Novel Aminoacridine Sensors Based on Molecularly Imprinted Hybrid Polymeric Membranes for Static and Hydrodynamic Drug Quality Control Monitoring


 ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Apparatus and Reagents
2.2. Synthesis of Molecularly Imprinted Polymers
2.3. Membrane Sensors and Potential Measurements
2.4. Binding Capacity of Aminoacridine Molecularly Imprinted Polymer
2.5. Selectivity Towards Other Cations
2.6. Determination of ACR in Biological Fluids
2.7. Determination of ACR in Pharmaceutical Samples
3. Results and Discussions
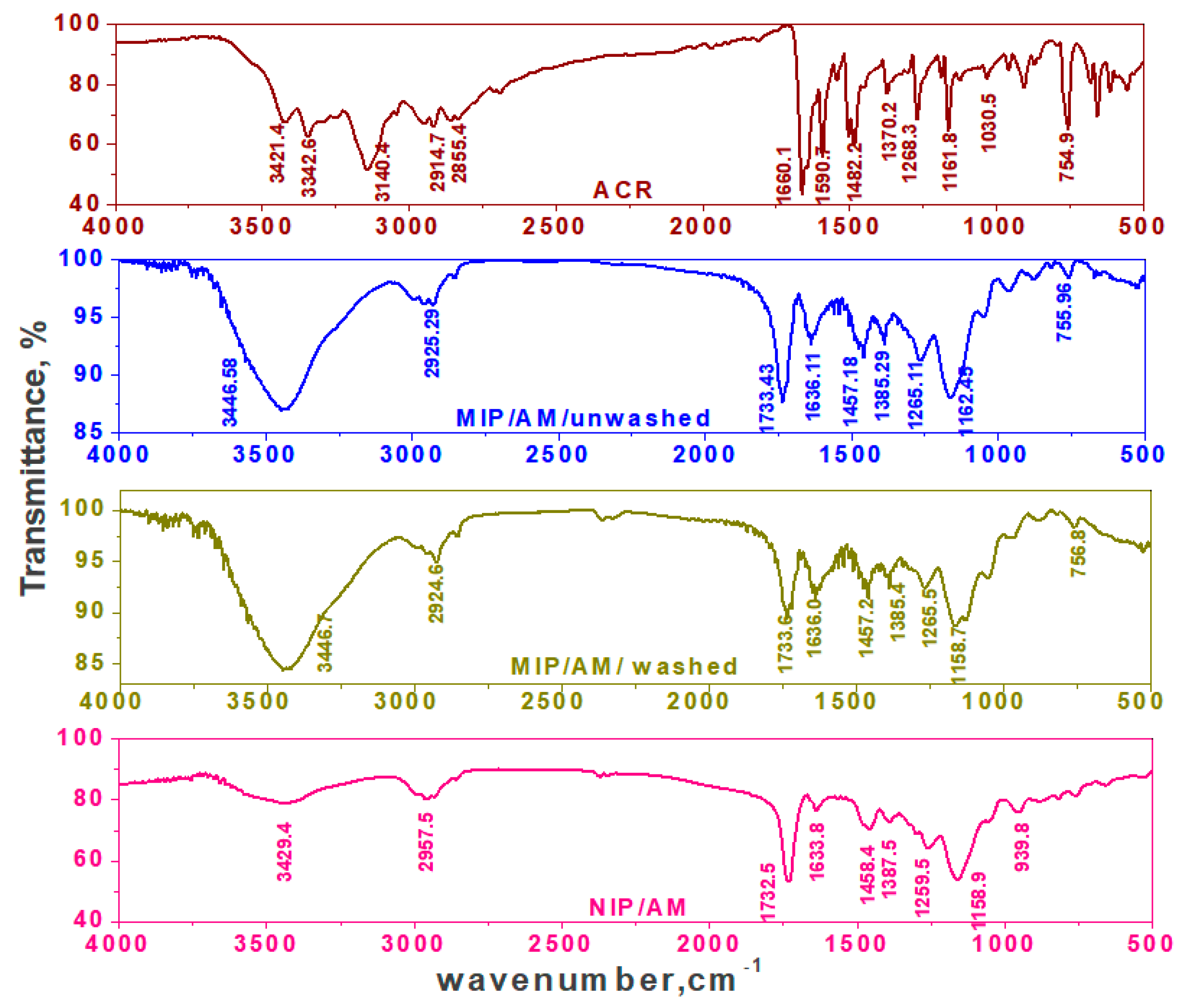
3.1. Characterization of the MIP Particles
3.2. Binding Capacity of MIPs and Scatchard Analysis
3.3. Sensors Performances
3.4. Sensors Selectivity
3.5. Hydrodynamic Monitoring of Aminoacridine
3.6. The Analytical Applications
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gilman, A.G.; Goodman, L.S.; Gilman, A. (Eds.) Goodman and Gilman’s the Pharmacological Basis of Therapeutics, 6th ed.; Macmillan Publishing Co., Inc.: New York, NY, USA, 1980; p. 980. [Google Scholar]
- Osol, A.; Hoover, J.E. (Eds.) Remington’s Pharmaceutical Sciences, 15th ed.; Mack Publishing Co.: Easton, PA, USA, 1975; p. 1102. [Google Scholar]
- Schmitz, J.P. 9-aminoacridine—Its present status and current recommendations for use as a surgical and endodontic irrigant in dentistry. Oral Surg. Oral Med. Oral Pathol. 1980, 50, 273–276. [Google Scholar] [CrossRef]
- Budavari, S. (Ed.) Medication (VET): Antiseptic. In The Merck Index—An Encyclopedia of Chemicals, Drugs, and Biologicals; Merck and Co., Inc.: Whitehouse Station, NJ, USA, 1996; p. 72. [Google Scholar]
- Guerrero, R.O.S.; Icardo, M.C.; Gómez, C.; Benito, J.M. FIA-Chemiluminescence determination of acridine yellow. Anal. Lett. 2003, 36, 1039–1049. [Google Scholar] [CrossRef]
- Naidu, P.R.; Krishnan, V.R. Determination of Acridine by Potentiometric Titration in Acetic Acid. Anal. Chem. 1961, 33, 497–498. [Google Scholar] [CrossRef]
- Rao, K.C.M.; Naidu, P.R. Determination of acridine in Non-aqueous Media. Proc. Indian Acad. Sci. Sect. A 1972, 75, 86–91. [Google Scholar] [CrossRef]
- Barek, J.; Matějka, J.; Zima, J. Determination of Acridine, Benz [c] acridine and Dibenz [a, h] acridine by Fast Scan Differential Pulse Voltammetry and Adsorptive Stripping Voltammetry. Collect. Czech. Chem. Commun. 1994, 59, 294–308. [Google Scholar] [CrossRef]
- Bebawy, L.I.; Elghobashy, M.R.; Abbas, S.S.; Shokry, R.F. Chromatographic Determination of Aminoacridine Hydrochloride, Lidocaine Hydrochloride and Lidocaine Toxic Impurity in Oral Gel. J Chromatogr. Sci. 2015, 54, 492–499. [Google Scholar] [CrossRef] [Green Version]
- Syed, A.A.; Silwadi, M.F.; Kirumakki, S.R.; Narasimiah, N. Gas chromatographic determination of residual acridine and iminodibenzyl in dibenzazepine drugs and related compounds. Acta Chromatogr. 2002, 12, 80–88. [Google Scholar]
- Bunch, E.A. Pectrophotometric determination of aminacrinehydrochloridein creams, jellies, and suppositories: Interlaboratory study. J. Assoc. Off. Anal. Chem. 1987, 70, 560–565. [Google Scholar]
- Calatayud, J.M.; Sampedro, A.S.; Civera, P.V.; Benito, C.G. Spectrophotometrtc and fluorimetric determination of 9-amino acridine by flow injection analysis. Anal. Lett. 1990, 23, 2315–2325. [Google Scholar] [CrossRef]
- Piletsky, S.A.; Alcock, S.; Turner, A.P. Molecular imprinting: At the edge of the third millennium. Trends Biotechnol. 2001, 19, 9–12. [Google Scholar] [CrossRef]
- Mayes, A.G.; Mosbach, K. Molecularly imprinted polymers: Useful materials for analytical chemistry? TrAC Trends Anal. Chem. 1997, 16, 321–332. [Google Scholar] [CrossRef]
- Haupt, K.; Mosbach, K. Molecularly Imprinted Polymers and Their Use in Biomimetic Sensors. Chem. Rev. 2000, 100, 2495–2504. [Google Scholar] [CrossRef] [PubMed]
- Al-Kindy, S.; Badía, R.; Suárez-Rodríguez, J.L.; Díaz-García, M.E. Molecularly Imprinted Polymers and Optical Sensing Applications. Crit. Rev. Anal. Chem. 2000, 30, 291–309. [Google Scholar] [CrossRef]
- Whitcombe, M.J.; Kirsch, N.; Nicholls, I.A. Molecular imprinting science and technology: A survey of the literature for the years 2004–2011. J. Mol. Recognit. 2014, 27, 297–401. [Google Scholar] [PubMed]
- Kamel, A.H.; Al Hamid, F.A.; Soror, T.Y.; Galal, H.R.; El Gendy, F.A. Solid contact biosensor based on man-tailored polymers for acetylcholine detection: Application to acetycholinesterase assay. Eur. Chem. Bull. 2016, 5, 266–273. [Google Scholar]
- Kamel, A.H.; Hassan, A.M.E. Solid Contact Potentiometric Sensors Based on Host-Tailored Molecularly Imprinted Polymers for Creatine Assessment. Int. J. Electrochem. Sci. 2016, 11, 8938–8949. [Google Scholar] [CrossRef]
- El-Naby, E.H.; Kamel, A.H. Potential transducers based man-tailored biomimetic sensors for selective recognition of dextromethorphan as an antitussive drug. Mater. Sci. Eng. C 2015, 54, 217–224. [Google Scholar] [CrossRef]
- Kamel, A.H.; Galal, H.R. MIP-Based Biomimetic Sensors for Static and Hydrodynamic Potentiometric Transduction of Sitagliptin in Biological fluids. Int. J. Electrochem. Sci. 2014, 9, 4361–4373. [Google Scholar]
- Kamel, A.H.; Soror, T.Y.; Al-Romian, F.M. Graphite Solid-Contact Mepiquat Potentiometric Sensors Based on Molecularly Imprinted Polymers and Their Application to Flow Through Analysis. Anal. Methods 2012, 4, 3007–3012. [Google Scholar] [CrossRef]
- Abd-Rabboh, H.S.M.; Kamel, A.H. Mimicking receptor for cyanide based on ion imprinting and their applications in potential transduction. Electroanalysis 2012, 24, 1409–1415. [Google Scholar] [CrossRef]
- Kamel, A.H.; Mahmoud, W.H.; Mostafa, M.S. Biomimetic ciprofloxacin sensors made of molecularly imprinted network receptors for potential measurements. Anal. Methods 2011, 3, 957–964. [Google Scholar] [CrossRef]
- Kamel, A.H.; Sayour, H.E.M. Miniaturized Potentiometric Sensors Based on Molecularly Imprinted Polymers for Flow-Through Assay of Quinine in Soft Drinks and Urine. Electroanalysis 2009, 21, 2701–2708. [Google Scholar] [CrossRef]
- El-Kosasy, A.M.; Kamel, A.H.; Hussin, L.H.; Ayad, M.F.; Fares, N.V. Mimicking new receptors based on molecular imprinting and their application to potentiometric assessment of 2, 4-dichlorophenol as a food taint. Food Chem. 2018, 250, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Lorenzo, C.; Concheiro, A. Handbook of Molecularly Imprinted Polymers; Smithers Group Company: London, UK, 2013. [Google Scholar]
- Hassan, S.S.M.; Elnemma, E.M.; Mahmoud, W.H.; Mohamed, A.H.K. Continuous Potentiometric monitoring of Sildenafil Citrate (Viagra) in Pharmaceutical Preparations using Novel membrane sensors. J. Appl. Electrochem. 2006, 36, 139–146. [Google Scholar] [CrossRef]
- Sellergren, B. Molecularly Imprinted Polymers; Elsevier: Amsterdam, The Netherlands, 2001. [Google Scholar]
- Buck, R.P.; Lindner, E. Recommendations for nomenclature of ion selective electrodes (IUPAC Recommendations 1994). Pure Appl. Chem. 1994, 66, 2527–2536. [Google Scholar] [CrossRef]
- Nørby, J.G.; Ottolenghi, P.; Jensen, J. Scatchard plot: Common misinterpretation of binding experiments. Anal. Biochem. 1980, 102, 318–320. [Google Scholar] [CrossRef]
- IUPAC. Recommendations for nomenclature of ion-selective electrodes. Pure Appl. Chem. 1976, 48, 129–132. [Google Scholar]
- Goodell, J.R.; Svensson, B.; Ferguson, D.M. Spectrophotometric Determination and Computational Evaluation of the Rates of Hydrolysis of 9-Amino-Substituted Acridines. J. Chem. Inf. Model. 2006, 46, 876–883. [Google Scholar] [CrossRef]
- Kupai, J.; Razali, M.; Buyuktiryaki, S.; Kecili, R.; Szekely, G. Long-term stability and reusability of molecularly imprinted polymers. Polym. Chem. 2017, 8, 666–673. [Google Scholar] [CrossRef]
- Bakker, E.; Pretsch, E. Potentiometry at trace levels. Trends Anal. Chem. 2001, 20, 11–19. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Scatchard Parameters | MIP/MAA/ACR | NIP/MAA/ACR | MIP/AM/ACR | NIP/AM/ACR |
|---|---|---|---|---|
| Kd1, μM | 1764.91 | - | 22.14 | 242.72 |
| Qmax1, μmol/g | 497.35 | - | 24.37 | 49.48 |
| Kd2, μM | 401.6 | 15.04 | 724.63 | 487.8 |
| Qmax2, μmol/g | 250.07 | 35.82 | 329.71 | 69.87 |
| Parameters | Sensor (I) MIP/MAA | Sensor (II) MIP/AM | Sensor (III) MIP/MAA/TPB | Sensor (IV) MIP/AM/TPB |
|---|---|---|---|---|
| Slope, (mV/decade) | 51.2 ± 1.3 | 50.5 ± 1.4 | 39.9 ± 0.9 | 41.0 ± 1.6 |
| Correlation coefficient, (r2) | 0.9997 | 0.9999 | 0.9998 | 0.9998 |
| Linear range, (M) | 5.2 × 10−6–1.0 × 10−3 | 5.2 × 10−6–1.0 × 10−3 | 5.2 × 10−6–1.0 × 10−3 | 5.2 × 10−6–1.0 × 10−3 |
| Detection limit, (M) | 2.5 × 10−7 | 8.7 × 10−7 | 1.0 × 10−6 | 8.9 × 10−7 |
| Working range, (pH) | 4.0–7.0 | 3.5–7.0 | 4.0–7.0 | 3.5–7.0 |
| Response time, (s) | 10 | 10 | 10 | 10 |
| Life span, (week) | 8 | 8 | 8 | 8 |
| Standard deviation, (mV) | 0.98 | 1.2 | 1.8 | 1.6 |
| Interfering Ion | Log Kpoti,j | |||
|---|---|---|---|---|
| Sensor (I) MIP/MAA | Sensor(II) MIP/AM | Sensor (III) MIP/MAA/TPB | Sensor (IV) MIP/AM/TPB | |
| ACR | 0 | 0 | 0 | 0 |
| Piperidine | −2.85 | −2.92 | −2.88 | −2.77 |
| Ethylendiamine | −2.73 | −2.86 | −2.86 | −2.89 |
| 3−Aminopyidine | −2.90 | −2.93 | −3.00 | −2.89 |
| Hydroxylamine | −2.89 | −2.9 | −3.00 | −2.92 |
| Histidine | −2.80 | −2.94 | −3.05 | −2.95 |
| Alanine | −2.85 | −3.01 | −3.07 | −2.96 |
| Imidazole | −2.88 | −2.95 | −3.14 | −2.95 |
| Methylamine | −3.04 | −3.11 | −3.10 | −2.98 |
| Hexamine | −2.94 | −3.00 | −3.05 | −3.02 |
| Amprolium HCl | −2.71 | −2.86 | −3.30 | −3.04 |
| Urea | −2.87 | −2.98 | −3.10 | −3.04 |
| Dimethylamine | −2.90 | −3.06 | −2.86 | −3.10 |
| Parameters | Sensor (I) MIP/MAA | Sensor (II) MIP/AM |
|---|---|---|
| Slope, (mV/decade) | 50.1 ± 0.3 | 50.7 ± 0.2 |
| Correlation coefficient, (r2) | 0.9988 | 0.9988 |
| Linear range, M | 10−5–10−2 | 10−5–10−2 |
| Detection limit, M | 1.0 × 10−6 | 1.0 × 10−5 |
| Working range, (pH) | 4.0–7.0 | 3.5–7.0 |
| Response time, (s) | 10 | 10 |
| Life span, (week) | 8 | 8 |
| Flow rate, mL/min | 3.5 | 3.5 |
| Sample rate/h | 18 | 22 |
| Sample | Added (μM) | Labeled (g) | Static Mode | Hydrodynamic Mode | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Sensor (I) | Sensor (II) | Sensor (I) | Sensor (II) | |||||||
| Found | RSD a | Found | RSD a | Found | RSD a | Found | RSD a | |||
| S1 (Urine) | 10 | - | 10.2 ± 0.04 | 102.0 ± 0.3 | - | - | 9.78 | 97.8 ± 0.4 | 9.77 | 97.7 ± 0.4 |
| S2 (Urine) | 50 | - | 50.3 ± 0.02 | 100.6 ± 0.14 | 51 ± 0.6 | 102.0 ± 1.2 | - | - | - | - |
| S3 (Urine) | 100 | - | 99.8 ± 0.01 | 99.8.1 ± 0.7 | 93.4 ± 0.7 | 93.4 ± 1.3 | 93.3 | 93.3 ± 0.8 | 95.5 | 95.5 ± 0.8 |
| S4 (Urine) | 1000 | - | - | - | 906 ± 0.5 | 90.6 ± 2.0 | 990 | 99.0 ± 0.5 | 1040 | 104.0 ± 0.5 |
| S5 (Plasma) | 10 | - | 10.2 ± 0.06 | 102.0 ± 1.3 | 10.5 ± 1.4 | 105 ± 2.5 | 9.64 | 96.4 ± 0.4 | 9.77 | 97.7 ± 0.4 |
| S6 (Plasma) | 100 | - | 94.4 ± 0.05 | 94.44 ± 2.0 | 93.3 ± 0.5 | 93.3 ± 1.2 | 97.7 | 97.7 ± 0.4 | 95.5 | 95.5 ± 0.4 |
| S7 (Plasma) | 1000 | - | 1090 ± 0.01 | 109 ± 1.3 | 934 ± 0.4 | 93.4 ± 1.5 | 1050 | 105.0 ± 0.3 | - | - |
| S8 (Septogel) | - | 0.01 | 0.01058 ± 0.01 | 105.8 ± 1.2 | 0.00998 ± 0.05 | 99.8 ± 1.8 | 0.00987 | 98.7 ± 0.1 | 0.00972 | 97.2 ± 0.3 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
S. M. Hassan, S.; E. Amr, A.E.-G.; Abd El-Naby, H.; El-Naggar, M.; H. Kamel, A.; M. Khalifa, N. Novel Aminoacridine Sensors Based on Molecularly Imprinted Hybrid Polymeric Membranes for Static and Hydrodynamic Drug Quality Control Monitoring. Materials 2019, 12, 3327. https://doi.org/10.3390/ma12203327
S. M. Hassan S, E. Amr AE-G, Abd El-Naby H, El-Naggar M, H. Kamel A, M. Khalifa N. Novel Aminoacridine Sensors Based on Molecularly Imprinted Hybrid Polymeric Membranes for Static and Hydrodynamic Drug Quality Control Monitoring. Materials. 2019; 12(20):3327. https://doi.org/10.3390/ma12203327
Chicago/Turabian StyleS. M. Hassan, Saad, Abd El-Galil E. Amr, Heba Abd El-Naby, Mohamed El-Naggar, Ayman H. Kamel, and Nagy M. Khalifa. 2019. "Novel Aminoacridine Sensors Based on Molecularly Imprinted Hybrid Polymeric Membranes for Static and Hydrodynamic Drug Quality Control Monitoring" Materials 12, no. 20: 3327. https://doi.org/10.3390/ma12203327