Theoretical Study of As2O3 Adsorption Mechanisms on CaO surface


Abstract
:1. Introduction
2. Methods and Modeling
2.1. Methods
2.2. Modeling
3. Results and Discussions
3.1. Stable Sorption Structures
3.1.1. Stable Physisorption Structures
3.1.2. Stable Chemisorption Structures
- Category I: As2O3’s As is located on the hollow site
- Category II: All of As2O3’s O is located on or close to superficial Ca top site
- Category III: As2O3 transforms into a spoon-shaped structure
- Category IV: All of As2O3’s As is located on two neighboring superficial O top site
3.2. Adsorption Process
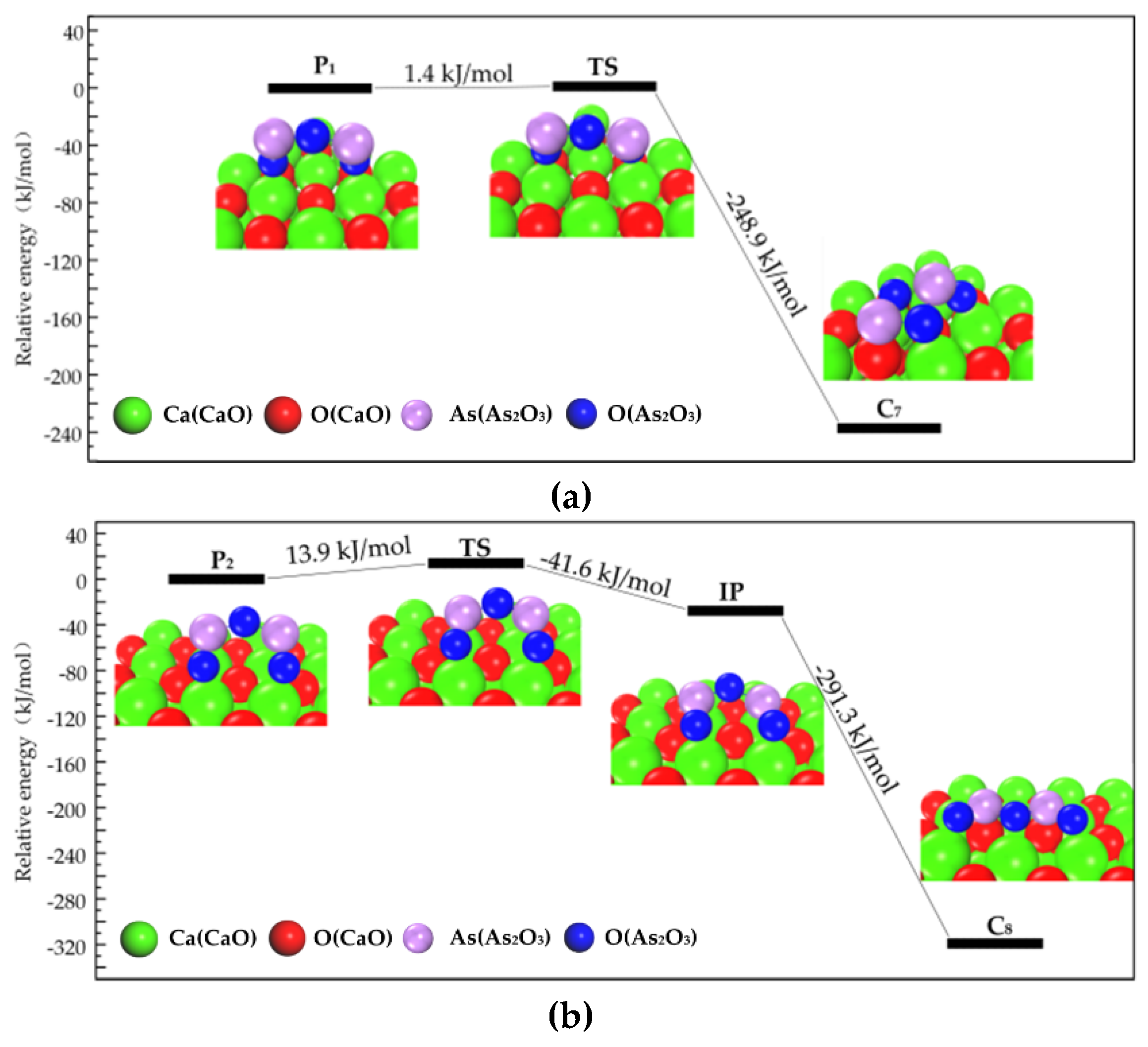
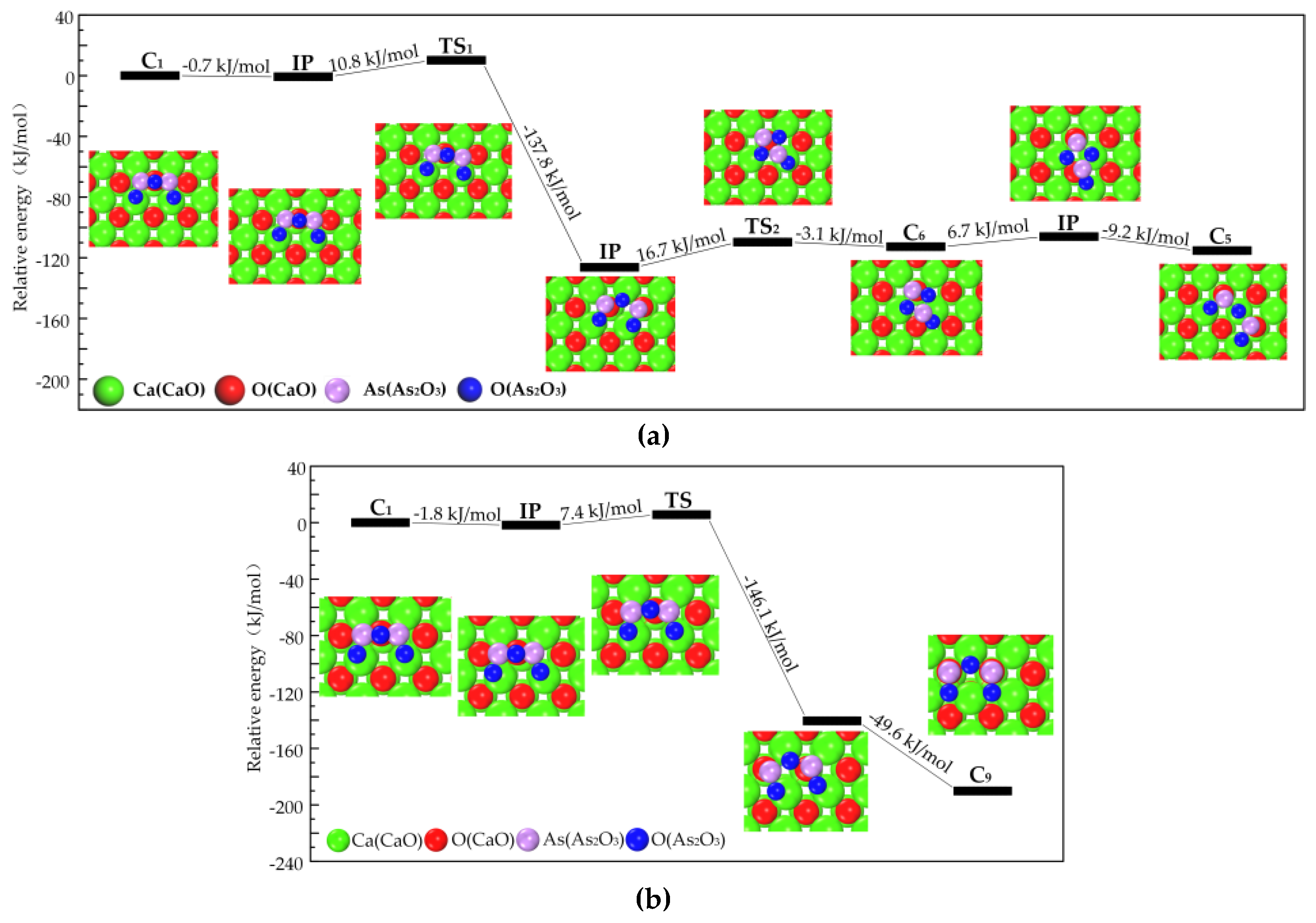
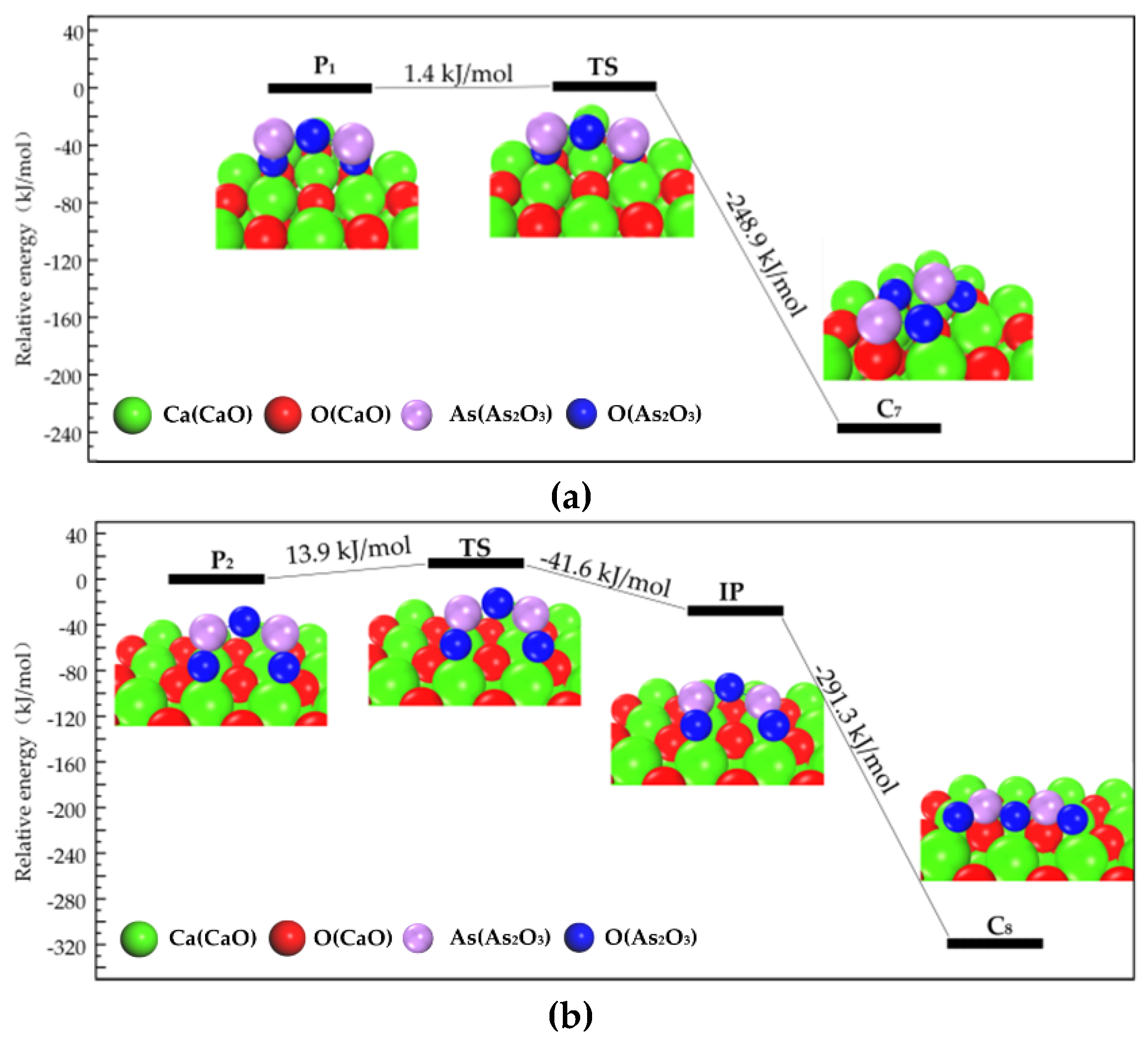
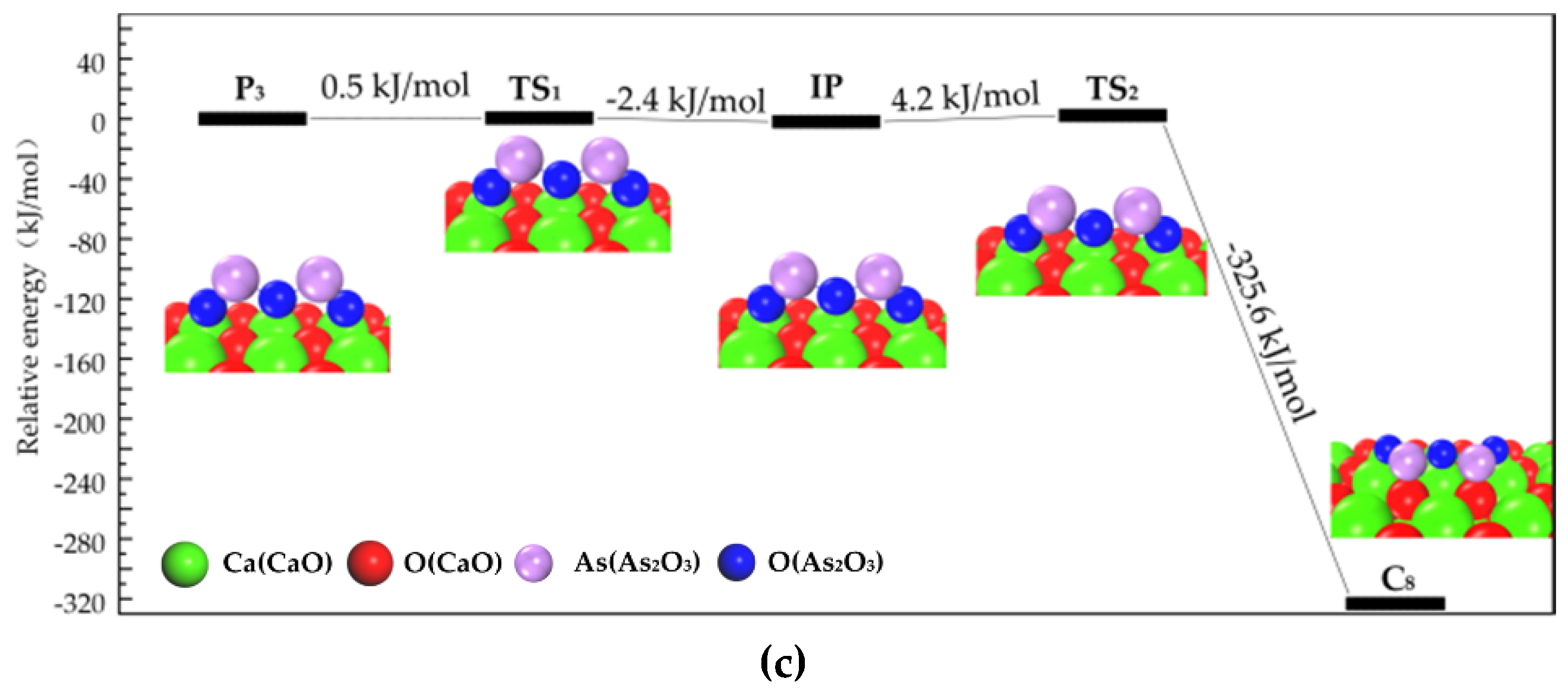
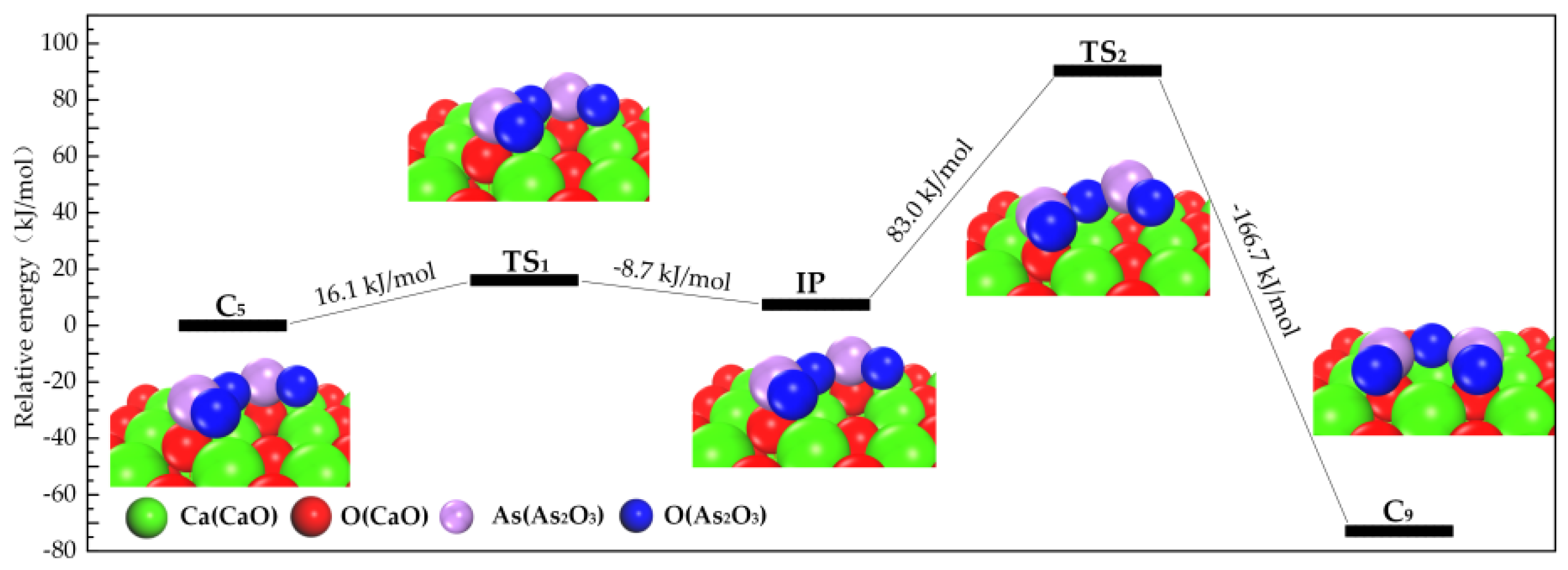
3.2.1. Transformation Process of Physisorption Structures to Chemisorption Structures
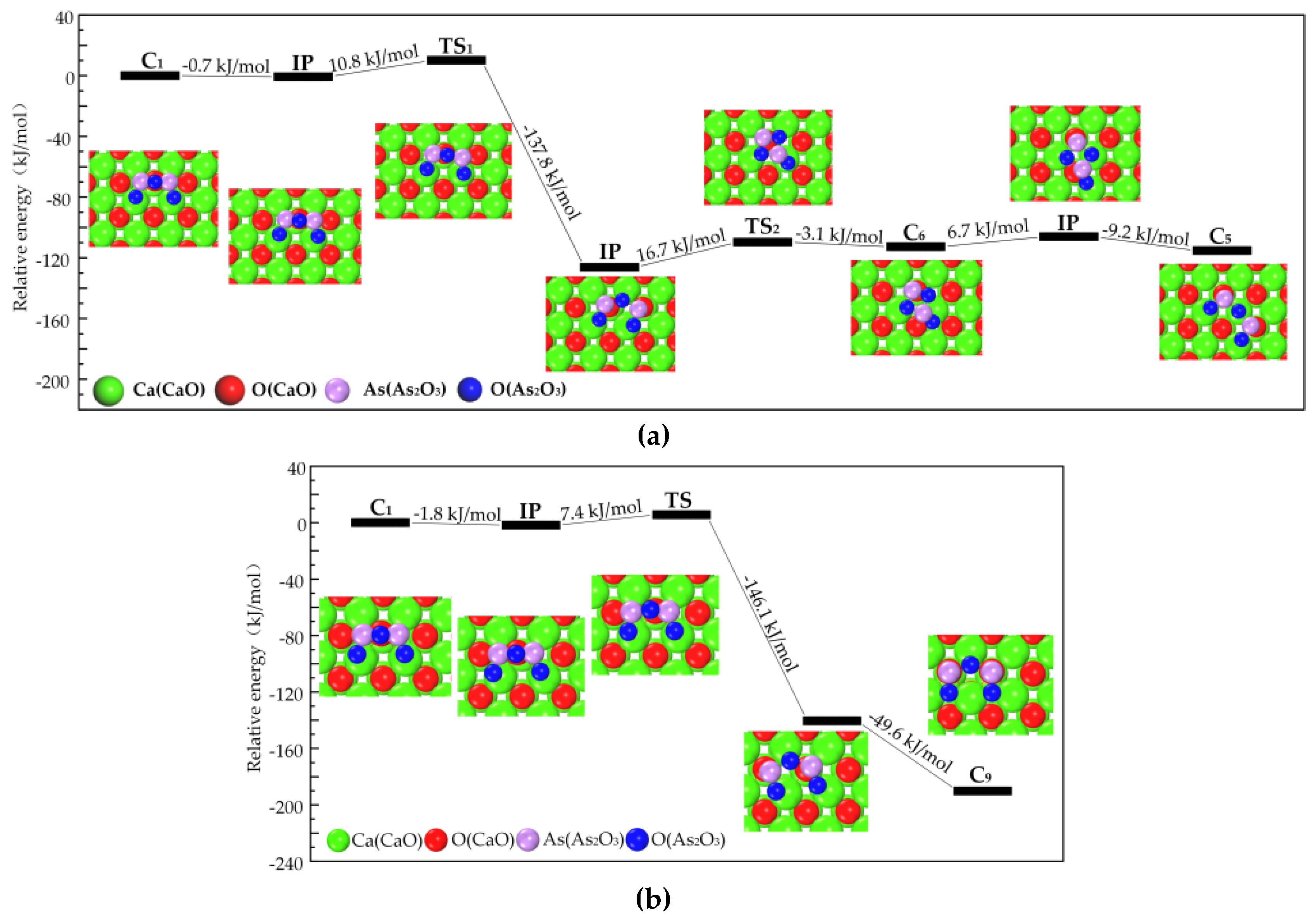
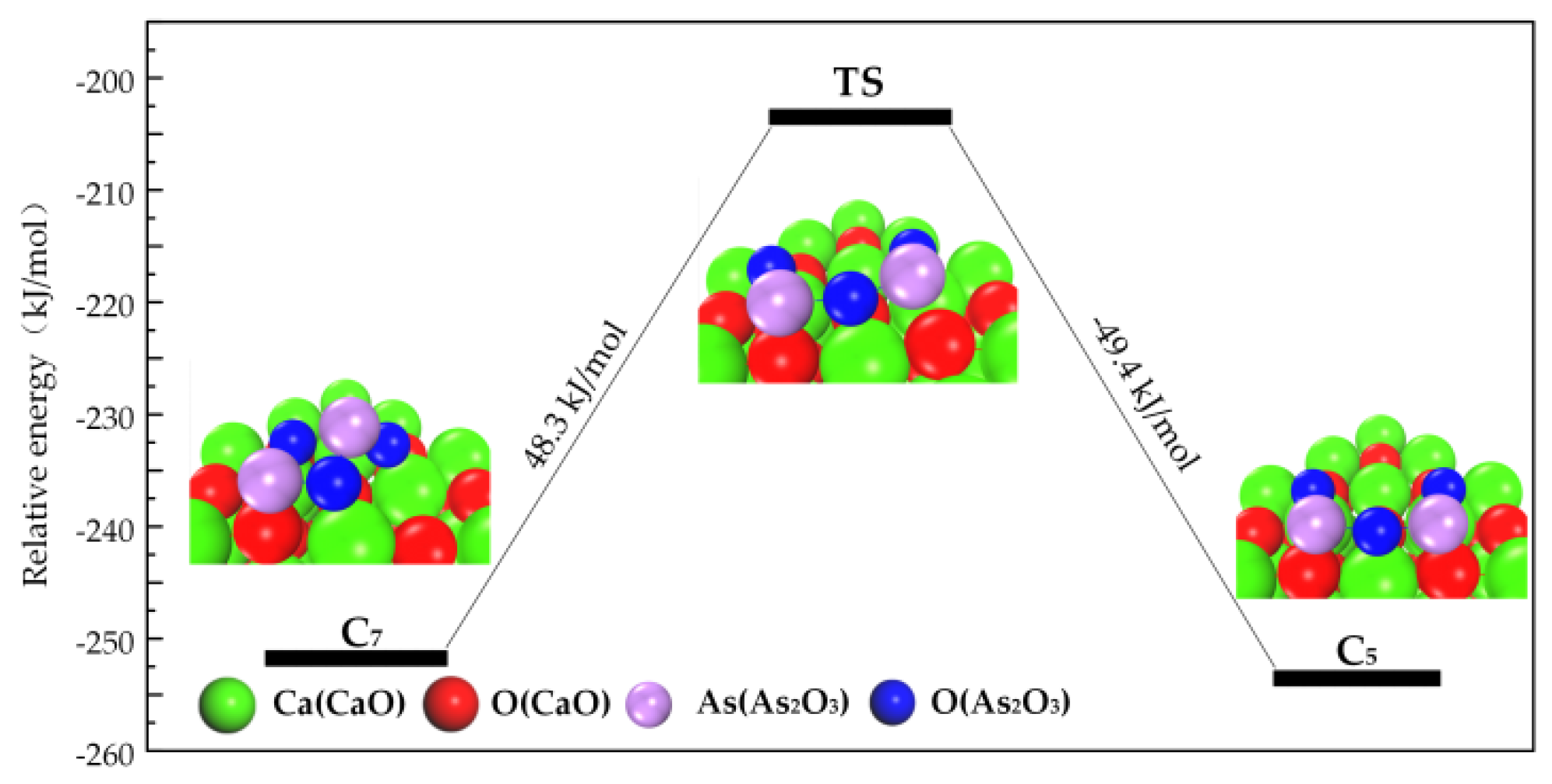
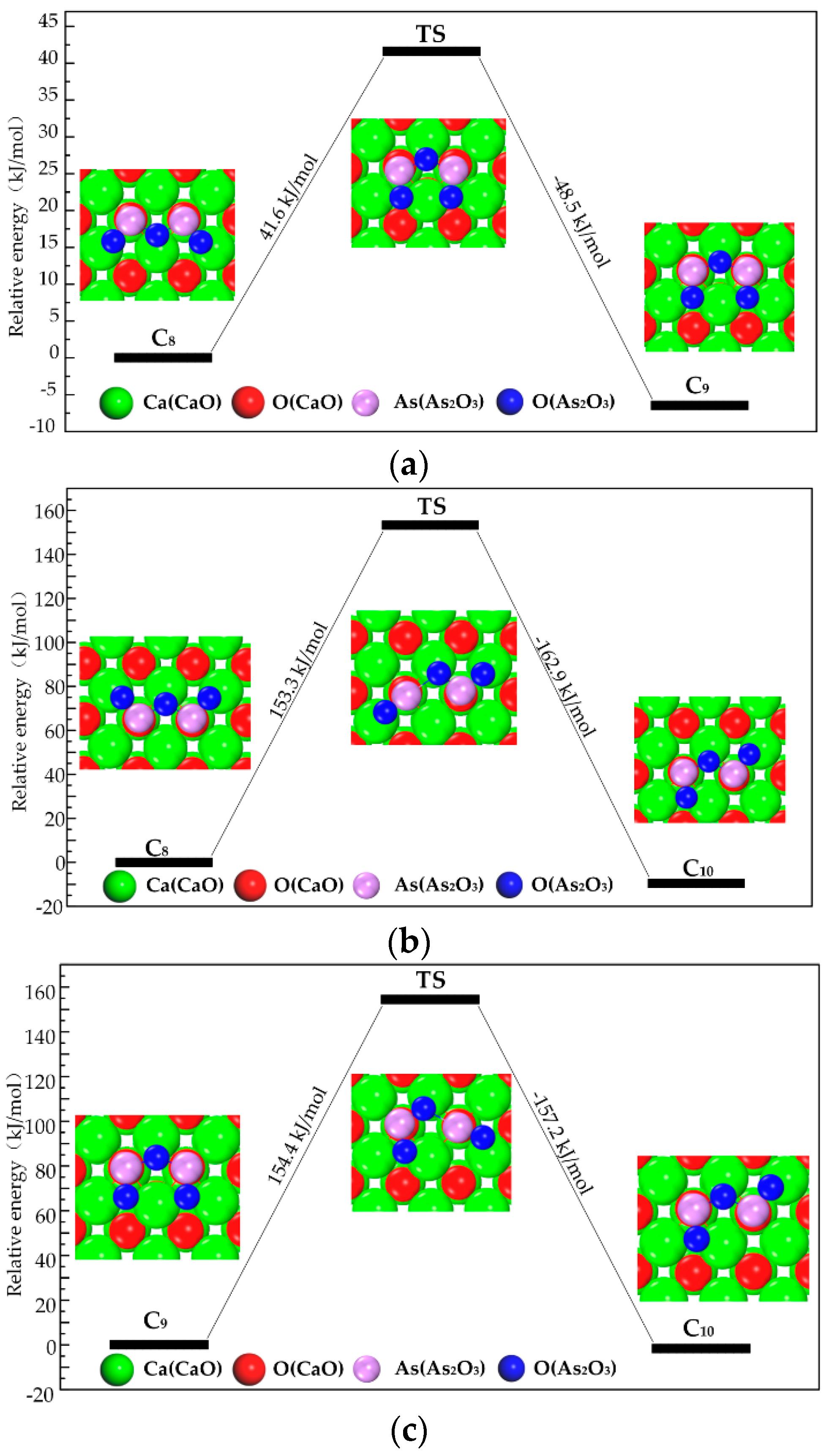
3.2.2. Transformation Process of Chemisorption Structures
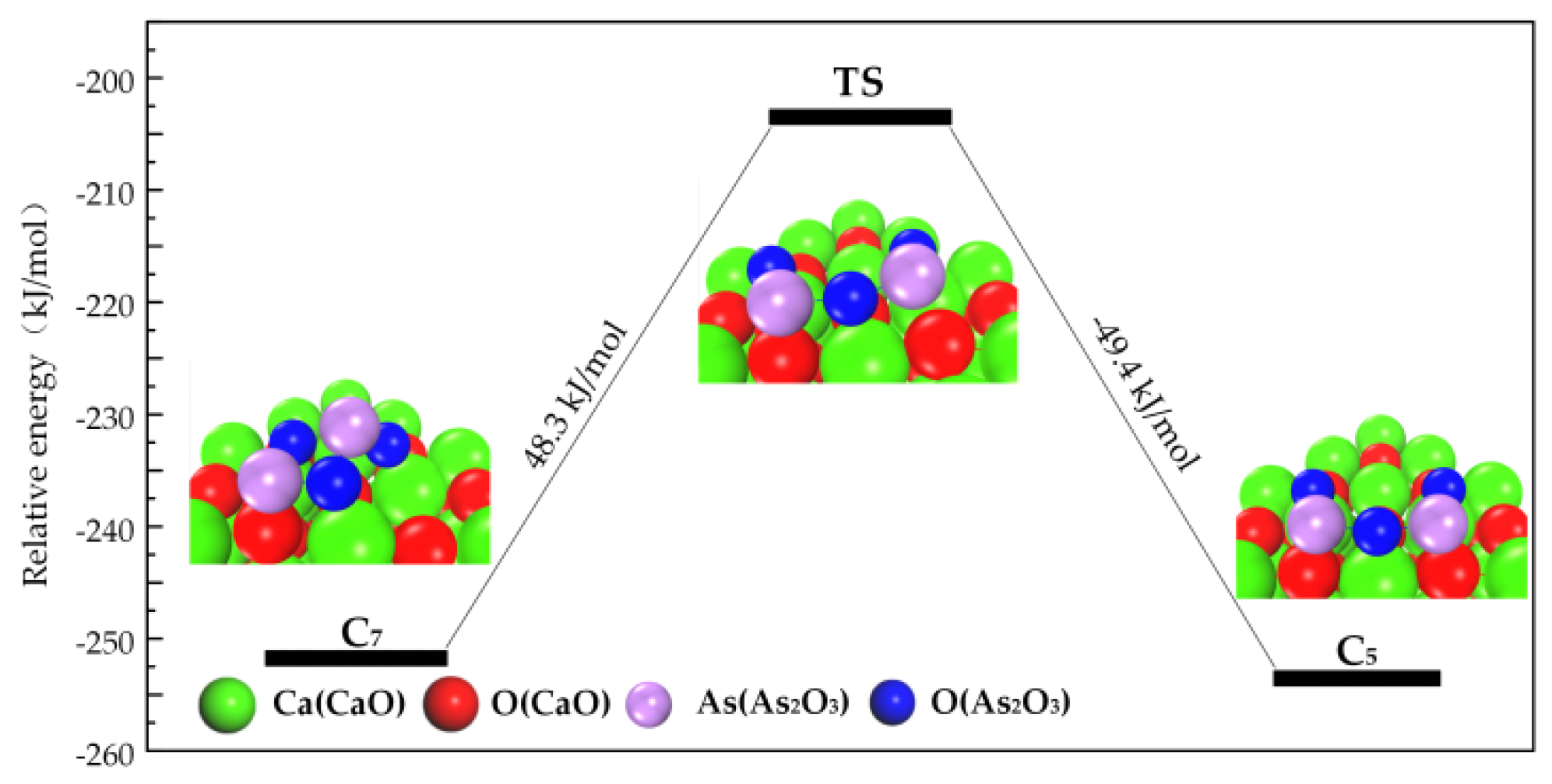
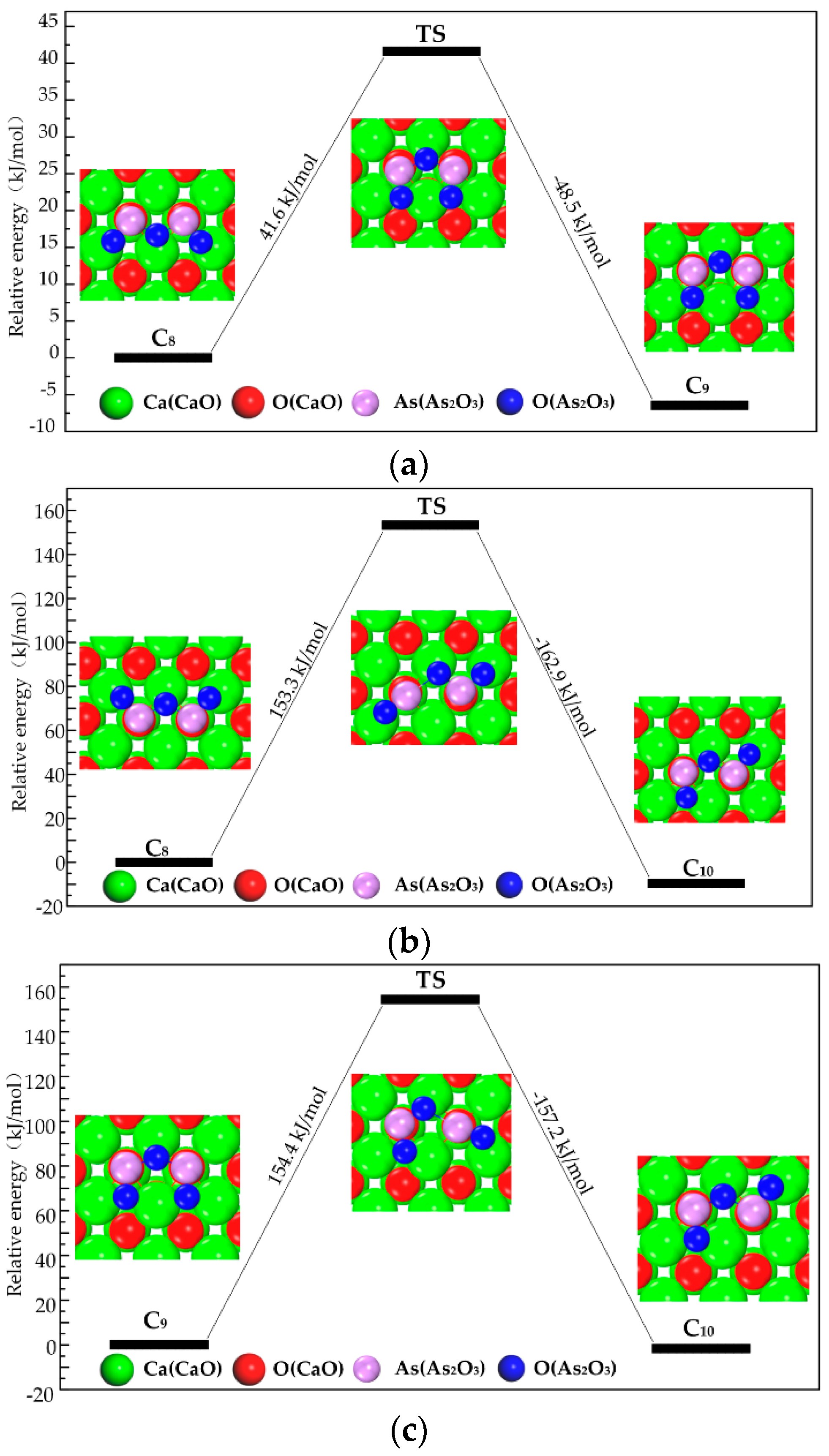
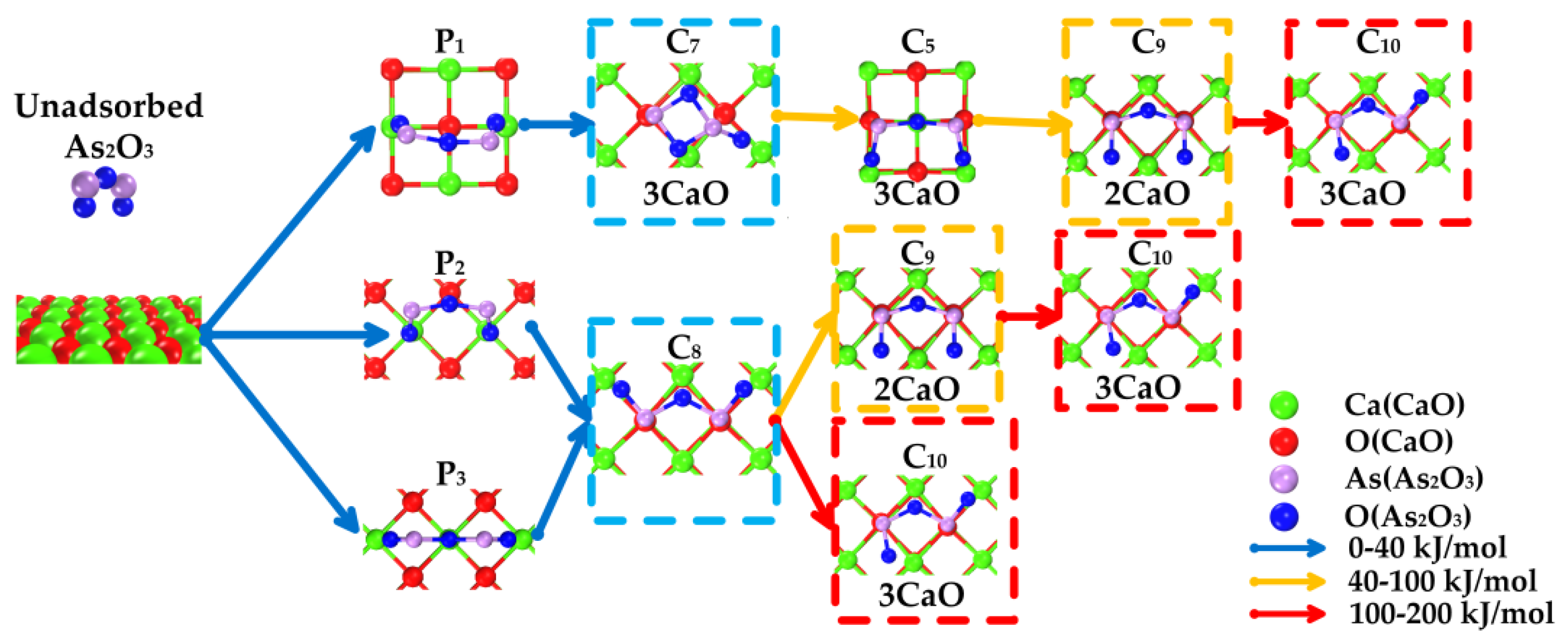
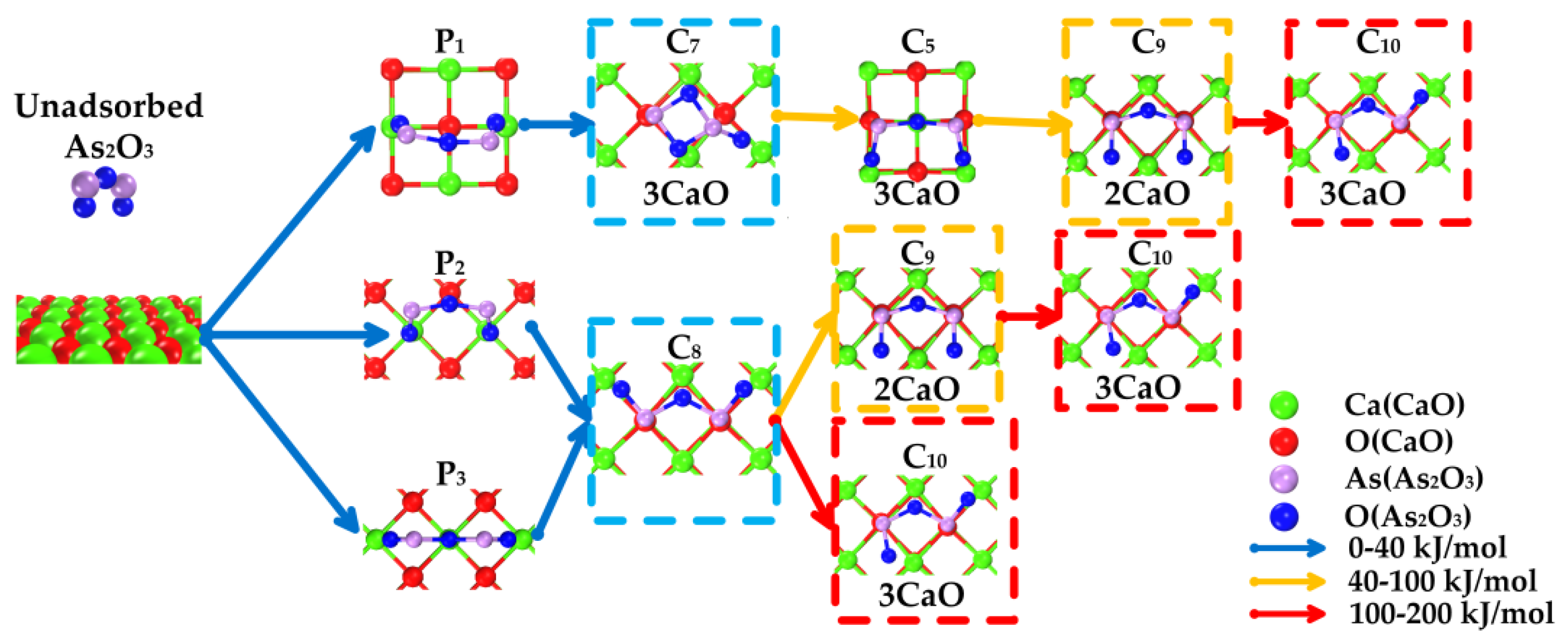
3.3. Path of the Reaction
- As2O3 → P1 → C7 → C9 →C10;
- As2O3 → P2 or P3 → C8 → C9 → C10;
- As2O3 → P2 or P3 → C8 → C10.
3.4. Partial Density of States (PDOS)
3.5. Influence of O2 on Adsorbed As2O3
4. Conclusions
- (1)
- Physisorption active sites are composed of superficial Ca atoms that interact with O of As2O3. Chemisorption active sites are superficial O atoms that interact with As of As2O3;
- (2)
- The adsorption process can be described as follows: the isolated As2O3 molecule is firstly adsorbed on the CaO surface by physisorption, and then physisorbed As2O3 will transform to chemisorbed As2O3. Due to thermal vibration, the chemisorbed As2O3 would overcome the energy barrier and transform to a more stable chemisorbed As2O3 state. The adsorption product is AsO33−;
- (3)
- The adsorption products of As2O3 under an O2-containing atmosphere are AsO43−. The adsorption product’s structure is influenced by the adsorption temperature. Under relatively low-temperature, the product is Ca3As2O8; under relatively medium-temperature, the product is Ca3As2O7; and under relatively high-temperature, the product is Ca3As2O8.
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Liu, R.; Yang, J.; Xiao, Y.; Liu, Z. Fate of Forms of arsenic in Yima coal during pyrolysis. Energy Fuels 2009, 23, 2013–2017. [Google Scholar] [CrossRef]
- Clarke, L.B.; Sloss, L.L. Trace Elements-Emissions from Coal Combustion and Gasification; IEA Coal Research: London, UK, 1992; Volume 8, pp. 1822–1826. [Google Scholar]
- Gao, J.; Yu, J.; Yang, L. Urinary arsenic metabolites of subjects exposed to elevated arsenic present in coal in Shaanxi province, China. Int. J. Environ. Res. Public Health 2011, 8, 1991–2008. [Google Scholar] [CrossRef] [PubMed]
- Kapaj, S.; Peterson, H.; Liber, K.; Bhattacharya, P. Human health effects from chronic arsenic poisoning—A review. J. Environ. Sci. Health Part A 2006, 41, 2399–2428. [Google Scholar] [CrossRef] [PubMed]
- 1990 Clean Air Act Amendment. Public Law; 1990. Available online: https://www.gpo.gov/fdsys/pkg/USCODE-2013-title42/html/USCODE-2013-title42-chap85-subchapI-partA-sec7412.htm (accessed on 11 February 2019).
- Duan, J.; Tan, J.; Hao, J.; Chai, F. Size distribution, characteristics and sources of heavy metals in haze episod in Beijing. J. Environ. Sci. 2014, 26, 189–196. [Google Scholar] [CrossRef]
- Pacyna, J.M.; Pacyna, E.G. An assessment of global and regional emissions of trace metals to the atmosphere from anthropogenic sources worldwide. Environ. Rev. 2001, 9, 269–298. [Google Scholar] [CrossRef]
- Tian, H.; Liu, K.; Zhou, J.; Lu, L.; Hao, J.; Qiu, P.; Gao, J.; Zhu, C.; Wang, K.; Hua, S. Atmospheric Emission Inventory of Hazardous Trace Elements from China’s Coal-Fired Power Plants Temporal Trends and Spatial Variation Characteristics. Environ. Sci. Technol. 2014, 48, 3575–3582. [Google Scholar] [CrossRef] [PubMed]
- Mercury and air toxics Standards (MATS). Public Law; 2013. Available online: https://www.epa.gov/mats (accessed on 11 February 2019).
- Helsen, L. Sampling technologies and air pollution control devices for gaseous and particulate arsenic: A review. Environ. Pollut. 2005, 137, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Sterling, R.O.; Helble, J.J. Reaction of arsenic vapor species with fly ash compounds: Kinetics and speciation of the reaction with calcium silicates. Chemosphere 2003, 51, 1111–1119. [Google Scholar] [CrossRef]
- Jadhav, R.A.; Fan, L. Capture of gas-phase arsenic oxide by lime: Kinetic and mechanistic studies. Environ. Sci. Technol. 2001, 35, 794–799. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Tong, H.; Zhuo, Y.; Li, Y.; Xu, X. Simultaneous removal of SO2 and trace As2O3 from flue gas: Mechanism, kinetics study, and effect of main gases on arsenic capture. Environ. Sci. Technol. 2007, 41, 2894–2900. [Google Scholar] [CrossRef] [PubMed]
- Li, Y. Experimental Study on Simultaneous Removal of Trace Selenium and Arsenic in Flue Gas Desulphurization within Medium Temperature Range; Tsinghua University: Beijing, China, 2006. [Google Scholar]
- López-Antón, M.A.; Díaz-Somoano, M.; Spears, D.A.; Martínez-Tarazona, M.R. Arsenic and selenium capture by fly ashes at low temperature. Environ. Sci. Technol. 2006, 40, 3947–3951. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.; Strezov, V.; Stevanov, C.; Nelson, P.F. Speciation of arsenic and selenium in coal combustion products. Energy Fuels 2007, 21, 506–512. [Google Scholar] [CrossRef]
- Huggins, F.E.; Senior, C.L.; Chu, P.; Ladwig, K.; Huffman, G.P. Selenium and arsenic speciation in fly ash from full-scale coal-burning utility plants. Environ. Sci. Technol. 2007, 41, 3284–3289. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; Gupta, R.; Zhao, Y.; Zhang, J. Release Behaviors of Arsenic in Fine Particles Generated from a Typical High-Arsenic Coal at a High Temperature. Energy Fuels 2016, 30, 6201–6209. [Google Scholar] [CrossRef]
- Jialin, Y.; Jingjing, X.; Qinfang, Z.; Binwen, Z.; Baolin, W. First-principles studies on the structural and electronic properties of As clusters. Materials 2018, 11, 1596. [Google Scholar]
- Li, Z.; Yangwen, W.; Jian, H.; Qiang, L.; Yongping, Y.; Laibao, Z. Mechanism of mercury adsorption and oxidation by oxygen over the CeO2 (111) surface: A DFT study. Materials 2018, 11, 485. [Google Scholar]
- Zhang, S.; Hu, X.; Lu, Q.; Zhang, J. Density functional theory study of arsenic and selenium adsorption on the CaO (001) surface. Energy Fuels 2011, 25, 2932–2938. [Google Scholar] [CrossRef]
- Segall, M.D.; Lindan, P.J.; Probert, M.A.; Pickard, C.J.; Hasnip, P.J.; Clark, S.J.; Payne, M.C. First-principles simulation: Ideas, illustrations and the CASTEP code. J. Phys. Condens. Matter 2002, 14, 2717. [Google Scholar] [CrossRef]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.I.; Refson, K.; Payne, M.C. First principles methods using CASTEP. Cryst. Mater. 2005, 220, 567–570. [Google Scholar] [CrossRef]
- Burke, K.; Ernzerhof, M.; Perdew, J.P. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar]
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B Condens. Matter 1990, 41, 7892. [Google Scholar] [CrossRef] [PubMed]
- Von Barth, U.; Hedin, L. A local exchange-correlation potential for the spin polarized case. i. J. Phys. C Solid State Phys. 1972, 5, 1629. [Google Scholar] [CrossRef]
- Press, W.H.; Teukolsky, S.A.; Vetterling, W.T.; Flannery, B.P. Numerical Recipes in C.; Cambridge University Press: Cambridge, UK, 1996; Volume 2. [Google Scholar]
- Halgren, T.A.; Lipscomb, W.N. The synchronous-transit method for determining reaction pathways and locating molecular transition states. Chem. Phys. Lett. 1977, 49, 225–232. [Google Scholar] [CrossRef]
- Henkelman, G.; Jónsson, H. Improved tangent estimate in the nudged elastic band method for finding minimum energy paths and saddle points. J. Chem. Phys. 2000, 113, 9978–9985. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B Condens. Matter 1976, 13, 5188. [Google Scholar] [CrossRef]
- Ghebouli, M.A.; Ghebouli, B.; Bouhemadou, A.; Fatmi, M.; Bouamama, K. Structural, electronic, optical and thermodynamic properties of SrxCa1−xO, BaxSr1−xO and BaxCa1−xO alloys. J. Alloys Compd. 2011, 509, 1440–1447. [Google Scholar] [CrossRef]
- Verbraeken, M.C.; Suard, E.; Irvine, J.T. Order and disorder in Ca2ND0.90H0.10–A structural and thermal study. J. Solid State Chem. 2011, 184, 2088–2096. [Google Scholar] [CrossRef]
- Da Hora, G.C.A.; Longo, R.L.; Da Silva, J.B.P. Calculations of structures and reaction energy profiles of As2O3 and As4O6 species by quantum chemical methods. Int. J. Quantum Chem. 2012, 112, 3320–3324. [Google Scholar] [CrossRef]
- Liu, H.; Xiang, H.; Gong, X.G. First principles study of adsorption of O2 on Al surface with hybrid functionals. J. Chem. Phys. 2011, 135, 214702. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Yao, J.G.; Zhang, Z.; Sceats, M.; Zhuo, Y.; Li, L.; Maitland, G.C.; Fennell, P.S. Pressurized calcium looping in the presence of steam in a spout-fluidized-bed reactor with dft analysis. Fuel Process. Technol. 2018, 169, 24–41. [Google Scholar] [CrossRef]
- Fan, Y.; Zhuo, Y.; Zhu, Z.; Du, W.; Li, L. Zerovalent selenium adsorption mechanisms on a CaO surface: DFT calculation and experimental study. J. Phys. Chem. A 2017, 121, 7385. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Zhuo, Y.; Li, L. SeO2 adsorption on a CaO surface: DFT and experimental study on the adsorption of multiple SeO2 molecules. Appl. Surf. Sci. 2017, 420, 465–471. [Google Scholar] [CrossRef]
- Sun, S.; Zhang, D.; Li, C.; Wang, Y.; Yang, Q. Density functional theory study of mercury adsorption and oxidation on CuO (111) surface. Chem. Eng. J. 2014, 258, 128–135. [Google Scholar] [CrossRef]
- Xiang, W.; Liu, J.; Chang, M.; Zheng, C. The adsorption mechanism of elemental mercury on CuO (110) surface. Chem. Eng. J. 2012, 200, 91–96. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Studt, F.; Abild-Pedersen, F.; Bligaard, T. Fundamental Concepts in Heterogeneous Catalysis; John Wiley & Sons: Hoboken, NJ, USA, 2014. [Google Scholar]
- Mahuli, S.; Agnihotri, R.; Chauk, S.; Ghosh-Dastidar, A.; Fan, L.-S. Mechanism of Arsenic Sorption by Hydrated Lime. Environ. Sci. Technol 1998, 31, 3226–3231. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substance | Previous Data | Simulated Data |
|---|---|---|
| CaO [31,32] | 4.836 Å/4.807 Å | 4.837 Å |
| As2O3 [33] | As–O bond 1.794 Å | As–O bond 1.814 Å |
| As–O bond 1.610 Å | As–O bond 1.622 Å | |
| O–As–O angle 106.3° | O–As–O angle 111.2° | |
| As–O–As angle 133.8° | As–O–As angle 141.8° | |
| O2 [34] | O–O 1.210 Å | O–O 1.240 Å |
| Name | Top View | Front View | Electron Density Cloud | Structure Details | Eads |
|---|---|---|---|---|---|
| P1 |  | 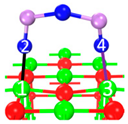 | 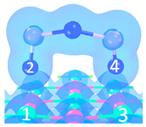 | Bond12: 2.450 Å Bond34: 2.469 Å | −65.8 kJ/mol |
| P2 |  | 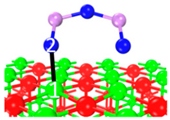 | 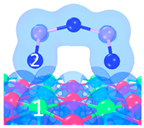 | Bond12: 2.380 Å | −62.6 kJ/mol |
| P3 | 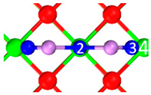 |  | 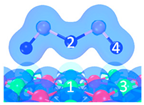 | Bond12: 2.876 Å Bond34: 2.539 Å | −58.4 kJ/mol |
 | |||||
| Category | Name | Top View | Front View | Electron Density Cloud | Structure Details | Eads |
|---|---|---|---|---|---|---|
| I | C1 | 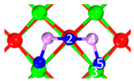 |  | 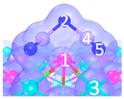 | Bond12: 2.635 Å Bond14: 2.086 Å Bond35: 2.360 Å | −198.5 kJ/mol |
| II | C2 | 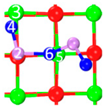 | 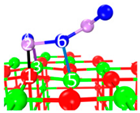 |  | Bond12: 1.858 Å Bond34: 2.360 Å Bond56: 2.386 Å | −222.1 kJ/mol |
| II | C3 | 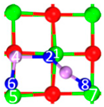 | 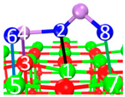 |  | Bond12: 2.424 Å Bond34: 1.815 Å Bond56: 2.314 Å Bond78: 2.490 Å | −274.4 kJ/mol |
| II | C4 | 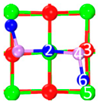 | 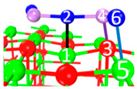 | 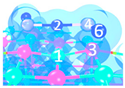 | Bond12: 2.269 Å Bond34: 1.949 Å Bond56: 2.391 Å | −292.0 kJ/mol |
| II | C5 | 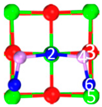 | 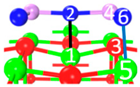 |  | Bond12: 2.293 Å Bond34: 1.943 Å Bond56: 2.298 Å | −315.1 kJ/mol |
| III | C6 | 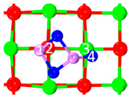 | 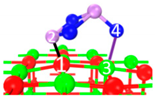 |  | Bond12: 1.815 Å Bond34: 2.355 Å | −302.3 kJ/mol |
| III | C7 | 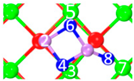 |  |  | Bond12: 1.788 Å Bond34: 2.528 Å Bond56: 2.514 Å Bond78: 2.422 Å | −314.0 kJ/mol |
| IV | C8 |  |  | 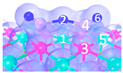 | Bond12: 2.357 Å Bond34: 1.901 Å Bond56: 2.298 Å | −381.7 kJ/mol |
| IV | C9 | 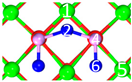 | 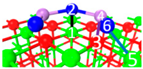 |  | Bond12: 2.472 Å Bond34: 1.869 Å Bond56: 2.503 Å | −388.6 kJ/mol |
| IV | C10 | 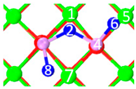 | 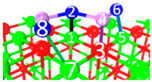 |  | Bond12: 2.382 Å Bond34: 1.894 Å Bond56: 2.299 Å Bond78: 2.392 Å | −391.4 kJ/mol |
| | ||||||
| Name | Top View | Front View | Electron Density Cloud | Structure Details | Eads |
|---|---|---|---|---|---|
| C5 under O2 | 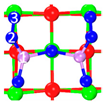 | 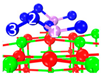 | 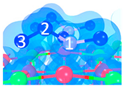 | Bond12: 1.764 Å Bond34: 1.452 Å Bond56: 2.263 Å | −165.2 kJ/mol |
| C8 under O2 |  | 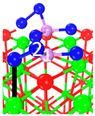 | 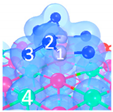 | Bond12: 1.763 Å Bond34: 1.599 Å Bond56: 2.427 Å | −174.4 kJ/mol |
 | |||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, Y.; Weng, Q.; Zhuo, Y.; Dong, S.; Hu, P.; Li, D. Theoretical Study of As2O3 Adsorption Mechanisms on CaO surface. Materials 2019, 12, 677. https://doi.org/10.3390/ma12040677
Fan Y, Weng Q, Zhuo Y, Dong S, Hu P, Li D. Theoretical Study of As2O3 Adsorption Mechanisms on CaO surface. Materials. 2019; 12(4):677. https://doi.org/10.3390/ma12040677
Chicago/Turabian StyleFan, Yaming, Qiyu Weng, Yuqun Zhuo, Songtao Dong, Pengbo Hu, and Duanle Li. 2019. "Theoretical Study of As2O3 Adsorption Mechanisms on CaO surface" Materials 12, no. 4: 677. https://doi.org/10.3390/ma12040677
APA StyleFan, Y., Weng, Q., Zhuo, Y., Dong, S., Hu, P., & Li, D. (2019). Theoretical Study of As2O3 Adsorption Mechanisms on CaO surface. Materials, 12(4), 677. https://doi.org/10.3390/ma12040677