1. Background
Abnormalities of cell structures and their functions are caused by external factors such as ultraviolet (UV) light, bacterial and virus infections, as well as obesity and an unhealthy lifestyle [
1]. The formation of cancer cells is also related to genetic mutations and abnormalities in hormone and immune conditions [
2]. These factors are responsible for cancer development, but also have an influence on the chemoresistance of cancer cells. The standard procedures of cancer treatments are based on surgical resection, radiotherapy, and chemotherapy. Despite increasing research efforts, the knowledge of the main mechanisms of developing chemoresistance in different types of cancer cells is not commonly used in clinical oncology. Inefficient drug administration into cancer cells is related to cancer resistance due to genetic mutation, including genes involved in drug transporting, metabolizing enzymes, and DNA damage repair [
3].
Nanobiotechnology has introduced a new perspective for using nanosized elements against cancer diseases. Nanoparticles, because of their size (<100 nm), have unique physiochemical features including a large surface-to-mass ratio, easy surface functionalization, quantum characteristics [
4,
5], and, consequently, novel biological properties. The cellular transport of nanoparticles is also significantly different from that of chemical compounds or drugs because they are able to move across cells, reach nuclear membranes, and target specific structures such as proteins or gene sequences [
6,
7]. Nanoparticles of graphene oxide (GO), the thinnest material, with the thickness of one carbon atom, have recently been evaluated as a useful biomaterial for cancer therapy and diagnosis. The GO nanosheets produced by the modified Hummer´s method are stable in water suspensions, mainly because of the presence of hydrophilic oxygenated groups on their surface as well as on the edges [
7]. According to a previous study, GO has a minor effect on the viability and morphology of different cells, including glioma U87 and U118 cells [
8]. On the other hand, Chang et al. [
9] reported that GO may generate oxidative stress, causing adverse effects on the viability of A549 cancer cells. The GO treatment led to the structural deformation of mitochondria, affecting the mitochondria membrane potential [
10]. Moreover, other examples of nanoparticles like gold nanoparticles, of which clinical application in head and neck cancer treatment was accepted by the FDA [
11], also showed in other studies harmful effects on mammal cell toxicity and induced long-term organ damage [
12]. However, because of minor harmful effects on cell viability, GO has been investigated [
13] as a potential delivery platform for drugs, bioactive molecules, DNA, RNA, and other oligonucleotides [
14].
The catalytic properties of platinum atoms are useful for cancer treatment. However, the lack of selectivity between cancer and non-cancer cells is the major drawback of Pt therapy [
15]. Moreover, the development of chemoresistance by cancer cells is the second largest challenge for treatment strategies. The use of Pt-based nanomaterials can be a solution for some of the side effects of platinum-based cancer therapy. Platinum nanoparticles (NP-Pt) have catalytic activity and a high reactivity, similar to Pt-based drugs, but their large surface-to-mass ratio is a unique physiochemical feature and have an impact on their size-dependent bioavailability [
15]. Previous studies have demonstrated that NP-Pt has anticancer activity against several cancer cell lines, e.g., glioma U87 [
16,
17] and U251 [
18], colorectal HT29 [
19], breast MCF-7, liver HepG-2 [
20], and lymphoma U937 [
21]. The genotoxic effect of NP-Pt was also observed in non-cancer cell lines, human bronchial epithelial cells HBEC3-kt, where NP-Pt increased micronucleus and DNA degradation [
22]. The NP-Pt mechanism of apoptosis activation is based on the direct interaction of Pt with the DNA of cancer cells [
16,
17,
18,
20] and additionally by Pt
+ ions released under H
2O
2 generation in endosomes [
15]. Brown et al. [
23] also showed that after intravenous injection of NP-Pt into mice with the HepG2 liver tumor, NP-Pt affected the tumor tissue, but were also accumulated in spleen and liver tissue. Moreover, NP-Pt circulating in the bloodstream were detected, which means that NP-Pt are able to cross membrane barriers and cause adverse effects. Zhang et al. [
24] showed that NP-Pt were efficiently deposited on the GO surface but did not show the anticancer activity against MCF-7 and SGC-7901 cell lines. We hypothesized that the use of GO as a carrier for Pt-NP can retain NP-Pt directly on the cancer cell surface, slow the release of Pt into cells, limit the possibility of uncontrolled circulation of NP-Pt, and, consequently, increase the anticancer efficiency. In this context, the objective of this study was the characterization of anticancer properties of GO-NP-Pt against morphologically, metabolically, and genotypically different cancer cell lines.
2. Materials and Methods
2.1. Preparation and Characterization of Nanocolloids
2.1.1. Platinum Nanoparticles and Graphene Oxide
The colloid of platinum nanoparticles was purchased from Nano-koloid (Warsaw, Poland). This material is produced by a patented electric non-explosive method (Polish patent 380649) from high-purity metal (99.9999%) and high-purity demineralized Milli-Q water. Graphene oxide powder (GO) (purity 99.99%) was purchased from CEZEMAT (Warsaw, Poland) and dispersed in ultrapure water to prepare a 1.0 mg/mL solution.
After 30 minutes of sonification, the hydrocolloids of GO-NP-Pt, NP-Pt, and GO were diluted to different concentrations with 1× Dulbecco’s modified Eagle’s culture medium (Sigma-Aldrich, St. Louis, MO, USA) immediately prior to exposure to cells.
2.1.2. Complex of Graphene Oxide and Platinum Nanoparticles
Suspensions of GO (200 μg/mL) and NP-Pt (100 μg/mL) were prepared in ultrapure water and used without additional purification and filtration. Ultrasonic coating of GO nanosheets for 30 min took place in a 50-mL glass flask.
2.1.3. Scanning Electron Microscopy
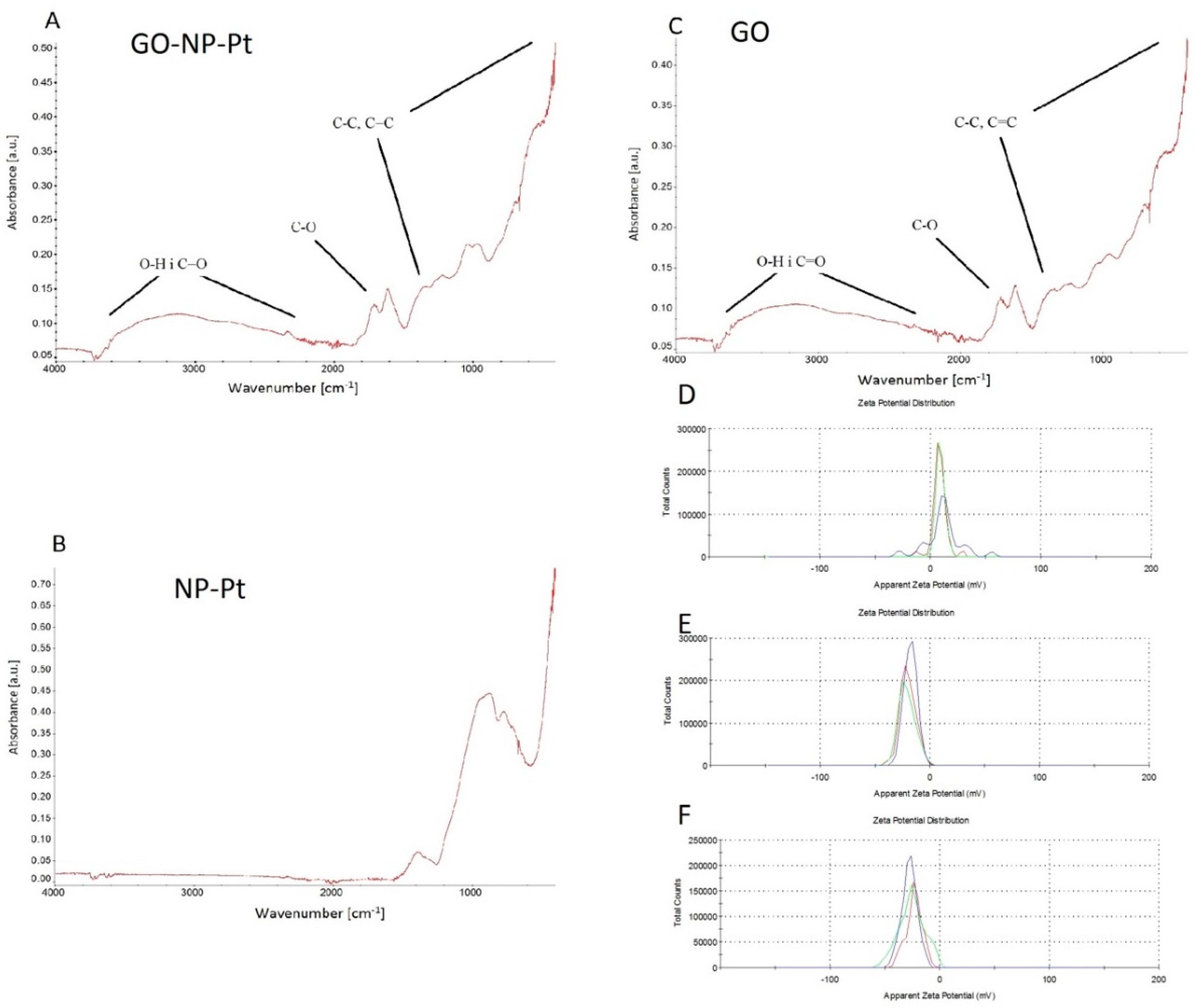
The shape and size of the NP-Pt GO and GO-NP-Pt complexes were inspected using a scanning transmission electron microscope. Electron microscope images were taken with the use of a Hitachi SU8230 ultra-high-resolution field emission scanning electron microscope (Hitachi High-Technologies Corporation, Tokyo, Japan), using the transmission mode at 30.0 kV accelerating voltage. Images were taken with the use of gold transmission electron microscopy (TEM) grids coated with Lacey carbon film, which were immersed in GO samples and dried prior to observation.
2.1.4. Fourier Transform Infrared (FTIR) Spectroscopy
The FTIR spectra of dry samples of NP-Pt, GO, and GO-NP-Pt were recorded using a Nicolet 6700 FTIR spectroscope with diamond ATR pickup (Thermo Scientific, Waltham, MA, USA). All samples were prepared by dropping 500 µL of sample suspension on a microscope glass and drying the suspension.
2.1.5. ζ-Potential Measurements
The ζ-potentials of NP-Pt, GO, and GO-NP-Pt were measured by the laser dynamic scattering electrophoretic method, using the Smoluchowski approximation with a Zetasizer Nano ZS90 (Malvern Instruments, Malvern, UK). Each sample was measured after stabilization at 25 °C for 120 s. All measurements were performed in triplicate.
2.2. Cell Cultures and Treatments
Based on previous unpublished data and cancer statistic [
25] the different cancer cell lines were selected for in vitro investigation. The cell lines MCF-7, LNCaP, Hela B, HepG2, SW480, HT29, HCT116, and Colo205 were obtained from the American Type Culture Collection (Manassas, VA, USA) and maintained in Dulbecco’s modified Eagle’s (MCF-7, Hela B, HepG2), Roswell Park Memorial Institute (LNCaP, Colo205, SW480), or McCoy’s 5A culture medium (HT29, HCT116), supplemented with 10% fetal bovine serum (Sigma-Aldrich), 1% penicillin, and streptomycin (Sigma-Aldrich) at 37 °C in a humidified atmosphere of 5% CO
2/95% air in a NuAire DH AutoFlow CO
2 Air-Jacketed Incubator (Plymouth, MN, USA).
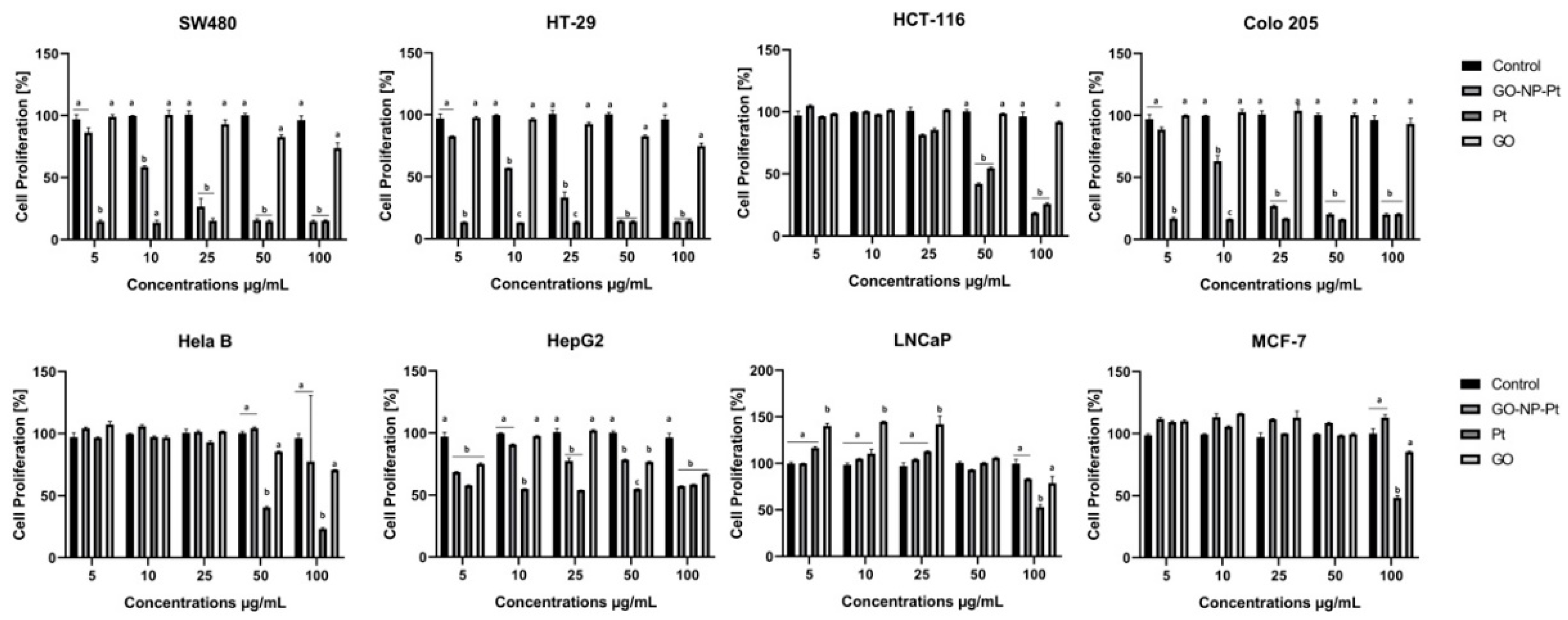
2.3. Proliferation Assay
Cell proliferation was evaluated using the bromodeoxyuridine (BrdU) incorporation assay (BrdU colorimetric) (Roche Applied Science, Indianapolis, IN, USA). The cell lines MCF-7, LNCaP, Hela B, HepG2, SW480, HT29, HCT116, and Colo205 (1 × 104) were placed in 96-well plates and cultured overnight. Next, the medium was removed and hydrocolloids of GO at concentrations of 5.0, 10.0, 25.0, 50.0, and 100.0 µg/mL, NP-Pt at concentrations of 0.1, 1.0, 5.0, 10.0, and 25.0 µg/mL, and GO-NP-Pt at mixed concentrations of GO 5.0: NP-Pt 0.1 µg/mL, GO 10.0: NP-Pt 1.0 µg/mL, GO 25.0: NP-Pt 5.0 µg/mL, GO 50.0: NP-Pt 10.0 µg/mL, and GO 100.0: NP-Pt 25.0 µg/mL were introduced to the cells for the following 24 h. Next, the BrdU labeling reagent was added and incubated with cells for 24 h. Subsequently, the culture media was removed, the cells were fixed, and the DNA was denatured in one step by adding FixDenat. In the following step, the cells were incubated with anti-BrdU- Fab fragments conjugated with peroxidase (POD) antibodies for 90 min at room temperature. After the removal of the antibody conjugate, the cells were washed, and the substrate solution was added. The reaction product was quantified by measuring absorbance using a scanning multi-well spectrophotometer (Infinite M200, Tecan, Durham, NC, USA) at 370 nm with a reference wavelength of 492 nm and finally expressed as the difference between BrdU-positive and -negative samples, expressed as optical density (OD). All measurements were performed in triplicate.
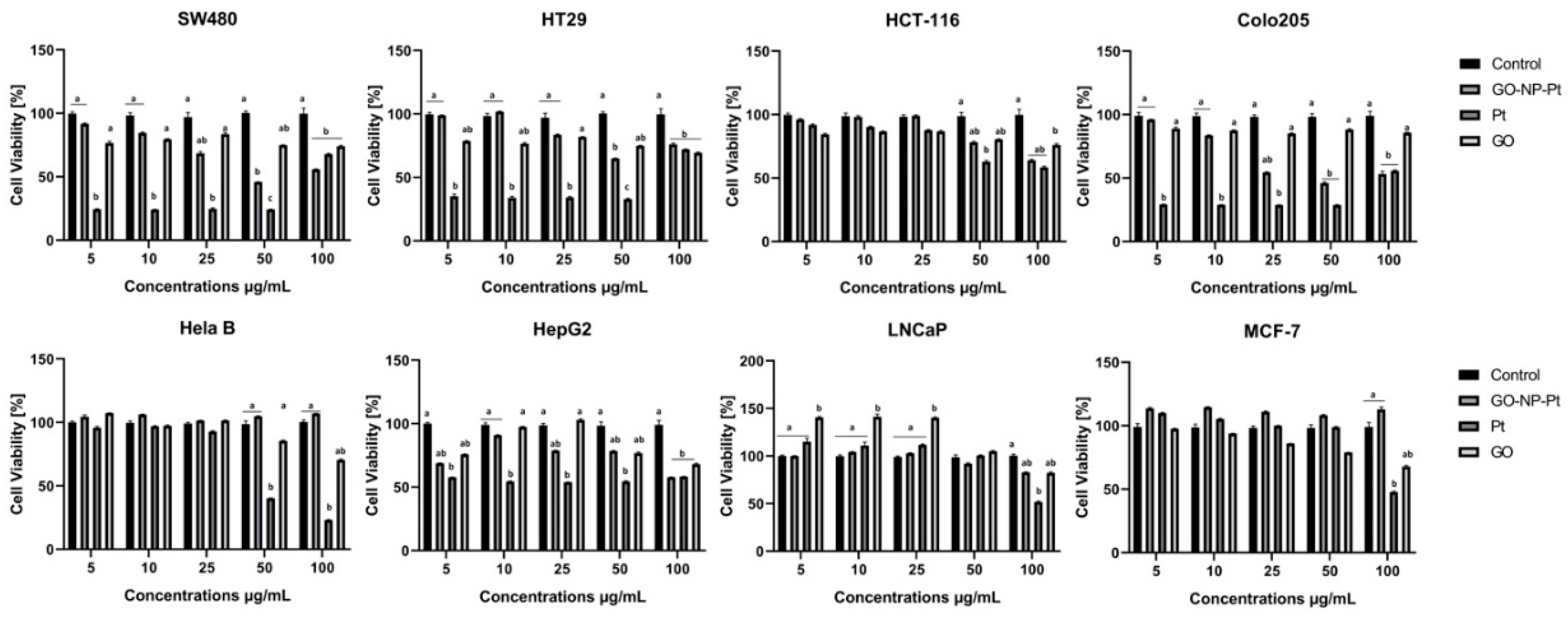
2.4. Cell Viability Assay
Cell viability was evaluated using a 2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxyanilide salt (XXT)-based cell viability assay kit (Life Technologies, Taastrup, Denmark). Cells MCF-7, LNCaP, Hela B, HepG2, SW480, HT29, HCT116, and Colo205 were incubated in 96-well plates (5 × 104 cells per well) with hydrocolloids of nanoparticles of GO, NP-Pt, and GO-NP-Pt at the same concentrations as for the proliferation assessment. In the subsequent step, the XTT solution was added to each well and incubated for an additional 3 h at 37 °C. The optical density (OD) of each well was recorded at 450 nm in a scanning multi-well spectrophotometer (Infinite M200, Tecan, Durham, NC, USA). Cell viability was expressed as a percentage (ODtest—ODblank)/(ODcontrol—ODblank), where “ODtest” is the optical density of cells exposed to NP-Pt, GO, and GO-NP-Pt, “ODcontrol” is the optical density of the control sample, and “ODblank” is the optical density of wells without cancer cells.
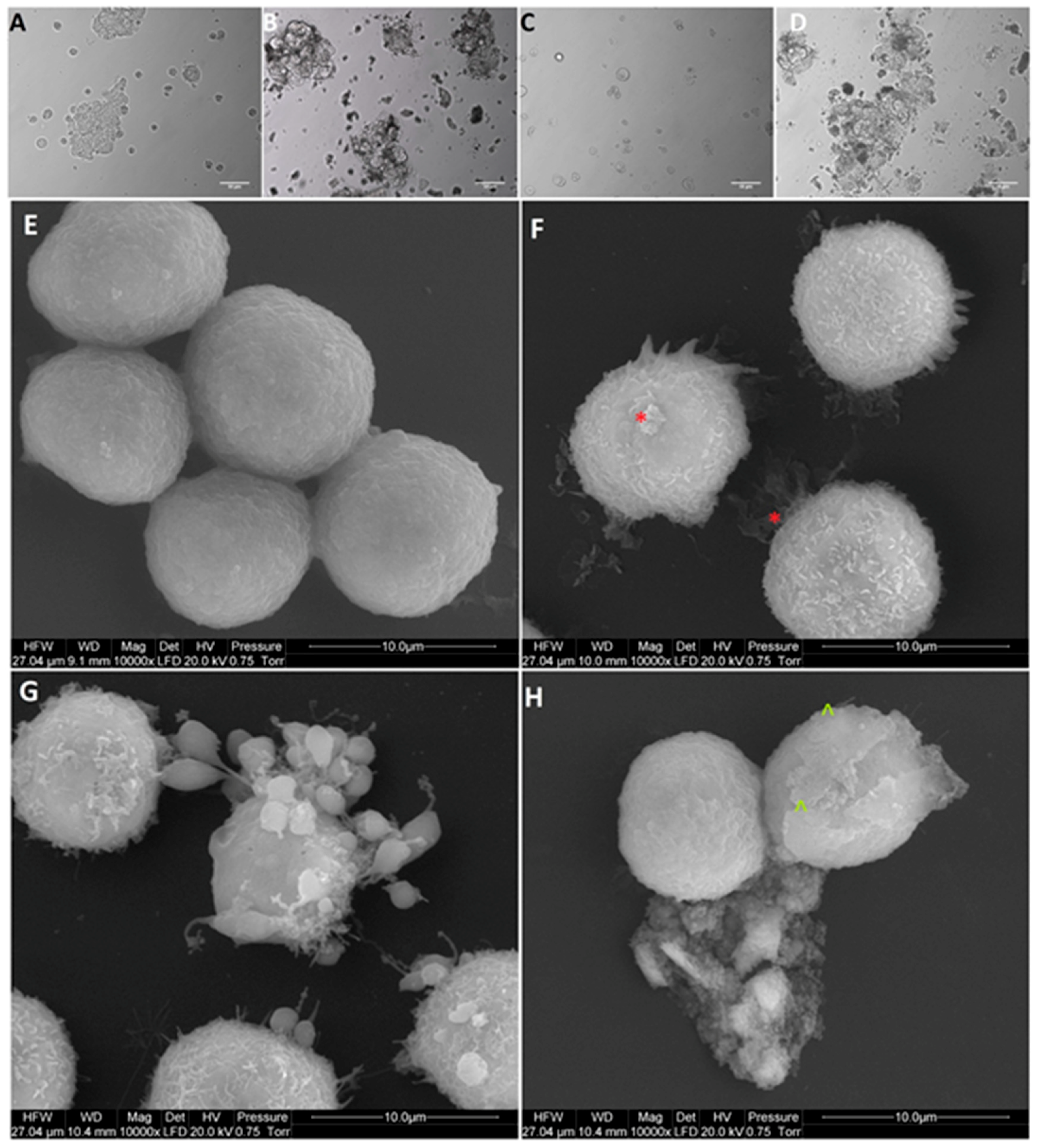
2.5. Cell Morphology
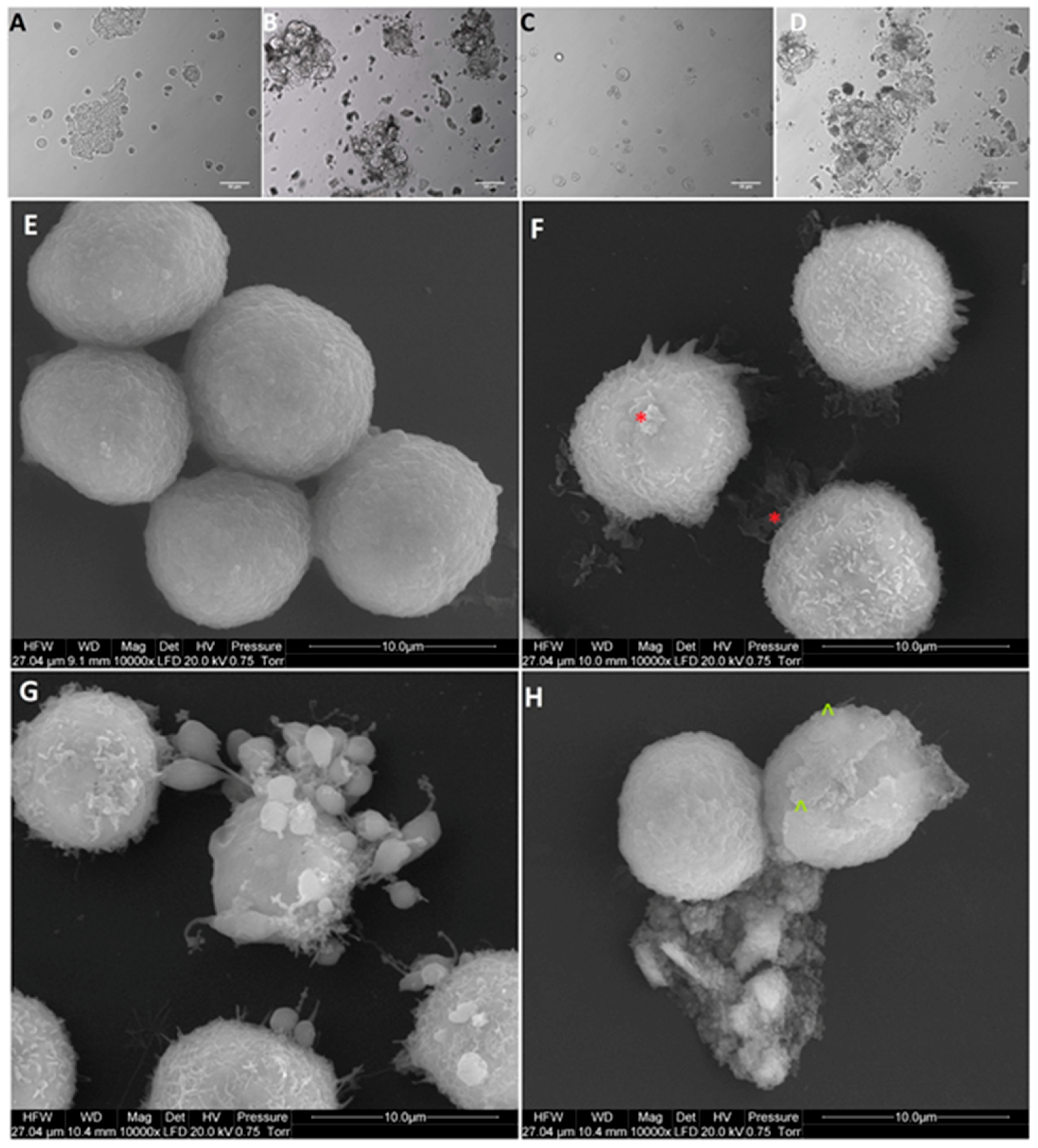
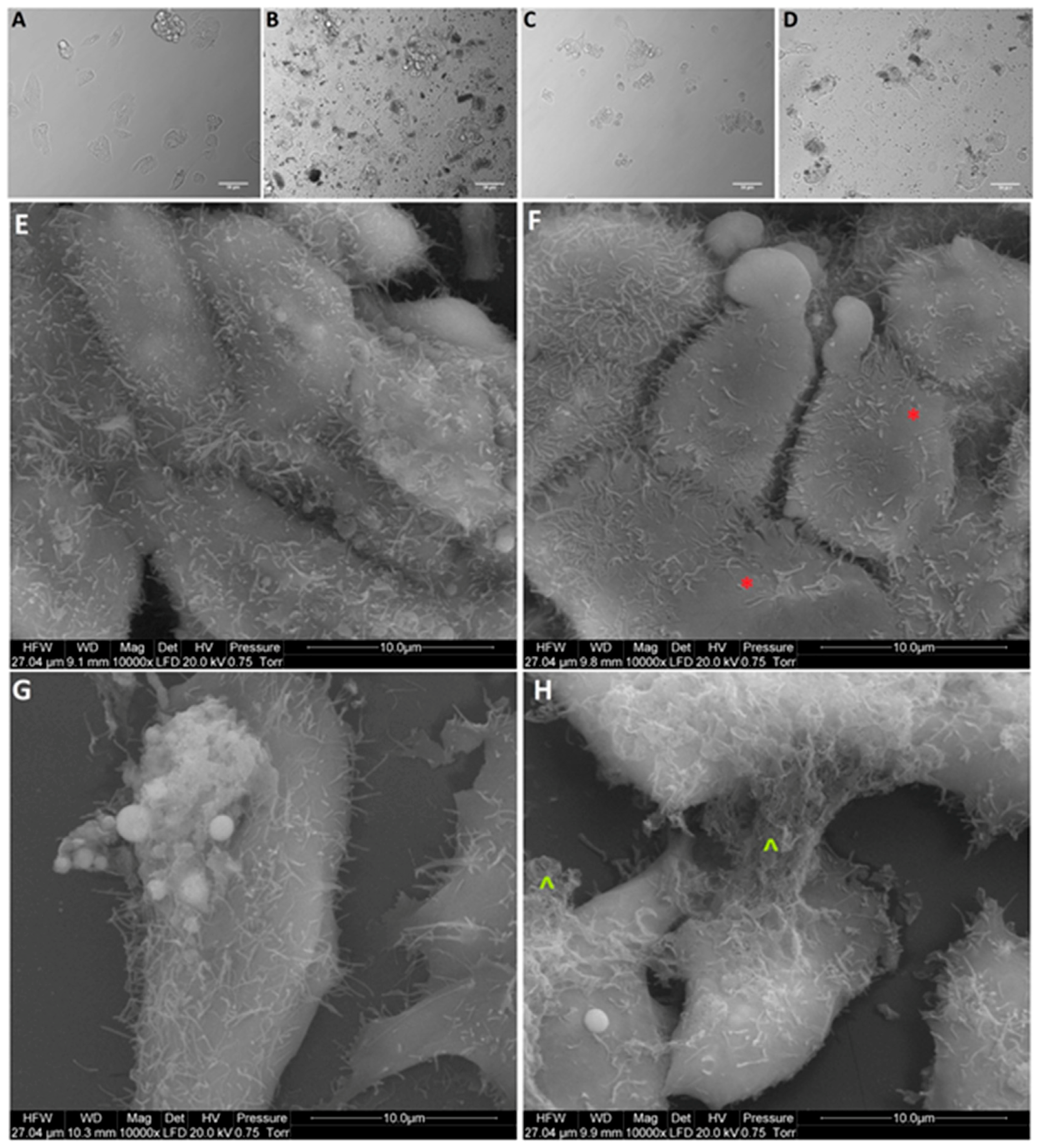
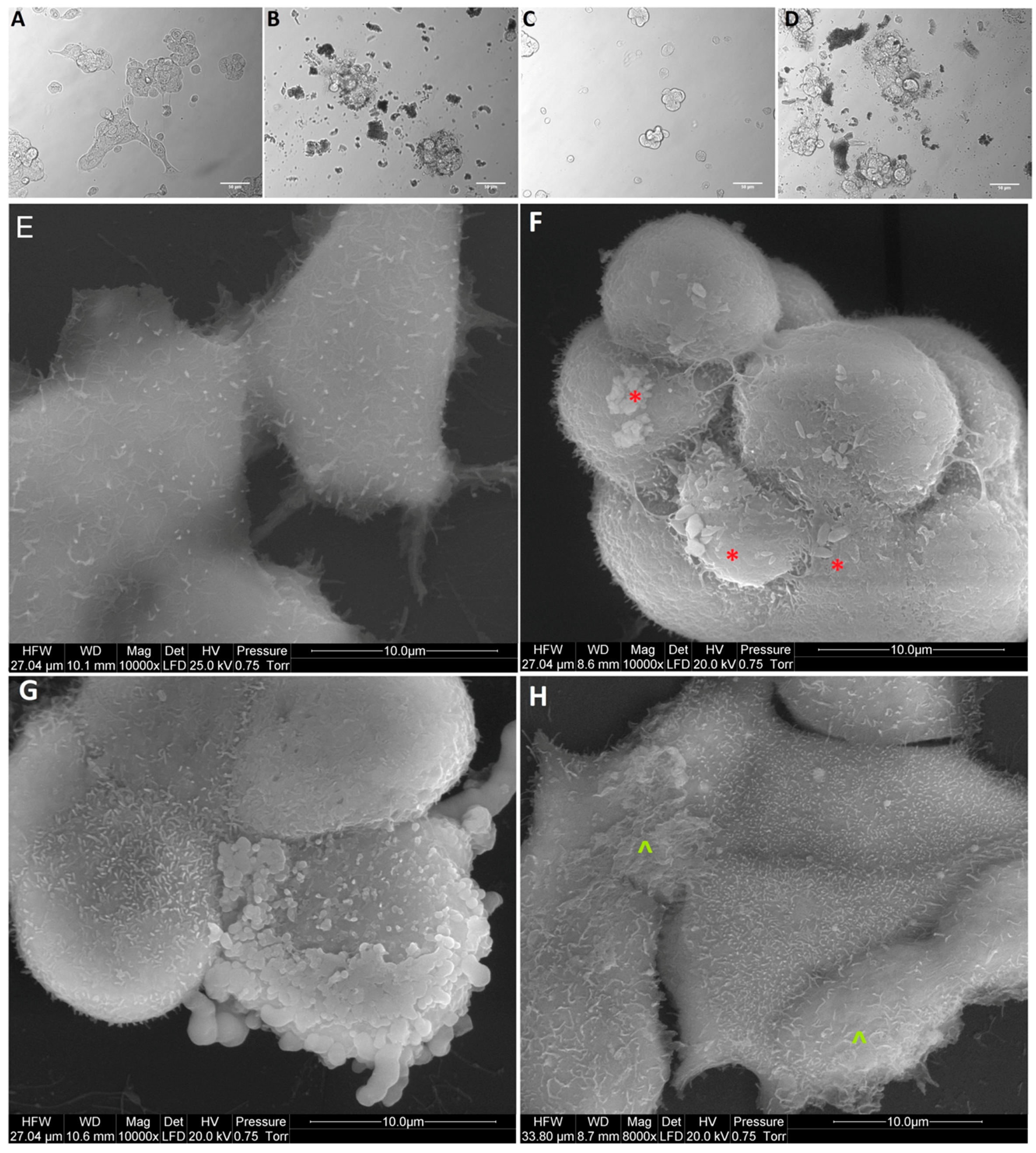
Based on obtained results of the viability and proliferation status of MCF-7, LNCaP, Hela B, HepG2, SW480, HT29, HCT116, and Colo205 cells for cell morphology investigation, a selection was made, and MCF-7, Colo205, and HepG2 were chosen as the most interesting results. Cells from the selected cells line were placed on 6-well plates (1 × 105 cells per well) and treated with NP-Pt (25.0 µg/mL), GO (100.0 µg/mL), and GO-NP-Pt (GO100:Pt25 µg/mL). After 24 h of incubation with nanoparticles, the cell morphology was recorded under an optical microscope (DM750; Leica Microsystems GmbH, Wetzlar, Germany), using the software package LAS EZ version 2.0. All measurements were performed in triplicate.
The scanning electron microscopy (SEM) analysis of MCF-7, Colo205, and HepG2 cancer cells, treated with (100 µg/mL), NP-Pt (25 µg/mL), and GO-NP-Pt (GO100:Pt25 µg/mL), was performed by means of a FEI QUANTA 200 electron microscope (Hillsboro, OR, USA). The cell samples were rinsed in phosphate buffered saline (PBS) (0.1 M, pH 7.2; P4417, Sigma), fixed in 2.5% glutaraldehyde (G5882, Sigma) for 1 h, washed twice in 0.1 M PBS (0 01 M, pH 7.2; P4417, Sigma), and placed on aluminum SEM stubs. The SEM stubs were kept in a moist atmosphere for 1 h, washed in PBS (0.1 M, pH 7.2; P4417, Sigma), post-fixed in 1% osmium tetroxide (75632, Sigma) for 1 h, rinsed in distilled water, and dehydrated in graded ethanol. After critical-point drying with liquid CO2 in a vacuum apparatus (Polaron CPD 7501, Quorum Technologies, Newhaven, East Sussex, UK) and coating with gold-palladium (JEE-4C, JEOL Ltd., Tokyo, Japan), the samples were inspected by SEM at 1 keV (FEI QUANTA 200).
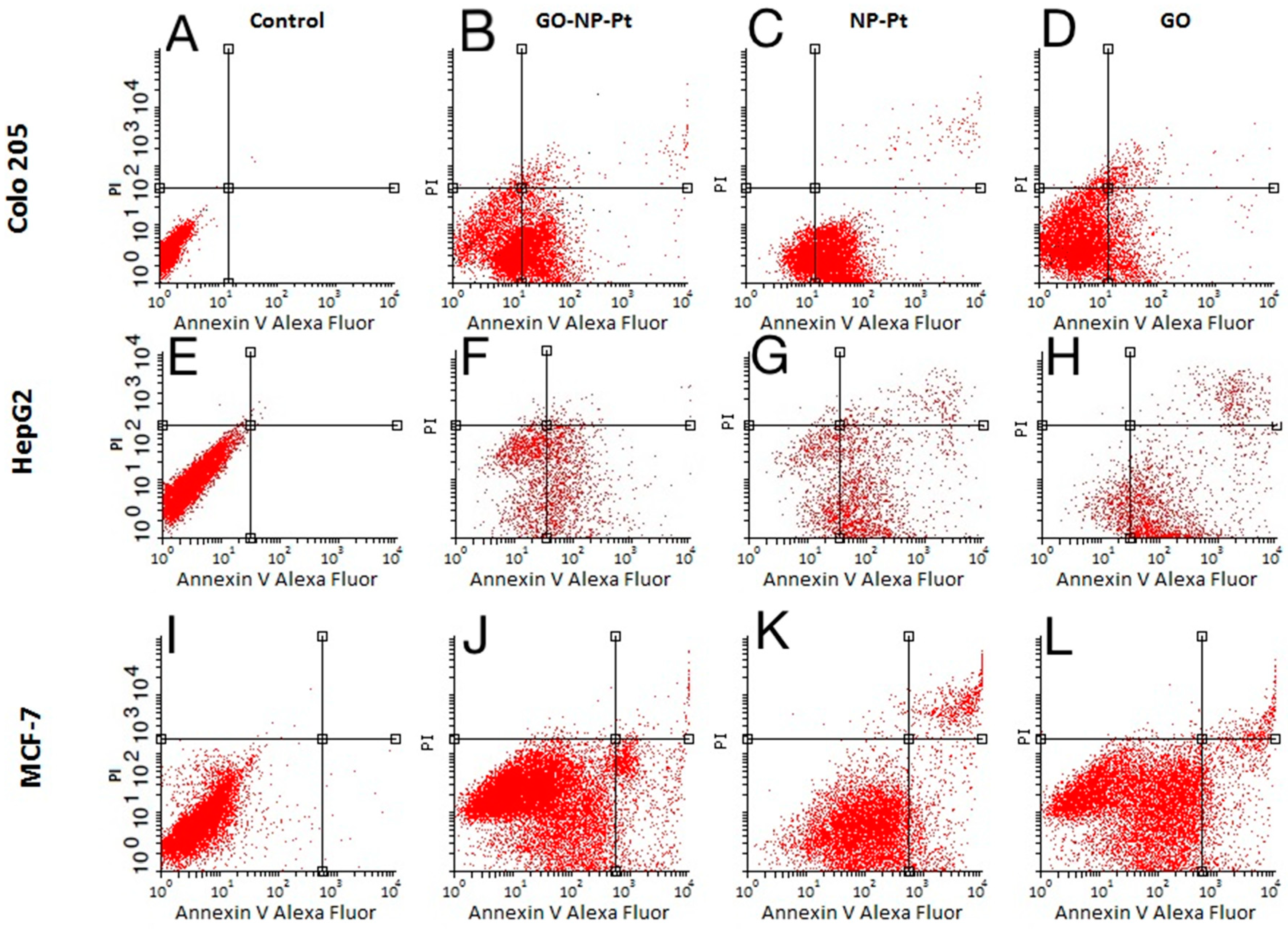
2.6. Apoptosis Assay
Apoptosis was evaluated using the Alexa Fluor
® 488 Annexin V/Dead Cell Apoptosis Kit with Alexa Fluor 488 Annexin V and propidium iodide (PI) for flow cytometry (Life Technologies, Carlsbad, CA, USA). The MCF-7, Colo205, and HepG2 cells (1 × 10
5 cells per well) were incubated for 24 h. Subsequently, the medium was removed, and GO (100 µg/mL), NP-Pt (25 µg/mL), and GO-NP-Pt (GO
100:Pt
25 µg/mL) in the culture medium were added to the cells and incubated for an additional 24 h. The positive control was prepared as described in Jaworski et al. [
26] After this, the MCF-7, Colo 205, and HepG2 cells were harvested, washed in cold PBS, transferred to tubes, and stained according to the Annexin V/PI staining protocol (Life Technologies). Cancer cells were analyzed by flow cytometry (FACSCalibur, Becton Dickinson, Franklin Lakes, NJ, USA), measuring the fluorescence emission at 530 nm and 575 nm (or equivalent), using excitation at 488 nm. Positive cells were identified on the basis of the fluorescence intensity of Annexin V-Alexa Fluor 488 (early stage of apoptosis) or PI (end stage of apoptosis and necrosis). Data were analyzed using the Cell Quest Pro software ver. 5.1 (Becton Dickinson), and the regions were set on the basis of positive and negative control samples.
2.7. Gene Expression
2.7.1. Isolation of Total RNA
For the isolation of total RNA, MCF-7, Colo205, and HepG2 cells (1 × 105 cells per well) were incubated for 24 h. Subsequently, the medium was removed, and GO (100 µg/mL), NP-Pt (25 µg/mL), and GO-NP-Pt (GO100:Pt25 µg/mL) in the culture medium were added to the cells and incubated for an additional 24 h. Total RNA was isolated using a PureLink® RNA Mini Kit (Ambion™ Life Technologies, Foster City, CA, USA). The resulting cell pellet was resuspended in lysis buffer containing 1% 2-mercaptoethanol, and subsequently, the frozen metal balls were added to the probe and homogenized in a TissueLyser ball mill (Qiagen, Germantown, MD, USA) for 5 min at 50 Hz. The homogenate was centrifuged at 12,000× g. The supernatant, containing total RNA, was transferred into a new tube, and one volume 70% ethanol was added into each volume of cell homogenate, following the manufacturer’s instructions. Total RNA was eluted in a volume of 50 µL RNase-free water and stored at −80 °C. The isolated RNA was measured using a NanoDrop 2000 spectrophotometer (Thermo Scientific, Wilmington, DE, USA). The cDNA was synthesized with a cDNA High Capacity Reverse Transcription Kit (AppliedBiosystems, Foster City, CA, USA) to reverse-transcript the mRNA to cDNA, using 2200 ng per reaction. The obtained cDNA was measured using a NanoDrop 2000 spectrophotometer and stored for further analysis at −20 °C.
2.7.2. Real-Time PCR
The ∆∆Ct method was used to determine the expression of mRNA, using real-time PCR:
The reaction was carried out using 48-well plates and the Luminaris Color HiGreen reagents qPCR Master Mix (Thermo Fisher Scientific, Waltham, MA, USA); 100 ng of cDNA were used for each reaction. The following genes were examined:
caspase-3 and
proliferating cell nuclear antigen (
PCNA). The primers used for this procedure are presented in
Table 1. Glyceraldehyde-3-phosphate dehydrogenase (GPDH) was used as the reference house-keeping gene. The reaction conditions were set as specified by the manufacturer, and each sample was analyzed in duplicate. The procedure was conducted using a StepOnePlus™ Real-Time PCR System.
2.8. Statistical Analysis
Data were analyzed using two-way analysis of variance Graph Pad Prism B ver. 8 (GraphPad Software, San Diego, CA, USA). Differences between groups were tested using Tukey’s multiple range tests. All mean values are presented with standard deviations.
4. Discussion
Nanotechnology provides advanced techniques for the detection of cancer cells, the delivery of anticancer drugs, or the activation of programmed cell death by nanoparticles. Nanoparticles have unique physiochemical properties such as nanometric size, large surface-to-mass ratio, and high reactivity with biological structures. The NP-Pt have been considered as an alternative to bulk Pt anticancer agents, showing cytotoxic activity against different types of cancer cells [
16,
17,
18,
19,
20,
21,
26]. The main mechanism of NP-Pt action is related to the high catalytic activity of Pt and their ability to interact with cell components, including genetic material [
17,
19,
20,
21,
27]. The NP-Pt induce programmed cell death by apoptosis after direct interaction with cancer cell DNA helix [
16,
18]. However, the possibility of aggregation of NP-Pt and uncontrolled biodistribution are the major drawback of its usage in anticancer therapy.
In the present work, the synthesized complex of GO and NP-Pt was composed of GO multilayers decorated with well-dispersed NP-Pt on the GO surface. The surface of graphene oxide and its functionalization are significant for its physicochemical features and thereby important in the anticancer assessment of the GO-NP-Pt complex. The present results may only be representative for the used complex. The complex of GO-NP-Pt was created by self-organization of GO and Pt, induced by sonication. The FTIR analysis of GO-NP-Pt did not show any changes in the chemical bonding on the surface of GO platelets coated with NP-Pt compared to the raw GO platelets, indicating that the type of the connection between GO platelets and NP-Pt is based on non-chemical bonding such as van der Waals forces. Wen et al. [
28] also reported that silver nanoparticles were able to impinge the cytoskeletal structures, not by chemical reactions, but by van der Waals forces. Our findings are consistent with the previously published data where negatively charged nanoparticles had stronger harmful effects on cells than cationic particles [
29]. The obtained
ζ-potential measurements of GO-NP-Pt showed that the noncomplex had a higher anionic charge than NP-Pt, which probably decreased its cytotoxic effect against cancer cells compared to NP-Pt treatment. The internalization of GO-NP-Pt is related to the membrane tension of cancer cells and the size of delivered nanoparticles [
30]. The size of investigated complex of GO-NP-Pt was close to the size of GO platelets. Regarding to previous published data by Linares et al. [
31], the main mechanism of cellular GO-NP-Pt internalization was the endocytosis, but to clarify this mechanism the future follow-up research is needed. These results are probably related to GO-NP-Pt electrical charge disruptions compared to NP-Pt, affecting the cell response to nanoparticles. The charge, size, shape, and the chemical composition have an impact on the cytotoxicity of nanoparticles [
32]. In previous studies, NP-Pt had a size-dependent toxicity to cancer and normal cells as well as tissues [
15,
33].
The goal of GO functionalization by NP-Pt was to enhance the anticancer activity of GO and NP-Pt. Zhang et al. [
24] also obtained NP-Pt-loaded GO nanostructures by placing well-dispersed NP-Pt onto folic acid-modified GO platelets, but the chemical reactions of this synthesis were more complicated and involved activity of poly (allylamine hydrochloride). The catalytic activity of NP-Pt could be limited by the addition of chemical or biological stabilizers to prevent NP-Pt agglomeration. However, the usage of GO as an anchoring platform helps to prevent agglomeration without losing its anticancer properties. The present study showed that the density of Pt coating was irregular, but NP-Pt located on the specific parts of GO platelets, especially on the edges as well as on the wrinkles of graphene oxide.
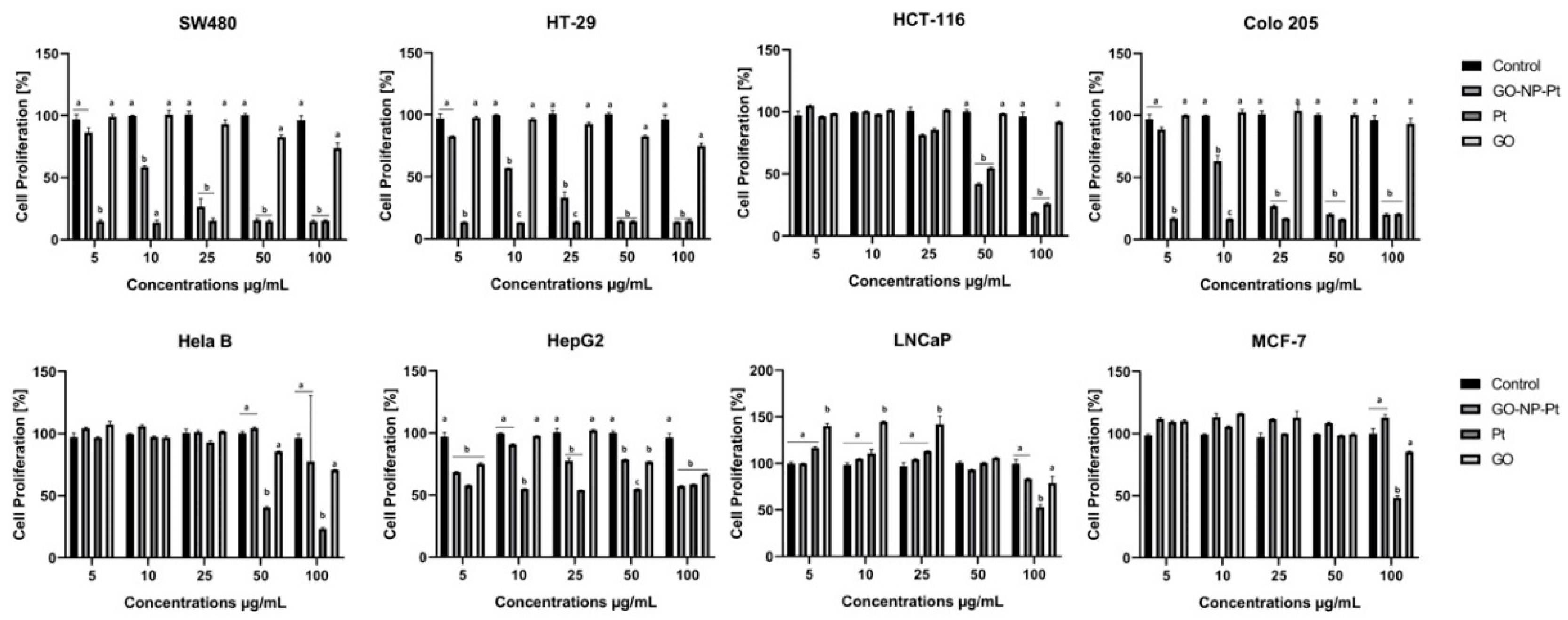
In the present study, we observed decreased proliferation and viability in the majority of the inspected cancer cell lines. The results of the viability assessment of GO-NP-Pt, NP-Pt, and GO showed that the best anticancer efficiency was observed after GO-NP-Pt at the concentration of GO
100:Pt
25 µg/mL against HepG2 and MCF-7 cells. The proliferation status of SW480, HT29, and Colo 205 cancer cell lines was significantly decreased after GO-NP-Pt treatment with the highest concentration (GO
100:Pt
25 µg/mL). However, the MFC-7 cell line was the most resistant one to GO-NP-Pt at all concentrations. The results, also showed that cytotoxicity of NP-Pt located on GO platelets, decreased compared to the bare NP-Pt. The synthesis of nanocomplexes of GO-NP-Pt was beneficial for limitation of uncontrolled released of NP-Pt in a biological system and helps to decrease the possible side effects of NP-Pt. Similar results were obtained by Zhang et al. [
24], where MCF-7 cells showed strong GO-NP-Pt resistance. In these studies, the authors also pointed out that GO acted bidirectionally as a stabilizer and reducing agent to obtain a high quality of well-dispersed NP-Pt on the GO surface. Moreover, GO platelets are biocompatible materials, with minor effects on cell structure and metabolism [
34], but still have an extremely high capacity for cell membrane attachment [
35]. The HepG2 cell line showed the insignificant changes in mRNA expression of
PCNA. The results indicated that the characteristic type of cell grow of HepG2 cell line, had an impact on a possible attachment of GO-NP-Pt to the cell membrane. The lower GO-NP-Pt attachment to the cell membrane reduced cytotoxic effect of GO-NP-Pt nanocomplex and had no impact on the mRNA expression of
PCNA. Furthermore, at the gene expression profile of HepG2 cell, the mRNA level of MRP2—multidrug resistance protein 2 is overexpressed and can lead to developing chemoresistance [
36] for anticancer activity of Pt atoms. The present results in terms of cell morphology showed that GO platelets had an extremely high affinity to cell membranes in all experimental groups, with a minor degree of deformation. On the other hand, NP-Pt treatment caused major deformations of Colo 205, HepG2, and MCF-7 cell structure with loss of plasma membrane integrity. Nanocomplexes of GO-NP-Pt at the highest concentration of GO
100:Pt
25 µg/mL showed a high affinity to cell membranes and also caused cell body deformation. Moreover, the high affinity of GO-NP-Pt to the cell membranes was characteristic for all types of inspected cell lines, which indicates that nanoparticles can inhibit cellular functions of any type of cancer cell lines. The main mechanism of interaction between GO-NP-Pt and the cell surface was probably based on interactions between nanoparticles and structural proteins through electrostatic forces. Our results demonstrate that the level of mRNA expression of
caspase-3 increased significantly after GO-NP-Pt treatment only in the Colo 205 cell line compared to the non-treated cells (control group). A previous study [
37] showed that GO combined with metal nanoparticles caused up-regulation of the mRNA levels of
caspase-3,
caspase-9, and
Bax genes and activated programmed cell death. Analysis of mRNA expression of
PCNA showed that GO-NP-Pt caused a significant reduction of the
PCNA gene expression level of the cell lines Colo 205 and MCF-7.
There are three main cancer treatment strategies: surgery, chemotherapy, and radiation. The major drawbacks of all these treatments are the lack of selectivity between cancer and non-cancer cells and/or incomplete eradication of cancer cells. Nanoparticles of GO and NP-Pt are of interest for the development of cancer treatments because of their unique physiochemical and biological properties [
8,
9,
10,
16,
17,
18,
19,
20,
21]. However, the accumulation of NP-Pt into cells and body tissue after in-vivo administration is also one of the drawbacks for future clinical applications. The results of biodistribution and toxicity evaluation showed that NP-Pt were accumulated in the liver and spleen, but small amounts of Pt were also detected in other tissues of mice 24 h after administration. However, after prolonged administration, NP-Pt were mainly retained in the liver and spleen, and only negligible amounts were found in the other tissues [
23]. Similar results were obtained after GO in-vivo administration by intravenous injection, where GO platelets were also accumulated in the liver [
34]. The results of biodistribution evaluation of GO also showed that GO platelets had a non-systemic toxicity, but a tendency to accumulate and aggregate in the vicinity of the injection sites [
38]. GO is a biocompatible material with surface that is easy to functionalize with nanoparticles or bioactive molecules [
13,
14,
15,
35]. The goal of using the drug deliver carriers is the effectiveness, selectivity and safety of drug administration. GO platelets are biocompatible, which means that GO is able to perform an appropriate host response without undesirable local or systemic effects. The biocompatibility of GO increases the local ability of NP-Pt to perform their anticancer activity. Moreover, GO has an ability to cross the cell membrane structure and delivered GO platelets into cancer cells, partly without involving transporting proteins [
8]. These features can limit the development of multiple drug resistance in cancer cells. The accumulation at injection sites may be useful for effective direct intratumor injection of GO functionalized with NP-Pt and to increase local anticancer activity of Pt.

 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}