Key Role of Precursor Nature in Phase Composition of Supported Molybdenum Carbides and Nitrides


Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Precursors
2.3. Synthesis of Molybdenum Carbides and Nitrides
- Heating (10 °C/min) of the precursor in the N2 flow (75 cm3/min) at 200 °C for 12 h;
- Heating to the desired temperature (of 10 °C/min) in a working gas flow (75 cm3/min). A mixture of 20 vol% H2 in N2 was used to prepare nitride samples and a mixture of 20 vol% CH4 in H2 for the carbide preparation. After reaching the desired temperature, the reaction was run for 3 h;
- Cooling to the room temperature in the working gas flow (75 cm3/min);
- Flushing the reactor with nitrogen (400 cm3/min) for 30 min;
- Passivation in 1 vol% O2 in Ar (75 cm3/min) for 2 h.
2.4. Characterisation
3. Results and Discussion
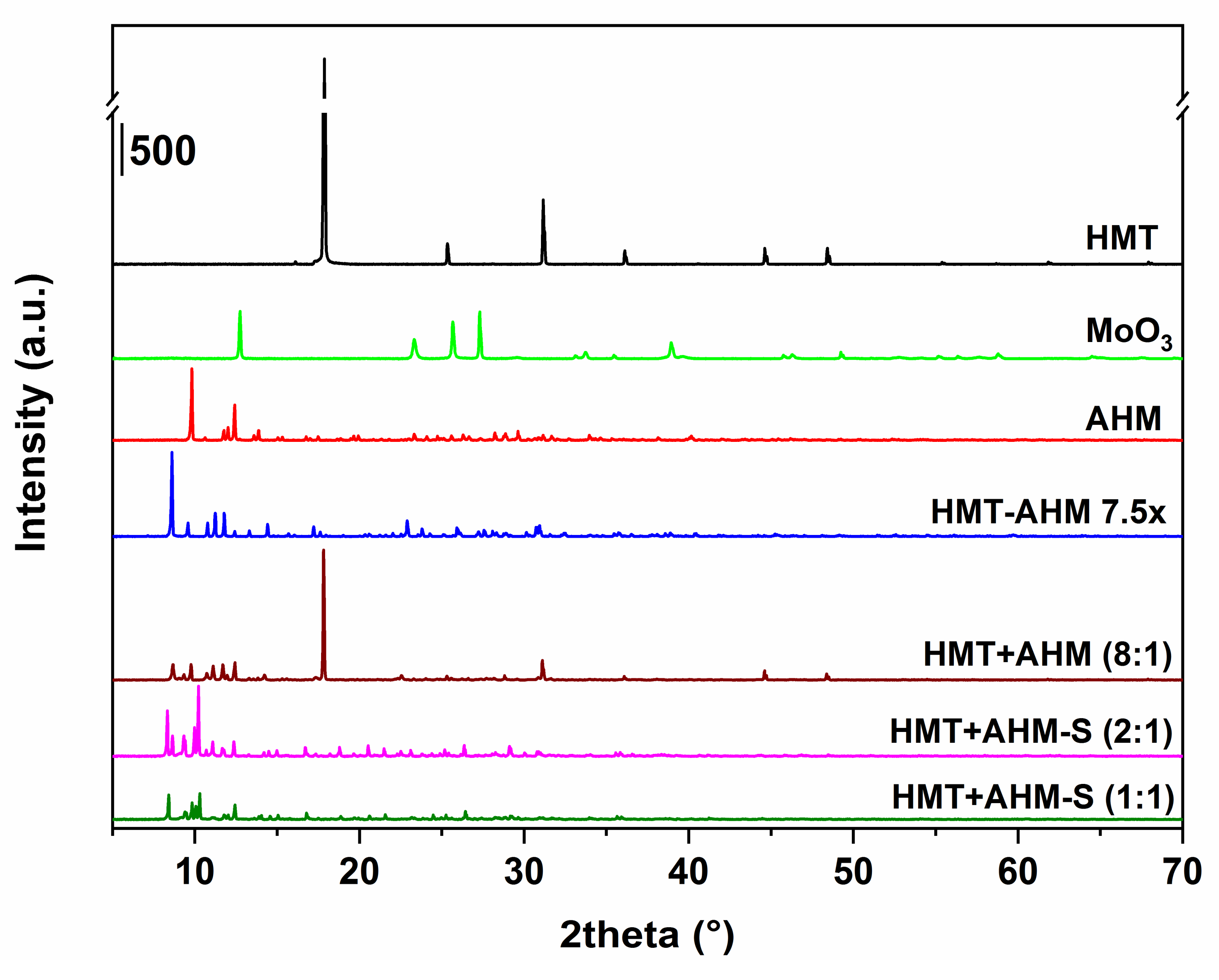
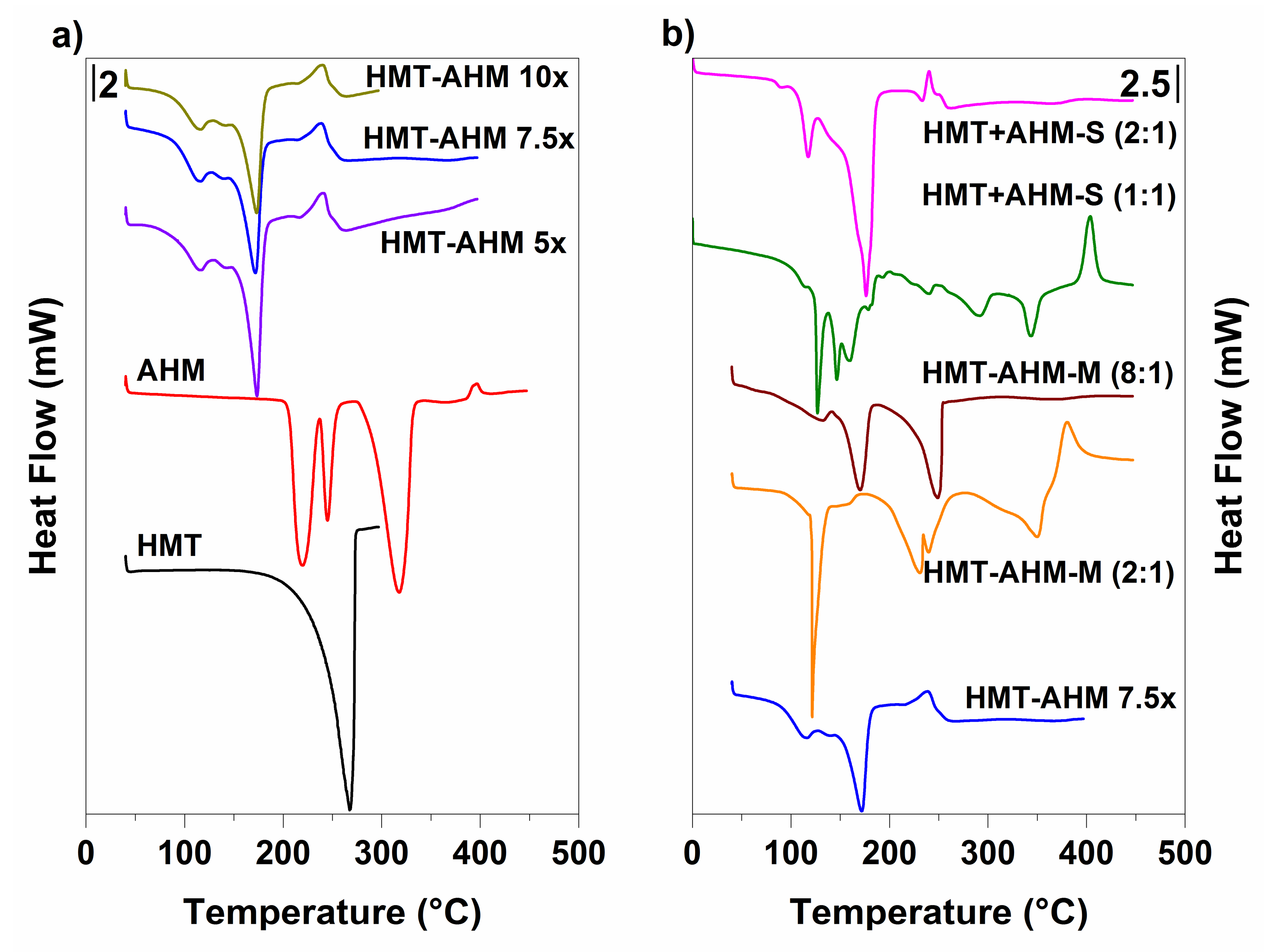
3.1. Precursors
3.2. Non-Supported MoCx
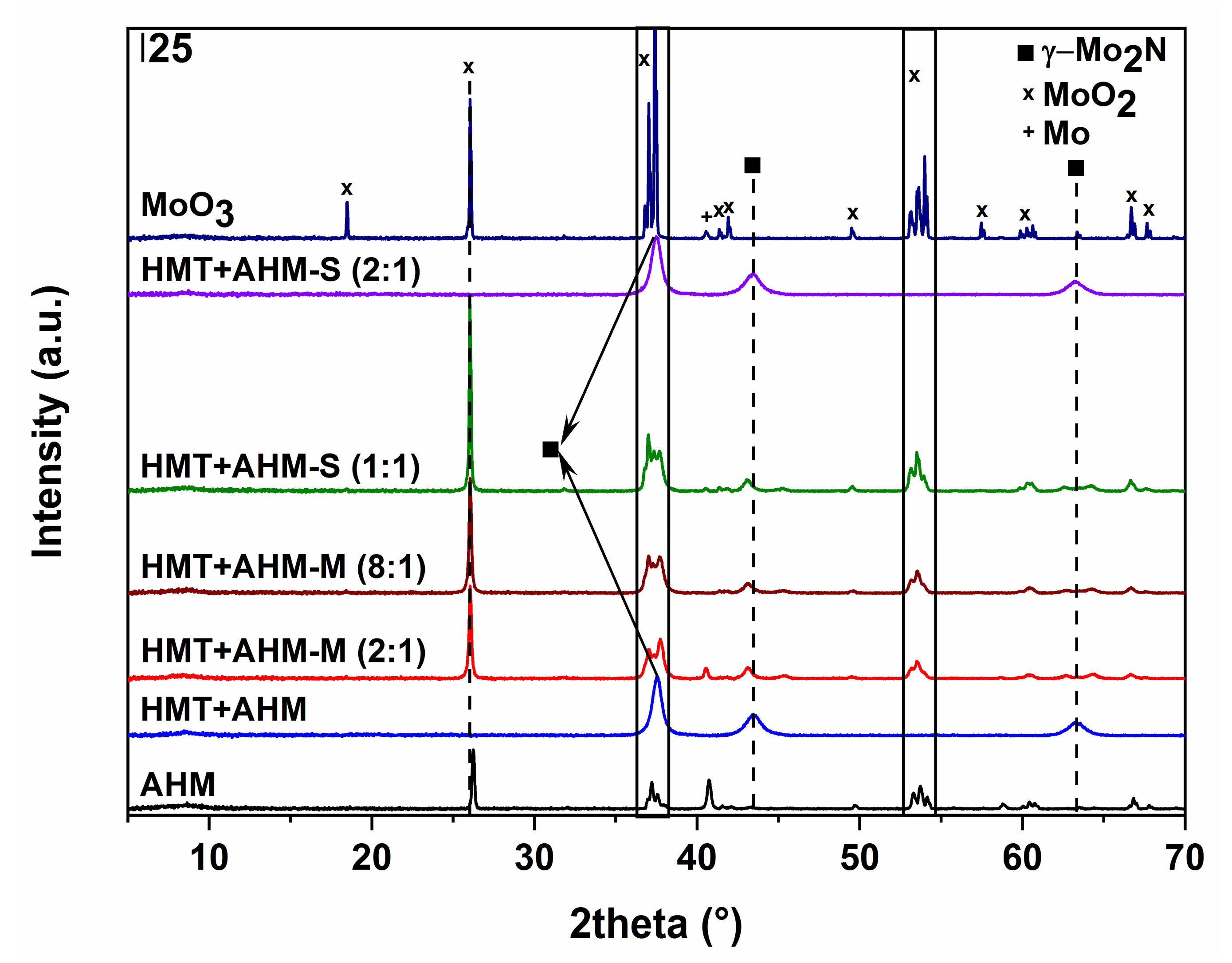
3.3. Non-Supported MoNx
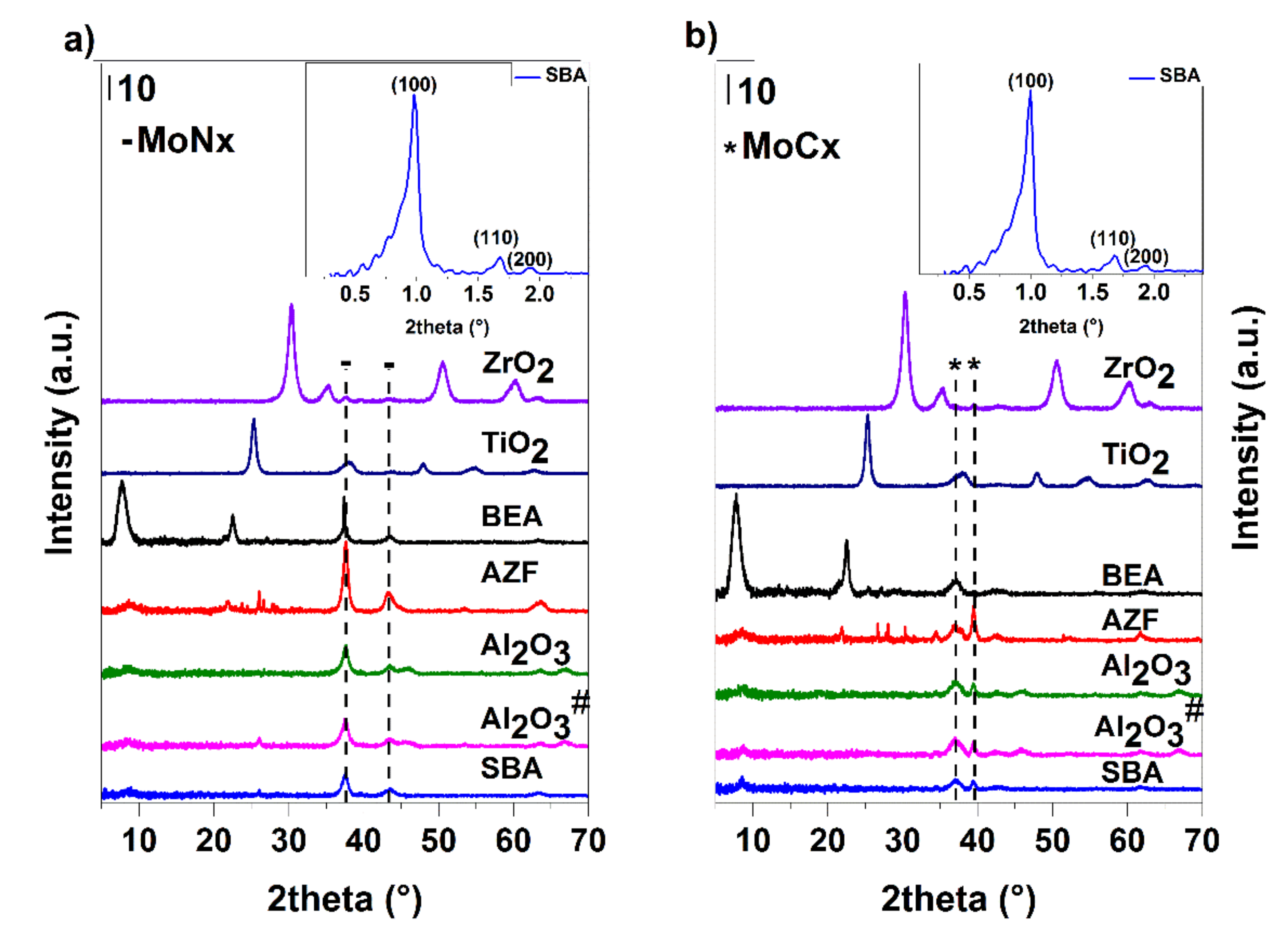
3.4. Supported Samples
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Calaisa, J.L. Band structure of transition metal compounds. Adv. Phys. 1977, 26, 847–885. [Google Scholar] [CrossRef]
- Wu, M.; Lin, X.; Hagfeldt, A.; Ma, T. Low-cost molybdenum carbide and tungsten carbide counter electrodes for dye-sensitized solar cells. Angew. Chem. Int. Ed. 2011, 50, 3520–3524. [Google Scholar] [CrossRef]
- Oyama, S.T. Preparation and catalytic properties of transition metal carbides and nitrides. Catal. Today 1992, 15, 179–200. [Google Scholar] [CrossRef]
- Furimsky, E. Metal carbides and nitrides as potential catalysts for hydroprocessing. Appl. Catal. A Gen. 2003, 240, 1–28. [Google Scholar] [CrossRef]
- Abe, H.; Cheung, T. Keung; Bell, A.T. The activity of transition metal nitrides for hydrotreating quinoline and thiophene. Catal. Lett. 1993, 21, 11–18. [Google Scholar] [CrossRef]
- Zhao, Y.; Kamiya, K.; Hashimoto, K.; Nakanishi, S. In situ CO2-emission assisted synthesis of molybdenum carbonitride nanomaterial as hydrogen evolution electrocatalyst. J. Am. Chem. Soc. 2015, 137, 110–113. [Google Scholar] [CrossRef] [PubMed]
- Neylon, M.K.; Choi, S.; Kwon, H.; Curry, K.E.; Thompson, L.T. Catalytic properties of early transition metal nitrides and carbides: N-butane Hydrogenolysis, dehydrogenation and isomerization. Appl. Catal. A Gen. 1999, 183, 253–263. [Google Scholar] [CrossRef]
- Al Shalwi, M. The Preparation of Molybdenum Carbonitrides by Single Source Routes and a Study of Their Lattice. Master’s Thesis, University of Glasgow, Glasgow, UK, May 2012. [Google Scholar]
- Oyama, S.T. Introduction to the chemistry of transition metal carbides and nitrides. In The Chemistry of Transition Metal Carbides and Nitrides; Springer: Berlin, Germany, 1996; pp. 1–27. [Google Scholar]
- Nakajima, T. Chemical vapor deposition of tungsten carbide, molybdenum carbide nitride, and molybdenum nitride films. J. Electrochem. Soc. 1997, 144, 2096. [Google Scholar] [CrossRef]
- Hyeon, T.; Fang, M.; Suslick, K.S. Nanostructured molybdenum carbide: Sonochemical synthesis and catalytic properties. J. Am. Chem. Soc. 1996, 118, 5492–5493. [Google Scholar] [CrossRef]
- Pang, M.; Li, C.; Ding, L.; Zhang, J.; Su, D.; Li, W.; Liang, C. Microwave-assisted preparation of Mo2C/CNTs nanocomposites as efficient electrocatalyst supports for oxygen reduction reaction. Ind. Eng. Chem. Res. 2010, 49, 4169–4174. [Google Scholar] [CrossRef]
- Bystrov, Y.A.; Vetrov, N.Z.; Lisenkov, A.A. Plasmachemical synthesis of carbide compounds in metal-containing plasma jet from vacuum arc discharge. Tech. Phys. Lett. 2008, 34, 734–736. [Google Scholar] [CrossRef]
- Merzhanov, A.G.; Borovinskaya, I.P. Self-propagated high-temperature synthesis of refractory inorganic compounds. Doklady Akademii Nauk SSSR 1972, 204, 366–369. [Google Scholar]
- Guil-López, R.; Nieto, E.; Botas, J.A.; Fierro, J.L.G. On the genesis of molybdenum carbide phases during reduction-carburization reactions. J. Solid State Chem. 2012, 190, 285–295. [Google Scholar] [CrossRef]
- Schaidle, J.A.; Schweitzer, N.M.; Ajenifujah, O.T.; Thompson, L.T. On the preparation of molybdenum carbide-supported metal catalysts. J. Catal. 2012, 289, 210–217. [Google Scholar] [CrossRef]
- Chai, S.H.; Schwartz, V.; Howe, J.Y.; Wang, X.; Kidder, M.; Overbury, S.H.; Dai, S.; Jiang, D.E. Graphitic mesoporous carbon-supported molybdenum carbides for catalytic hydrogenation of carbon monoxide to mixed alcohols. Microporous Mesoporous Mater. 2013, 170, 141–149. [Google Scholar] [CrossRef]
- Tang, C.; Sun, A.; Xu, Y.; Wu, Z.; Wang, D. High specific surface area Mo2C nanoparticles as an efficient electrocatalyst for hydrogen evolution. J. Power Sources 2015, 296, 18–22. [Google Scholar] [CrossRef]
- Vitale, G.; Guzmán, H.; Frauwallner, M.L.; Scott, C.E.; Pereira-Almao, P. Synthesis of nanocrystalline molybdenum carbide materials and their characterization. Catal. Today 2015, 250, 123–133. [Google Scholar] [CrossRef]
- Preiss, H.; Meyer, B.; Olschewski, C. Preparation of molybdenum and tungsten carbides from solution derived precursors. J. Mater. Sci. 1998, 33, 713–722. [Google Scholar] [CrossRef]
- Patel, M.; Subrahmanyam, J. Synthesis of nanocrystalline molybdenum carbide (Mo2C) by solution route. Mater. Res. Bull. 2008, 43, 2036–2041. [Google Scholar] [CrossRef]
- Baklanova, O.N.; Vasilevich, A.V.; Lavrenov, A.V.; Drozdov, V.A.; Muromtsev, I.V.; Arbuzov, A.B.; Trenikhin, V.; Sigaeva, S.S.; Temerev, V.L.; Gorbunova, O.V.; et al. Molybdenum carbide synthesized by mechanical activation an inert medium. J. Alloys Compd. 2017, 698, 1018–1027. [Google Scholar] [CrossRef]
- Torabi, O.; Golabgir, M.H.; Tajizadegan, H.; Torabi, H. A study on mechanochemical behavior of MoO3-Mg-C to synthesize molybdenum carbide. Int. J. Refract. Met. Hard Mater. 2014, 47, 18–24. [Google Scholar] [CrossRef]
- Saghafi, M.; Ataie, A.; Heshmati-Manesh, S. Effects of mechanical activation of MoO3/C powder mixture in the processing of nano-crystalline molybdenum. Int. J. Refract. Met. Hard Mater. 2011, 29, 419–423. [Google Scholar] [CrossRef]
- Khabbaz, S.; Honarbakhsh-Raouf, A.; Ataie, A.; Saghafi, M. Effect of processing parameters on the mechanochemical synthesis of nanocrystalline molybdenum carbide. Int. J. Refract. Met. Hard Mater. 2013, 41, 402–407. [Google Scholar] [CrossRef]
- Choi, J.-G.; Brenner, J.R.; Colling, C.W.; Demczyk, B.G.; Dunning, J.L.; Thompson, L.T. Synthesis and characterization of molybdenum nitride hydrodenitrogenation catalysts. Catal. Today 1992, 15, 201–222. [Google Scholar] [CrossRef]
- Wolden, C.A.; Pickerell, A.; Gawai, T.; Parks, S.; Hensley, J.; Way, J.D. Synthesis of β-Mo2C thin films. ACS Appl. Mater. Interfaces 2011, 3, 517–521. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Liu, X.; Wang, S.; Huo, C.; Li, Y.W.; Wang, J.; Jiao, H. Stability of β-Mo2C facets from Ab initio atomistic thermodynamics. J. Phys. Chem. C 2011, 115, 22360–22368. [Google Scholar] [CrossRef]
- Wang, T.; Wang, S.; Li, Y.W.; Wang, J.; Jiao, H. Adsorption equilibria of CO coverage on β-Mo2C surfaces. J. Phys. Chem. C 2012, 116, 6340–6348. [Google Scholar] [CrossRef]
- Zhu, Q.; Chen, Q.; Yang, X.; Ke, D. A new method for the synthesis of molybdenum carbide. Mater. Lett. 2007, 61, 5173–5174. [Google Scholar] [CrossRef]
- Vitale, G.; Frauwallner, M.L.; Hernandez, E.; Scott, C.E.; Pereira-Almao, P. Low temperature synthesis of cubic molybdenum carbide catalysts via pressure induced crystallographic orientation of MoO3 precursor. Appl. Catal. A Gen. 2011, 400, 221–229. [Google Scholar] [CrossRef]
- Xiao, T.C.; York, A.P.E.; Williams, V.C.; Al-Megren, H.; Hanif, A.; Zhou, X.Y.; Green, M.L.H. Preparation of molybdenum carbides using butane and their catalytic performance. Chem. Mater. 2000, 12, 3896–3905. [Google Scholar] [CrossRef]
- Bouchy, C.; Derouane-Abd Hamid, S.B.; Derouane, E.G. A new route to the metastable FCC molybdenum carbide α-MoC. Chem. Commun. 2000, 125–126. [Google Scholar] [CrossRef]
- Bouchy, C.; Schmidt, I.; Anderson, J.; Jacobsen, C.J.; Derouane, E.; Derouane-Abd Hamid, S. Metastable fcc α-MoC1−x supported on HZSM5: Preparation and catalytic performance for the non-oxidative conversion of methane to aromatic compounds. J. Mol. Catal. A Chem. 2000, 163, 283–296. [Google Scholar] [CrossRef]
- Miyao, T.; Shishikura, I.; Matsuoka, M.; Nagai, M.; Oyama, S.T. Preparation and characterization of alumina-supported molybdenum carbide. Appl. Catal. A Gen. 1997, 165, 419–428. [Google Scholar] [CrossRef]
- Koós, Á.; Solymosi, F. Production of CO-free H2 by formic acid decomposition over Mo2C/carbon catalysts. Catal. Lett. 2010, 138, 23–27. [Google Scholar] [CrossRef]
- Leclercq, L.; Provost, M.; Pastor, H.; Grimblot, J.; Hardy, A.M.; Gengembre, L.; Leclercq, G. Catalytic properties of transition metal carbides. I. Preparation and physical characterization of bulk mixed carbides of molybdenum and tungsten. J. Catal. 1989, 117, 371–383. [Google Scholar] [CrossRef]
- Ma, Y.; Guan, G.; Shi, C.; Zhu, A.; Hao, X.; Wang, Z.; Kusakabe, K.; Abudula, A. Low-temperature steam reforming of methanol to produce hydrogen over various metal-doped molybdenum carbide catalysts. Int. J. Hydrogen Energy 2014, 39, 258–266. [Google Scholar] [CrossRef]
- Bej, S.K.; Bennett, C.A.; Thompson, L.T. Acid and base characteristics of molybdenum carbide catalysts. Appl. Catal. A Gen. 2003, 250, 197–208. [Google Scholar] [CrossRef]
- Lee, W.S.; Wang, Z.; Wu, R.J.; Bhan, A. Selective vapor-phase hydrodeoxygenation of anisole to benzene on molybdenum carbide catalysts. J. Catal. 2014, 319, 44–53. [Google Scholar] [CrossRef]
- Lee, J.S.; Oyama, S.T.; Boudart, M. Molybdenum carbide catalysts. I. Synthesis of unsupported powders. J. Catal. 1987, 106, 125–133. [Google Scholar] [CrossRef]
- Roy, A.; Serov, A.; Artyushkova, K.; Brosha, E.L.; Atanassov, P.; Ward, T.L. Facile synthesis of high surface area molybdenum nitride and carbide. J. Solid State Chem. 2015, 228, 232–238. [Google Scholar] [CrossRef]
- Afanasiev, P. New single source route to the molybdenum nitride Mo2N. Inorg. Chem. 2002, 41, 5317–5319. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.-Q.; Zhang, Z.-B.; Zhang, M.-H. The efficient synthesis of a molybdenum carbide catalyst via H2-thermal treatment of a Mo(VI)-hexamethylenetetramine complex. Dalton Trans. 2011, 40, 1098–1104. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.M.; Wang, X.H.; Zhang, M.H.; Du, X.Y.; Li, W.; Tao, K.Y. Synthesis of bulk and supported molybdenum carbide by a single-step thermal carburization method. Chem. Mater. 2007, 19, 1801–1807. [Google Scholar] [CrossRef]
- Huo, X.; Wang, Z.; Huang, J.; Zhang, R.; Fang, Y. Bulk Mo and Co-Mo carbides as catalysts for methanation. Catal. Commun. 2016, 79, 39–44. [Google Scholar] [CrossRef]
- Kovács, T.N.; Hunyadi, D.; de Lucena, A.L.A.; Szilágyi, I.M. Thermal decomposition of ammonium molybdates. J. Therm. Anal. Calorim. 2016, 124, 1013–1021. [Google Scholar] [CrossRef]
- Chouzier, S.; Czeri, T.; Roy-Auberger, M.; Pichon, C.; Geantet, C.; Vrinat, M.; Afanasiev, P. Decomposition of molybdatehexamethylenetetramine complex: One single source route for different catalytic materials. J. Solid State Chem. 2011, 184, 2668–2677. [Google Scholar] [CrossRef]
- Stellwagen, D.R.; Bitter, J.H. Structure–performance relations of molybdenum- and tungsten carbide catalysts for deoxygenation. Green Chem. 2015, 17, 582–593. [Google Scholar] [CrossRef]
- Alexander, A.-M.; Hargreaves, J.S.J. Alternative catalytic materials: Carbides, nitrides, phosphides and amorphous boron alloys. Chem. Soc. Rev. 2010, 39, 4388–4401. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.M.; Du, X.Y.; Zhang, M.H.; Li, W.; Tao, K.Y. Synthesis of bulk and alumina-supported γ-Mo2N catalysts by a single-step complex decomposition method. Catal. Today 2008, 131, 156–161. [Google Scholar] [CrossRef]
- Wang, H.; Li, W.; Zhang, M. New approach to the synthesis of bulk and supported bimetallic molybdenum nitrides. Chem. Mater. 2005, 17, 3262–3267. [Google Scholar] [CrossRef]
- Wang, X.H.; Zhang, M.H.; Li, W.; Tao, K.Y. A simple synthesis route and characterisation of Co3Mo3C. Dalt. Trans. 2007, 5165–5170. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.S.H.; Mäki-Arvela, P.; Akhmetzyanova, U.; Tišler, Z.; Hachemi, I.; Rudnäs, A.; Smeds, A.; Eränen, K.; Aho, A.; Kumar, N.; et al. Direct hydrodeoxygenation of algal lipids extracted from Chlorella alga. J. Chem. Technol. Biotechnol. 2017, 92, 741–748. [Google Scholar] [CrossRef]
- Yao, Z.W. Exploration on synthesis of activated carbon supported molybdenum carbide, nitride and phosphide via carbothermal reduction route. J. Alloys Compd. 2009, 475, L38–L41. [Google Scholar] [CrossRef]
- Pielaszek, J.; Mierzwa, B.; Medjahdi, G.; Marêché, J.F.; Puricelli, S.; Celzard, A.; Furdin, G. Molybdenum carbide catalyst formation from precursors deposited on active carbons: XRD studies. Appl. Catal. A Gen. 2005, 296, 232–237. [Google Scholar] [CrossRef]
- Wang, X.H.; Hao, H.L.; Zhang, M.H.; Li, W.; Tao, K.Y. Synthesis and characterization of molybdenum carbides using propane as carbon source. J. Solid State Chem. 2006, 179, 538–543. [Google Scholar] [CrossRef]
- Wu, Q.; Christensen, J.M.; Chiarello, G.L.; Duchstein, L.D.L.; Wagner, J.B.; Temel, B.; Grunwaldt, J.D.; Jensen, A.D. Supported molybdenum carbide for higher alcohol synthesis from syngas. Catal. Today 2013, 215, 162–168. [Google Scholar] [CrossRef]
- Darujati, A.R.S.; Thomson, W.J. Stability of supported and promoted-molybdenum carbide catalysts in dry-methane reforming. Appl. Catal. A Gen. 2005, 296, 139–147. [Google Scholar] [CrossRef]
- Du, X.; France, L.J.; Kuznetsov, V.L.; Xiao, T.; Edwards, P.P.; AlMegren, H.; Bagabas, A. Dry reforming of methane over ZrO2-supported Co–Mo carbide catalyst. Appl. Petrochem. Res. 2014, 4, 137–144. [Google Scholar] [CrossRef]
- Vo, D.V.N.; Adesina, A.A. Fischer-Tropsch synthesis over alumina-supported molybdenum carbide catalyst. Appl. Catal. A Gen. 2011, 399, 221–232. [Google Scholar] [CrossRef]
- McCrea, K.R.; Logan, J.W.; Tarbuck, T.L.; Heiser, J.L.; Bussell, M.E. thiophene hydrodesulfurization over alumina-supported molybdenum carbide and nitride catalysts: Effect of Mo loading and phase. J. Catal. 1997, 171, 255–267. [Google Scholar] [CrossRef]
- Boullosa-Eiras, S.; Lødeng, R.; Bergem, H.; Stöcker, M.; Hannevold, L.; Blekkan, E.A. Catalytic hydrodeoxygenation (HDO) of phenol over supported molybdenum carbide, nitride, phosphide and oxide catalysts. Catal. Today 2014, 223, 44–53. [Google Scholar] [CrossRef]
- Brungs, A.J.; York, A.P.E.; Claridge, J.B.; Márquez-Alvarez, C.; Green, M.L.H. Dry reforming of methane to synthesis gas over supported molybdenum carbide catalysts. Catal. Lett. 2000, 70, 117–122. [Google Scholar] [CrossRef]
- Nagai, M.; Miyao, T.; Tuboi, T. Hydrodesulfurization of dibenzothiophene on alumina-supported molybdenum nitride. Catal. Lett. 1993, 18, 9–14. [Google Scholar] [CrossRef]
- Colling, C.W.; Thompson, L.T. The structure and function of supported molybdenum nitride hydrodenitrogenation catalysts. J. Catal. 1994, 146, 193–203. [Google Scholar] [CrossRef]
- Zhong, H.; Zhang, H.; Liu, G.; Liang, Y.; Hu, J.; Yi, B. A novel non-noble electrocatalyst for PEM fuel cell based on molybdenum nitride. Electrochem. Commun. 2006, 8, 707–712. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, T.; Xia, L.; Li, T.; Zheng, M.; Wu, Z.; Wang, X.; Wei, Z.; Xin, Q.; Li, C. Catalytic decomposition of hydrazine over supported molybdenum nitride catalysts in a monopropellant thruster. Catal. Lett. 2002, 79, 21–25. [Google Scholar] [CrossRef]
- Tyrone Ghampson, I.; Sepúlveda, C.; Garcia, R.; García Fierro, J.L.; Escalona, N.; Desisto, W.J. Comparison of alumina- and SBA-15-supported molybdenum nitride catalysts for hydrodeoxygenation of guaiacol. Appl. Catal. A Gen. 2012, 435–436, 51–60. [Google Scholar] [CrossRef]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
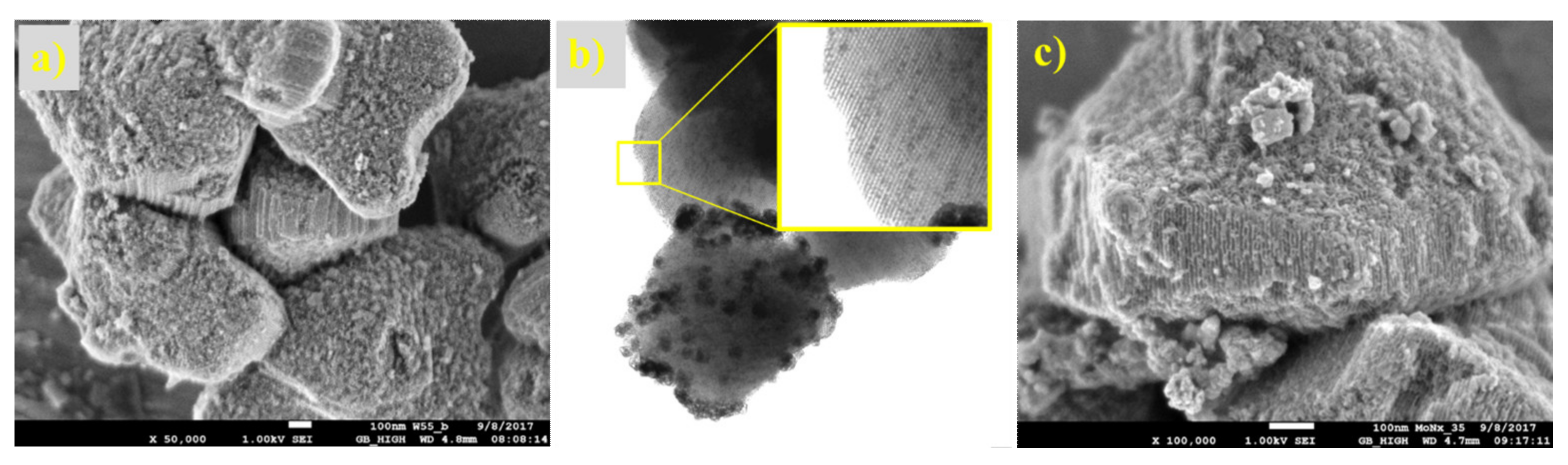{kind=link}
| Sample | SBET (m2/g) | Elemental Analysis (wt%) | Content of Crystalline Phase (%)/Crystallite Size D (nm) | |||
|---|---|---|---|---|---|---|
| C | N | β-Mo2C | α-Mo2C | MoO2 | ||
| AHM-600 | 3.9 | 1.64 | <0.05 | - | 12/18.2 | 88/- |
| AHM-700 | 7.4 | 5.41 | <0.05 | 34/- | 66/14.3 | - |
| AHM-800 | 5.3 | 6.32 | <0.05 | - | 100/40.2 | - |
| HMT-AHM-600 * | 19.4 | 5.49 | <0.05 | 100/4.9 | - | - |
| HMT-AHM-700 * | 25.3 | 6.52 | <0.05 | 32/- | 68/14.6 | - |
| HMT-AHM-800 * | 14.1 | 8.34 | <0.05 | - | 100/21.3 | - |
| HMT+AHM-S (1:1) ** | 16.2 | 4.96 | <0.05 | - | 100/18.4 | - |
| HMT+AHM-S (2:1) ** | 51.8 | 4.43 | <0.05 | - | 100/15.1 | - |
| HMT+AHM-M (2:1) ** | 8.7 | 5.66 | <0.05 | - | 100/21.3 | - |
| HMT+AHM-M (8:1) ** | 18.3 | 6.27 | <0.05 | - | 100/19.8 | - |
| MoO3 ** | 0.0 | 5.81 | <0.05 | - | 100/18.5 | - |
| Sample | SBET (m2/g) | Elemental Analysis (wt%) | Content of Crystalline Phase (%)/Crystallite Size D (nm) | |||||
|---|---|---|---|---|---|---|---|---|
| C | N | β-Mo2N | γ-Mo2N | Mo3N2 | Mo | MoO2 | ||
| AHM-700 | 0.0 | <0.05 | 0.37 | 19/27.4 | - | - | 36/- | 45/- |
| AHM-800 | 0.0 | 0.05 | <0.05 | - | - | - | 57/- | 43/- |
| AHM-900 | 0.1 | <0.05 | <0.05 | - | - | - | 69/- | 31/- |
| HMT-AHM-700 * | 25.0 | 0.45 | 5.68 | - | 100/13.8 | - | - | - |
| HMT-AHM-800 * | 11.5 | <0.05 | 5.96 | 100/26.0 | - | - | - | - |
| HMT-AHM-900 * | 0.0 | <0.05 | <0.05 | - | - | - | 100/- | - |
| HMT+AHM-S (1:1) ** | 16.2 | 0.15 | 2.44 | 14/18.3 | - | - | - | 86/- |
| HMT+AHM-S (2:1) ** | 13.2 | 0.54 | 5.95 | - | - | 100/11.7 | - | - |
| HMT+AHM-M (2:1) ** | 12.2 | 0.15 | 2.32 | 78/17.6 | - | - | 1/- | 21/- |
| HMT+AHM-M (8:1) ** | 29.0 | 0.09 | 0.97 | 76/17.5 | - | - | - | 24/- |
| MoO3 ** | 0.3 | <0.05 | <0.05 | - | - | - | 2/- | 98/- |
| Support | Initial SBET (m2/g) | Supported Nitrides | Supported Carbides | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Chemical Composition (wt%) | SBET (m2/g) | Chemical Composition (wt%) | SBET (m2/g) | ||||||||||
| Mo | X * | Si | C | N | Mo | X * | Si | C | N | ||||
| Al2O3 | 192 | 22.5 | 33.9 | 0.0 | 0.08 | 1.03 | 123.6 | 22.1 | 38.3 | 0.0 | 1.46 | <0.05 | 118.8 |
| Al2O3 # | 23.2 | 38.4 | 0.0 | 0.19 | 0.86 | 120.1 | 23.0 | 37.0 | 0.0 | 1.48 | <0.05 | 131.5 | |
| TiO2 | 167 | 17.7 | 43.9 | 0.0 | 0.42 | 0.53 | 103.7 | 16.5 | 44.5 | 0.0 | 1.06 | 0.16 | 102.1 |
| ZrO2 | 143 | 8.2 | 57.8 | 0.0 | 0.16 | 0.21 | 117.5 | 7.8 | 57.5 | 0.0 | 0.49 | <0.05 | 107.1 |
| AZF | 120 | 36.1 | 1.4 | 22.8 | 0.22 | 2.38 | 28.1 | 38.4 | 1.31 | 20.1 | 2.57 | <0.05 | 16.8 |
| SBA | 743 | 25.7 | 0.0 | 22.6 | 0.45 | 1.92 | 339.0 | 36.8 | 0.0 | 22.6 | 2.07 | <0.05 | 340.1 |
| BEA | 680 | 28.3 | 1.9 | 27.5 | 0.31 | 1.47 | 331.7 | 24.8 | 1.81 | 25.9 | 1.07 | <0.05 | 330.5 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tišler, Z.; Velvarská, R.; Skuhrovcová, L.; Pelíšková, L.; Akhmetzyanova, U. Key Role of Precursor Nature in Phase Composition of Supported Molybdenum Carbides and Nitrides. Materials 2019, 12, 415. https://doi.org/10.3390/ma12030415
Tišler Z, Velvarská R, Skuhrovcová L, Pelíšková L, Akhmetzyanova U. Key Role of Precursor Nature in Phase Composition of Supported Molybdenum Carbides and Nitrides. Materials. 2019; 12(3):415. https://doi.org/10.3390/ma12030415
Chicago/Turabian StyleTišler, Zdeněk, Romana Velvarská, Lenka Skuhrovcová, Lenka Pelíšková, and Uliana Akhmetzyanova. 2019. "Key Role of Precursor Nature in Phase Composition of Supported Molybdenum Carbides and Nitrides" Materials 12, no. 3: 415. https://doi.org/10.3390/ma12030415
APA StyleTišler, Z., Velvarská, R., Skuhrovcová, L., Pelíšková, L., & Akhmetzyanova, U. (2019). Key Role of Precursor Nature in Phase Composition of Supported Molybdenum Carbides and Nitrides. Materials, 12(3), 415. https://doi.org/10.3390/ma12030415