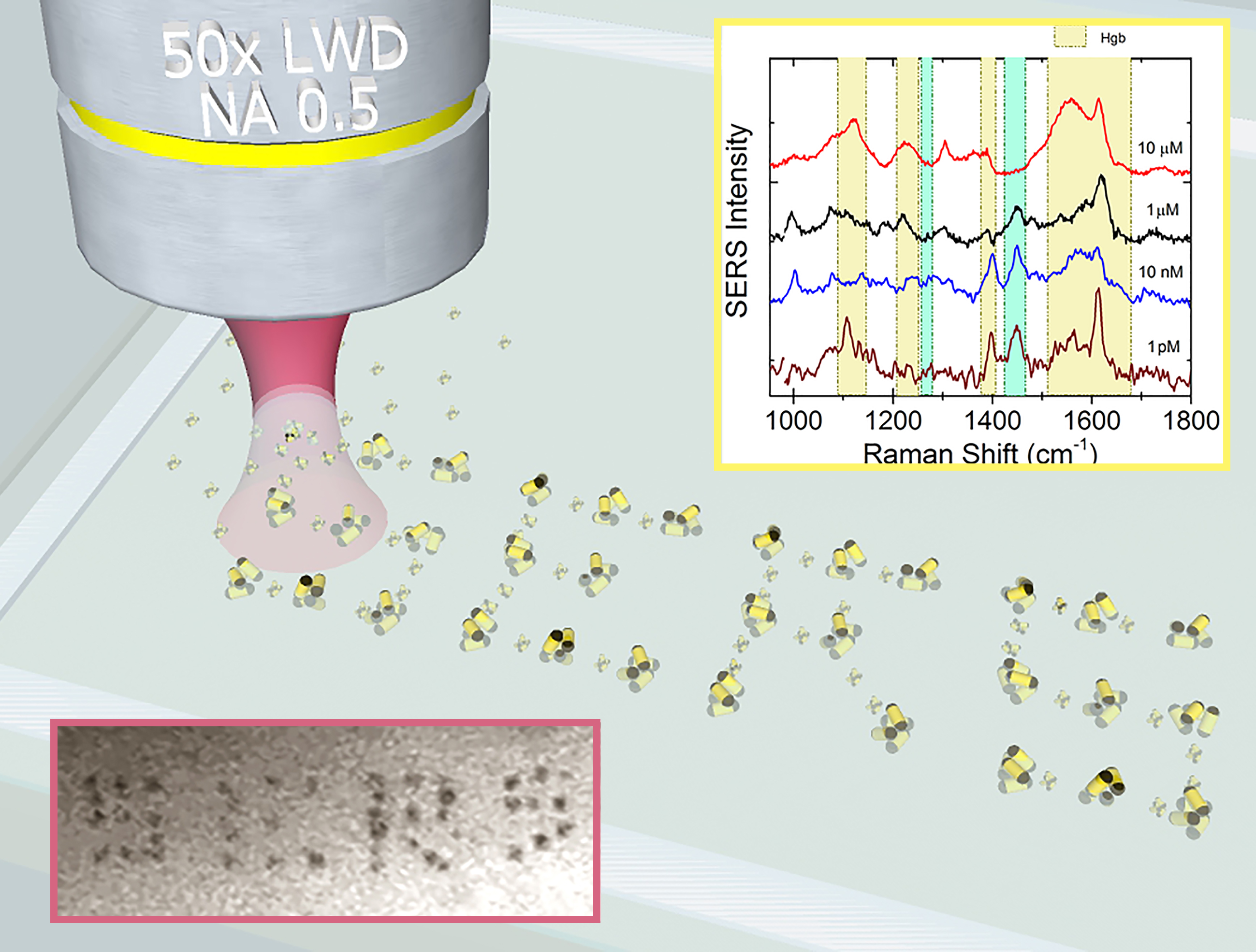
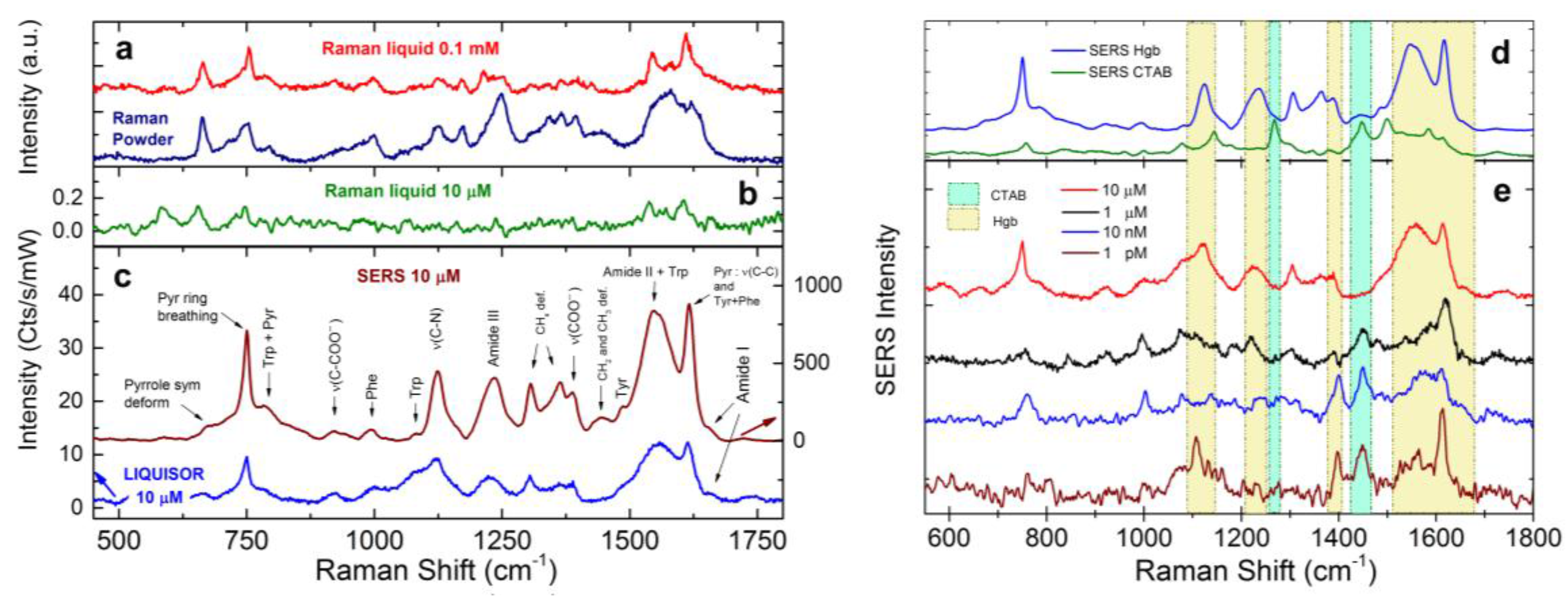
3.4. LIQUISOR Detection of Hemeproteins
3.4.1. Hemoglobin
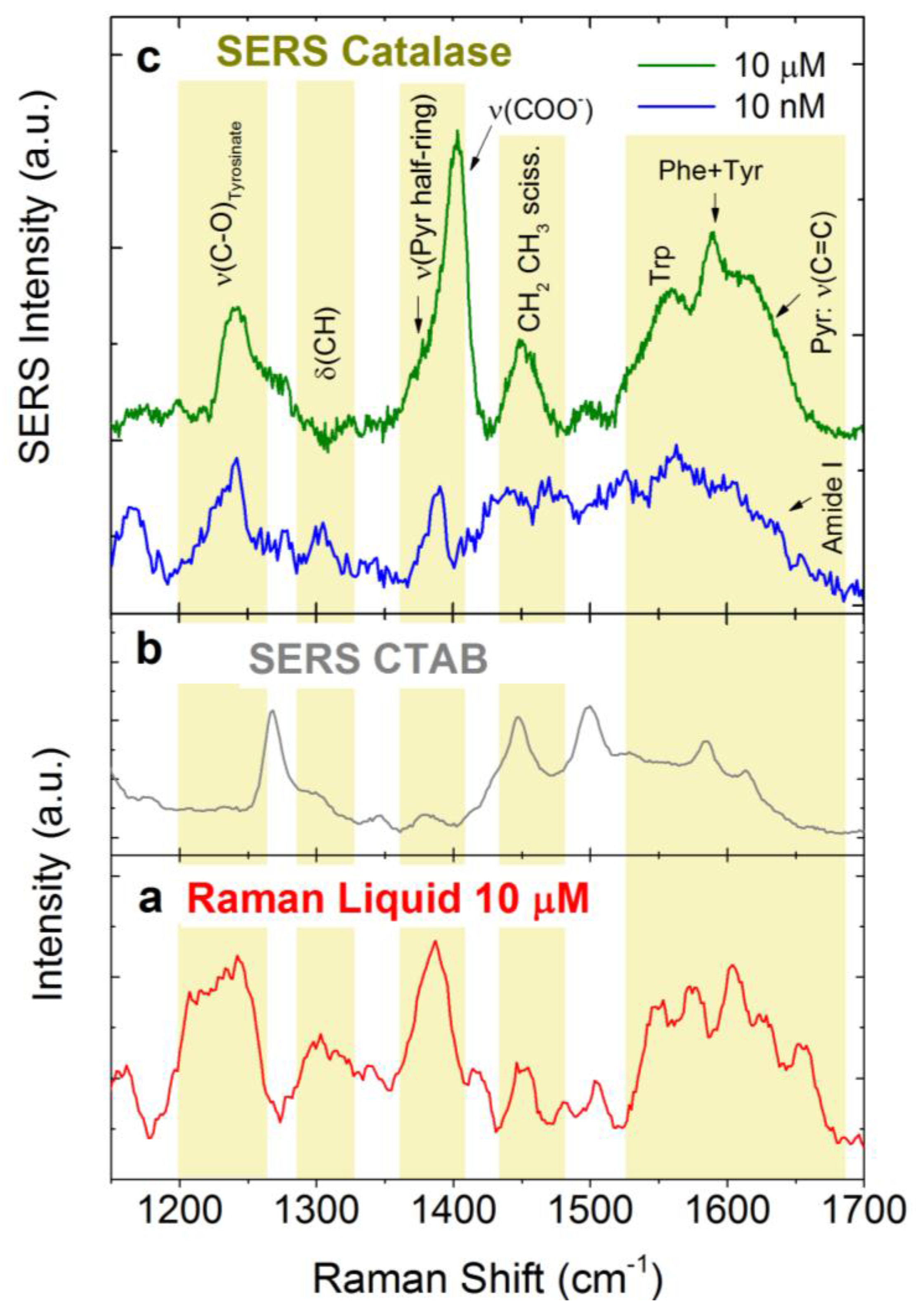
3.4.2. Catalase
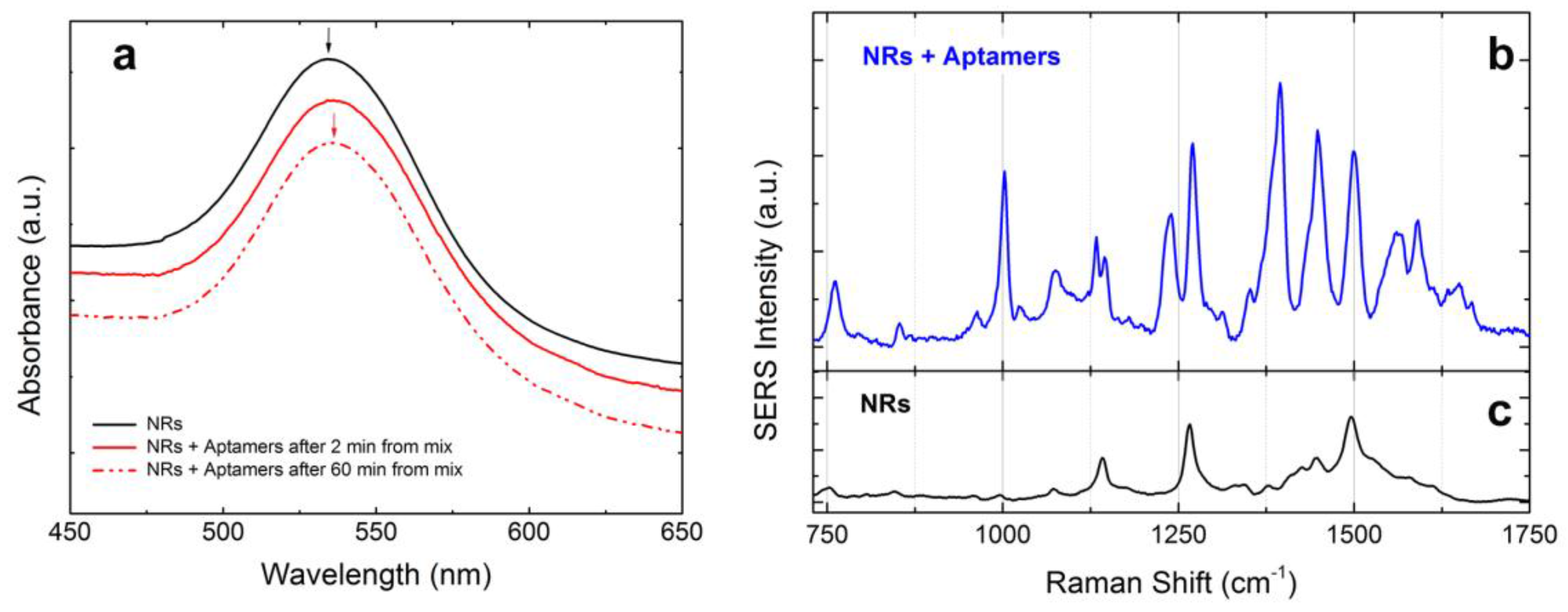
3.5. LIQUISOR Detection of Ochratoxin A Mediated by Aptamers
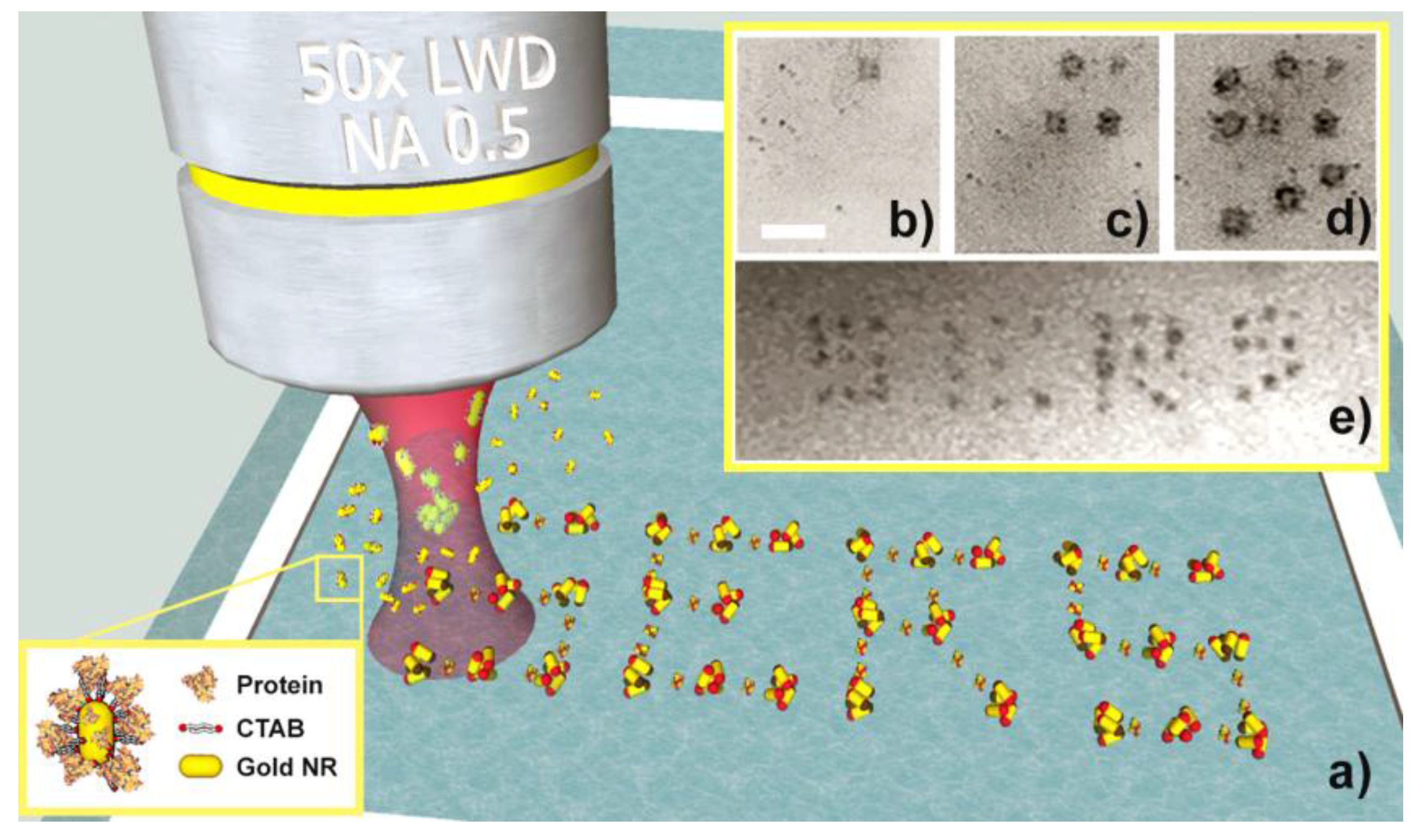
- Functionalization of the gold NRs with bioreceptors through the substitution of the CTAB layer with thiolated aptamers. This is obtained during incubation of CTAB-coated NRs with aptamers in a PB solution (10 mM). PB destabilizes the CTAB, fostering the interaction between the NRs and the thiol groups of the aptamers.
- Incubation of the functionalized NRs with the target molecules. The bioreceptors on the NRs surface will selectively capture the target proteins in the solution, creating BIO-NRCs surrounded by a double layer including the aptamers and the captured protein.
- Normal operation of the LIQUISOR procedure (λ = 660 nm, Objective 60×, NA 0.7). Laser irradiation will produce aggregates in which the target molecules located at the hot spots will experience an enhanced Raman scattering. This latter signal shows up and is compared to the SERS contribution from the aptamers.
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Le Ru, E.; Etchegoin, P.G. Principles of Surface Enhanced Raman Spectroscopy; Elsevier: Amsterdam, The Netherlands, 2009; ISBN 978-0-444-52779-0. [Google Scholar]
- Schluecker, S. Surface-Enhanced Raman Spectroscopy: Concepts and Chemical Applications. Angew. Chem. Int. Ed. 2014, 53, 4756–4795. [Google Scholar] [CrossRef] [PubMed]
- Han, X.X.; Huang, G.G.; Zhao, B.; Ozaki, Y. Label-free highly sensitive detection of proteins in aqueous solutions using surface-enhanced Raman scattering. Anal. Chem. 2009, 81, 3329–3333. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Gu, C.; Qian, F.; Li, Y.; Zhang, Y.L.; Gu, C.; Qian, F.; Li, Y.; Zhang, Y.L. Highly sensitive detection of proteins and bacteria in aqueous solution using surface-enhanced Raman scattering and optical fibers. Anal. Chem. 2011, 83, 5888–5894. [Google Scholar] [CrossRef] [PubMed]
- Schluecker, S. SERS Microscopy: Nanoparticle Probes and Biomedical Applications. Chemphyschem 2009, 10, 1344–1354. [Google Scholar] [CrossRef] [PubMed]
- Maier, S.A. Plasmonics: Fundamentals and Applications; Springer: New York, NY, USA, 2007; ISBN 978-0-387-37825-1. [Google Scholar]
- Amendola, V.; Pilot, R.; Frasconi, M.; Maragò, O.M.; Iatì, A. Surface plasmon resonance in gold nanoparticles: A review. J. Phys. Condens. Matter 2017, 29, 203002. [Google Scholar] [CrossRef] [PubMed]
- Kneipp, K.; Haka, A.S.; Kneipp, H.; Badizadegan, K.; Yoshizawa, N.; Boone, C.; Shafer-Peltier, K.E.; Motz, J.T.; Dasari, R.R.; Feld, M.S. Surface-Enhanced Raman Spectroscopy in Single Living Cells Using Gold Nanoparticles. Appl. Spectrosc. 2002, 56, 150–154. [Google Scholar] [CrossRef]
- Feng, M.; Tachikawa, H. Surface-enhanced resonance Raman spectroscopic characterization of the protein native structure. J. Am. Chem. Soc. 2008, 130, 7443–7448. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.J.; Zong, C.; Zheng, X.-S.; Hu, P.; Feng, J.-M.; Ren, B. Label free detection of Native proteins by surface enhanced Raman spectroscopy Using iodide modified Nanoparticles. Anal. Chem. 2014, 86, 2238–2245. [Google Scholar] [CrossRef] [PubMed]
- Amendola, V.; Meneghetti, M. Laser ablation synthesis in solution and size manipulation of noble metal nanoparticles. Phys. Chem. Chem. Phys. 2009, 11, 3805–3821. [Google Scholar] [CrossRef] [PubMed]
- Zakel, S.; Rienitz, O.; Güttler, B.; Stosch, R. Double isotope dilution surface-enhanced Raman scattering as a reference procedure for the quantification of biomarkers in human serum. Analyst 2011, 136, 3956–3961. [Google Scholar] [CrossRef] [PubMed]
- Kessentini, S.; Barchiesi, D.; D’Andrea, C.; Toma, A.; Guillot, N.; Di Fabrizio, E.; Fazio, B.; Maragò, O.M.; Gucciardi, P.G.; Lamy de la Chapelle, M. Gold dimer nanoantenna with slanted gap for tunable LSPR and improved SERS. J. Phys. Chem. C 2014, 118, 3209–3219. [Google Scholar] [CrossRef]
- Liu, H.L.; Yang, Z.; Meng, L.; Sun, Y.; Wang, J.; Yang, L.; Liu, J.; Tian, Z. Three-dimensional and time-ordered surface-enhanced Raman scattering hotspot matrix. J. Am. Chem. Soc. 2014, 136, 5332–5341. [Google Scholar] [CrossRef] [PubMed]
- Savage, K.; Hawkeye, M.M.; Esteban, R.; Borisov, A.G.; Aizpurua, J.; Baumberg, J.J. Revealing the quantum regime in tunnelling plasmonics. Nature 2012, 491, 574. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Crozier, K.B. Quantum mechanical limit to plasmonic enhancement as observed by surface enhanced Raman scattering. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- De Angelis, F.; Gentile, F.; Mecarini, F.; Das, G.; Moretti, M.; Candeloro, P.; Coluccio, M.L.; Cojoc, G.; Accardo, A.; Liberale, C.; et al. Breaking the diffusion limit with super-hydrophobic delivery of molecules to plasmonic nanofocusing SERS structures. Nat. Photonics 2011, 5, 682. [Google Scholar] [CrossRef]
- Yang, S.; Dai, X.; Stogin, B.B.; Wong, T.S. Ultrasensitive surface-enhanced Raman scattering detection in common fluids. Proc. Nat. Acad. Sci. USA 2016, 113, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Jana, N.R. Silver coated gold nanoparticles as new surface enhanced Raman substrate at low analyte concentration. Analyst 2003, 128, 954–956. [Google Scholar] [CrossRef]
- Messina, E.; Cavallaro, E.; Cacciola, A.; Saija, R.; Borghese, F.; Denti, P.; Fazio, B.; D’Andrea, C.; Gucciardi, P.G.; Iatì, A.M.; et al. Manipulation and raman spectroscopy with optically trapped metal nanoparticles obtained by pulsed laser ablation in liquids. J. Phys. Chem. C 2011, 115, 5115–5122. [Google Scholar] [CrossRef]
- Sivanesan, A.; Ly, H.K.; Kozuch, J.; Sezer, M.; Kuhlmann, U.; Fisher, A.; Weidinger, I.M. Functionalized Ag nanoparticles with tunable optical properties for selective protein analysis. Chem. Commun. 2011, 47, 3553–3555. [Google Scholar] [CrossRef] [PubMed]
- Bjerneld, E.J.; Svedberg, F.; Käll, M. Laser-induced growth and deposition of noble-metal nanoparticles for surface-enhanced raman scattering. Nano Lett. 2003, 3, 593–596. [Google Scholar] [CrossRef]
- Lee, S.J.; Piorek, B.D.; Meinhart, C.D.; Moskovits, M. Photoreduction at a Distance: Facile, nonlocal photoreduction of ag ions in solution by plasmon-mediated photoemitted electrons. Nano Lett. 2010, 10, 1329–1334. [Google Scholar] [CrossRef] [PubMed]
- Toussaint, K.C.; Liu, M.; Pelton, M.; Pesic, J.; Guffey, M.J.; Guyot-Sionnest, P.; Scherer, N.F. Plasmon resonance based optical trapping of single and multiple Au NPs. Opt. Express 2007, 15, 12018. [Google Scholar] [CrossRef]
- Liu, Y.; Huang, Z.; Zhou, F.; Lei, X.; Yao, B.; Meng, G.; Mao, Q. Highly sensitive fibre surface enhanced Raman scattering probes fabricated using laser induced self assembly in a meniscus. Nanoscale 2016, 8, 10607–10614. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Peng, X.; Wang, M.; Scarabelli, L.; Mao, Z.; Liz-Marzán, L.M.; Becker, M.F.; Zheng, Y. Light-directed reversible assembly of plasmonic nanoparticles using plasmon-enhanced thermophoresis. ACS Nano 2016, 10, 9659–9668. [Google Scholar] [CrossRef] [PubMed]
- Ding, T.; Valev, V.K.; Salmon, A.R.; Forman, C.J.; Smoukov, S.K.; Scherman, O.A.; Frenkel, D.; Baumberg, J.J. Light-induced actuating nanotransducers. Proc. Nat. Acad. Sci. USA 2016, 113, 201524209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baffou, G.; Quidant, R. Thermo-plasmonics: Using metallic nanostructures as nano-sources of heat. Laser Photonics Rev. 2013, 7, 171–187. [Google Scholar] [CrossRef]
- Ashkin, A. Acceleration and trapping of particles by radiation pressure. Phys. Rev. Lett. 1970, 24, 156. [Google Scholar] [CrossRef]
- Jones, P.; Maragó, O.M.; Volpe, G. Optical Tweezers: Principles and Applications; Cambridge University Press: Cambridge, UK, 2015; ISBN 9781107279711. [Google Scholar]
- Svedberg, F.; Käll, M. On the importance of optical forces in surface-enhanced Raman scattering (SERS). Faraday Discuss. 2006, 132, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Maragò, O.M.; Jones, P.H.; Gucciardi, P.G.; Volpe, G.; Ferrari, A.C. Optical trapping and manipulation of nanostructures. Nat. Nanotechnol. 2013, 8, 807–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Messina, E.; Cavallaro, E.; Cacciola, A.; Iatì, M.A.; Gucciardi, P.G.; Borghese, F.; Denti, P.; Saija, R.; Compagnini, G.; Meneghetti, M.; et al. Plasmon-enhanced optical trapping of gold nanoaggregates with selected optical properties. ACS Nano 2011, 5, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Patra, P.P.; Chikkaraddy, R.; Tripathi, R.P.N.; Dasgupta, A.; Kumar, P.G.V. Plasmofluidic single-molecule surface-enhanced Raman scattering from dynamic assembly of plasmonic nanoparticles. Nat. Commun. 2014, 5, 4357. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Lohmueller, T.; Feldmann, J. Optical injection of gold nanoparticles into living cells. Nano Lett. 2015, 15, 770–775. [Google Scholar] [CrossRef] [PubMed]
- Arias-González, J.R.; Nieto-Vesperinas, M. Optical forces on small particles: Attractive and repulsive nature and plasmon-resonance conditions. J. Opt. Soc. Am. A 2003, 20, 1201–1209. [Google Scholar] [CrossRef]
- Selhuber-Unkel, C.; Zins, I.; Schubert, O.; Sönnichsen, C.; Oddershede, L.B. Quantitative optical trapping of single gold nanorods. Nano Lett. 2008, 8, 2998–3003. [Google Scholar] [CrossRef] [PubMed]
- Brzobohatý, O.; Šiler, M.; Trojek, J.; Chvátal, L.; Karásek, V.; Paták, A.; Pokorná, Z.; Mika, F.; Zemánek, P. Three-dimensional optical trapping of a plasmonic nanoparticle using low numerical aperture optical tweezers. Sci. Rep. UK 2015, 5, 8106. [Google Scholar] [CrossRef] [PubMed]
- Guffey, M.J.; Scherer, N.F. All-optical patterning of Au nanoparticles on surfaces using optical traps. Nano Lett. 2010, 10, 4302–4308. [Google Scholar] [CrossRef] [PubMed]
- Gargiulo, J.; Violi, I.L.; Cerrota, S.; Chvátal, L.; Cortés, E.; Perassi, E.M.; Diaz, F.; Zemánek, P.; Stefani, F.D. Accuracy and Mechanistic Details of Optical Printing of Single Au and Ag Nanoparticles. ACS Nano 2017, 11, 9678–9688. [Google Scholar] [CrossRef] [PubMed]
- Svedberg, F.; Li, Z.; Xu, H.; Käll, M. Creating hot nanoparticle pairs for surface-enhanced Raman spectroscopy through optical manipulation. Nano Lett. 2006, 6, 2639–2641. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Yoshikawa, H.; Itoh, T.; Ishikawa, M. Surface Enhanced Raman Scattering from Pseudoisocyanine on Ag Nanoaggregates Produced by Optical Trapping with a Linearly Polarized Laser Beam. J. Phys. Chem. C 2009, 113, 11856. [Google Scholar] [CrossRef]
- Bálint, S.; Kreuzer, M.P.; Rao, S.; Badenes, G.; Miškovský, P.; Petrov, D. Simple route for preparing optically trappable probes for surface-enhanced Raman scattering. J. Phys. Chem. C 2009, 113, 17724–17729. [Google Scholar] [CrossRef]
- Liu, Z.; Hung, W.H.; Aykol, M.; Valley, D.; Cronin, S.B. Optical manipulation of plasmonic nanoparticles, bubble formation and patterning of SERS aggregates. Nanotechnology 2010, 21, 105304. [Google Scholar] [CrossRef] [PubMed]
- Tong, T.; Righini, M.; Gonzalez, M.U.; Quidant, R.; Käll, M. Optical aggregation of metal nanoparticles in a microfluidic channel for surface-enhanced Raman scattering analysis. Lab. Chip. 2009, 9, 193–195. [Google Scholar] [CrossRef] [PubMed]
- Fazio, B.; D’Andrea, C.; Foti, A.; Messina, E.; Irrera, A.; Donato, M.G.; Villari, V.; Micali, N.; Maragò, O.M.; Gucciardi, P.G. SERS detection of Biomolecules at Physiological pH via aggregation of Gold Nanorods mediated by Optical Forces and Plasmonic Heating. Sci. Rep. UK 2016, 6, 26952. [Google Scholar] [CrossRef] [PubMed]
- Guttler, B.; Zackel, S.; Wundrack, S.; Stosch, R. Isotope-Dilution Surface-Enhanced Raman Spectroscopy: Metrologically Traceable Reference Measurements at the Highest Precision Level and Their Application in Clinical Chemistry. In Handbook of Enhanced Spectroscopy; Pan Stanford Publishing Pte. Ltd.: Singapore, 2016; pp. 307–328. ISBN 9789814613323. [Google Scholar]
- Maerz, A.K.; Ackermann, R.; Malsch, D.; Bocklitz, T.; Henkel, T.; Popp, J. Towards a quantitative SERS approach—Online monitoring of analytes in a microfluidic system with isotope-edited internal standards. J. Biophotonics 2009, 2, 232–242. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Lin, X.; Jiang, C.; Li, C.; Lin, H.; Huang, J.; Wang, S.; Liu, G.; Yan, X.; Zhong, Q.; et al. Reliable quantitative SERS analysis facilitated by core–shell nanoparticles with embedded internal standards. Angew. Chem. Int. Ed. 2015, 54, 7308–7312. [Google Scholar] [CrossRef] [PubMed]
- Nima, Z.; Mahmood, M.; Xu, Y.; Mustafa, T.; Watanabe, F.; Nedosekin, D.A.; Juratli, M.A.; Fahmi, T.; Galanzha, E.I.; Nolan, J.P.; et al. Circulating tumor cell identification by functionalized silver-gold nanorods with multicolor, super-enhanced SERS and photothermal resonances. Sci. Rep. UK 2014, 4, 4752. [Google Scholar] [CrossRef] [PubMed]
- Tombelli, S.; Minunni, M.; Mascini, M. Analytical applications of aptamers. Biosens. Bioelectron. 2005, 20, 2424–2434. [Google Scholar] [CrossRef] [PubMed]
- Fabris, L.; Dante, M.; Nguyen, T.-Q.; Tok, J.B.-H.; Bazan, G.C. SERS Aptatags: New responsive metallic nanostructures for heterogeneous protein detection by surface enhanced raman spectroscopy. Adv. Funct. Mater. 2008, 18, 2518–2525. [Google Scholar] [CrossRef]
- O’sullivan, C.K.; Guilbault, G.G. Commercial quartz crystal microbalances—theory and applications. Biosens. Bioelectron. 1999, 14, 663–670. [Google Scholar] [CrossRef]
- Lequin, R.M. Enzyme Immunoassay (Eia)/Enzyme-Linked Immunosorbent Assay (Elisa). Clin. Chem. 2005, 51, 2415–2418. [Google Scholar] [CrossRef] [PubMed]
- Hoa, X.D.; Kirk, A.G.; Tabrizian, M. Towards integrated and sensitive surface plasmon resonance biosensors: A review of recent progress. Biosens. Bioelectron. 2007, 23, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Cottat, M.; D’Andrea, C.; Yasukuni, R.; Malashikhina, N.; Grinyte, R.; Lidgi-Guigui, N.; Fazio, B.; Sutton, A.; Oudar, O.; Charnaux, N.; et al. High Sensitivity, High selectivity SERS detection of MnSOD using optical nanoantennas functionalized with Aptamers. J. Phys. Chem. C 2015, 119, 15532–15540. [Google Scholar] [CrossRef]
- Galarreta, B.C.; Tabatabaei, M.; Guieu, V.; Peyrin, E.; Lagugné-Labarthet, F. Microfluidic channel with embedded SERS 2D platform for the aptamer detection of ochratoxin A. Anal. Bioanal. Chem. 2013, 405, 1613–1621. [Google Scholar] [CrossRef] [PubMed]
- Neumann, O.; Zhang, D.; Tam, F.; Lai, S.; Wittung-Stafshede, P.; Halas, N.J. Direct optical detection of aptamer conformational changes induced by target molecules. Anal. Chem. 2009, 81, 10002–10006. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Aguado, J.A.; Penner, G. Fluorescence polarization based displacement assay for the determination of small molecules with aptamers. Anal. Chem. 2008, 80, 8853–8855. [Google Scholar] [CrossRef] [PubMed]
- Villari, V.; Mazzaglia, A.; Darcy, R.; O’Driscoll, C.M.; Micali, N. Nanostructures of cationic amphiphilic cyclodextrin complexes with DNA. Biomacromolecules 2013, 14, 811–817. [Google Scholar] [CrossRef] [PubMed]
- D’Andrea, C.; Irrera, A.; Fazio, B.; Foti, A.; Messina, E.; Maragò, O.M.; Kessentini, S.; David, C.; Gucciardi, P.G. Red shifted spectral dependence of the SERS enhancement in a random array of gold nanoparticles covered with a silica shell: Extinction versus scattering. J. Opt. 2015, 17, 114016. [Google Scholar] [CrossRef]
- Shemetov, A.A.; Nabiev, I.; Sukhanova, A. Molecular interaction of proteins and peptides with nanoparticles. ACS Nano 2012, 6, 4585–4602. [Google Scholar] [CrossRef] [PubMed]
- Kah, J.C.Y.; Zubieta, A.; Saavedra, R.A.; Hamad-Schifferli, K. Stability of gold nanorods passivated with amphiphilic ligands. Langmuir 2012, 28, 8834–8844. [Google Scholar] [CrossRef] [PubMed]
- Tsai, D.H.; Del Rio, F.W.; Keene, A.M.; Tyner, K.M.; Mac Cuspie, R.I.; Cho, T.J.; Zachariah, M.R.; Hackley, V.A. Adsorption and conformation of serum albumin protein on gold nanoparticles investigated using dimensional measurements and in situ spectroscopic methods. Langmuir 2011, 27, 2464–2477. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, J.; Pan, J.; Jiang, X.; Ji, Y.; Li, Y.; Qu, Y.; Zhao, Y.; Wu, X.; Chen, C. Revealing the binding structure of the protein corona on gold nanorods using synchrotron radiation-based techniques: Understanding the reduced damage in cell membranes. J. Am. Chem. Soc. 2013, 135, 17359–17368. [Google Scholar] [CrossRef] [PubMed]
- Zijlstra, P.; Pedro, M.R.P.; Orrit, M. Optical detection of single non-absorbing molecules using the surface plasmon resonance of a gold nanorod. Nat. Nanotechnol. 2012, 7, 379–382. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Neumann, O.; Wang, H.; Yuwono, V.M.; Barhoumi, A.; Perham, M.; Hartgerink, J.D.; Wittung-Stafshede, P.; Halas, N.J. Gold nanoparticles can induce the formation of protein-based aggregates at physiological pH. Nano Lett. 2009, 9, 666–671. [Google Scholar] [CrossRef] [PubMed]
- Dubois, L.H.; Nuzzo, R.G. Synthesis, structure, and properties of model organic surfaces. Ann. Rev. Phys. Chem. 1992, 43, 437. [Google Scholar] [CrossRef]
- Thioune, N.; Lidgi-Guigui, N.; Cottat, M.; Gabudean, A.M.; Focsan, M.; Benoist, H.M.; Astilean, S.; Lamy de la Chapelle, M. Study of gold nanorods–protein interaction by localized surface plasmon resonance spectroscopy. Gold Bull. 2013, 46, 275–281. [Google Scholar] [CrossRef]
- Sheehan, P.E.; Whitman, L.J. Detection Limits for Nanoscale Biosensors. Nano Lett. 2005, 5, 803–807. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Joshi, P.; Shanker, V.; Ansari, Z.A.; Singh, S.P.; Chakrabarti, P. Contrasting effect of gold nanoparticles and nanorods with different surface modifications on the structure and activity of bovine serum albumin. Langmuir 2011, 27, 7722. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Gupta, A.; Verma, N.C.; Nandi, C.K. Kinetics of protein adsorption on gold nanoparticle with variable protein structure and nanoparticle size. J. Chem. Phys. 2015, 143, 164709. [Google Scholar] [CrossRef] [PubMed]
- Donato, M.G.; Messina, E.; Foti, A.; Smart, T.; Jones, P.H.; Iatì, M.A.; Saija, R.; Gucciardi, P.G.; Maragò, O.M. Optical trapping and optical force positioning of two-dimensional materials. Nanoscale 2018, 10, 1245–1255. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Sun, M.; Ma, W.; Kuang, H.; Xu, C. Biological molecules-governed plasmonic nanoparticle dimers with tailored optical behaviors. J. Phys. Chem. Lett. 2017, 8, 5633. [Google Scholar] [CrossRef] [PubMed]
- Lehninger, A.L. Biochemistry, 2nd ed.; Worth Publishers, Inc.: New York, NY, USA, 1975; 111p, ISBN 978-0879010478. [Google Scholar]
- De Luca, A.C.; Rusciano, G.; Ciancia, R.; Martinelli, V.; Pesce, G.; Rotoli, B.; Selvaggi, L.; Sasso, A. Spectroscopical and mechanical characterization of normal and thalassemic red blood cells by Raman Tweezers. Opt. Express 2008, 16, 7943–7957. [Google Scholar] [CrossRef] [PubMed]
- Brunner, H.; Sussner, H. Resonance Raman scattering on haemoglobin. Biochim. Biophys. Acta 1973, 310, 20–31. [Google Scholar] [CrossRef]
- Xu, H.; Bjerneld, E.J.; Käll, M.; Börjesson, L. Spectroscopy of single hemoglobin molecules by surface enhanced raman scattering. Phys. Rev. Lett. 1999, 83, 4357–4360. [Google Scholar] [CrossRef]
- Drescher, D.; Büchner, T.; McNaughton, D.; Kneipp, J. SERS reveals the specific interaction of silver and gold nanoparticles with hemoglobin and red blood cell components. Phys. Chem. Chem. Phys. 2013, 15, 5364. [Google Scholar] [CrossRef] [PubMed]
- Kaminska, A.; Forster, R.J.; Kayes, T.E. The impact of adsorption of bovine pancreatic trypsin inhibitor on CTAB-protected gold nanoparticle arrays: A Raman spectroscopic comparison with solution denaturation. J. Raman Spectrosc. 2010, 41, 130–135. [Google Scholar] [CrossRef]
- Dendramis, A.L.; Schwinn, E.W.; Sperline, R.P. SERS study of CTAB on Copper. Surf. Sci. 1983, 134, 675–688. [Google Scholar] [CrossRef]
- Bergmeyer, H.U. Methods of Enzymatic Analysis, 2nd ed.; Academic Press: New York, NY, USA, 1974; Volume 2, pp. 438–439. [Google Scholar]
- Sánchez-Rodríguez, R.; Torres-Mena, J.E.; del Pozo Yauner, L.; Pérez-Carreón, J.I. Biomarkers of the Antioxidant Response: A Focus on Liver Carcinogenesis in Biomarkers in Liver Disease; Springer: Amsterdam, The Netherlands, 2016; pp. 1–24. ISBN 978-94-007-7742-2. [Google Scholar]
- Vlahogianni, T.; Dassenakis, M.; Scoullos, M.J.; Valavanidis, A. Integrated use of biomarkers (superoxide dismutase, catalase and lipid peroxidation) in mussels Mytilus galloprovincialis for assessing heavy metals’ pollution in coastal areas from the Saronikos Gulf of Greece. Mar. Pollut. Bull. 2007, 54, 1361–1371. [Google Scholar] [CrossRef] [PubMed]
- Chuang, W.-J.; Johnson, S.; Van Wart, H.E. Resonance raman spectra of bovine liver catalase: enhancement of proximal tyrosinate vibrations. J. Inorg. Biochem. 1988, 34, 201–219. [Google Scholar] [CrossRef]
- Zezza, F.; Longobardi, F.; Pascale, M.; Eremin, S.A.; Visconti, A. Fluorescence polarization immunoassay for rapid screening of ochratoxin A in red wine. Anal. Bioanal. Chem. 2009, 395, 1317–1323. [Google Scholar] [CrossRef] [PubMed]
- Kabak, B. Ochratoxin A in cereal-derived products in Turkey: Occurrence and exposure assessment. Food Chem. Toxicol. 2009, 47, 348–352. [Google Scholar] [CrossRef] [PubMed]
- O'Brien, E.; Dietrich, D.R. Ochratoxin A: The continuing enigma. Crit. Rev. Toxicol. 2005, 35, 33–60. [Google Scholar] [CrossRef] [PubMed]
- Ganbold, E.-O.; Lee, C.M.; Cho, E.-M.; Son, S.J.; Kim, S.; Joo, S.-W.; Yang, S.I. Subnanomolar detection of ochratoxin A using aptamer-attached silver nanoparticles and surface-enhanced Raman scattering. Anal. Methods UK 2014, 6, 3573. [Google Scholar] [CrossRef]
- Gillibert, G.; Triba, M.; Lamy de la Chapelle, M. Surface enhanced raman scattering sensor for highly sensitive and selective detection of Ochratoxin A. Analyst 2018, 143, 339. [Google Scholar] [CrossRef] [PubMed]
- Indrasekara, A.; Wadams, R.; Fabris, L. Ligand exchange on gold nanorods: going back to the future. Part. Part. Syst. Charact. 2014, 31, 819–838. [Google Scholar] [CrossRef]
- Le Ru, E.C.; Grand, J.; Sow, I.; Somerville, W.R.C.; Etchegoin, P.G.; Treguer-Delapierre, M.; Charron, G.; Félidj, N.; Lévi, G.; Aubard, J. A scheme for detecting every single target molecule with surface-enhanced raman spectroscopy. Nano Lett. 2011, 11, 5013–5019. [Google Scholar] [CrossRef] [PubMed]
- Messina, E.; Donato, M.G.; Zimbone, M.; Saija, R.; Iatì, M.A.; Calcagno, L.; Fragalà, M.E.; Compagnini, G.; D’Andrea, C.; Foti, A.; et al. Optical trapping of silver nanoplatelets. Opt. Express 2015, 23, 8720–8730. [Google Scholar] [CrossRef] [PubMed]
- Schutz, M.; Steinigeweg, D.; Salehi, M.; Kompe, K.; Schlucker, S. Hydrophilically stabilized gold nanostars as SERS labels for tissue imaging of the tumor suppressor p63 by immuno-SERS microscopy. Chem. Commun. 2011, 47, 4216–4218. [Google Scholar] [CrossRef] [PubMed]
- Messina, E.; D’Urso, L.; Fazio, E.; Satriano, C.; Donato, M.G.; D’Andrea, C.; Maragò, O.M.; Gucciardi, P.G.; Compagnini, G.; Neri, F. Tuning the structural and optical properties of gold/silver nano-alloys prepared by laser ablation in liquids for optical limiting, ultra-sensitive spectroscopy, and optical trapping. J. Quant. Spectrosc. Radiat. Trans. 2012, 113, 220–228. [Google Scholar] [CrossRef]
- Spadaro, D.; Iatí, M.A.; Pérez-Piñeiro, J.; Vázquez-Vázquez, J.; Correa-Duarte, M.A.; Donato, M.G.; Gucciardi, P.G.; Raija, R.; Strangi, G.; Maragò, O.M. Optical Trapping of Plasmonic Mesocapsules: Enhanced Optical Forces and SERS. J. Phys. Chem. C 2017, 121, 691–700. [Google Scholar] [CrossRef]










© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Foti, A.; D’Andrea, C.; Villari, V.; Micali, N.; Donato, M.G.; Fazio, B.; Maragò, O.M.; Gillibert, R.; Lamy de la Chapelle, M.; Gucciardi, P.G. Optical Aggregation of Gold Nanoparticles for SERS Detection of Proteins and Toxins in Liquid Environment: Towards Ultrasensitive and Selective Detection. Materials 2018, 11, 440. https://doi.org/10.3390/ma11030440
Foti A, D’Andrea C, Villari V, Micali N, Donato MG, Fazio B, Maragò OM, Gillibert R, Lamy de la Chapelle M, Gucciardi PG. Optical Aggregation of Gold Nanoparticles for SERS Detection of Proteins and Toxins in Liquid Environment: Towards Ultrasensitive and Selective Detection. Materials. 2018; 11(3):440. https://doi.org/10.3390/ma11030440
Chicago/Turabian StyleFoti, Antonino, Cristiano D’Andrea, Valentina Villari, Norberto Micali, Maria Grazia Donato, Barbara Fazio, Onofrio M. Maragò, Raymond Gillibert, Marc Lamy de la Chapelle, and Pietro G. Gucciardi. 2018. "Optical Aggregation of Gold Nanoparticles for SERS Detection of Proteins and Toxins in Liquid Environment: Towards Ultrasensitive and Selective Detection" Materials 11, no. 3: 440. https://doi.org/10.3390/ma11030440
APA StyleFoti, A., D’Andrea, C., Villari, V., Micali, N., Donato, M. G., Fazio, B., Maragò, O. M., Gillibert, R., Lamy de la Chapelle, M., & Gucciardi, P. G. (2018). Optical Aggregation of Gold Nanoparticles for SERS Detection of Proteins and Toxins in Liquid Environment: Towards Ultrasensitive and Selective Detection. Materials, 11(3), 440. https://doi.org/10.3390/ma11030440