A Quantum–Mechanical Study of Clean and Cr–Segregated Antiphase Boundaries in Fe3Al


Abstract
:1. Introduction
2. Materials and Methods
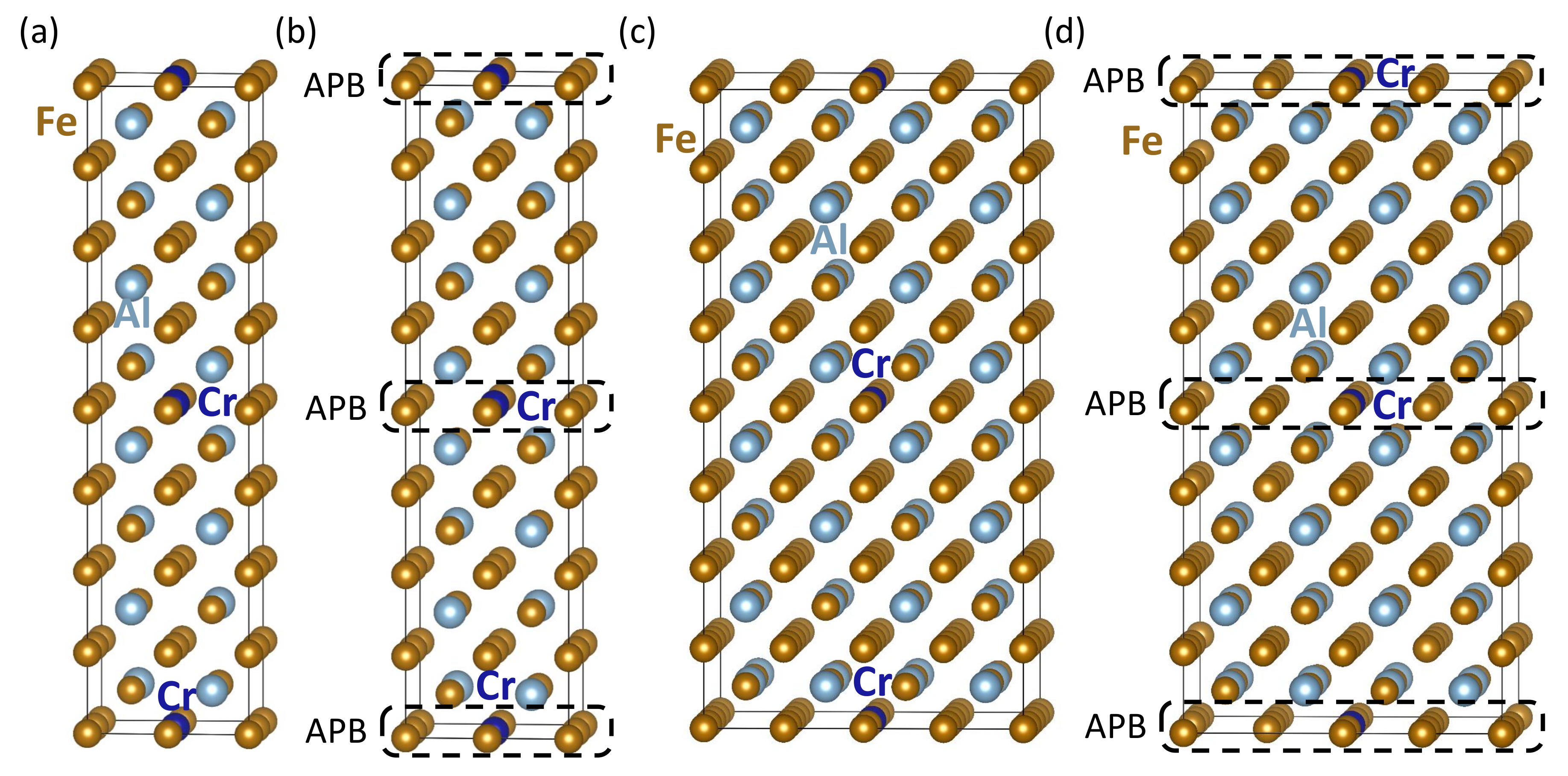
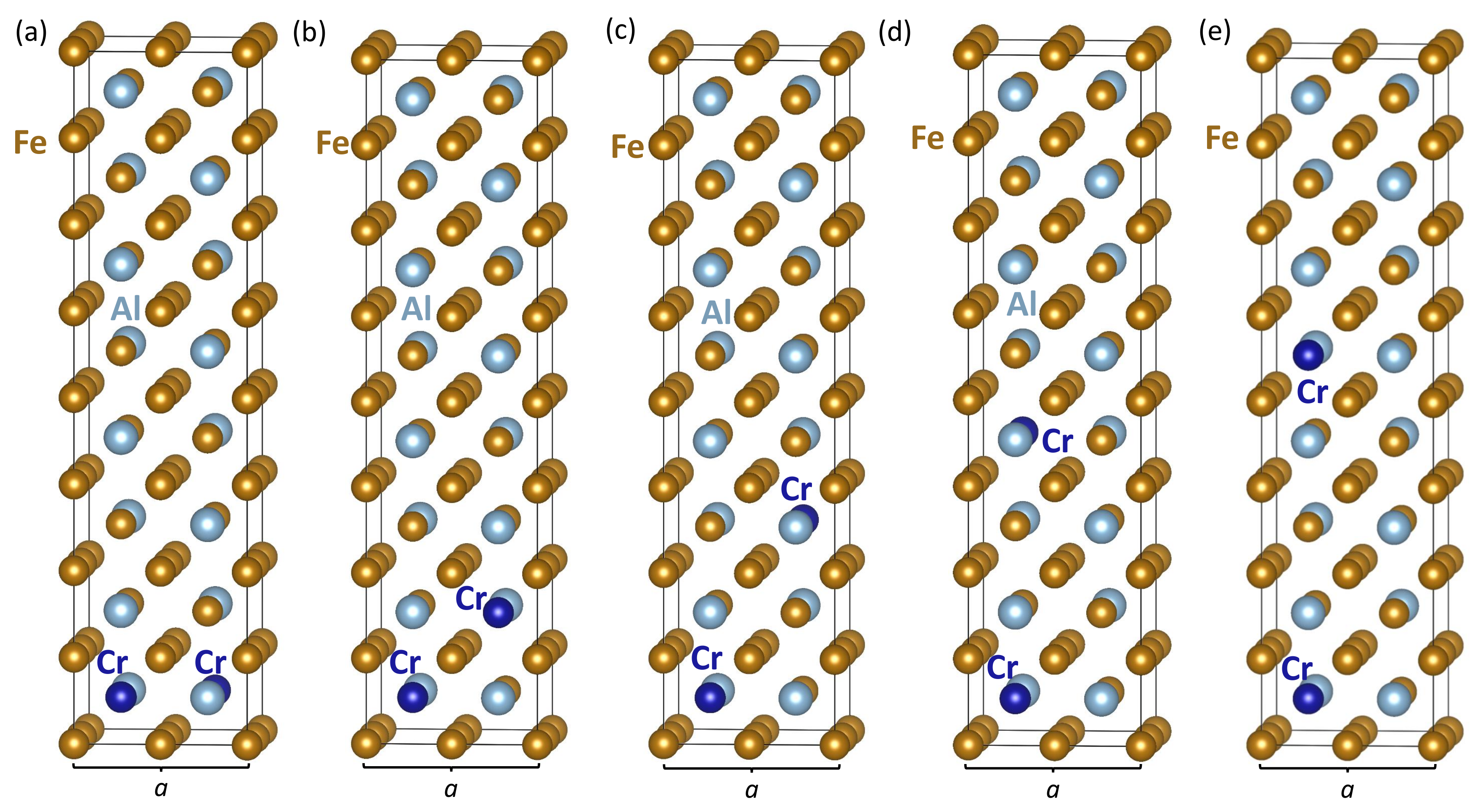
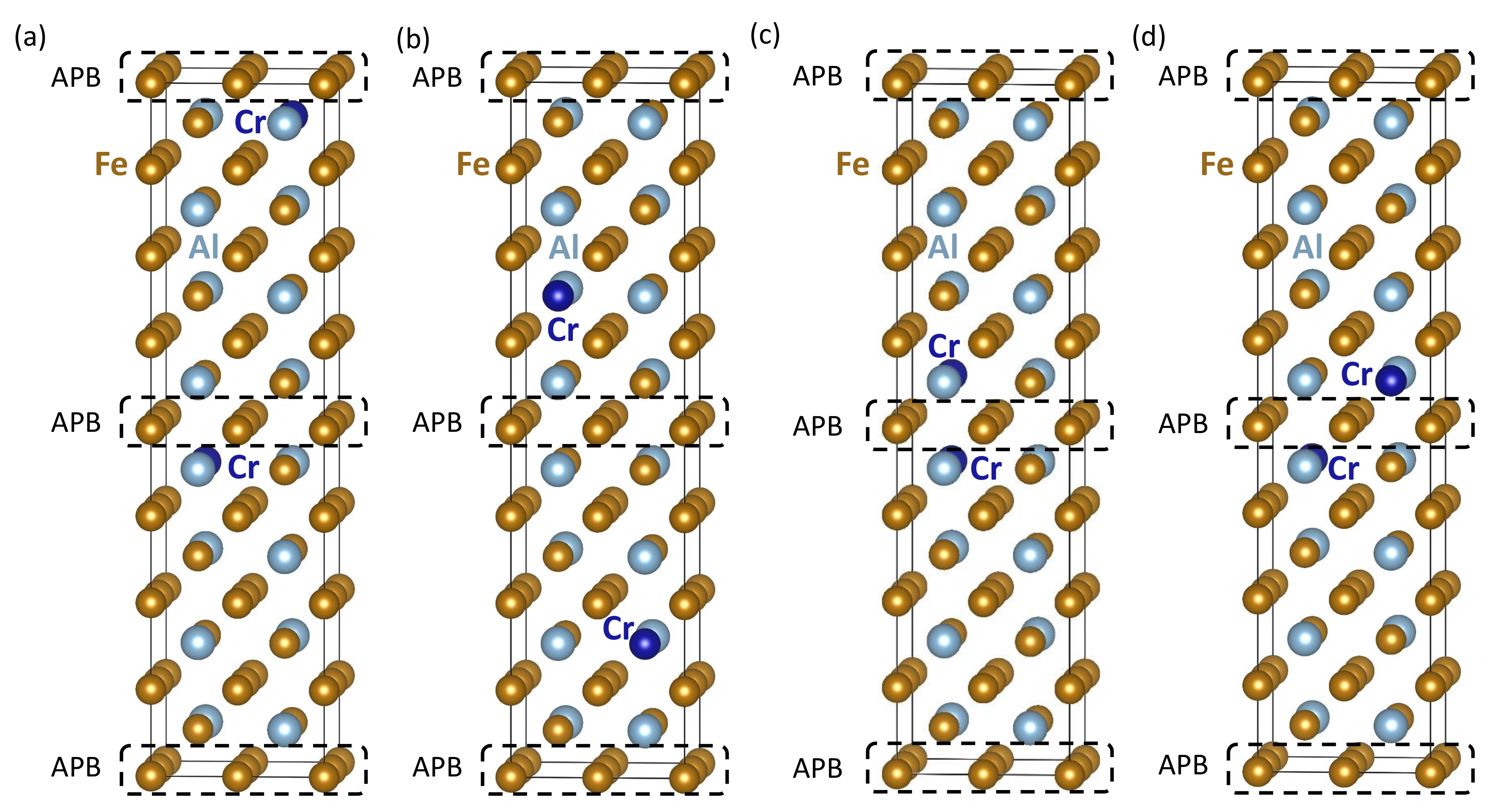
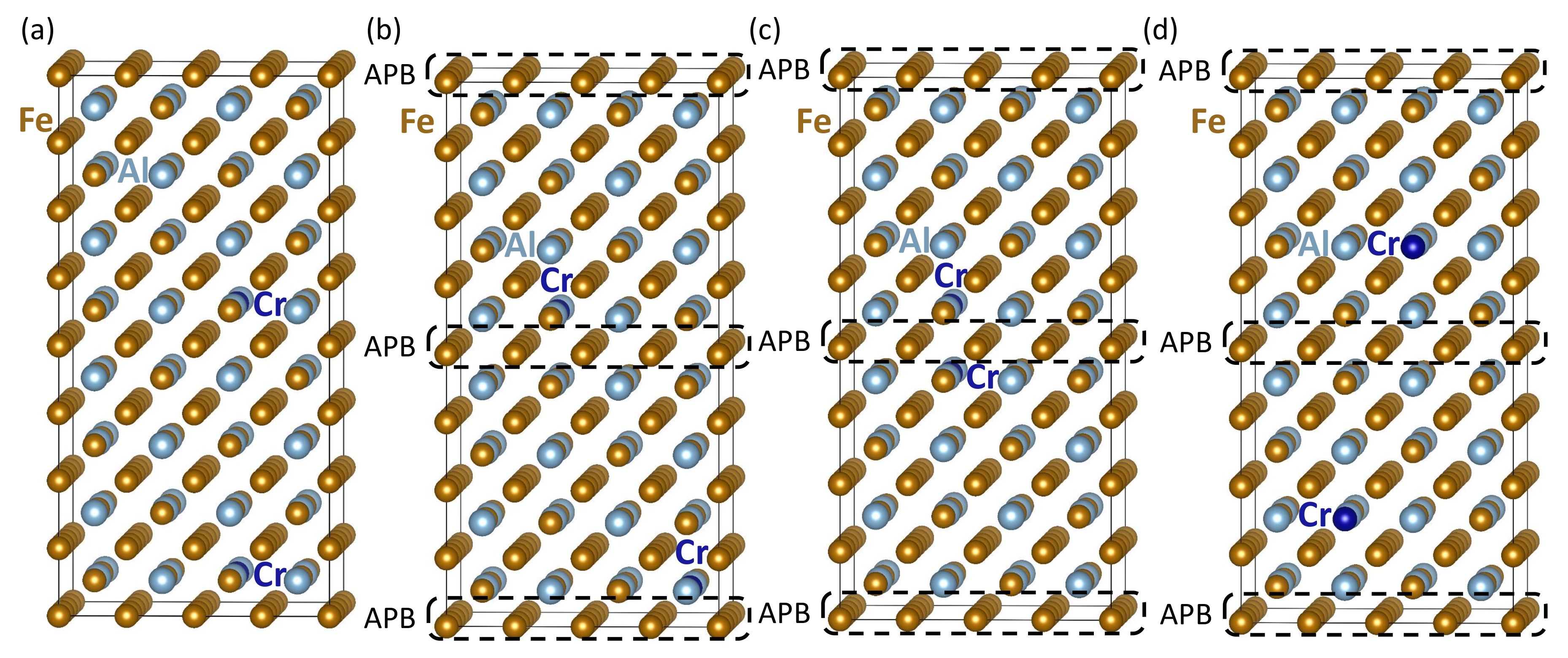
3. Results
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
References
- Sauthoff, G. Intermetallics; VCH Verlagsgesellschaft: Weinheim, Germany, 1995. [Google Scholar]
- Liu, C.T.; Stringer, J.; Mundy, J.N.; Horton, L.L.; Angelini, P. Ordered intermetallic alloys: An assessment. Intermetallics 1997, 5, 579–596. [Google Scholar] [CrossRef]
- Stoloff, N.S. Iron aluminides: Present status and future prospects. Mater. Sci. Eng. A 1998, 258, 1–14. [Google Scholar] [CrossRef]
- Liu, C.T.; Lee, E.H.; McKamey, C.G. An environmental-effect as the major cause for room-temperature embrittlement in FeAl. Scr. Metall. Mater. 1989, 23, 875–880. [Google Scholar] [CrossRef]
- Lynch, R.J.; Heldt, L.A.; Milligan, W.W. Effects of alloy composition on environmental embrittlement of B2 ordered iron aluminides. Scr. Metall. Mater. 1991, 25, 2147–2151. [Google Scholar] [CrossRef]
- Liu, C.T.; McKamey, C.G.; Lee, E.H. Environmental-effects on room-temperature ductility and fracture in Fe3Al. Scr. Metall. Mater. 1990, 24, 385–389. [Google Scholar] [CrossRef]
- Lynch, R.J.; Gee, K.A.; Heldt, L.A. Environmental embrittlement of single-crystal and thermomechanically processed B2-ordered iron aluminides. Scr. Metall. Mater. 1994, 30, 945–950. [Google Scholar] [CrossRef]
- Kattner, U.; Burton, B. Al-Fe (Aluminium-Iron). In Phase Diagrams of Binary Iron Alloys; Okamoto, H., Ed.; ASM International: Materials Park, OH, USA, 1993; pp. 12–28. [Google Scholar]
- Palm, M.; Inden, G.; Thomas, N. The Fe-Al-Ti system. J. Phase Equilibria 1995, 16, 209–222. [Google Scholar] [CrossRef]
- Palm, M.; Lacaze, J. Assessment of the Al-Fe-Ti system. Intermetallics 2006, 14, 1291–1303. [Google Scholar] [CrossRef]
- Palm, M.; Sauthoff, G. Deformation behaviour and oxidation resistance of single-phase and two-phase L21-ordered Fe-Al-Ti alloys. Intermetallics 2004, 12, 1345–1359. [Google Scholar] [CrossRef]
- Sundman, B.; Ohnuma, I.; Dupin, N.; Kattner, U.R.; Fries, S.G. An assessment of the entire Al-Fe system including D0(3) ordering. Acta Mater. 2009, 57, 2896–2908. [Google Scholar] [CrossRef]
- Jirásková, Y.; Pizúrová, N.; Titov, A.; Janičkovič, D.; Friák, M. Phase separation in Fe-Ti-Al alloy—Structural, magnetic, and Mössbauer study. J. Magn. Magn. Mater. 2018, 468, 91–99. [Google Scholar] [CrossRef]
- Dobeš, F.; Dymáček, P.; Friák, M. Force-to-Stress Conversion Methods in Small Punch Testing Exemplified by Creep Results of Fe-Al Alloy with Chromium and Cerium Additions. IOP Conf. Ser. Mater. Sci. Eng. 2018, 461, 012017. [Google Scholar] [CrossRef]
- Dobeš, F.; Dymáček, P.; Friák, M. Small punch creep of Fe-Al-Cr alloy with Ce addition and its relation to uniaxial creep tests. Kov. Mater. Met. Mater. 2018, 56, 205. [Google Scholar] [CrossRef]
- Dymáček, P.; Dobeš, F.; Jirásková, Y.; Pizúrová, N.; Friák, M. Tensile, creep and fracture testing of prospective Fe-Al-based alloys using miniature specimens. Theor. Appl. Fract. Mech. 2019, 99, 18–26. [Google Scholar] [CrossRef]
- Dobeš, F.; Dymáček, P.; Friák, M. The Influence of Niobium Additions on Creep Resistance of Fe-27 at. % Al Alloys. Metals 2019, 9, 739. [Google Scholar] [CrossRef]
- Watson, R.E.; Weinert, M. Transition-metal aluminide formation: Ti, V, Fe, and Ni aluminides. Phys. Rev. B 1998, 58, 5981–5988. [Google Scholar] [CrossRef]
- Gonzales-Ormeno, P.; Petrilli, H.; Schon, C. Ab-initio calculations of the formation energies of BCC-based superlattices in the Fe-Al system. Calphad-Comput. Coupling Ph. Diagrams Thermochem. 2002, 26, 573. [Google Scholar] [CrossRef]
- Friák, M.; Neugebauer, J. Ab initio study of the anomalous volume-composition dependence in Fe-Al alloys. Intermetallics 2010, 18, 1316–1321. [Google Scholar] [CrossRef]
- Amara, H.; Fu, C.C.; Soisson, F.; Maugis, P. Aluminum and vacancies in α-iron: Dissolution, diffusion, and clustering. Phys. Rev. B 2010, 81, 174101. [Google Scholar] [CrossRef]
- Liu, S.; Duan, S.; Ma, B. First-principles calculation of vibrational entropy for Fe-Al compounds. Phys. Rev. B 1998, 58, 9705–9709. [Google Scholar]
- Kulikov, N.I.; Postnikov, A.V.; Borstel, G.; Braun, J. Onset of magnetism in B2 transition-metal aluminides. Phys. Rev. B 1999, 59, 6824–6833. [Google Scholar] [CrossRef]
- Fähnle, M.; Drautz, R.; Lechermann, F.; Singer, R.; Diaz-Ortiz, A.; Dosch, H. Thermodynamic properties from ab-initio calculations: New theoretical developments, and applications to various materials systems. Phys. Status Solidi B-Basic Solid State Phys. 2005, 242, 1159–1173. [Google Scholar] [CrossRef]
- Friák, M.; Deges, J.; Krein, R.; Frommeyer, G.; Neugebauer, J. Combined ab initio and experimental study of structural and elastic properties of Fe3Al-based ternaries. Intermetallics 2010, 18, 1310. [Google Scholar] [CrossRef]
- Kirklin, S.; Saal, J.E.; Hegde, V.I.; Wolverton, C. High-throughput computational search for strengthening precipitates in alloys. Acta Mater. 2016, 102, 125–135. [Google Scholar] [CrossRef]
- Airiskallio, E.; Nurmi, E.; Heinonen, M.H.; Vayrynen, I.J.; Kokko, K.; Ropo, M.; Punkkinen, M.P.J.; Pitkanen, H.; Alatalo, M.; Kollar, J.; et al. High temperature oxidation of Fe-Al and Fe-Cr-Al alloys: The role of Cr as a chemically active element. Corros. Sci. 2010, 52, 3394–3404. [Google Scholar] [CrossRef]
- Medvedeva, N.I.; Park, M.S.; Van Aken, D.C.; Medvedeva, J.E. First-principles study of Mn, Al and C distribution and their effect on stacking fault energies in fcc Fe. J. Alloy. Compd. 2014, 582, 475–482. [Google Scholar] [CrossRef]
- Čížek, J.; Lukáč, F.; Procházka, I.; Kužel, R.; Jirásková, Y.; Janičkovič, D.; Anwand, W.; Brauer, G. Characterization of quenched-in vacancies in Fe-Al alloys. Physica B 2012, 407, 2659–2664. [Google Scholar] [CrossRef]
- Ipser, H.; Semenova, O.; Krachler, R. Intermetallic phases with D0(3)-structure: A statistical-thermodynamic model. J. Alloy. Compd. 2002, 338, 20–25. [Google Scholar] [CrossRef]
- Lechermann, F.; Welsch, F.; Elsässer, C.; Ederer, C.; Fähnle, M.; Sanchez, J.; Meyer, B. Density-functional study of Fe3Al: LSDA versus GGA. Phys. Rev. B 2002, 65, 132104. [Google Scholar] [CrossRef]
- Connetable, D.; Maugis, P. First principle calculations of the kappa-Fe3AlC perovskite and iron-aluminium intermetallics. Intermetallics 2008, 16, 345–352. [Google Scholar] [CrossRef]
- Lechermann, F.; Fähnle, M.; Meyer, B.; Elsässer, C. Electronic correlations, magnetism, and structure of Fe-Al subsystems: An LDA+U study. Phys. Rev. B 2004, 69, 165116. [Google Scholar] [CrossRef]
- Kellou, A.; Grosdidier, T.; Raulot, J.M.; Aourag, H. Atomistic study of magnetism effect on structural stability in Fe3Al and Fe3AlX (X = H, B, C, N, O) alloys. Phys. Status Solidi B-Basic Solid State Phys. 2008, 245, 750–755. [Google Scholar] [CrossRef]
- Šesták, P.; Friák, M.; Holec, D.; Všianská, M.; Šob, M. Strength and brittleness of interfaces in Fe-Al superalloy nanocomposites under multiaxial loading: An ab initio and atomistic study. Nanomaterials 2018, 8, 873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friák, M.; Slávik, A.; Miháliková, I.; Holec, D.; Všianská, M.; Šob, M.; Palm, M.; Neugebauer, J. Origin of the low magnetic moment in Fe2AlTi: An Ab initio study. Materials 2018, 11, 1732. [Google Scholar] [CrossRef] [Green Version]
- Miháliková, I.; Friák, M.; Jirásková, Y.; Holec, D.; Koutná, N.; Šob, M. Impact of Nano-Scale Distribution of Atoms on Electronic and Magnetic Properties of Phases in Fe-Al Nanocomposites: An Ab Initio Study. Nanomaterials 2018, 8, 1059. [Google Scholar] [CrossRef] [Green Version]
- Friák, M.; Holec, D.; Šob, M. Quantum-Mechanical Study of Nanocomposites with Low and Ultra-Low Interface Energies. Nanomaterials 2018, 8, 1057. [Google Scholar] [CrossRef] [Green Version]
- Miháliková, I.; Friák, M.; Koutná, N.; Holec, D.; Šob, M. An Ab Initio Study of Vacancies in Disordered Magnetic Systems: A Case Study of Fe-Rich Fe-Al Phases. Materials 2019, 12, 1430. [Google Scholar] [CrossRef] [Green Version]
- Marcinkowski, M.; Brown, N. Theory and direct observation of dislocations in the Fe3Al superlattices. Acta Metall. 1961, 9, 764–786. [Google Scholar] [CrossRef]
- Marcinkowski, M.J.; Brown, N. Direct Observation of Antiphase Boundaries in the Fe3Al Superlattice. J. Appl. Phys. 1962, 33, 537–552. [Google Scholar] [CrossRef]
- McKamey, C.G.; Horton, J.A.; Liu, C.T. Effect of chromium on properties of Fe3Al. J. Mater. Res. 1989, 4, 1156–1163. [Google Scholar] [CrossRef]
- Morris, D.; Dadras, M.; Morris, M. The influence of cr addition on the ordered microstructure and deformation and fracture-behavior of a fe-28-percent-al intermetallic. Acta Metall. Mater. 1993, 41, 97–111. [Google Scholar] [CrossRef]
- Kral, F.; Schwander, P.; Kostorz, G. Superdislocations and antiphase boundary energies in deformed Fe3Al single crystals with chromium. Acta Mater. 1997, 45, 675–682. [Google Scholar] [CrossRef]
- Allen, S.; Cahn, J. Microscopic theory for antiphase boundary motion and its application to antiphase domain coarsening. Acta Metall. 1979, 27, 1085–1095. [Google Scholar] [CrossRef]
- Wang, K.; Wang, Y.; Cheng, Y. The Formation and Dynamic Evolution of Antiphase Domain Boundary in FeAl Alloy: Computational Simulation in Atomic Scale. Mater. Res. Ibero Am. J. Mater. 2018, 21. [Google Scholar] [CrossRef] [Green Version]
- Balagurov, A.M.; Bobrikov, I.A.; Sumnikov, V.S.; Golovin, I.S. Antiphase domains or dispersed clusters? Neutron diffraction study of coherent atomic ordering in Fe3Al-type alloys. Acta Mater. 2018, 153, 45–52. [Google Scholar] [CrossRef]
- Murakami, Y.; Niitsu, K.; Tanigaki, T.; Kainuma, R.; Park, H.S.; Shindo, D. Magnetization amplified by structural disorder within nanometre-scale interface region. Nat. Commun. 2014, 5, 4133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oguma, R.; Matsumura, S.; Eguchi, T. Kinetics of B2-and D03 type ordering and formation of domain structures in Fe-Al alloys. J. Phys. Condens. Matter 2008, 20, 275225. [Google Scholar] [CrossRef]
- McKamey, C.; Horton, J.; Liu, C. Effect of chromium on room-temperature ductility and fracture mode in Fe3Al. Scr. Metall. 1988, 22, 1679–1681. [Google Scholar] [CrossRef]
- Culbertson, G.; Kortovich, C.S. AFWAL-TR-4155; Air Force Wright Aeronautical Laboratories, Wright-Patterson Air Force Base: Dayton, OH, USA, 1986. [Google Scholar]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. B 1964, 136, B864–B871. [Google Scholar] [CrossRef] [Green Version]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. A 1965, 140, A1133–A1138. [Google Scholar] [CrossRef] [Green Version]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Perdew, J.P.; Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B 1992, 45, 13244–13249. [Google Scholar] [CrossRef] [PubMed]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Holec, D.; Mayrhofer, P.H. First-principles study of elastic properties of cubic Cr1−xAlxN alloys. J. Appl. Phys. 2013, 113, 043511. [Google Scholar] [CrossRef] [Green Version]
- Friák, M.; Buršíková, V.; Pizúrová, N.; Pavlů, J.; Jirásková, Y.; Homola, V.; Miháliková, I.; Slávik, A.; Holec, D.; Všianská, M.; et al. Elasticity of Phases in Fe-Al-Ti Superalloys: Impact of Atomic Order and Anti-Phase Boundaries. Crystals 2019, 9, 299. [Google Scholar] [CrossRef] [Green Version]
- Friák, M.; Lago, D.; Koutná, N.; Holec, D.; Rebok, T.; Šob, M. Multi-phase ELAStic Aggregates (MELASA) software tool for modeling anisotropic elastic properties of lamellar composites. Comput. Phys. Commun. 2020, 247, 106863. [Google Scholar] [CrossRef]
- Friák, M.; Všianská, M.; Holec, D.; Zelený, M.; Šob, M. Tensorial elastic properties and stability of interface states associated with Σ5(210) grain boundaries in Ni3(Al,Si). Sci. Technol. Adv. Mater. 2017, 18, 273. [Google Scholar] [CrossRef]
- Friák, M.; Všianská, M.; Holec, D.; Šob, M. Quantum-mechanical study of tensorial elastic and high-temperature thermodynamic properties of grain boundary states in superalloy-phase Ni3Al. IOP Conf. Ser. Mater. Sci. Eng. 2017, 219, 012019. [Google Scholar] [CrossRef] [Green Version]
- Friák, M.; Zelený, M.; Všianská, M.; Holec, D.; Šob, M. An ab initio study of connections between tensorial elastic properties and chemical bonds in Σ5(210) grain boundaries in Ni3Si. Materials 2018, 11, 2263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimsditch, M.; Nizzoli, F. Effective elastic constants of superlattices of any symmetry. Phys. Rev. B 1986, 33, 5891–5892. [Google Scholar] [CrossRef] [PubMed]
- Zamanzade, M.; Barnoush, A.; Motz, C. A Review on the Properties of Iron Aluminide Intermetallics. Crystals 2016, 6, 10. [Google Scholar] [CrossRef] [Green Version]
- Tang, M.; Carter, W.C.; Cannon, R.M. Diffuse interface model for structural transitions of grain boundaries. Phys. Rev. B 2006, 73, 024102. [Google Scholar] [CrossRef] [Green Version]
- Rohrer, G.S. Grain boundary energy anisotropy: A review. J. Mater. Sci. 2011, 46, 5881–5895. [Google Scholar] [CrossRef] [Green Version]
- Cantwell, P.R.; Tang, M.; Dillon, S.J.; Luo, J.; Rohrer, G.S.; Harmer, M.P. Grain boundary complexions. Acta Mater. 2014, 62, 1–48. [Google Scholar] [CrossRef]
- Rohrer, G.S. Measuring and Interpreting the Structure of Grain-Boundary Networks. J. Amer. Ceram. Soc. 2011, 94, 633–646. [Google Scholar] [CrossRef]
- Kuzmina, M.; Herbig, M.; Ponge, D.; Sandlöbes, S.; Raabe, D. Linear complexions: Confined chemical and structural states at dislocations. Science 2015, 349, 1080–1083. [Google Scholar] [CrossRef]
- Dillon, S.J.; Harmer, M.P.; Luo, J. Grain Boundary Complexions in Ceramics and Metals: An Overview. JOM 2009, 61, 38–44. [Google Scholar] [CrossRef]
- Shi, X.; Luo, J. Developing grain boundary diagrams as a materials science tool: A case study of nickel-doped molybdenum. Phys. Rev. B 2011, 84. [Google Scholar] [CrossRef] [Green Version]
- Kundu, A.; Asl, K.M.; Luo, J.; Harmer, M.P. Identification of a bilayer grain boundary complexion in Bi-doped Cu. Scr. Mater. 2013, 68, 146–149. [Google Scholar] [CrossRef]
- Bojarski, S.A.; Ma, S.; Lenthe, W.; Harmer, M.P.; Rohrer, G.S. Changes in the Grain Boundary Character and Energy Distributions Resulting from a Complexion Transition in Ca-Doped Yttria. Metall. Mater. Trans. A 2012, 43A, 3532–3538. [Google Scholar] [CrossRef]
- Rickman, J.M.; Chan, H.M.; Harmer, M.P.; Luo, J. Grain-boundary layering transitions in a model bicrystal. Surf. Sci. 2013, 618, 88–93. [Google Scholar] [CrossRef]
- Bojarski, S.A.; Harmer, M.P.; Rohrer, G.S. Influence of grain boundary energy on the nucleation of complexion transitions. Scrip. Mater. 2014, 88, 1–4. [Google Scholar] [CrossRef]
- Frazier, W.E.; Rohrer, G.S.; Rollett, A.D. Abnormal grain growth in the Potts model incorporating grain boundary complexion transitions that increase the mobility of individual boundaries. Acta Mater. 2015, 96, 390–398. [Google Scholar] [CrossRef]
- Zhou, N.; Luo, J. Developing grain boundary diagrams for multicomponent alloys. Acta Mater. 2015, 91, 202–216. [Google Scholar] [CrossRef]
- Moghadam, M.M.; Rickman, J.M.; Harmer, M.P.; Chan, H.M. The role of boundary variability in polycrystalline grain-boundary diffusion. J. App. Phys. 2015, 117, 045311. [Google Scholar] [CrossRef] [Green Version]
- Momma, K.; Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Computed Properties | Figure 3a | Figure 3b | Figure 3c | Figure 3d | Figure 3e |
|---|---|---|---|---|---|
| E (meV per 64 atoms) | 0 | 40 | 73 | 82 | 105 |
| ( per 64 atoms) | 81.0 | 81.8 | 82.6 | 82.6 | 82.4 |
| () | −0.9 | −1.1 | −1.3 | −1.3 | −1.3 |
| () Fe | 14 × (2.3–2.5) | 14 × (2.3–2.5) | 14 × (2.3–2.5) | 14 × (2.3-2.5) | 14 × (2.3–2.5) |
| () Fe no Cr in 1NN | 24 × (1.8–1.9) | 20 × (1.8–1.9) | 16 × (1.8–1.9) | 16 × (1.8-1.9) | 16 × (1.8–1.9) |
| Fe with 1 Cr in 1NN | 8 × 1.5 | 16 × (1.5–1.6) | 16 × 1.5 | 16 × 1.5 | |
| Fe with 2 Cr in 1NN | 8 × 0.9 | 4 × 0.8 |
| Computed Properties | Figure 4a | Figure 4b | Figure 4c | Figure 4d |
|---|---|---|---|---|
| E (meV per 64 atoms) | 0 | −20 | −189 | −30 |
| ( per 64 atoms) | 82.4 | 82.5 | 80.8 | 77.2 |
| () | −1.2 | −1.2 | −0.9 | −1.3 |
| () Fe | 14 × (2.3–2.6) | 14 × (2.3–2.5) | 14 × (2.3–2.6) | 14 × (2.3–2.4) |
| () Fe no Cr in 1NN | 16 × (1.8–1.9) | 16 × (1.8–1.9) | 20 × (1.8–1.9) | 20 × (1.8–1.9) |
| Fe with 1 Cr in 1NN | 16 × (1.4–1.6) | 16 × 1.5 | 8 × (1.5–1.6) | 8 × 1.4 |
| Fe with 2 Cr in 1NN | 4 × 0.5 | 4 × 0.0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Friák, M.; Všianská, M.; Šob, M. A Quantum–Mechanical Study of Clean and Cr–Segregated Antiphase Boundaries in Fe3Al. Materials 2019, 12, 3954. https://doi.org/10.3390/ma12233954
Friák M, Všianská M, Šob M. A Quantum–Mechanical Study of Clean and Cr–Segregated Antiphase Boundaries in Fe3Al. Materials. 2019; 12(23):3954. https://doi.org/10.3390/ma12233954
Chicago/Turabian StyleFriák, Martin, Monika Všianská, and Mojmír Šob. 2019. "A Quantum–Mechanical Study of Clean and Cr–Segregated Antiphase Boundaries in Fe3Al" Materials 12, no. 23: 3954. https://doi.org/10.3390/ma12233954